

Abstract
Background
Malan syndrome is a recently introduced overgrowth disorder described in a limited number of individuals. Haploinsufficiency and also point mutations of NFIX gene have been proposed as its leading causative mechanism, however, due to the limited number of cases and different deletion sizes, genotype/phenotype correlations are still limited.
Methods
Here, we report the first Brazilian case of Malan syndrome caused by a 990 kb deletion in 19p13.2p13.12, focusing on clinical and behavioral aspects of the syndrome.
Results
The patient presented with macrocephaly, facial dysmorphisms, hypotonia, developmental delay, moderate thoracolumbar scoliosis, and seizures. The intellectual and behavioral assessments showed severe cognitive, language, and adaptive functions impairments. The 19p deleted region of our patient encompasses NFIX, CACNA1A, which seems to be related to a higher frequency of seizures among individuals with microdeletions in 19p13.2, and 15 other coding genes, including CC2D1A and NACC1, both known to be involved in neurobiological process and pathways.
Conclusion
Deletions involving NFIX gene should be considered in patients with overgrowth during childhood, macrocephaly, developmental delay, and seizures, as well as severe intellectual disability.
Keywords: 19p13.2 microdeletion, CACNA1A, Malan syndrome, NFIX, overgrowth disorder
Malan syndrome is an overgrowth disorder described in a limited number of individuals and, due to the limited number of cases and different deletion sizes, genotype/phenotype correlations are still limited. We report a new case of Malan syndrome caused by a 990 kb deletion in 19p13.2p13.12, and revise the previous reports discussing possible genotype–phenotype correlations within the deleted region.

1. INTRODUCTION
Malan syndrome (OMIM #614753), also named Sotos‐like syndrome or Sotos syndrome 2, characterized by overgrowth, facial dysmorphism, intellectual disability and behavior problems, is a recently introduced clinical condition described in a limited number of individuals. So far, descriptions of patients in the literature comprise cases of haploinsufficiency of NFIX gene, due to chromosomal microdeletions in 19p13.2 region (Auvin, Holder‐Espinasse, Lamblin, & Andrieux, 2009; Bonaglia et al., 2010; Dolan et al., 2010; Dong et al., 2016; Hino‐Fukuyo et al., 2015; Jezela‐Stanek et al., 2016; Jorge, Silva, Águeda, Dória, & Leão, 2015; Karmarkar, Amarillo, & Larsen, 2014; Klaassens et al., 2015; Kuroda et al., 2017; Lyon et al., 2015; Lysy et al., 2009; Malan et al., 2010; Natiq et al., 2014; Nimmakayalu et al., 2013; Priolo et al., 2018; Shimojima et al., 2015; Welham et al., 2015), and also point mutations in NFIX (Gurrieri et al., 2015; Jezela‐Stanek et al., 2016; Klaassens et al., 2015; Lu et al., 2017; Malan et al., 2010; Martinez et al., 2015; Oshima et al., 2017; Priolo et al., 2012, 2018; Rai, Narayanan, & Phadke, 2018; Yoneda et al., 2012).
Here we report a new case of Malan Syndrome caused by a 990 kb deletion in 19p13.2p13.12. This is the first Brazilian case and it highlights clinical and behavioral aspects of the syndrome.
2. CLINICAL REPORT
The patient was the first child of nonconsanguineous and healthy parents after a preterm and uneventful pregnancy. His measurements, corrected for birth at 34 1/7 weeks, were as follows: birth weight 2.645 g (0.37 SD), birth length 47 cm (1.9 SD), and head circumference 35 cm (3.9 SD). At 20 months of age, his height was 87 cm (1 SD), weight was 12 kg (0.50 SD), and head circumference was 50 cm (1.72 SD). He showed dysmorphic features including relative macrocephaly, long face, frontal bossing, frontal flat hemangioma, down‐slanting palpebral fissures, deep‐set eyes, long eyelashes, open mouth appearance, pointed chin, long hands, slender habitus, and severe hypotonia.
