

Abstract
Background
MODY‐NEUROD1 is a rare form of monogenic diabetes caused by mutations in Neuronal differentiation 1 (NEUROD1). Until now, only a few cases of MODY‐NEUROD1 have been reported worldwide and the real contribution of mutations in NEUROD1 in monogenic diabetes and its clinical impact remain unclear.
Methods
Genomic DNA was isolated from peripheral blood lymphocytes of 25 unrelated Brazilians patients with clinical characteristics suggestive of monogenic diabetes and the screening of the entire coding region of NEUROD1 was performed by Sanger sequencing.
Results
We identified one novel frameshift deletion (p.Phe256Leufs*2) in NEUROD1 segregating in an autosomal dominant inheritance fashion. Almost 20 years after the first report of NEUROD1‐MODY, only a few families in Europe and Asia had shown mutations in NEUROD1 as the cause of monogenic diabetes.
Conclusion
To our knowledge, we described the first case of NEUROD1‐MODY in a Latin American family.
Keywords: diabetes mellitus, MODY, MODY6, monogenic diabetes, NEUROD1
In the present study, one novel frameshift deletion (p.Phe256Leufs*2) was identified in NEUROD1 segregating in a diabetic family. This is the first case of MODY‐NEUROD1 reported in Latin America.

1. INTRODUCTION
Neuronal differentiation 1 (NEUROD1 – Gene ID: 4760 – OMIM *601724), also known as BETA2, encodes a basic helix‐loop‐helix (bHLH) transcription factor that heterodimerizes with the ubiquitous bHLH protein E47 and regulates insulin gene (INS) expression through binding to its E‐box motif promoter (Naya, Stellrecht, & Tsai, 1995). In 1999, mutations in NEUROD1 were associated to early onset type 2 diabetes mellitus (DM) in two European descendent families inherited in an autosomal dominant fashion for the first time (Malecki et al., 1999), being further classified as maturity‐onset diabetes of the young type 6 (MODY6; OMIM #606394) (Fajans, Bell, & Polonsky, 2001). Almost 20 years after the first report, only a few families in Europe (Ağladıoğlu et al., 2016; Gonsorčíková et al., 2008; Kristinsson et al., 2001; Szopa et al., 2016) and Asia (Ang et al., 2016; Chapla et al., 2015; Doddabelavangala Mruthyunjaya et al., 2017; Horikawa et al., 2018; Liu et al., 2007) had shown mutations in NEUROD1 as the cause of diabetes.
Epidemiological studies of monogenic diabetes are scarce in multiethnic populations, such as Brazilian. This population is very diverse and comprises individuals of multiple ethnic backgrounds and racial admixture, especially Caucasians and Afro‐descendants. Studies aiming the rare forms of monogenic diabetes are needed in a mixed population and, to the best of our knowledge, this is the first case to report a NEUROD1 mutation in a Latin American family.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The Ethics and Research Committee of the Clementino Fraga Filho University Hospital approved this study protocol (No. 70238). All participants were informed about the aim of this study and provided verbal and written consent according to Helsinki Declaration.
2.2. Patients
In this cross‐sectional observational study, we selected 25 unrelated Brazilian probands negative for GCK or HNF1A, HNF4A, and HNF1B mutations. The inclusion criteria were age at onset ≤35 years, BMI (body mass index) <30 kg/m2 or 95th percentile at the diagnosis; a positive family history of diabetes in at least two generations; negative β‐cells anti‐glutamic acid descarboxylase (anti‐GAD) and anti‐IA‐2 autoantibodies. We excluded patients with type 1 diabetes, history of diabetic ketoacidosis, clinical signs of insulin resistance, and presence of secondary causes of diabetes.
2.3. Molecular screening
Genomic DNA was isolated from peripheral blood leukocytes using QIAamp DNA Blood Mini Kit (Qiagen) and from the family members who accepted to participate in this study, we extracted genomic DNA from buccal epithelial cells (Aidar & Line, 2007). The screening of the entire coding region of NEUROD1 was carried out by amplification of two overlapping fragments using two pairs of primers (primers available upon request; RefSeq NG_011820.2, NM_002500.4 and NP_002491.2). Polymerase chain reaction (PCR) products were purified by Clean Sweep PCR Purification Reagent (Applied Biosystems) and bidirectional Sanger sequencing was performed using the Big Dye Terminator Kit v3.1 (Applied Biosystems), conducted on an ABI 3130 Automatic Genetic Analyzer (Applied Biosystems).
