

Abstract
Background
One of the most important steps taken by Beyond Batten Disease Foundation in our quest to cure juvenile Batten (CLN3) disease is to understand the State of the Science. We believe that a strong understanding of where we are in our experimental understanding of the CLN3 gene, its regulation, gene product, protein structure, tissue distribution, biomarker use, and pathological responses to its deficiency, lays the groundwork for determining therapeutic action plans.
Objectives
To present an unbiased comprehensive reference tool of the experimental understanding of the CLN3 gene and gene product of the same name.
Methods
BBDF compiled all of the available CLN3 gene and protein data from biological databases, repositories of federally and privately funded projects, patent and trademark offices, science and technology journals, industrial drug and pipeline reports as well as clinical trial reports and with painstaking precision, validated the information together with experts in Batten disease, lysosomal storage disease, lysosome/endosome biology.
Results
The finished product is an indexed review of the CLN3 gene and protein which is not limited in page size or number of references, references all available primary experiments, and does not draw conclusions for the reader.
Conclusions
Revisiting the experimental history of a target gene and its product ensures that inaccuracies and contradictions come to light, long‐held beliefs and assumptions continue to be challenged, and information that was previously deemed inconsequential gets a second look. Compiling the information into one manuscript with all appropriate primary references provides quick clues to which studies have been completed under which conditions and what information has been reported. This compendium does not seek to replace original articles or subtopic reviews but provides an historical roadmap to completed works.
Keywords: Batten, CLN3, JNCL, juvenile Batten, neuronal ceroid lipofuscinosis
The CLN3 gene, its regulation, gene product, CLN3 protein structure, tissue distribution, biomarker use, and pathological responses to its deficiency.
1. INTRODUCTION
Basic knowledge of the expression, regulation, structure, and function transmembrane‐bound and other proteins, enables the discovery of compounds to modulate their behavior. Along with analyses of disease‐causing mutations, investigators pursue creative approaches to restore protein function(s) and their associated pathways. Therefore, it is critically important that academicians, pharmaceutical investigators, and clinician scientists, be provided with a complete, easy‐to‐access, and an up‐to‐date State of the Science. Historically, one relied on review articles designed to summarize current thinking. However, an information explosion fueled by advances in molecular biology, genetic engineering, and new animal models, coupled with competing hypotheses of authors and size limits of review articles has resulted in the production of irregular, nonsystematic review articles in disease research leading to unintentional bias and widening knowledge gaps. To combat this problem, investigators new to the field must spend hundreds of hours sifting, reading, and evaluating original publications; defeating the purpose for which review articles were created.
Beyond Batten Disease Foundation (BBDF) has taken a lead to help support new and existing researchers in their quest for experimentally proven, unbiased, information. This manuscript is part of a larger strategic plan to advance research in Batten disease by fueling the creation of key physical and informational resources. The foundation worked with Thomson Reuters to gather referenced CLN3 gene and CLN3 protein information and with painstaking attention to detail, validated the information together with experts in CLN3 disease, lysosomal storage disease, and lysosome/endosome biology. The resultant indexed review of CLN3 and CLN3 is not limited in size, focuses on information from original articles, is reviewed by in‐area experts and inclusion in validated databases, and does not draw conclusions. By collecting all of the available information into a single, searchable reference manual, this review saves valuable time and ensures all topic areas are covered; however, readers will still need to review original literature cited here.
The information found within this reference tool is cultivated by: (a) MetaBase™ (version 6.20), a systems biology database, a former product of GeneGo, IntegritySM, (b) a drug and pipeline information database, Cortellis™, and (c) a drug and clinical trial information database, Thomson Innovation™, including patent information from around the world and public databases (December 2014 version), such as NCBI, Ensembl, dbSNP, UniProt, MGI and others. The information cultivated from these databases was then traced back its original source and rigorously reviewed. If the experiment was conducted more than once, all references were added.
The manual includes a research history of the CLN3 gene, discussion of gene regulation, protein structure, tissue distribution, co‐regulated gene expression, biomarker use, and pathological responses to CLN3 protein deficiencies in yeast through humans. Supplementary materials include a list of CLN3 research tools and their associated first‐published reports.
The authors note that following data extraction from the information systems mentioned above, the information gathered here was verified and the associated primary literature was cited along with the experimental methods used. We believe bioinformatics databases such as those mentioned above offer scientists the opportunity to access and cross‐reference a wide variety of biologically relevant data providing new insights and further means to validate their discoveries. However, the authors would like to stress that the databases listed here were used only to provide a framework for the document. The rapid release of new data from various –omics and other programs annotated using computational analysis sometimes leads to misinformation, which we found to be the case for the CLN3 gene and protein. Therefore, the authors worked to provide their readership with direct access to primary, experimentally proven data. Finally, the authors diligently tried to avoid summarizing the information herein in support of one hypothesis over another or drawing any conclusions for the reader. This comprehensive agnostic presentation of experimental findings is meant to complement and not overlap investigator‐driven primary and review literature.