The first seizure episode was at 10 months of age and it developed into tonic seizures that were controlled initially by phenobarbital, and then by clonazepam plus valproate. Follow‐up EEG showed bilateral frontotemporal sharp waves, and a cranial MRI‐scan showed a reduction of the periventricular white matter with a compensatory dilation of the lateral ventricles and the third ventricle. At 9 years and 8 months of age, his measurements were: height 135 cm (−0.24 SD), weight 35 kg (0.73 SD) with a BMI‐for‐age 19.2 (1.08 SD) and head circumference 56.5 cm (>3 SD). In addition, he presented moderate thoracolumbar scoliosis, pectus carinatum, exotropia, and pale retina. At 19 years and 7 months of age (Figure 1), his measurements were: height 155 cm (−3.01 SD), weight 45 kg (−3.45 SD) with a BMI‐for‐age 18.7 (1.79 SD) and head circumference 57.5 cm (2 SD).
Figure 1.
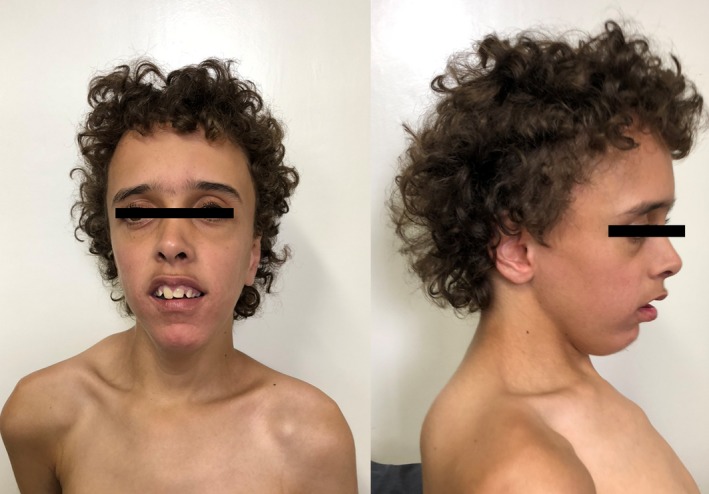
Patient at 19 years and 7 months of age showing macrocephaly, long face, frontal bossing, down‐slanting palpebral fissures, deep‐set eyes, long eyelashes, anteverted nares, open mouth appearance, and pointed chin
3. INTELLECTUAL AND BEHAVIORAL ASSESSMENTS
The patient was submitted to an intellectual performance assessment, which included the Brazilian version of the Wechsler Abbreviated Intelligence Scale—WASI (Trentini, Yates, & Heck, 2014) and The Vineland Adaptive Behavioral Scale—VABS‐II (Sparrow, Cicchetti, & Balla, 2005). Additionally, parents answered the Child Behavior Checklist—CBCL (Achenbach & Resorta, 2001), by which presence of behavioral problems was investigated. A neuropsychologist conducted the assessment in individual sessions and appropriate rooms of a neurodevelopmental outpatient clinic from the Universidade Federal de São Paulo, Brazil.
At the time of assessment, the patient was 19 years old. Analysis of parents’ answers to the VABS‐II indicated low performance (standard‐score < 40) in all adaptive domains: Communication (receptive, expressive, and written), Daily Life Skills (personal, domestic, and community), Socialization (interpersonal relationships, play and leisure time, and coping skills) and Motor Skills (Table S1). He presented a limited understanding of verbal instructions and communicated only by vocalizations and a few meaningful gestures. Therefore, an accurate assessment of intellectual quotient (IQ) was not possible. Quantitative analysis of parents’ answers to the CBCL did not indicate the presence of any internalizing (e.g., anxiety or depression) or externalizing (e.g., aggression) behavioral problems at clinical levels (t‐score < 65).
4. CYTOGENETIC AND MOLECULAR STUDIES
This research was approved by the Ethics Committee of UNIFESP. Informed consent for clinical and genetic analyses was obtained from the patient's parents.
G‐banding karyotype was performed on peripheral blood lymphocyte cultures, according to standard procedures and revealed normal results. Array assay using the CytoScan 750K Array (Affymetrix, Santa Clara, CA, USA) showed a 990 kb 19p13.2p13.12 deletion. The final result was given as 46,XY. arr[GRCh37/hg19] 19p13.2p13.12(13068720_14053305)×1.
