

Abstract
Background
Retinoblastoma (Rb) is a rare intraocular malignant tumor in children with high overall survival. Predisposition to Rb is linked to RB1 germline mutations with high penetrance, but rare RB1 low‐penetrance variants are also known. Rb survivors are at risk of second primary malignancies (SPMs), mostly osteosarcoma and soft‐tissue sarcoma. Nevertheless, the risk of primary osteosarcoma developing without prior Rb has not been reported in RB1 germline mutation carriers.
Methods
We report a patient in whom osteosarcoma developed at age 17 as a first primary malignancy within a family context of sarcoma.
Results
Unexpectedly, genetic testing identified a low‐penetrance germline mutation in RB1 [NM_000321.2: c.45_76dup; p.(Pro26Leufs*50)]. In eight additional similar cases from published and unpublished reports of families, first primary osteosarcomas and sarcomas mostly developed in RB1 low‐penetrance mutation carriers without prior Rb.
Conclusion
We propose that first primary sarcoma and osteosarcoma could be a novel clinical presentation of a RB1‐related hereditary predisposition syndrome linked to RB1 low‐penetrance germline mutations. In these families, careful screening of primary non‐Rb cancer and SPMs is required by maintaining enhanced clinical vigilance. Implementing lifelong periodic whole‐body MRI screening might be a complementary strategy for unaffected carrier relatives in these families.
Keywords: cancer screening, low penetrance, Osteosarcoma, RB1, Sarcoma, whole‐body MRI

The risk of primary osteosarcoma developing without prior retinoblastoma has not been reported in RB1 germline mutation carriers. We report a patient in whom osteosarcoma developed at age 17 as a first primary malignancy within a family context of sarcoma. Unexpectedly, genetic testing identified a low‐penetrance germline mutation in RB1 [NM_000321.2: c.45_76dup, p.(Pro26Leufs*50)]. We propose that first primary sarcoma and osteosarcoma could be a novel clinical presentation of a RB1‐related hereditary predisposition syndrome linked to RB1 low‐penetrance germline mutations and discuss the screening strategy to implement for unaffected carrier relatives in these families.
1. INTRODUCTION
Retinoblastoma (Rb) is a rare intraocular malignant tumor in children that is due to biallelic inactivation of the tumor suppressor gene, RB1, in the developing retina. Both autosomal‐dominant hereditary and nonhereditary forms coexist. Hereditary Rb is linked to RB1 germline mutations with an autosomal‐dominant mode of inheritance and high penetrance. Hence, germline carriage most often leads to bilateral Rb, with a median‐age of 1 year at diagnosis. First‐line treatment depends on the phenotypic presentation and includes surgery (enucleation), local and systemic adjuvant treatment, and various conservative treatments, reaching a 95% overall survival in industrialized countries. External beam radiotherapy is now avoided because it maximizes the risk of second primary malignancies (SPMs, see below) in the field of irradiation (Marees et al., 2008; Temming et al., 2017).
RB1 germline carriers in whom Rb develops are also at risk of SPMs (i.e., 20% at age 30 and 50% at age 50 [Marees et al., 2008]). The spectrum of SPMs classically includes, in decreasing order of frequency, osteosarcoma, soft‐tissue sarcoma, melanoma and epithelial carcinoma (mostly lung and bladder). SPM screening is a well‐known but still unsolved top priority objective in clinical management of Rb survivors because SPMs are the major cause of death in adulthood (Aerts et al., 2004). Classically, SPM risk management includes medical advice about additional cancer risk factors such as avoiding smoking, excessive UV exposure, and other DNA‐damaging agents. It also includes enhanced awareness by patients and health providers of any symptom or sign that might signal a non‐Rb cancer, such as an unexplained lump or inflammatory bone pain. Routine screening by imaging of non‐Rb SPM is neither recommended in international guidelines nor currently implemented.