2. GENERAL INFORMATION
2.1. Description of the CLN3 gene
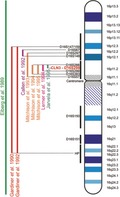
The official name of this gene is “ceroid‐lipofuscinosis, neuronal 3” and official symbol is CLN3. Less commonly used terms include BATTENIN, BTS, JNCL (Juvenile Neuronal Ceroid Lipofuscinosis), and MGC102840. CLN3 was discovered using linkage analysis in search for the disease causing gene (mutation) in 48 children with progressive vision loss, seizures, decline of intellect and loss of motor ability (Eiberg, Gardiner, & Mohr, 1989). Researchers found a linkage between CLN3 disease and haptoglobin (140100) on chromosome 16q22 revealing the location of the CLN3 gene (Eiberg et al., 1989; Gardiner et al., 1990). Linkage studies in larger groups of families followed by physical mapping of markers by mouse/human hybrid cell analysis and fluorescence in situ hybridization refined the coordinates of CLN3 to the interval between D16S288 and D16S298 (see Figure 1: LinkageMapping, (Callen et al., 1991, 1992; Järvelä, Mitchison, Callen, et al., 1995; Järvelä, Mitchison, O'rawe, et al., 1995; Lerner et al., 1994; Mitchison, O'Rawe, Lerner, et al., 1995; Mitchison, O’Rawe, Taschner, et al., 1995; Mitchison et al., 1994; Mitchison, Williams, et al., 1993; Mitchison, Thompson, et al., 1993) The final pieces of the puzzle were added by the International Batten Disease Consortium in 1995 (Batten Disease Consortium, 1995). Today, we know that the cytogenetic location of CLN3 is on the short arm of chromosome 16 at position 12.1 at Genomic coordinates chr16:28,466,653–28,492,302 [OMIM 607042] (NCBI gene entry, 1,201 (Järvelä, Mitchison, O'rawe, et al., 1995; Järvelä, Mitchison, Callen, et al., 1995; Mitchison, O'Rawe, Lerner, et al., 1995; Mitchison, Williams, et al., 1993).
Figure 1.
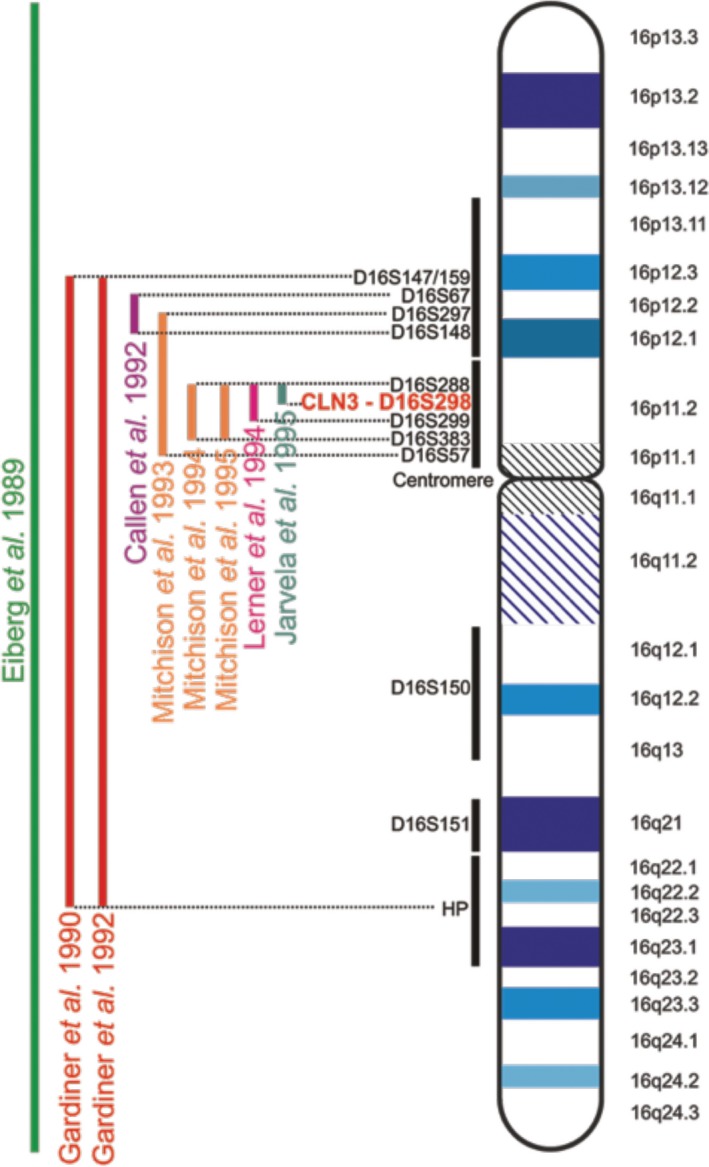
Linkage Mapping. The location of the gene responsible for JNCL was mapped to a region on chromosome 16. Initial findings placed the gene on the long arm of chromosome 16, due to its linkage with the haptoglobin (HP) locus. Later, the location of CLN3 was narrowed down to markers tagging the 16p11.2 region. The dinucleotide marker D16S298 is located in an intron of the CLN3 gene and thus represents the true location of CLN3
In 1997, Mitchison and colleagues reported the genomic structure and complete nucleotide sequence of CLN3, with an estimated number of 15 exons that span 15 kilobases (kb), (Mitchison et al., 1997). Sequence comparisons between CLN3 and homologous expressed sequence tags suggest alternative splicing of the gene and at least 1 additional upstream exon. Marker loci in strong allelic association with the disease loci have been identified (Mole & Gardiner, 1991).