5. DISCUSSION
We identified a 990 kb deletion including the NFIX gene in a patient with Malan syndrome. NFIX haploinsufficiency or point mutations, clustered mostly in exon 2 (Priolo et al., 2018), are the leading causative mechanism in Malan syndrome (Gurrieri et al., 2015; Klaassens et al., 2015; Malan et al., 2010). NFIX (OMIM * 164005), a member of the nuclear factor I family of transcription factors, is essential for normal brain and skeletal development and Nfix deficiency in mice leads to brain malformations including ventriculomegaly and partial agenesis of the corpus callosum (Driller et al., 2007; Malan et al., 2010). To date, together with the present study, 35 patients were described with Malan syndrome phenotype, all of them presenting deletions involving NFIX gene, with variable breakpoints (Auvin et al., 2009; Bonaglia et al., 2010; Dolan et al., 2010; Dong et al., 2016; Hino‐Fukuyo et al., 2015; Jezela‐Stanek et al., 2016; Jorge et al., 2015; Karmarkar et al., 2014; Klaassens et al., 2015; Kuroda et al., 2017; Lyon et al., 2015; Lysy et al., 2009; Malan et al., 2010; Natiq et al., 2014; Nimmakayalu et al., 2013; Priolo et al., 2018; Shimojima et al., 2015; Welham et al., 2015) (Figure 2).
Figure 2.

Genome map of the overlapping deletions of our patient (gray bar) and previously reported cases (black bars), and known genes in the chromosome 19p13 region. The patients` rearrangements were organized based on their initial deletion coordinates. Data were uploaded to UCSC genome browser, NCBI Build 37, February 2009, hg19 Assembly (http://www.genome.ucsc.edu)
Apart from overgrowth and intellectual disability/global developmental delay, the most consistent clinical feature of patients with Malan syndrome is facial dysmorphism. Priolo et al. (2018) refined genotype–phenotype correlations in individuals with Malan syndrome and found that all patients presented with typical facial features that include a long or triangular face (which become more elongated with age), prominent forehead, depressed nasal bridge, deep‐set eyes, down‐slanting palpebral fissures, short nose with anteverted nares and upturned tip, long philtrum, small mouth that is often held open, with a thin upper vermillion in a cupid bow shape, an everted lower lip, and a prominent chin. Our patient presented with the main facial dysmorphic features of patients with Malan syndrome and also, moderate thoracolumbar scoliosis, pectus carinatum, exotropia, and pale retina. According to Priolo et al. (2018), visual impairments are common and skeletal anomalies are frequent in patients with Malan syndrome, especially a slender habitus, scoliosis, pectus carinatum, or excavatum, which occur at varying frequencies.
Despite being considered an overgrowth syndrome, neither weight nor length at birth are above the 2 SD in the majority of patients (Priolo et al., 2018). Comparing all the patients described in the literature who present 19p13 microdeletion encompassing the NFIX gene, 48.5% (15/31) presented with postnatal overgrowth in height, and in only 9% (2/22) the birth length is above 2SD (Table S2). Macrocephaly, on the other hand, seems to be an important and more frequent clinical feature in patients with 19p13 microdeletion. Among patients with NFIX haploinsufficiency, we found that 36% (9/25) present head circumference above 2 SD at birth and 58% (18/31) in the postnatal period (Table S2). Our patient presented macrocephaly at birth and during infancy, but at 19 years and 7 months of age, his head circumference was 57.5 cm (2 SD), with a height of 155 cm (−3.01 SD), suggesting a relative macrocephaly. It is important to observe that moderate scoliosis also compromises his height.