Still, atypical Rb families displaying low penetrance (unaffected germline carriers) and variable expressivity (bilateral, unilateral, and retinoma cases) have been described (Harbour, 2001). These families carry rare RB1 low‐penetrance pathogenic variants. The SPM risk in these carriers is controversial. Indeed, Dommering et al. suggested a lesser risk of SPMs, whereas Chaussade et al. reported similar SPM frequency in both low‐ and high‐penetrance variant carriers (Chaussade et al., 2018; Dommering et al., 2012). Regardless, the significant risk of an osteosarcoma developing as an SPM in Rb survivors is well established, whereas the risk of such a cancer developing as a first primary malignancy without prior Rb in RB1 germline mutation carriers has not been delineated (Temming et al., 2017).
Here, we report a patient in whom osteosarcoma developed at age 17 as a first primary malignancy within a family context of sarcoma. Multistep genetic testing based on this peculiar family medical history and a review of similar hidden cases led us to highlight sarcoma as a potential first clinical expression of an RB1 low‐penetrance germline mutation.
2. CLINICAL REPORT
At age 17, the proband (IV.1, Figure 1) presented subacute left‐knee pain and was diagnosed with a first primary malignancy: high‐grade osteosarcoma of the left lower end of the femur. Because his maternal uncle (III.5) died from osteosarcoma at age 12 in the 1970s, the proband was eligible for Li‐Fraumeni syndrome testing. TP53‐germline targeted sequencing was rapidly performed and the results were negative. Further family medical history exploration revealed early‐onset unilateral Rb in his first‐degree female cousin (IV.4) at age 4 months in the 1990s. The cousin was then referred to the oncogenetics department at age 18. Subsequent cancer gene panel sequencing (including RB1, TP53, CDKN2A, ATM, BRCA2, and MLH1 among others) revealed a germline heterozygous deleterious frameshift variant in exon 1 of RB1 [NM_000321.2: c.45_76dup, p.(Pro26Leufs*50)]. The proband (IV.1) carried the RB1 mutation identified. In the meantime, cousin (IV.4), an Rb survivor, was diagnosed with a SPM (osteosarcoma of the leg).
Figure 1.
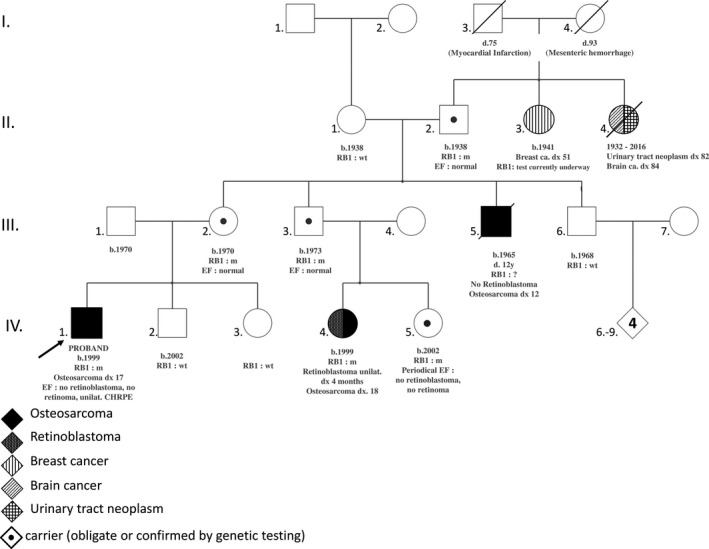
Pedigree of reported patient. RB1 m: carrier of the pathogenic variant in RB1 (NM_000321.2: c.45_76dup, p.(Pro26Leufs*50)). RB1 wt: RB1 wild type; RB1 ?: RB1 sequencing not performed; TP53 wt: TP53 wild type. b., year of birth; ca., cancer; CHRPE, congenital hypertrophy of the retinal pigment epithelium; d., age at death; dx xx, cancer diagnosis at age xx years old; EF, eye fundoscopy; unilat., unilateral
Genetic status for the RB1 family germline mutation could not be assessed in the deceased uncle who had an osteosarcoma (III.5). The proband's maternal grandfather (II.2), mother (III.2), maternal uncle (III.3) and cousin (IV.5) underwent targeted genetic testing, which confirmed that they were the unaffected carriers of the family germline mutation. Eye fundoscopy performed in the proband (IV.1), his mother (III.2), his grandfather (II.2), and his unaffected carrier uncle (III.3), at ages 18, 47, 80, and 27, respectively, and periodically in his unaffected carrier female cousin (IV.5) did not reveal any active Rb and excluded retinoma.