3. RESULTS
The entire coding region of NEUROD1 was screened in a cohort of 25 unrelated Brazilian patients with clinical suspicious of monogenic diabetes (10 males and 15 females; average age at diagnosis: 22.68 ± 8.9 years; BMI average: 24.8 ± 4.54 kg/m2), previously tested and negative for mutation in GCK, HNF1A, HNF4A, and HNF1B (data not published).
We detected one novel frameshift deletion of two thymine resulting in a change from phenylalanine to leucine in the position 256 of the protein, followed by a premature stop codon (p.Phe256Leufs*2). This deletion was not seen in the remaining 24 patients and in ClinVar, dbSNP, HGMD®, ExAC Browser, GnomAD, 1000 Genomes project databases, and in the literature. Mutation Taster predicted p.Phe256Leufs*2 to be the disease‐causing mutation (score 1). The mutation is located in the transactivation domain, a highly conserved domain across several species (Figure 1).
Figure 1.
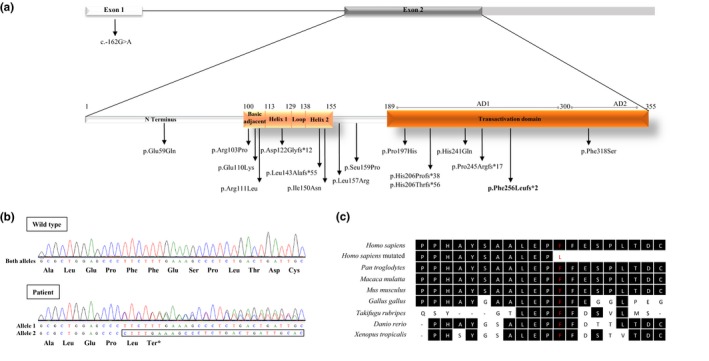
NEUROD1 mutations previously identified in diabetic patients and the novel p.Phe256Leufs*2 mutation. (a) Diagrammatic representation of NEUROD1 and NEUROD1 protein structure. NEUROD1 presents two exons, being exon 1 noncoding. NEUROD1 protein contains two domains: basic helix‐loop‐helix domain, which is divided in basic adjacent, helix 1, loop and helix 2; and transactivation domain, which has two activating domains (AD1 and AD2). Arrows indicate the position of mutations described in the literature and in this study (bold). (b) Electropherograms of NEUROD1 exon 2 wild type (above) and p.Phe256Leufs*2 identified in the patient DM24 (below). (c) Alignment of NEUROD1 across species shows amino acid 256 (in red) evolutionary conserved across species
The p.Phe256Leufs*2 mutation was found in a Brazilian family segregating in an autosomal dominant pattern from the maternal side (Figure 2). The index case is a 33 years old man who was diagnosed with diabetes at 25 years of age after some months of polyuria, polydipsia, and weight loss of approximately 10 kg (BMI at diagnostic: 28.9 kg/m2). He was included in this study at 30 years with mean fasting glucose of 137 mg/dl and glycated hemoglobin (HbA1c‐HPLC) of 7.5% (HbA1c at diagnosis: 6.5%). He presented no diabetic microvascular complications such as retinopathy (normal fundoscopy), diabetic renal disease (normal renal ultrasound and negative microalbuminuria), and neuropathy after 8 years of manifestation of disease.
Figure 2.