The clinical features observed in individuals with deletions of NFIX and a variable number of other genes do not show significant differences in comparison with individuals who present NFIX point mutations. The main difference seems to be related to the frequency of seizures/epilepsy and EEG abnormalities among individuals with microdeletions (Kuroda et al., 2017; Priolo et al., 2018), which may be explained by the presence of a contiguous gene disorder. Located 109 kb from NFIX, CACNA1A has been associated with such increased prevalence of seizures (Auvin et al., 2009; Marangi et al., 2012; Natiq et al., 2014; Priolo et al., 2018). CACNA1A gene (OMIM *601011) encodes a voltage‐dependent calcium channel subunit expressed in neuronal tissue and mutations in this gene have been associated with epilepsy (Kors et al., 2004; Zamponi, Lory, & Perez‐Reyes, 2010) and chronic neurological disorders (Ducros et al., 2001; Ophoff et al., 1996; Wan et al., 2011). Of 27 individuals reported with deletion encompassing NFIX and CACNA1A genes, including our patient, 12 presented with seizures (Auvin et al., 2009; Bonaglia et al., 2010; Hino‐Fukuyo et al., 2015; Kuroda et al., 2017; Lyon et al., 2015; Natiq et al., 2014; Nimmakayalu et al., 2013; Priolo et al., 2018; Shimojima et al., 2015). On the other hand, four of eight patients have been reported with seizures and 19p13.2 deletions not involving CACNA1A (Dolan et al., 2010; Jezela‐Stanek et al., 2016; Klaassens et al., 2015; Table S2). Since 12 of 27 (44,5%) cases presenting seizures and CACNA1A deletions is not significantly different from 4 of 8 (50%) cases presenting seizures and non‐CACNA1A‐containing deletions, we could not confirm, based on the cases described so far, that CACNA1A haploinsufficiency is the only factor responsible for the increased prevalence of seizures among patients with 19p deletion.
Previous studies describing patients with Malan syndrome reported intellectual disability varying from moderate to severe levels, as well as behavioral profiles marked by anxiety symptoms (Priolo et al., 2018). Welham et al. (2015) studied behavioral characteristics of a group of 10 patients with 19p13.2 microdeletions. The authors found a high frequency of autism spectrum disorders characteristics, repetitive, and challenging behaviors (such as aggression and self‐injury) and they concluded that the characteristics might be associated with 19p13.2 microdeletions. However, in only three cases, the deleted region included NFIX.
Our patient showed severe overall cognitive and adaptive deficits, with an expressive language repertoire restricted to few words and meaningful gestures. During the clinical interview, the mother reported signs of anxiety, but only in new situations or unfamiliar contexts. The clinical geneticist also identified signs of anxiety in routine medical consultations. On the other hand, different from Welham et al. (2015) findings, the parents’ answers to the CBCL did not indicate the presence of internalization or externalization, or even autistic‐like, behavioral problems.
Besides NFIX and CACNA1A genes, the 19p deleted region of our patient encompasses 15 other coding genes (Figure 2). Among them, deletions of CC2D1A gene (OMIM *610055), which regulates the expression of serotonin receptors in neuronal tissue and have been associated with non‐syndromic intellectual disability (Basel‐Vanagaite et al., 2006), and NACC1 gene (OMIM *610672), which encodes a protein that works as a transcriptional regulator and is associated with neurodevelopmental disorder characterized by epilepsy, cataracts, feeding difficulties, and delayed brain myelination (Schoch et al., 2017), might be able to contribute to the severe cognitive and adaptive functions impairments found in our patient, since they are involved in neurobiological pathways and processes. However, it should be noted that CC2D1A is a recessive intellectual disability gene, while NACC1 is associated with a dominant disease caused by a recurrent missense variant, so detailed studies about the involvement of this genes, as well as the other genes located in the deleted 19p region of our patient, besides NFIX, could accurately define the genotype–phenotype correlations linked to 19p deletions.
In conclusion, we suggest that deletions involving NFIX gene should be considered in patients with overgrowth during childhood, macrocephaly, developmental delay, and seizures, as well as severe intellectual disability. Our cytogenomic, clinical, and behavioral assessment data contribute to a better understanding of the syndrome for an accurate diagnosis, prognosis, and genetic counseling of Malan syndrome patients.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
Supporting information
ACKNOWLEDGMENTS
The authors have no conflict of interest to declare.
Bellucco FT, de Mello CB, Meloni VA, Melaragno MI. Malan syndrome in a patient with 19p13.2p13.12 deletion encompassing NFIX and CACNA1A genes: Case report and review of the literature. Mol Genet Genomic Med. 2019;7:e997 10.1002/mgg3.997
REFERENCES
- Achenbach, T. M. , & Resorta, L. A. (2001). Manual for ASEBA school‐age forms and profiles. In Burlington, VT: Research Center for Children, Youth, and Families, University of Vermont.