In the proband (IV.1), a single round flat lesion was observed on the far superior and temporal peripheral retina of the right eye (Figure 2a, white arrows). This depigmented lesion was well demarcated by a ring or “halo” around its margin. The lesion was fully in line with a benign tumor of the retinal pigment epithelium (i.e., a solitary congenital hypertrophy of the retinal pigment epithelium) and not suggestive of familial adenomatous polyposis.
Figure 2.
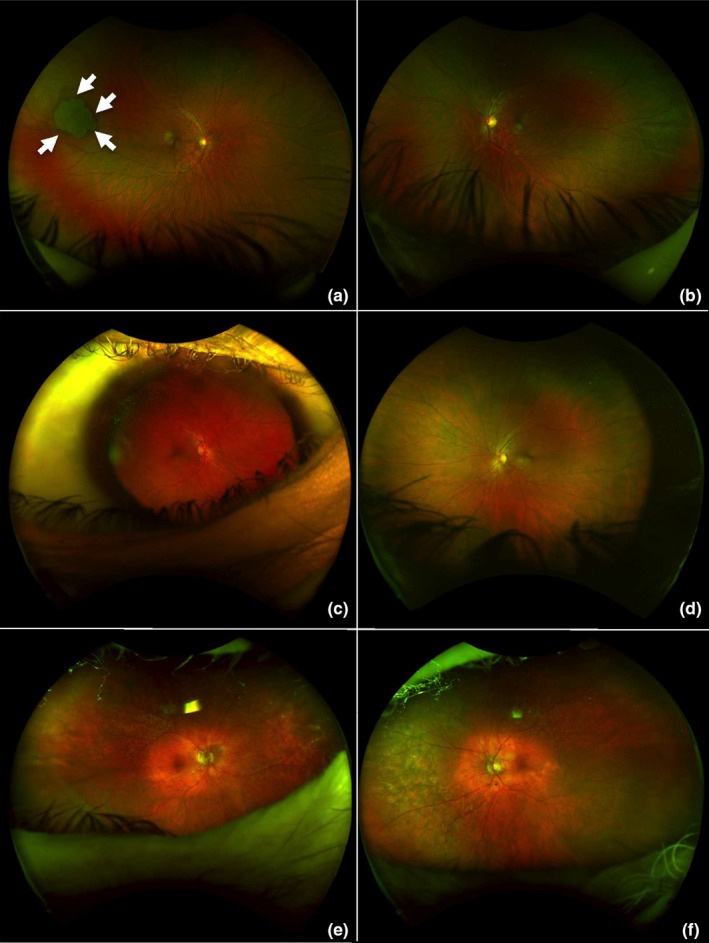
Ultra‐widefield retinal imaging (200° with single capture acquired with Optos device) of the proband (IV.1 [in Figure 1], a and b), his mother (III.2, c and d) and his maternal grandfather (II.2, e and f), all carrying the mutation in RB1 [NM_000321.2: c.45_76dup, p.(Pro26Leufs*50)]. (a and b) Ophthalmologic examination of the proband at age 18, in whom osteosarcoma developed at age 17: no history of leukocoria or strabismus. Visual acuity was 20/20 in both eyes. There was no retinoblastoma or retinoma. A single round flat lesion appeared on the far superior and temporal peripheral retina in the right eye (a, white arrows). This depigmented lesion is well demarcated with a ring or “halo” around its margin and agrees with a benign tumor of the retinal pigment epithelium (i.e., solitary congenital hypertrophy of the retinal pigment epithelium [CHRPE]). (c–f) No malignancy developed in the 47‐year‐old mother (c and d) or the 80‐year‐old grandfather (e and f) of the proband. Ultra‐widefield photographs did not reveal any active or spontaneously regressed retinoblastoma
The osteosarcoma in both the proband and his cousin (IV.4) was treated by induction chemotherapy, resection surgery, and adjuvant chemotherapy. At the time of writing, 13 months had elapsed since the treatment ended for the proband, and adjuvant chemotherapy was still ongoing for the cousin: both were alive and did not show malignant relapse or other SPMs.