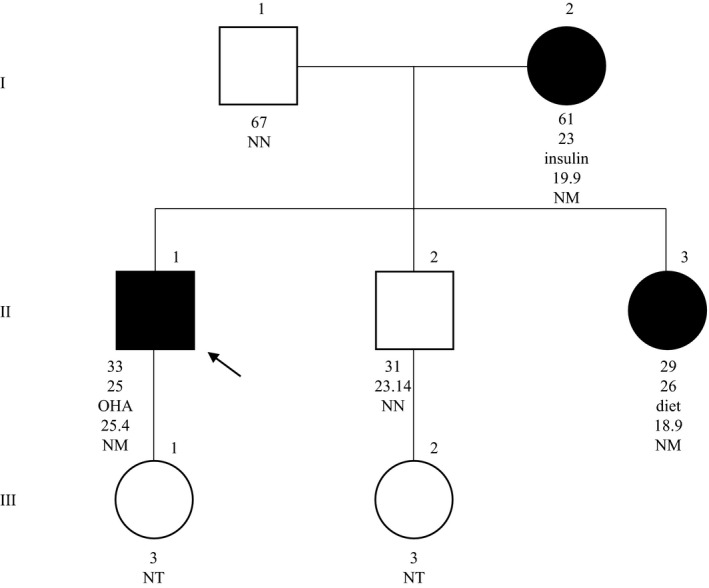
Pedigree, clinical characteristics, and genotype of family 24. Filled symbols and empty symbols represent diabetic patients and healthy individuals, respectively. The present age of the individuals is shown below the symbols, followed by the age at diagnosis, the most recent treatment, body mass index (kg/m2) and genotype interpretation. OHA, oral hypoglycemic agents; Genotypes are expressed by normal allele (N) and mutated allele (M); NT, not tested. An arrow indicates the index case
At diagnosis, the index case was treated with metformin 2 g/day. One month later, since he presented poor glycemic control, he started being treated with NPH insulin (0.2 UI/kg). After 4 years with the same low dose of NPH insulin, a detectable basal C‐peptide and a good glycemic control with insulin (HbA1c: 6%), his treatment was changed to glimepiride and metformin, since he presented some hypoglycemic episodes during these years. Nowadays he presents good glycemic control using glimepiride 6 mg/day and metformin 2 g/day (HbA1c: 6.6%), and fewer episodes of hypoglycemia. The only comorbidity is hypertension and it is well controlled with nitrendipine 20 mg/day, losartan 100 mg/day, and atenolol 25 mg/day and the major causes of secondary hypertension were excluded (Cushing's syndrome, pheochromocytoma, primary hyperaldosteronism, and renal‐artery stenosis). He never had diabetic ketoacidosis and was negative for anti‐GAD and anti‐IA‐2 autoantibodies.
The patient's mother (BMI: 19.9 kg/m2) was diagnosed with diabetes at the age of 23 years with a mean fasting glucose of 330 mg/dl and since then, she had been on basal insulin therapy (mean fasting glucose: 113 mg/dl; HbA1c: 9.4%). She developed diabetic retinopathy and nephropathy. The index case's sister (BMI 18.9 kg/m2) was diagnosed with accidental hyperglycemia at the age of 26 years, with mean fasting glucose of 250 mg/dl; During the first 2 years she was initiated with NPH insulin, then with gliclazide 30 mg/day and lastly, with 29 years, her glucose levels were managed only with low‐carbohydrate diet and exercise (mean fasting glucose: 97 mg/dl; HbA1c: 6.2%; C‐peptide: 2.3 ng/ml). To this moment, the index case and his family members did not present neurological abnormalities.
The mutation p.Phe256Leufs*2 was not present in the proband's brother, a normal weight (BMI: 23.14 kg/m2) man of 31 years. Levels of glucose and HbA1c were 113 mg/dl and 5.6%, respectively.
4. DISCUSSION
The genetic diagnosis of MODY in Brazil has been mostly limited to GCK and HNF1A, after a selection for testing guided by clinical criteria (Giuffrida et al., 2017). For this reason, rare forms are poorly studied. In this work, we screened all coding region of NEUROD1 in patients with clinical phenotype of monogenic diabetes, negative for mutations in GCK, HNF1A, HNF4A and HNF1B.