- Auvin, S. , Holder‐Espinasse, M. , Lamblin, M.‐D. , & Andrieux, J. (2009). Array‐CGH detection of a de novo 0.7‐Mb deletion in 19p13.13 including CACNA1A associated with mental retardation and epilepsy with infantile spasms. Epilepsia, 50(11), 2501–2503. 10.1111/j.1528-1167.2009.02189.x [DOI] [PubMed] [Google Scholar]
- Basel‐Vanagaite, L. , Attia, R. , Yahav, M. , Ferland, R. J. , Anteki, L. , Walsh, C. A. , … Shohat, M. (2006). The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non‐syndromic mental retardation. Journal of Medical Genetics, 43(3), 203–210. 10.1136/jmg.2005.035709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia, M. C. , Marelli, S. , Novara, F. , Commodaro, S. , Borgatti, R. , Minardo, G. , … Zuffardi, O. (2010). Genotype–phenotype relationship in three cases with overlapping 19p13.12 microdeletions. European Journal of Human Genetics, 18(12), 1302–1309. 10.1038/ejhg.2010.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan, M. , Mendelsohn, N. J. , Pierpont, M. E. , Schimmenti, L. A. , Berry, S. A. , & Hirsch, B. (2010). A novel microdeletion/microduplication syndrome of 19p13.13. Genetics in Medicine, 12(8), 503–511. 10.1097/GIM.0b013e3181e59291 [DOI] [PubMed] [Google Scholar]
- Dong, H.‐Y. , Zeng, H. , Hu, Y.‐Q. , Xie, L. I. , Wang, J. , Wang, X.‐Y. , … Tan, Z.‐P. (2016). 19p13.2 Microdeletion including NFIX associated with overgrowth and intellectual disability suggestive of Malan syndrome. Molecular . Cytogenetics, 9(1), 71 10.1186/s13039-016-0282-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driller, K. , Pagenstecher, A. , Uhl, M. , Omran, H. , Berlis, A. , Grunder, A. , & Sippel, A. E. (2007). Nuclear Factor I X Deficiency causes brain malformation and severe skeletal defects. Molecular and Cellular Biology, 27(10), 3855–3867. 10.1128/mcb.02293-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros, A. , Denier, C. , Joutel, A. , Cecillon, M. , Lescoat, C. , Vahedi, K. , … Tournier‐Lasserve, E. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. New England Journal of Medicine, 345(1), 17–24. 10.1056/NEJM200107053450103 [DOI] [PubMed] [Google Scholar]
- Gurrieri, F. , Cavaliere, M. L. , Wischmeijer, A. , Mammì, C. , Neri, G. , Pisanti, M. A. , … Priolo, M. (2015). NFIX mutations affecting the DNA‐binding domain cause a peculiar overgrowth syndrome (Malan syndrome): A new patients series. European Journal of Medical Genetics, 58(9), 488–491. 10.1016/j.ejmg.2015.06.009 [DOI] [PubMed] [Google Scholar]
- Hino‐Fukuyo, N. , Kikuchi, A. , Arai‐Ichinoi, N. , Niihori, T. , Sato, R. , Suzuki, T. , … Kure, S. (2015). Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Human Genetics, 10.1007/s00439-015-1553-6 [DOI] [PubMed] [Google Scholar]
- Jezela‐Stanek, A. , Kucharczyk, M. , Falana, K. , Jurkiewicz, D. , Mlynek, M. , Wicher, D. , … Krajewska‐Walasek, M. (2016). Malan syndrome (Sotos syndrome 2) in two patients with 19p13.2 deletion encompassing NFIX gene and novel NFIX sequence variant. Biomedical Papers, 160(1), 161–167. 10.5507/bp.2016.006 [DOI] [PubMed] [Google Scholar]
- Jorge, R. , Silva, C. , Águeda, S. , Dória, S. , & Leão, M. (2015). Intellectual disability and overgrowth‐A new case of 19p13.13 microdeletion syndrome with digital abnormalities. American Journal of Medical Genetics Part A, 167(11), 2839–2843. 10.1002/ajmg.a.37280 [DOI] [PubMed] [Google Scholar]
- Karmarkar, S. , Amarillo, I. , & Larsen, D. (2014). Soto‐like syndrome and cortical malformation associated with chromosome 19p13.2 microdeletion including nuclear factor‐1X (NFIX). Neurology, 82(10 Supplement).