3. DISCUSSION
Here, we report for the first time a patient with an RB1 germline mutation in whom a first primary osteosarcoma developed without prior Rb. The initial reason for the referral to the oncogenetics department was the aggregation of osteosarcomas in two second‐degree relatives, which was not directly suggestive of RB1 involvement. Indeed, the initial pattern of family medical data was misleading and steered genetic exploration toward TP53 sequencing, especially considering its positivity in up to 10% of children with osteosarcomas (Shaul et al., 2018). Additional medical data from one third‐degree relative with early‐onset unilateral Rb and a second primary osteosarcoma eventually led to the relevant diagnosis of RB1‐related hereditary predisposition syndrome (HPS). Of note, the absence of pathogenic variants in known or suspected sarcoma genes included in the panel testing strengthened RB1 involvement in this family (Ballinger et al., 2016; Chan et al., 2017; Jouenne et al., 2017; Tlemsani, Leroy, et al., 2018; Tlemsani, Pasmant, et al., 2018). On the other hand, we could not rule out that the osteosarcoma developed in the proband's uncle might have occurred independently of the germline RB1 mutation (Taylor et al., 2007 ). In any case, these unusual diagnostic steps could be considered instructive considering that in clinical practice, pedigree presentation varies greatly among families.
A literature review retrieved four cases of primary malignancy without prior Rb (sarcoma and nonsarcoma cancers but no osteosarcoma) in families with RB1 low‐penetrance hereditary mutations. Sarcoma was diagnosed in a female carrier of a pathogenic RB1 splice variant (c.2520+6T>C) at age 64 (Dommering et al., 2014), leiomyosarcoma in a male carrier of the missense variant p.(Arg661Trp) at age 44 (Dommering et al., 2014); melanoma with no personal history of Rb in a male carrier of a deletion of exon 4 of RB1 (Dryja, Rapaport, McGee, Nork, & Schwartz, 1993); and leiomyosarcoma in a 69‐year‐old female carrying a missense p.(His483Tyr) variant, showing sebaceous cell carcinoma of the eyelid 3 years later (Serrano et al., 2011).
A retrospective collection of similar unpublished data was initiated within the Institut Curie and a French network for Rb: four additional families with RB1 mutations and at least one mutation carrier with sarcoma without prior Rb were found (pedigrees depicted in Figure S1). In family A, osteosarcoma as a first primary malignancy was diagnosed at age 65 in the father of the proband [c.55del, p.(Glu19Asnfs*48)] and bilateral Rb was diagnosed at age 2 in the proband. The proband's father (S1A: II.4) had been considered an unaffected obligate carrier since metastatic bilateral Rb was diagnosed in his sister (S1A: II.3) before she was 1 year old. In family B, the proband (S1B: IV.4) had been diagnosed with unilateral Rb at age 18 months, and a germline RB1 mutation [c.1981C>T; p.(Arg661Trp)] was detected. At age 30, his carrier sister (S1B: IV.3) was diagnosed with a first primary leiomyosarcoma. Considering the co‐occurrence of synovial sarcoma in one maternal aunt (S1B: III.14), TP53 sequencing was performed in patient IV.3, but findings were normal, whereas RB1 sequencing revealed a paternally inherited mutation. In family C, the proband (S1C: IV.7) was diagnosed with unilateral Rb at age 2 years (c.264+5G>A mutation). His paternal grandfather (S1C: II.3) was a carrier and soft‐tissue sarcoma developed at age 79. In family D, harboring a c.1981C>T; p.(Arg661Trp) variant, the proband (S1D: VI.4) was diagnosed with unilateral Rb at age 20 months. One of his fourth‐degree relatives (S1D: V.4), a 12‐year‐old girl without genetic testing, whose sister (S1D: V.5) was diagnosed with bilateral Rb and carried the RB1 germline mutation, died from osteosarcoma without prior Rb.