Haplotype analysis identified a homozygous deletion mutation of 966 base pairs (bps) in 73% of 200 affected JNCL patients from 16 different countries (Mitchison, O'Rawe, Lerner, et al., 1995; Mitchison, Thompson, et al., 1993). This deletion mutation was originally believed to stretch 1.02 kb (Batten Disease Consortium, 1995). As a result, many reviews and primary articles refer to this deletion as the “1.02 kb” deletion. However, it was later confirmed that the common deletion spans 966 bps and is therefore more appropriately called the “1 kb” deletion. Cultured fibroblasts from a JNCL patient homozygous for the common 1 kb deletion have been shown to express a major transcript of 521 bps and a minor transcript of 408 bps (Kitzmüller, Haines, Codlin, Cutler, & Mole, 2008). The major transcript contains exon 6 spliced to exon 9 and is thought to encode a truncated CLN3 protein containing the first 153 amino acids of CLN3 plus an additional 28 novel amino acids resulting from an out‐of‐frame RNA sequence at the novel splice site. This gives rise to the following mutant protein sequence:
| 1 | MGGCAGSRRRFSDSEGEETVPEPRLPLLDHQGAHWKNAVGFWLLGLCNNFSYVVMLSAA | 60 |
| 61 | DILSHKRTSGNQSHVDPGPTPIPHNSSSRFDCNSVSTAAVLLADILPTLVIKLLAPLGLH | 120 |
| 121 | LLPYSPRVLVSGICAAGSFVLVAFSHSVGTSLCAISCCSHLLRPRTLEGKKKQRAQPGSP | 180 |
| 181 | S | 181 |
(Underlined letters indicate truncated first 153 AAs of CLN3 followed by 28 novel AAs due to a frameshift at the novel splice site). It is important to note that there is an S written in error at position 166 in some GenBank entries (GenBank: EF587245/1 and publications (Kitzmüller et al., 2008). This should be a “T” as shown above in yellow highlight (GenBank accession no AF077964 and AF077968, which are consistent with genomic sequence NG 008654.2).
2.2. CLN3 gene details
The full name of the gene is ceroid‐lipofuscinosis, neuronal 3, also known by the following symbols (CLN3, BTS, and JNCL). Gene product names include Batten disease protein, Batten, and Battenin. CLN3 gene product deficiency results in a rare, fatal inherited disorder of the nervous system that typically begins in childhood. The first symptom is usually progressive vision loss in previously healthy children followed by personality changes, behavioral problems and slow learning. Seizures commonly appear within 2–4 years of vision loss. However, seizures and psychosis can appear at any time during the course of the disease. Progressive loss of motor functions (movement and speech) start with clumsiness, stumbling and Parkinson‐like symptoms; eventually, those affected become wheelchair‐bound, are bedridden, and die prematurely. [see “CLN3‐Clinical Data.” The neuronal ceroid lipofuscinoses (Batten disease). Ed. Mole, S.E., Williams R.E., Goebel H.H., Machado da Silva G., Cary, North Carolina, Oxford University Press, 2011. Pages 117–119. Print].
The most common name for the disease is Batten disease which was first used to describe the juvenile and presumably the CLN3 form (prior to the discovery of the gene). Today the term Batten is widely used in the US and UK to refer to all 13 forms of neuronal ceroid lipofuscinosis. The disease has also been called Batten‐Mayou disease, Batten‐Spielmeyer‐Vogt disease, CLN3‐related neuronal ceroid‐lipofuscinosis, juvenile Batten disease, Juvenile cerebroretinal degeneration, juvenile neuronal ceroid lipofuscinosis, Spielmeyer‐Vogt disease and the term adopted in 2012, CLN3 disease. Using the Basic Local Alignment Search (BLAST) tool to align regions of similarity between known biological sequences, investigators discovered that the CLN3 gene is highly conserved amongst Homo sapiens, Canis Lupus familiaris, Mus musculus, Danio rerio, Drosophila melanogaster, Caenorhabditis elegans, Schizosaccharomyces cerevisiae, and Schizosaccharomyces pombe (Altschul et al., 1997; Katz et al., 1999; Mitchell, Porter, Kuwabara, & Mole, 2001; Pearce & Sherman, 1997). The National Center for Biotechnology Information (NCBI) Gene lists 179 orthologs discovered through comparison of known sequences.
2.3. Exon and Intron structure and alternative splice variants of CLN3
The CLN3 gene on the p‐arm of chromosome 16 spanning bases 28,466,653 to 28,492,302 http://genome.ucsc.edu/cgi-bin/hgGene?db=hg19%26hgg_gene=CLN3 (Haeussler et al., 2019; Kent et al., 2002) https://www.ncbi.nlm.nih.gov/variation/view/ (version 1.5.6 last update July 17, 2017). The CLN3 protein coding sequence begins on exon two at base 544 and ends at the final exon of the mRNA.