After almost two decades past from the initial report of NEUROD1‐MODY (Malecki et al., 1999), only a small numbers of Asian and European families were identified (Ağladıoğlu et al., 2016; Ang et al., 2016; Chapla et al., 2015; Doddabelavangala Mruthyunjaya et al., 2017; Gonsorčíková et al., 2008; Horikawa et al., 2018; Kristinsson et al., 2001; Liu et al., 2007; Szopa et al., 2016). In these populations, NEUROD1 mutations range from low frequencies as in Poland (0.64%; Szopa et al., 2016) to high frequencies as in India (7.14%; Chapla et al., 2015). In our study, we found a frequency of 4%, similar to that observed in Turkish (4.65%; Ağladıoğlu et al., 2016; Table 1).
Table 1.
Clinical characteristics of known and novel NEUROD1 mutations associated to monogenic diabetes
| Protein level | c.DNA level | Accession number | Mutation type | Domain | Origin | Sample (%) | Sample type | Met. | Age (years) | AOD (years) | DT | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p.Arg111Leu | c.332G>T | rs104893649 | Missense | Basic adjacent | European descendent | 2/94 (2.12) | Type 2 DM | Sanger sequencing | 65 | 40 | Insulin | (Malecki et al., 1999) |
| p.His206Profs*38 | c.616_617ins | rs387906384 | Frameshift | AD1 | 74 | 33 | Insulin | |||||
| p.Glu110Lys | c.328C>A | rs763092306 | Missense | Basic adjacent | Iceland | 1/3 | MODY Families | Sanger sequencing | N/A | N/A | N/A | (Kristinsson et al., 2001) |
| p.Ser159Pro | c.475T>C | N/A | Missense | N/A | China | 1/85 (1.17) | Type 2 DM | Sanger sequencing | 27 | 27 | OHA | (Liu et al., 2007) |
| p.His241Gln | c.723C>G | rs561017686 | Missense | AD1 | Czech Republic | 2/30 (6.66) | MODY | Sanger sequencing | 44a | 20 | Insulin | (Gonsorčíková et al., 2008) |
| 39 | 30 | OHA + insulin | ||||||||||
| p.Asp122Glyfs*12 | c.364_365ins | N/A | Frameshift | Helix 1 | Pakistan | 2/44 (4.54) | PNDM | Sanger sequencing | N/A | 8b | N/A | (Rubio‐Cabezas et al., 2010) |
| p.Leu143Alafs*55 | c.427_428del | rs1485945978 | Frameshift | Helix 2 | Hungary | N/A | 4b | N/A | ||||
| p.Pro197His | c.590C>A | rs8192556 | Missense | AD1 | Turkish | 2/43 (4.65) | MODY | NGS panel | 15 | 14 | Diet | (Ağladıoğlu et al., 2016) |
| 13 | 12 | Diet | ||||||||||
| p.His241Gln | c.723C>G | rs561017686 | Missense | AD1 | India | 4/56 (7.14) | MODY | NGS panel | 47 | 28 | OHA | (Chapla et al., 2015) |
| 35 | 24 | OHA + insulin | ||||||||||
| p.Glu59Gln | c.175G>C | rs553756272 | Missense | N terminus | 30 | 30 | OHA | |||||
| NA | c.−162G>A | rs537184640 | NA | 5ʹUTR | 30 | 28 | OHA | |||||
| p.Arg103Pro | c.308G>C | N/A | Missense | Basic adjacent | Poland | 1/156 (0.64) | MODY | NGS panel | 66 | 23 | Insulin | (Szopa et al., 2016) |
| p.Pro197His | c.590C>A | rs8192556 | Missense | AD1 | Asian | 1/84 (1.19) | MODY | NGS panel | N/A | N/A | N/A | (Ang et al., 2016) |
| p.Glu59Gln | c.175G>C | rs553756272 | Missense | N terminus | India | 2/50 (4) | GDM | NGS panel | 36 | 36 | OHA | (Doddabelavangala Mruthyunjaya et al., 2017) |
| p.Phe318Ser | c.953T>C | N/A | Missense | AD2 | 29 | 27 | OHA | |||||
| p.His206Profs*38 | c.616_617ins | rs387906384 | Frameshift | AD1 | Japan | 4/275 (1.45) | MODY | Sanger sequencing | 17 | 14 | Insulin | (Horikawa et al., 2018) |
| p.Pro245Argfs*17 | c.734_734del | N/A | Frameshift | AD1 | 25 | 11 | Insulin | |||||
| p.Leu157Arg | c.470T>G | N/A | Missense | N/A | 24 | 10 | OHA + insulin | |||||
| p.His206Thrfs*56 | c.616_616del | N/A | Frameshift | AD1 | 15 | 12 | OHA + insulin | |||||
| p.Ile150Asn | c.449T>A | N/A | Missense | Helix 2 | Turkish | 1 | PNDM family | NGS panel | 13.4 | 9b | Insulin | (Demirbilek et al., 2018) |