In all these families, mutations were missenses, splice mutations leaving pocket domains intact, frameshifts in exon 1 or large intragenic deletions, all commonly known as low‐penetrance mutations (Harbour, 2001).
Overall, we propose first primary sarcoma and osteosarcoma, regardless of family history of Rb, as a novel phenotypic indication for RB1 sequencing.
Interestingly, all individuals diagnosed with first primary sarcoma carried RB1 low‐penetrance alleles. Hence, we questioned if the parent of origin effect demonstrating a higher Rb risk with paternally inherited low‐penetrance alleles holds true with sarcoma risk (Eloy et al., 2016). A paternal inheritance was observed (3 out of 3 cases) in families with p.Arg661Trp carriers affected by first primary sarcoma or melanoma without prior Rb (Dommering et al., 2014, family S1B, family S1D) but the number of families is too small to draw any conclusion.
The main puzzling question is how to explain why the retinal tissue was spared from tumorigenesis whereas aggressive cancerogenesis was enabled in the bone or soft tissues, with osteosarcoma or sarcoma development decades later. Hypothetically, nongenetic factors could be involved. In addition, a theoretical possibility is somatic mosaicism with postzygotic reversion of the inherited mutation sparing retinal tissue (Hirschhorn, 2003). Moreover, a digenic or oligogenic model involving modifier genes such as MDM2 and a tissue‐dependent factor and gene network seems relevant (Castéra et al., 2010). This type of model could agree with an apparently permissive state for tumorigenesis of retinal or bone or soft‐tissue cells induced by RB1 low‐penetrance mutations. This permissive state could be insufficient to drive tumorigenesis by itself but combined with another oncogenic cellular phenomenon might lead to cancer.
All adults newly diagnosed as carriers but not affected by Rb in the family we report underwent ophthalmologic examination with eye fundoscopy, which did not reveal any retinoma. This observation could be a first clue suggesting that retinal tissue was not the location of poorly aggressive tumorigenesis and was completely spared.
Further investigations are required to shed light on mechanisms explaining the apparent tissue‐ and individual‐dependent phenomena underlying carcinogenesis in nonretinal tissues in these RB1‐mutated patients without prior Rb.
Besides further molecular understanding, clinical management is a major issue. In the main family we report, in addition to the classical ophthalmologic and oncologic posttreatment monitoring, follow‐up of the proband, his female cousin and above all their unaffected carrier relatives was under cautious discussion. Considering family member preferences together with a multidisciplinary medical opinion, annual lifelong whole‐body MRI was decided in all carrier individuals, beginning as early as the genetic carrier status was known. Generalization of this option to all unaffected RB1 low‐penetrance germline mutation carriers remains to be cautiously assessed. Indeed, how to implement (methods and scheduling) the most efficient and nonharmful screening of non‐Rb cancer in individuals carrying an RB1 reduced‐penetrance germline mutation even if not affected by Rb in childhood is a crucial and challenging medical issue. In addition to enhanced awareness of any suspicious symptom such as a lump or bone pain, routine periodic imaging might be an option, in light of the nine family medical histories we describe. Nevertheless, at what age such screening should start and its detection yield, cost‐effectiveness, harmlessness and acceptability remain to be ascertained.