The NCBI Reference Sequence (RefSeq) database reports six transcript variants for the CLN3 gene suggesting that alternative splicing affects exons and the 3′ and 5′ UTRs (Untranslated Region;(Kent et al., 2002; Pruitt et al., 2014, https://www.ncbi.nlm.nih.gov/variation/view/; version 1.5.6 last update July 17, 2017) of the genomic sequence. Most of the six isoforms contain between 13 and 16 exons; however, shorter isoforms are also reported in the Consensus CDS Protein Set. CLN3 isoform a consists of 438 amino acids (aa), and is encoded by the longest transcript variant 1 (NM_001042432.1) (Barnett, Pickle, & Elting, 1990) and 2 (NM_000086.2), 1915 bp and 1879 bp, respectively. Transcript variant 3 (NM_001286104.1) encodes for isofom b (NP_001273033.1) which lacks an alternate in‐frame exon resulting in a shorter protein of 414 aas. Variant 4 (NM_001286105.1) encodes for isoform c (NP_001273034.1) which begins with a downstream AUG (start codon) in the 5′UTR resulting in a different N‐terminus. Isoform c also lacks two exons, which gives rise to a truncated version of the protein consisting of only 338 aas. Isoform d (NP_001273038.1) encoded by transcript variant 5 (NM_001286109.1) contains variations in both 5′ and 3′ UTRs and lacks an in‐frame exon resulting in translation initiation at a downstream AUG and resultant protein of 360 aas. Similarly, isoform e (NP_001273039.1) encoded by variant 6 (NM_001286110.1) harbors alterations in the 5′ UTR and lacks an in‐frame exon leading to a delay in the initiation of translation and produces a protein of 384 aas.
RefSeq records are generated when there is experimental or published evidence in support of the full‐length product, whereas transcript alignments to the assembled genome indicate the possibility of a gene product. RefSeq records list 6 transcripts whereas Ensembl, which is not limited to proven transcripts, lists 64 potential transcripts for the CLN3 gene, providing additional clues to CLN3’s spatiotemporal patterns of expression. However, neither tissue, development, nor disease‐specific studies have been completed to indicate which transcripts are expressed under various conditions (Zerbino et al., 2018).
2.3.1. Impact of splice variants and Single Nucleotide Polymorphisms on the protein domain structure of CLN3
Interestingly, mapping the mutations causative of Batten Disease on a CLN3 topological model reveals that most of these mutations face the luminal side of the intracellular compartments. Moreover, evolutionarily constrained analysis of the aa sequence revealed that luminal loop 2, is the most highly conserved domain across species (Gachet, Codlin, Hyams, & Mole, 2005; Muzaffar & Pearce, 2008). Of particular note, the most common mutation found in CLN3 disease patients, the “1kb” deletion in which exons 7 and 8 are excised, maps within this loop. Similar to the second loop, the predicted amphipathic helix on the luminal face between the fifth and sixth transmembrane helices contains several missense mutations (Figure 2) (Kousi, Lehesjoki, & Mole, 2012; Nugent, Mole, & Jones, 2008). The clustering of a majority of missense mutations in these two luminal regions strongly suggests that they are critical sites for CLN3 protein interaction and function (Cotman & Staropoli, 2012).
Figure 2.
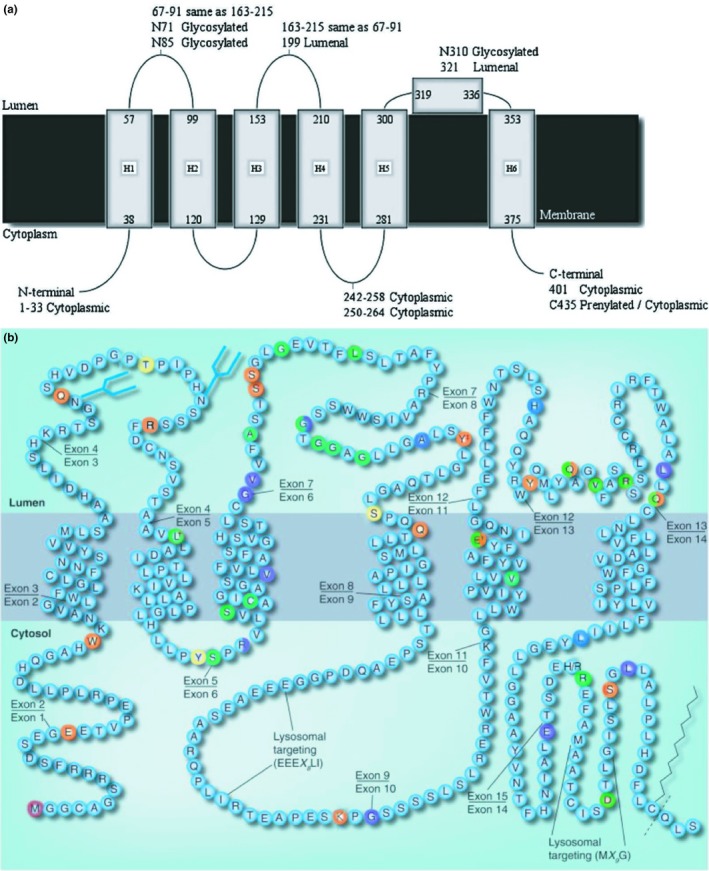
2a and 2b: 2a Schematic model for human CLN3 showing the six transmembrane helices, proposed amphipathic helix and experimentally determined loop locations. 2b The predicted topology of CLN3 is depicted. Sites for post‐translational modifications are shown as blue forked lines for N‐glycosylation, zigzags for prenylation, and dotted lines for potential cleavage sites following prenylation. Disease‐causing mutations are colored in red, orange, yellow, green, blue, and violet. If the mutation covers multiple residues, only the first residue is marked. (NB‐All disease‐causing mutations are not shown. For a completes list see Table 1)
Mutations in CLN3 are classically associated with CLN3 disease where retinal degeneration is followed by mental and physical deterioration and premature death. Recent studies show that CLN3 mutations may also result in a nonsyndromic retinal degeneration whose onset differs considerably from classical Batten disease. CLN3 joins a growing number of genes such as TTC8, BBS2, and USH2A, whose mutations are commonly associated with syndromic diseases that include a subset of patients with isolated vision loss. Some mutations, as in the case of CLN3, can result in either phenotype (see Table 1) (Goyal, Jäger, Robinson, & Vanita, 2016; Rivolta, Sweklo, Berson, & Dryja, 2000; Shevach et al., 2015).