| p.Phe256Leufs*2 | c.766_767del | N/A | Frameshift | AD1 | Brazil | 1/25 (4) | MODY | Sanger sequencing | 30 | 25 | OHA | c |
Abbreviations: AD, activating domain, AOD, age of diagnosis; DM, diabetes mellitus; DT, diabetes treatment; GDM, gestational diabetes mellitus; Met, methodology; N/A, not available/not applicable; OHA, oral hypoglycemic agents; PNDM, permanent neonatal diabetes mellitus; Ref., references; UTR, untranslated.
Age at death.
Weeks.
Novel mutation identified in this study.
So far, 20 index cases with monogenic diabetes were previously reported with heterozygous mutations in NEUROD1 and the data collection shows an average age of diagnosis (AOD) of 23.37 ± 9 years (ranging 10–40 years), similar to our patient's AOD (25 years). The treatment varied among patients, the use of oral hypoglycemic agents (OHA) was the most frequent (36.8%), followed by insulin (31.6%), OHA + insulin (21.1%) and diet (10.5%). Interestingly, all probands with frameshift mutations were treated with insulin (Table 1).
NEUROD1 is a transcription factor expressed in pancreatic cells (Naya et al., 1997), and mutations that cause a disturbance in this protein lead to the hyperglycemia that was observed in all index cases. It is also expressed in neuronal cells (Naya et al., 1997), which could explain the neuronal manifestations observed in two patients with p.Pro197His mutation in heterozygosity described with pituitary hypoplasia, growth hormone deficiency and epilepsy (Ağladıoğlu et al., 2016). Mental retardation was observed in a female patient and her mother, both carrying p.Pro245Argfs*17 (Horikawa et al., 2018). Besides, Rubio‐Cabezas et al. (2010) identified two probands with p.Asp122Glyfs*12 and p.Leu143Alafs*55 frameshift mutations in homozygosity leading to the syndrome of permanent neonatal DM (PNDM) and neurological abnormalities including developmental delay, sensorineural deafness, and visual impairment (Rubio‐Cabezas et al., 2010). Demirbilek et al. (2018) recently described a novel case of PNDM in a 13‐year‐old girl with a homozygous missense mutation (p.Ile150Asn) showing a similar clinical presentation from the previously reported PNDM cases (Demirbilek et al., 2018; Rubio‐Cabezas et al., 2010).
Additionally, Horikawa and Enya (2019) observed that there are two times more affected female patients than affected male patients among the cases described and that the majority of the cases inherited the mutation from their mother, which was also observed in this study. Further analysis on the pathophysiology of NEUROD1 will help to clarify the reason of this discrepancy in an autosomal disease (Horikawa & Enya, 2019).
To the best of our knowledge, this is the first case reported to have a NEUROD1‐MODY mutation in a Latin American cohort. The novel p.Phe256Leufs*2 mutation in the activating domain 1 has similar structural effects in the protein as those caused by the first reported p.His206Profs*38 mutation (Malecki et al., 1999). It leads to the loss of 60% of the transactivation domain, and likely that p.His206Profs*38, probably has a compromised biological activity since the transactivation domain is required for the ligation of NEUROD1 with the coactivator p300 (Qiu, Sharma, & Stein, 1998). The majority of mutations found in NEUROD1 associated with monogenic diabetes (13 cases [54.2%]) is located in the transactivation domain, comprising four frameshift mutations (Horikawa et al., 2018; Malecki et al., 1999) and three missense mutations (Ağladıoğlu et al., 2016; Ang et al., 2016; Chapla et al., 2015; Doddabelavangala Mruthyunjaya et al., 2017; Gonsorčíková et al., 2008; Figure 1).