Lifelong routine screening of solid malignancies issue is analogous to the management of at‐risk individuals diagnosed with Li‐Fraumeni syndrome. Indeed, patients carrying germline mutations in TP53 are at high risk of a wide range of malignancies, including sarcoma. In these child and adult patients, early presymptomatic detection of malignancies improves overall survival (Villani et al., 2016). Hence, guidelines for surveillance of Li‐Fraumeni patients now include annual rapid whole‐body MRI, which is lifelong, specifically aimed at screening sarcoma (Kratz et al., 2017). Furthermore, whole‐body MRI has been found the best means of imaging screening for cancers in children in terms of its safety and diagnostic performance (Friedman et al., 2014; Greer, Voss, & States, 2017; Guimarães et al., 2017). However, in hereditary Rb survivors, whole‐body MRI monitoring assessed in a retrospective pilot study involving 25 patients was not sufficiently sensitive (Friedman et al., 2014).
Further prospective assessment of this type of screening by imaging over a longer period of time, with a larger cohort, and including a number of RB1 carriers who are not Rb survivors is needed to address the challenge of optimizing long‐term non‐Rb follow‐up of such carriers.
Although data from somatic sequencing of different types of sarcoma are rapidly accumulating and show RB1 monoallelic or biallelic tumor genomic alterations commonly detected in sarcomas (Cancer Genome Atlas Research Network, 2017; Chen et al., 2014), little is known about the RB1 germline mutation in patients with primary sarcoma and osteosarcoma. The data we report advocate for considering germline sequencing of RB1 in patients with unexplained sarcoma, especially germline TP53‐negative osteosarcoma, regardless of the personal or family history of Rb.
To conclude, the patients and pedigrees we report shed light on an unusual pattern of expressivity and low‐penetrance germline variants in RB1 and raise the question of expanding the tumor spectrum of RB1‐related HPS with first primary sarcoma and osteosarcoma. This would require further investigations to unravel the underlying mechanisms. Most importantly, we suggest that sarcoma and osteosarcoma, regardless of Rb family history, might be a novel clinical presentation requiring RB1 germline testing. We also highlight screening of nonretinal cancer and SPM as major issues to address in these families. It also prompts a watchful medical, especially genetic, management of apparently unaffected carriers of RB1 low‐penetrance germline mutation.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
ACKNOWLEDGMENTS
The authors thank the patients and their family for authorizing to share their medical data with scientific community and RETINOSTOP for their continued support. The authors thank Laura Smales for valuable editorial assistance.
Imbert‐Bouteille M, Gauthier‐Villars M, Leroux D, et al. Osteosarcoma without prior retinoblastoma related to RB1 low‐penetrance germline pathogenic variants: A novel type of RB1‐related hereditary predisposition syndrome? Mol Genet Genomic Med. 2019;7:e913 10.1002/mgg3.913
Funding information
Internal institutional public funding.
REFERENCES
- Aerts, I. , Pacquement, H. , Doz, F. , Mosseri, V. , Desjardins, L. , Sastre, X. , … Quintana, E. (2004). Outcome of second malignancies after retinoblastoma: A retrospective analysis of 25 patients treated at the Institut Curie. European Journal of Cancer, 40, 1522–1529. 10.1016/j.ejca.2004.03.023 [DOI] [PubMed] [Google Scholar]