Table 1.
Representation of reported disease‐causing mutations, their associated regions of the DNA (i.e. promoter, coding, noncoding), cDNA change, genomic DNA change, protein change, type of mutation, human phenotype (CLN3 disease or nonsyndromic retinitis pigmentosa). Adapted from https://www.ucl.ac.uk/ncl/CLN3mutationtable.htm
| Amino Acid position | Mutation location | cDNA Change | Genomic DNA change | Protein change | Type of mutation | Phenotype | References |
|---|---|---|---|---|---|---|---|
| Promoter | c.‐1101C>T | g.28504365G>A | p.(=) | Sequence variant | Kousi et al. (2012) | ||
| Promoter | c.‐681_‐676delTGAAGC | g.28503756_28503761delGCTTCA | p.(?) | Sequence variant | Kousi et al. (2012) | ||
| 1 | Exon 1 | c.1A>C | g.28503080T>G | p.? | Missense or aberrant start | CLN3 Disease | Kousi et al. (2012) |
| 17 | Exon 2 | c.49G>T | g.28502879C>A | p.(Glu17*) | Nonsense | Kwon et al. (2005) | |
| 35 | Exon 2 | c.105G>A | g.28502823C>T | p.(Trp35*) | Nonsense | CLN3 Disease | Kousi et al. (2012) |
| Exon 2 | c.125+1G>C | g.28502794C>G | p.? | splice defect | Non‐syndromic retinal disease | Wang et al. (2014) | |
| Intron 2 | c.125+5G>A | g.28502798C>T | p.? | splice defect | CLN3 Disease | Kousi et al. (2012) | |
| Intron 2 | c.126‐1G>A | g.28500708C>T | p.? | splice defect | CLN3 Disease | Mole et al. (2001) | |
| 72 | Exon 3 | c.214C>T | g.28500619G>A | p.(Gln72*) | Nonsense | CLN3 Disease | Kousi et al. (2012) |
| Intron 3 | c.222+2T>G | g.28500609A>C | p.? | Splice site | CLN3 Disease | Kousi et al. (2012) | |
| Intron 3 | c.222+5G>C | g.28500606C>G | p.? | Splice site | CLN3 Disease | Kousi et al. (2012) | |
| 80 | Exon 4 | c.233_234insG | g.28499972_28499973insC | p.(Thr80Asnfs*12) | Frameshift | CLN3 Disease | Kousi et al. (2012) |
| 89 | Exon 4 | c.265C>T | g.28499941G>A | p.(Arg89*) | Nonsense | CLN3 Disease | Pérez‐Poyato et al. (2011) |
| 101 | Exon 5 | c.302T>C | g.28499055A>G | p.(Leu101Pro) | Missense | CLN3 Disease, protracted | Munroe et al. (1997) |
| 124 | Exon 5 | c.370dupT | g.28498987dup | p.(Tyr124Leufs*36) | Frameshift, introducing a premature stop codon | CLN3 Disease | Kousi et al. (2012) |
| Exon 5 intron or Exon 6 | c.375‐3C>G | p.(?) | splice | Non‐syndromic retinal disease | Ku et al. (2017) | ||
| 127 | Exon 6 | c.378_379dupCC | g.28498856_28498857dup | p.(Arg127Profs*55) | 1‐bp deletion and 2bp insertion | CLN3 Disease | Kousi et al. (2012); Munroe et al. (1997) |
| 127 | Exon 6 | c.379delC | g.28498858delG | p.(Arg127Glyfs*54) | 1‐bp deletion | Kousi et al. (2012) | |
| 131 | Exon 6 | c.391A>C | g.28498828T>G | p.(Ser131Arg) | Missense | Non‐syndromic retinal disease | Wang et al. (2014) |
| 134 | Exon 6 | c.400T>C | g.28498837A>G | p.(Cys134Arg) | Missense | CLN3 Disease | Kousi et al. (2012) |
| 142 | Exon 6 | c.424delG | g.28498813delC | p.(Val142Leufs*39) | 1‐bp deletion | CLN3 Disease | Bensaoula et al. (2000), Kousi et al. (2012) and Munroe et al. (1997) |
| maximum deletion of intron 5–15, Minimum deletion of exon 8–15; | c.432+?_1350‐?del | g.28488804_28498805del | 6‐kb deletion | CLN3 Disease | Mitchison, O’Rawe, et al. (1995) | ||
| Intron 6 | c.461‐1G>C | g.28497972C>G | p.? | Splice defect | CLN3 Disease | Kousi et al. (2012) | |
| Intron 6 | c.461‐3C>G | p.(?) | splice | Non‐syndromic retinal disease | Ku et al. (2017) | ||
| 154 | Intron 6 ‐ Intron 8 | c.461‐280_677+382del966 | g.28497286_28498251del | p.[Gly154Alafs*29, Val155_Gly264del] | (966 b deletion) | Batten Disease Consortium (1995), Munroe et al., (1997), Wisniewski, Zhong, et al. (1998), Zhong et al. (1998), Lauronen et al. (1999), Eksandh et al., 2000, Bensaoula et al., 2000, Teixeira et al. (2003), de los Reyes et al. (2004), Leman, Pearce, and Rothberg (2005), Kwon et al. (2005), Moore et al. (2008), Pérez‐Poyato et al. (2011), Kousi et al. (2012) and Ku et al. (2017) | |