- Klaassens, M. , Morrogh, D. , Rosser, E. M. , Jaffer, F. , Vreeburg, M. , Bok, L. A. , … Scott, R. H. (2015). Malan syndrome: Sotos‐like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. European Journal of Human Genetics, 23(5), 610–615. 10.1038/ejhg.2014.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors, E. E. , Melberg, A. , Vanmolkot, K. R. J. , Kumlien, E. , Haan, J. , Raininko, R. , … Van Den Maagdenberg, A. M. J. M. (2004). Childhood epilepsy, familial hemiplegic migraines, cerebellar ataxia and a new CACNA1A mutation. Neurology, 63(6), 1136‐1137. 10.1212/01.WNL.0000138571.48593.FC [DOI] [PubMed] [Google Scholar]
- Kuroda, Y. , Mizuno, Y. , Mimaki, M. , Oka, A. , Sato, Y. , Ogawa, S. , & Kurosawa, K. (2017). Two patients with 19p13.2 deletion (Malan syndrome) involving NFIX and CACNA1A with overgrowth, developmental delay, and epilepsy. Clinical Dysmorphology, 26(4), 224–227. 10.1097/MCD.0000000000000185 [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Chong, P. , Kira, R. , Seto, T. , Ondo, Y. , Shimojima, K. , & Yamamoto, T. (2017). Mutations in NSD1 and NFIX in Three Patients with Clinical Features of Sotos Syndrome and Malan Syndrome. Journal of Pediatric Genetics, 06(04), 234–237. 10.1055/s-0037-1603194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon, S. M. , Waggoner, D. , Halbach, S. , Thorland, E. C. , Khorasani, L. , & Reid, R. R. (2015). Syndromic craniosynostosis associated with microdeletion of chromosome 19p13.12‐19p13.2. Genes & Diseases, 2(4), 347–352. 10.1016/j.gendis.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysy, P. A. , Ravoet, M. , Wustefeld, S. , Bernard, P. , Nassogne, M.‐C. , Wyns, E. , & Sibille, C. (2009). A new case of syndromic craniosynostosis with cryptic 19p13.2‐p13.13 deletion. American Journal of Medical Genetics Part A, 149A(11), 2564–2568. 10.1002/ajmg.a.33056 [DOI] [PubMed] [Google Scholar]
- Malan, V. , Rajan, D. , Thomas, S. , Shaw, A. C. , Louis dit Picard, H. , Layet, V. , … Cormier‐Daire, V. (2010). Distinct Effects of Allelic NFIX Mutations on Nonsense‐Mediated mRNA Decay Engender Either a Sotos‐like or a Marshall‐Smith Syndrome. The American Journal of Human Genetics, 87(2), 189–198. 10.1016/j.ajhg.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi, G. , Orteschi, D. , Vigevano, F. , Felie, J. , Walsh, C. A. , Manzini, M. C. , & Neri, G. (2012). Expanding the spectrum of rearrangements involving chromosome 19: A mild phenotype associated with a 19p13.12‐p13.13 deletion. American Journal of Medical Genetics, Part A, 158A(4), 888–893. 10.1002/ajmg.a.35254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, F. , Marín‐Reina, P. , Sanchis‐Calvo, A. , Perez‐Aytés, A. , Oltra, S. , Roselló, M. , … Orellana, C. (2015). Novel mutations of NFIX gene causing Marshall‐Smith syndrome or Sotos‐like syndrome: One gene, two phenotypes. Pediatric Research, 78(5), 533–539. 10.1038/pr.2015.135 [DOI] [PubMed] [Google Scholar]
- Natiq, A. , Elalaoui, S. , Miesch, S. , Bonnet, C. , Jonveaux, P. , Amzazi, S. , & Sefiani, A. (2014). A new case of de novo 19p13.2p13.12 deletion in a girl with overgrowth and severe developmental delay. Molecular Cytogenetics, 7(1), 40 10.1186/1755-8166-7-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmakayalu, M. , Horton, V. K. , Darbro, B. , Patil, S. R. , Alsayouf, H. , Keppler‐Noreuil, K. , & Shchelochkov, O. A. (2013). Apparent Germline Mosaicism for a Novel 19p13.13 Deletion Disrupting NFIX and CACNA1A. American Journal of Medical Genetics Part A, 161(5), 1105–1109. 10.1002/ajmg.a.35790 [DOI] [PubMed] [Google Scholar]