- Ballinger, M. L. , Goode, D. L. , Ray‐Coquard, I. , James, P. A. , Mitchell, G. , & Niedermayr, E. , … Thomas, D. M. (2016). Monogenic and polygenic determinants of sarcoma risk: An international genetic study. The Lancet Oncology, 17, 1261–1271. 10.1016/s1470-2045(16)30147-4 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address: elizabeth.demicco@sinaihealthsystem.ca, Cancer Genome Atlas Research Network . (2017). Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell, 171, 950–965.e28. 10.1016/j.cell.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castéra, L. , Sabbagh, A. , Dehainault, C. , Michaux, D. , Mansuet‐Lupo, A. , Patillon, B. , … Houdayer, C. (2010). MDM2 as a modifier gene in retinoblastoma. Journal of the National Cancer Institute, 102, 1805–1808. 10.1093/jnci/djq416 [DOI] [PubMed] [Google Scholar]
- Chan, S. H. , Lim, W. K. , Ishak, N. D. B. , Li, S. T. , Goh, W. L. , Tan, G. S. , … Ngeow, J. (2017). Germline mutations in cancer predisposition genes are frequent in sporadic sarcomas. Scientific Reports, 7, 10660 10.1038/s41598-017-10333-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaussade, A. , Millot, G. , Wells, C. , Brisse, H. , Laé, M. , Savignoni, A. , … Houdayer, C. (2018). Correlation between RB1germline mutations and second primary malignancies in hereditary retinoblastoma patients treated with external beam radiotherapy. European Journal of Medical Genetics, 62(3), 217–223. 10.1016/j.ejmg.2018.07.017 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Bahrami, A. , Pappo, A. , Easton, J. , Dalton, J. , & Hedlund, E. , …, Dyer, M. A. ; St. Jude Children’s Research Hospital‐Washington University Pediatric Cancer Genome Project . (2014). Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Reports, 7, 104–112. 10.1016/j.celrep.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommering, C. J. , Marees, T. , van der Hout, A. H. , Imhof, S. M. , Meijers‐Heijboer, H. , Ringens, P. J. , … Moll, A. C. (2012). RB1 mutations and second primary malignancies after hereditary retinoblastoma. Familial Cancer, 11, 225–233. 10.1007/s10689-011-9505-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommering, C. J. , Mol, B. M. , Moll, A. C. , Burton, M. , Cloos, J. , Dorsman, J. C. , … van der Hout, A. H. (2014). RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. Journal of Medical Genetics, 51, 366–374. 10.1136/jmedgenet-2014-102264 [DOI] [PubMed] [Google Scholar]
- Dryja, T. P. , Rapaport, J. , McGee, T. L. , Nork, T. M. , & Schwartz, T. L. (1993). Molecular etiology of low‐penetrance retinoblastoma in two pedigrees. American Journal of Human Genetics, 52, 1122–1128. [PMC free article] [PubMed] [Google Scholar]
- Eloy, P. , Dehainault, C. , Sefta, M. , Aerts, I. , Doz, F. , Cassoux, N. , … Houdayer, C. (2016). A parent‐of‐origin effect impacts the phenotype in low penetrance retinoblastoma families segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genetics, 12, e1005888 10.1371/journal.pgen.1005888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, D. N. , Lis, E. , Sklar, C. A. , Oeffinger, K. C. , Reppucci, M. , Fleischut, M. H. , … Dunkel, I. J. (2014). Whole‐body magnetic resonance imaging (WB‐MRI) as surveillance for subsequent malignancies in survivors of hereditary retinoblastoma: A pilot study. Pediatric Blood & Cancer, 61, 1440–1444. 10.1002/pbc.24835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer, M.‐L.‐C. , Voss, S. D. , & States, L. J. (2017). Pediatric cancer predisposition imaging: Focus on whole‐body MRI. Clinical Cancer Research, 23, e6–e13. 10.1158/1078-0432.ccr-17-0515 [DOI] [PubMed] [Google Scholar]