| 154 | Intron 6 ‐ Intron 8 | c.461‐280_677+382del966 | g.28497286_28498251del | p.[Gly154Alafs*29, Val155_Gly264del] | (966 b deletion) | Non‐syndromic retinal disease | Ku et al. (2017) |
| 155 | Intron 6 | c.461‐13G>C | g.28497984C>G | p.[=, Val155Profs*2] | Aberrant splicing that removes exon 7 | CLN3 Disease | Munroe et al. (1997) |
| 158 | Exon 7 | c.472G>C | g.28497960C>G | p.(Ala158Pro) | Missense | CLN3 Disease | Kousi et al. (2012) |
| 161 | Exon 7 | c.482C>G | g.28497950G>C | p.(Ser161*) | Nonsense | Munroe et al. (1997) | |
| 162 | Exon 7 | p.(Ser162*) | Nonsense | Munroe et al. (1997) | |||
| 165 | Exon 7 | c.494G>A | p.(Gly165Glu) | Missense | Cortese et al. (2014) | ||
| 170 | Exon 7 | c.509T>C | g.28497923A>G | p.(Leu170Pro) | Missense | CLN3 Disease, protracted | Munroe et al. (1997) |
| Intron 7 | c.533+1G>C | g.28497898C>G | p.? | Splice defect/frameshift | CLN3 Disease, protracted | Batten Disease Consortium (1995), Lauronen et al. (1999) and Munroe et al. (1997) | |
| Intron 7 | c.533+1G>A | g.28497898C>T | p.? | Splice site | CLN3 Disease | Kousi et al. (2012) | |
| 187 | Exon 8 | c.558_559delAG | g.28497786_28497787delCT | p.(Gly187Aspfs*48) | 2‐bp deletion and Missense | CLN3 Disease | Munroe et al. (1997) |
| 187 | Exon 8 | c.560G>C | g.28497785C>G | p.(Gly187Ala) | Missense | CLN3 Disease | Mole et al. (2001) and Kousi et al. (2012) |
| 189 | Exon 8 | c.565G>C | g.28497780C>G | p.(Gly189Arg) | Missense | CLN3 Disease and Non‐syndromic retinal dystrophy | Kousi et al. (2012) and Wang et al. (2014) |
| 189 | Exon 8 | p.(Gly189Arg) | Missense | CLN3 Disease and Non‐syndromic retinal dystrophy | Kousi et al. (2012); Wang et al. (2014) | ||
| 192 | Exon 8 | c.575G>A | g.28497770C>T | p.(Gly192Glu) | Missense | CLN3 Disease | Pérez‐Poyato et al. (2011) |
| Exon 8 | c.582G>T | g.28497763C>A | p.(=) | Sequence variant | CLN3 Disease, protracted | Sarpong et al. (2009) | |
| 196 | Exon 8 | c.586dupG or c.586‐587insG | g.28497759dup | p.(Ala196Glyfs*40) | 1‐bp insertion | CLN3 Disease | Munroe et al. (1997) |
| 199 | Exon 8 | c.597C>A | g.28497748G>T | p.(Tyr199*) | Nonsense | CLN3 Disease, protracted | Sarpong et al. (2009), Kousi et al. (2012) and Mole et al. (2001) |
| 208 | Exon 8 | c.622dupT | g.28497723dup | p.(ser208Phefs*28) | Frameshift, introducing a premature stop codon | Kousi et al. (2012) and Pérez‐Poyato et al. (2011) | |
| 211 | Exon 8 | c.631C>T | g.28497714G>A | p.(Gln211*) | Nonsense | Munroe et al. (1997) | |
| Intron 8 | c.678‐?_1317+?del | g.28488837‐?_28495439+?del | p.? | partly characterised deletion | CLN3 Disease | Kousi et al. (2012) | |
| 262 | Exon 9 | c.784A>T | p.(Lys262*) | Nonsense | CLN3 Disease | Coppieters et al. (2014) | |
| Intron 9 | c.790+3A>C | g.28495324T>G | p.? | Sequence Variant | CLN3 Disease | Kousi et al. (2012) | |
| 264 | Intron 9 ‐ Intron 13 | c. 791_1056del | g.28491981_28494795del2815 | p.(Gly264Valfs*29) | 2.8‐kb deletion | CLN3 Disease, protracted | Batten Disease Consortium (1995), Munroe et al. (1997), Lauronen et al. (1999), Kousi et al. (2012) and Mole et al. (2001) |
| 155 | Exon 10 | c.831G>A | g.28493953C>T | p.[Val155_Gly264del, Gly280_Leu302del] | Sequence variant | CLN3 Disease | Zhong et al. (1998) |
| Exon 10 intron or Exon 11 | c.837+5G>A | p.(?) | splice | CLN3 Disease and Non‐syndromic retinal dystrophy | Ku et al. (2017) | ||
| 285 | Exon 11 | g.28493851T>C | p.(Ile285Val) | missense | CLN3 Disease and Non‐syndromic retinal dystrophy | Carss et al. (2017) and Ku et al. (2017) | |
| 290 | Exon 11 | c.868G>T | p.(Val290Leu) | Missense | Non‐syndromic retinal disease | Wang et al. (2014) | |