- Ophoff, R. A. , Terwindt, G. M. , Vergouwe, M. N. , van Eijk, R. , Oefner, P. J. , Hoffman, S. M. G. , … Frants, R. R. (1996). Familial Hemiplegic Migraine and Episodic Ataxia Type‐2 Are Caused by Mutations in the Ca2+ Channel Gene CACNL1A4. Cell, 87(3), 543–552. 10.1016/S0092-8674(00)81373-2 [DOI] [PubMed] [Google Scholar]
- Oshima, T. , Hara, H. , Takeda, N. , Hasumi, E. , Kuroda, Y. , Taniguchi, G. O. , … Komuro, I. (2017). A novel mutation of NFIX causes Sotos‐like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection. Human Genome Variation, 4, 17022 10.1038/hgv.2017.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priolo, M. , Grosso, E. , Mammì, C. , Labate, C. , Naretto, V. G. , Vacalebre, C. , … Laganà, C. (2012). A peculiar mutation in the DNA‐binding/dimerization domain of NFIX causes Sotos‐like overgrowth syndrome: A new case. Gene, 511(1), 103–105. 10.1016/j.gene.2012.08.040 [DOI] [PubMed] [Google Scholar]
- Priolo, M. , Schanze, D. , Tatton‐Brown, K. , Mulder, P. A. , Tenorio, J. , Kooblall, K. , … Hennekam, R. C. (2018). Further delineation of Malan syndrome. Human Mutation, 39(9), 1226–1237. 10.1002/humu.23563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai, A. , Narayanan, D. L. , & Phadke, S. R. (2018). Malan syndrome: Extension of genotype and phenotype spectrum. American Journal of Medical Genetics Part A, 176(12), 2896–2900. 10.1002/ajmg.a.40663 [DOI] [PubMed] [Google Scholar]
- Schoch, K. , Meng, L. , Szelinger, S. , Bearden, D. R. , Stray‐Pedersen, A. , Busk, O. L. , … Cogan, J. D. (2017). A Recurrent De Novo Variant in NACC1 Causes a Syndrome Characterized by Infantile Epilepsy, Cataracts, and Profound Developmental Delay. The American Journal of Human Genetics, 100(2), 343–351. 10.1016/j.ajhg.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima, K. , Okamoto, N. , Tamasaki, A. , Sangu, N. , Shimada, S. , & Yamamoto, T. (2015). An association of 19p13.2 microdeletions with Malan syndrome and Chiari malformation. American Journal of Medical Genetics Part A, 167(4), 724–730. 10.1002/ajmg.a.36959 [DOI] [PubMed] [Google Scholar]
- Sparrow, S. S. , Cicchetti, D. V. , & Balla, D. A. (2005). The Vineland Adaptive Behavior Scales (2nd ed). In Major psychological assessment instruments.
- Trentini, C. M. , Yates, D. B. , & Heck, V. S. (2014). Escala de Inteligência Wechsler Abreviada (WASI): Manual Profissional. In São Paulo, SP: Casa do Psicólogo.
- Wan, J. , Mamsa, H. , Johnston, J. L. , Spriggs, E. L. , Singer, H. S. , Zee, D. S. , … Jen, J. C. (2011). Large genomic deletions in CACNA1A cause episodic ataxia type 2. Frontiers in Neurology, 2, 51 10.3389/fneur.2011.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welham, A. , Singla, G. , Moss, J. , Penhallow, J. , Sheth, K. , Wilde, L. , … Oliver, C. (2015). Behavioral characteristics associated with 19p13.2 microdeletions. American Journal of Medical Genetics, Part A, 167(10), 2334–2343. 10.1002/ajmg.a.37180 [DOI] [PubMed] [Google Scholar]
- Yoneda, Y. , Saitsu, H. , Touyama, M. , Makita, Y. , Miyamoto, A. , Hamada, K. , … Matsumoto, N. (2012). Missense mutations in the DNA‐binding/dimerization domain of NFIX cause Sotos‐like features. Journal of Human Genetics, 57(3), 207–211. 10.1038/jhg.2012.7 [DOI] [PubMed] [Google Scholar]
- Zamponi, G. W. , Lory, P. , & Perez‐Reyes, E. (2010). Role of voltage‐gated calcium channels in epilepsy. Pflugers Archive, 460(2), 395–403. 10.1007/s00424-009-0772-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.