- Guimarães, M. D. , Noschang, J. , Teixeira, S. R. , Santos, M. K. , Lederman, H. M. , Tostes, V. , … Marchiori, E. (2017). Whole‐body MRI in pediatric patients with cancer. Cancer Imaging, 17, 6 10.1186/s40644-017-0107-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J. W. (2001). Molecular basis of low‐penetrance retinoblastoma. Archives of Ophthalmology, 119, 1699–1704. [DOI] [PubMed] [Google Scholar]
- Hirschhorn, R. (2003). In vivo reversion to normal of inherited mutations in humans. Journal of Medical Genetics, 40, 721–728. 10.1136/jmg.40.10.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouenne, F. , Chauvot de Beauchene, I. , Bollaert, E. , Avril, M. F. , Caron, O. , Ingster, O. , … Bressac‐de Paillerets, B. (2017). Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. Journal of Medical Genetics, 54, 607–612. 10.1136/jmedgenet-2016-104402 [DOI] [PubMed] [Google Scholar]
- Kratz, C. P. , Achatz, M. I. , Brugières, L. , Frebourg, T. , Garber, J. E. , Greer, M.‐L.‐C. , … Malkin, D. (2017). Cancer screening recommendations for individuals with Li‐Fraumeni syndrome. Clinical Cancer Research, 23, e38–e45. 10.1158/1078-0432.CCR-17-0408 [DOI] [PubMed] [Google Scholar]
- Marees, T. , Moll, A. C. , Imhof, S. M. , de Boer, M. R. , Ringens, P. J. , & van Leeuwen, F. E. (2008). Risk of second malignancies in survivors of retinoblastoma: More than 40 years of follow‐up. Journal of the National Cancer Institute, 100, 1771–1779. 10.1093/jnci/djn394 [DOI] [PubMed] [Google Scholar]
- Serrano, C. , Alonso, J. , Gómez‐Mariano, G. , Aguirre, E. , Diez, O. , Gadea, N. , … Graña, B. (2011). Low penetrance hereditary retinoblastoma in a family: What should we consider in the genetic counselling process and follow up? Familial Cancer, 10, 617–621. 10.1007/s10689-011-9445-y [DOI] [PubMed] [Google Scholar]
- Shaul, E. , Roth, M. , Lo, Y. , Geller, D. S. , Hoang, B. , Yang, R. , … Gill, J. (2018). Pediatric oncologist willingness to offer germline TP53 testing in osteosarcoma. Cancer, 124, 1242–1250. 10.1002/cncr.31212 [DOI] [PubMed] [Google Scholar]
- Taylor, M. , Dehainault, C. , Desjardins, L. , Doz, F. , Levy, C. , Sastre, X. , … Gauthier‐Villars, M. (2007). Genotype‐phenotype correlations in hereditary familial retinoblastoma. Human Mutation, 28, 284–293. 10.1002/humu.20443 [DOI] [PubMed] [Google Scholar]
- Temming, P. , Arendt, M. , Viehmann, A. , Eisele, L. , Le Guin, C. H. D. , Schündeln, M. M. , … Lohmann, D. R. (2017). Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatric Blood & Cancer, 64, 71–80. 10.1002/pbc.26193 [DOI] [PubMed] [Google Scholar]
- Tlemsani, C. , Leroy, K. , Gimenez‐Roqueplo, A.‐P. , Mansuet‐Lupo, A. , Pasmant, E. , Larousserie, F. , … Laurent‐Puig, P. (2018). Chemoresistant pleomorphic rhabdomyosarcoma: Whole exome sequencing reveals underlying cancer predisposition and therapeutic options. Journal of Medical Genetics, pii: jmedgenet-2018-105594. 10.1136/jmedgenet-2018-105594 [DOI] [PubMed] [Google Scholar]
- Tlemsani, C. , Pasmant, E. , Boudou‐Rouquette, P. , Bellesoeur, A. , Even, J. , Larousserie, F. , … Goldwasser, F. (2018). BRCA2 loss‐of‐function and high sensitivity to cisplatin‐based chemotherapy in a patient with a pleomorphic soft tissue sarcoma: effect of genomic medicine. The American Journal of the Medical Sciences, 356, 404–407. 10.1016/j.amjms.2018.04.015 [DOI] [PubMed] [Google Scholar]
- Villani, A. , Shore, A. , Wasserman, J. D. , Stephens, D. , Kim, R. H. , Druker, H. , … Malkin, D. (2016). Biochemical and imaging surveillance in germline TP53 mutation carriers with Li‐Fraumeni syndrome: 11 year follow‐up of a prospective observational study. The Lancet Oncology, 17, 1295–1305. 10.1016/S1470-2045(16)30249-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.