| 295 | Exon 11 | c.883G>A | g.28493821C>T | p.Glu295Lys | Missense and Nonsense | CLN3 Disease, protracted | Lauronen et al. (1999), Munroe et al. (1997), Wang et al. (2014) and Zhong et al. (1998) |
| 295 | Exon 11 | c.883G>T | g.28493821C>A | p.(Glu295*) | nonsense | CLN3 Disease and Non‐syndromic retinal dystrophy | Kousi et al. (2012), Ku et al. (2017) and Mole et al. (2001) |
| Intron 11 | c.906+5G>A | g.28493793C>T | splice defect | Splice site | CLN3 Disease | Kousi et al. (2012) | |
| 306 | Exon 12 | c.917T>A | p.(Leu306His) | missense | Non‐syndromic retinal disease | Ku et al. (2017) | |
| Exon 12 | c.944‐945insA | CLN3 Disease | Munroe et al. (1997) | ||||
| 313 | Exon 12 | c.954_962+18del27 | g.28493630_28493656del27 | p.Leu313_Trp321del/ splice defect | Splice defect | CLN3 Disease | Kousi et al. (2012) |
| 315 | Exon 12 | c.944dupA | g.28493666dup | p.(His315Glnfs*67) | 1‐bp insertion | CLN3 Disease | Licchetta et al. (2015) |
| Exon 12 | c.10633‐10660del | Frameshift after Q318, possible aberrant splicing | Mole et al. (2001) | ||||
| Exon12‐Intron12 | c.963‐1G>T | g.28493520C>A | p.? | Splice defect | CLN3 Disease | Kousi et al. (2012) | |
| 322 | c.966C>G | p.(Tyr322*) | Nonsense | Non‐syndromic retinal disease | Wang et al. (2014) | ||
| 330 | Exon 13 | c.988G>A | g.28493494C>T | p.(Val330Phe) | Missense | Non‐syndromic retinal disease | Carss et al. (2017) and Ku et al. (2017) |
| 334 | Exon 13 | c.1000C>T | g.28493482G>A | p.Arg334Cys | Missense | CLN3 Disease | Kousi et al. (2012) and Munroe et al. (1997) |
| 334 | Exon 13 | c.1001G>A | g.28493481C>T | p.Arg334His | Missense | CLN3 Disease, protracted | Batten Disease Consortium (1995), Kousi et al. (2012), Munroe et al. (1997) and Pérez‐Poyato et al. (2011) |
| 349 | Exon 13 | c.1045_1050del | p.(Ala349_Leu350del) | 6‐bp deletion | CLN3 Disease | Licchetta et al. (2015) | |
| 350 | Exon 13 | c.1048delC | g.28493434delG | p.Leu350CysfsX27 | 1‐bp deletion | CLN3 Disease | Kousi et al. (2012) |
| 352 | Exon 13 | c.1054C>T | g.28493428G>A | p.Gln352X | Nonsense | CLN3 Disease | Kousi et al. (2012) and Munroe et al. (1997) |
| Intron 13 | c.1056+3A>C | g.28493423T>G | p.? | Splice site | CLN3 Disease | Kousi et al. (2012) | |
| 379 | Exon 14 | c.1135_1138delCTGT | p.(Leu379Metfs*11) | 4‐bp insertion | CLN3 Disease | Drack, Miller, and Pearce (2013) | |
| 399 | Exon 14 | c.1195G>T | g.28489060C>A | p.(Glu399*) | 27‐bp deletion | CLN3 Disease | Kousi et al. (2012) |
| 400 | Intron 14 | c.1198‐1G>T | g.28488957C>A | p.Thr400* | Splice site | CLN3 Disease | Munroe et al. (1997); Mole et al. (2001) |
| 404 | Exon 15 | c.1211A>G | g.28488943T>C | p.(His404Arg) | Polymorphism | CLN3 Disease | Munroe et al. (1997) and Mole et al. (2001) |
| 405 | Exon 15 | c.1213C>T | g.28488945G>A | p.(Arg405Trp) | Missense | Non‐syndromic retinal disease | Carss et al. (2017), Ku et al. (2017), Wang et al. (2014) and Weisschuh et al. (2016) |
| 416 | Exon 15 | c.1247A>G | g.28488907T>C | p.(Asp416Gly) | Missense | CLN3 Disease | Kousi et al. (2012) |
| 423 | Exon 15 | c.1268C>A | g.28488886G>T | p.(Ser423*) | Nonsense | CLN3 Disease | Kousi et al. (2012) |
| 425 | Exon 15 | c.1272delG | g.28488882delC | p.(Leu425Serfs*87) | 1‐bp deletion | CLN3 Disease | Munroe et al. (1997) |
| g.28497285‐28498251del | NA | large deletion | Non‐syndromic retinal disease | Carss et al. (2017) |
2.4. CLN3 gene regulation