This study has some limitations. First, our sample size was small and may not show the real frequency of NEUROD1 mutations as cause of monogenic diabetes in our population. In addition, we did not analyze the possible presence of copy number variations that could also be compromising NEUROD1 function and would not be observed in our sequencing method.
5. CONCLUSION
In conclusion, we described a Brazilian family with a novel mutation in NEUROD1 segregating with diabetes in an autosomal dominant pattern of inheritance.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENTS
The authors are grateful to the patients and their families. This work was supported by National Council for Scientific and Technological Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), and Oswaldo Cruz Institute (IOC).
Abreu GDM, Tarantino RM, Cabello PH, et al. The first case of NEUROD1‐MODY reported in Latin America. Mol Genet Genomic Med. 2019;7:e989 10.1002/mgg3.989
REFERENCES
- Ağladıoğlu, S. Y. , Aycan, Z. , Çetinkaya, S. , Baş, V. N. , Önder, A. , Peltek Kendirci, H. N. , … Ceylaner, S. (2016). Maturity onset diabetes of youth (MODY) in Turkish children: Sequence analysis of 11 causative genes by next generation sequencing. Journal of Pediatric Endocrinology and Metabolism, 29(4), 487–496. 10.1515/jpem-2015-0039 [DOI] [PubMed] [Google Scholar]
- Aidar, M. , & Line, S. R. (2007). A simple and cost‐effective protocol for DNA isolation from buccal epithelial cells. Brazilian Dental Journal, 18(2), 148–152. 10.1007/BF01811861 [DOI] [PubMed] [Google Scholar]
- Ang, S. F. , Lim, S. C. , Tan, C. S. H. , Fong, J. C. W. , Kon, W. Y. C. , Lian, J. X. , … Sum, C. F. (2016). A preliminary study to evaluate the strategy of combining clinical criteria and next generation sequencing (NGS) for the identification of monogenic diabetes among multi‐ethnic Asians. Diabetes Research and Clinical Practice, 119, 13–22. 10.1016/j.diabres.2016.06.008 [DOI] [PubMed] [Google Scholar]
- Chapla, A. , Mruthyunjaya, M. D. , Asha, H. S. , Varghese, D. , Varshney, M. , Vasan, S. K. , … Thomas, N. (2015). Maturity onset diabetes of the young in India – A distinctive mutation pattern identified through targeted next‐generation sequencing. Clinical Endocrinology, 82(4), 533–542. 10.1111/cen.12541 [DOI] [PubMed] [Google Scholar]
- Demirbilek, H. , Hatipoglu, N. , Gul, U. , Tatli, Z. U. , Ellard, S. , Flanagan, S. E. , … Kurtoglu, S. (2018). Permanent neonatal diabetes mellitus and neurological abnormalities due to a novel homozygous missense mutation in NEUROD1. Pediatric Diabetes, 19(5), 898–904. 10.1111/pedi.12669 [DOI] [PubMed] [Google Scholar]
- Doddabelavangala Mruthyunjaya, M. , Chapla, A. , Hesarghatta Shyamasunder, A. , Varghese, D. , Varshney, M. , Paul, J. , … Thomas, N. (2017). Comprehensive maturity onset diabetes of the young (MODY) gene screening in pregnant women with diabetes in India. PLoS ONE, 12(1), e0168656 10.1371/journal.pone.0168656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajans, S. , Bell, G. , & Polonsky, K. (2001). Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. The New England Journal of Medicine, 345(13), 971–980. 10.1056/NEJMra002168 [DOI] [PubMed] [Google Scholar]
- Giuffrida, F. M. A. , Moises, R. S. , Weinert, L. S. , Calliari, L. E. , Manna, T. D. , Dotto, R. P. , … Reis, A. F. (2017). Maturity‐onset diabetes of the young (MODY) in Brazil: Establishment of a national registry and appraisal of available genetic and clinical data. Diabetes Research and Clinical Practice, 123, 134–142. 10.1016/j.diabres.2016.10.017 [DOI] [PubMed] [Google Scholar]
- Gonsorčíková, L. , Průhová, Š. , Cinek, O. , Ek, J. , Pelikánová, T. , Jørgensen, T. , … Lebl, J. (2008). Autosomal inheritance of diabetes in two families characterized by obesity and a novel H241Q mutation in NEUROD1. Pediatric Diabetes, 9(4pt2), 367–372. 10.1111/j.1399-5448.2008.00379.x [DOI] [PubMed] [Google Scholar]
- Horikawa, Y. , & Enya, M. (2019). Genetic dissection and clinical features of MODY6 (NEUROD1‐MODY). Current Diabetes Reports, 19(3), 12 10.1007/s11892-019-1130-9 [DOI] [PubMed] [Google Scholar]
- Horikawa, Y. , Enya, M. , Mabe, H. , Fukushima, K. , Takubo, N. , Ohashi, M. , … Takeda, J. (2018). NEUROD1‐deficient diabetes (MODY6): Identification of the first cases in Japanese and the clinical features. Pediatric Diabetes, 19(2), 236–242. 10.1111/pedi.12553 [DOI] [PubMed] [Google Scholar]
- Kristinsson, S. Y. , Talseth, B. , Steingrimsson, E. , Thorsson, A. V. , Helgason, T. , Hreidarsson, A. B. , … Arngrimsson, R. (2001). MODY in Iceland is associated with mutations in HNF‐1alpha and a novel mutation in NeuroD1. Diabetologia, 44(11), 2098–2103. 10.1007/s001250100016 [DOI] [PubMed] [Google Scholar]
- Liu, L. , Furuta, H. , Minami, A. , Zheng, T. , Jia, W. , Nanjo, K. , & Xiang, K. (2007). A novel mutation, Ser159Pro in the NeuroD1/BETA2 gene contributes to the development of diabetes in a Chinese potential MODY family. Molecular and Cellular Biochemistry, 303(1–2), 115–120. 10.1007/s11010-007-9463-0 [DOI] [PubMed] [Google Scholar]
- Malecki, M. T. , Jhala, U. S. , Antonellis, A. , Fields, L. , Doria, A. , Orban, T. , … Krolewski, A. S. (1999). Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nature Genetics, 23(3), 323–328. 10.1038/15500 [DOI] [PubMed] [Google Scholar]
- Naya, F. J. , Huang, H.‐P. , Qiu, Y. , Mutoh, H. , DeMayo, F. J. , Leiter, A. B. , & Tsai, M.‐J. (1997). Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD‐deficient mice. Genes & Development, 11(18), 2323–2334. 10.1101/gad.11.18.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya, F. J. , Stellrecht, C. M. M. , & Tsai, M. J. (1995). Tissue‐specific regulation of the insulin gene by a novel basic helix‐loop‐helix transcription factor. Genes & Development, 9(8), 1009–1019. 10.1101/gad.9.8.1009 [DOI] [PubMed] [Google Scholar]
- Qiu, Y. , Sharma, A. , & Stein, R. (1998). p300 mediates transcriptional stimulation by the basic helix‐loop‐helix activators of the insulin gene. Molecular and Cellular Biology, 18(5), 2957–2964. 10.1128/MCB.18.5.2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio‐Cabezas, O. , Minton, J. A. L. , Kantor, I. , Williams, D. , Ellard, S. , & Hattersley, A. T. (2010). Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes, 59(9), 2326–2331. 10.2337/db10-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szopa, M. , Ludwig‐Galezowska, A. H. , Radkowski, P. , Skupien, J. , Machlowska, J. , Klupa, T. , … Malecki, M. T. (2016). A family with the Arg103Pro mutation in the NEUROD1 gene detected by next‐generation sequencing ‐ Clinical characteristics of mutation carriers. European Journal of Medical Genetics, 59(2), 75–79. 10.1016/j.ejmg.2016.01.002 [DOI] [PubMed] [Google Scholar]