The CLN3 gene was sequenced in 1997 following the discovery of the protein. Sequencing began 1.1 kb upstream of the transcription start site and proceeded to 0.3 kb downstream of the polyadenylation site. Sequencing led to the conclusion that CLN3 is organized into at least 15 exons spanning 15 kb ranging from 47 to 356 bps in length (Mitchison et al., 1997). CLN3 also has 14 introns that vary from 80 to 4,227 bps in length. There are a total of 12 Alu repeats in the forward orientation and 9 in reverse orientation present within the introns and 5′‐ and 3′‐untranslated regions. The 5′ region of the CLN3 gene contains several potential transcription regulatory elements. Although there are two (TA)n repetitive motifs at nt 135 and 335 in the sequence, there is no consensus TATA‐1 box evident, suggesting that CLN3 is constitutively expressed (Mitchison et al., 1997). In addition, using the transcription factor DNA binding site database, putative cis‐acting regulatory elements were found in the 5′ flanking sequence, including potential transcription factor binding sites for AP‐1, AP‐2, and Sp1, two motifs for the erythroid‐specific transcription factor GATA‐1 and three potential CCAAT boxes (Mitchison et al., 1997).
2.4.1. CLN3 promotor and transcription factor binding analysis
Several regulatory elements have been found in the 5′ region of the CLN3 gene; however, the endogenous promoter of CLN3 has not been definitively characterized yet. To this end, Eliason and colleagues created transgenic CLN3 mice by knocking‐in a DNA sequence encoding for nuclear‐targeted bacterial β‐gal. This was achieved via homologous recombination of a targeting construct into embryonic stem cells, such that β‐gal transcription was controlled by a native sequence 5′ to the CLN3 coding region. This resulted in the replacement of most of exon 1 and all of exons 2–8, creating an effective null mutation (Ding, Tecedor, Stein, & Davidson, 2011; Eliason et al., 2007). In these studies, the authors demonstrated that CLN3 is ubiquitously expressed and that a regulatory and functional region at the 5′ of the gene is promoting its expression. Other studies have described a putative promoter region and identified predicted transcription factor binding sites (see Table 2 for complete list). Moreover, a multitude of transcription factors have been reported to regulate CLN3 expression pattern by direct interaction (physical binding) with the “promoter” region or by indirect interaction through protein partners. Indeed, TFEB has been shown to bind to CLEAR elements on the proximal promoter of CLN3, which increases CLN3 transcription (Palmieri et al., 2011; Sardiello et al., 2009). TFEB binding sites were first mapped to −24 (AGCACGTGAT) and +6 (GTCACGTGAT) on the promoter of CLN3 (Sardiello et al., 2009) and physical binding of TFEB was then further experimentally verified by ChIP‐seq (Palmieri et al., 2011). Table 2 reports nonredundant transcription factor interactor of CLN3 that regulate its expression profile. Additional information on each interaction, that is, the type of experimental support and key functional statement, is provided below.
Table 2.
Transcriptional regulators of CLN3
| Putative Transcriptional Regulator | Protein details | Putative effect | Methods used to identify putative transcriptional regulator | References |
|---|---|---|---|---|
| AP‐2A | Putative CLN3 promoter has an AP‐2A binding site. | Putative CLN3 promoter has an AP‐2A binding site. | The authors developed a computational genomics strategy termed ChIPModules which begins with experimentally‐determined binding sites and integrates those with positional weight matrices constructed from transcription factor binding sites, comparative genomics, and statistical learning methods with the purpose of identifying transcriptional regulatory modules. They identify that the putative CLN3 promoter contains an AP‐2alpha binding site | Jin, Rabinovich, Squazzo, Green, and Farnham (2006) |
| E2F1 | E2F1_HUMAN | Putative CLN3 promoter has a putative E2F1 binding site. | Researchers studied the binding of E2F1 to promoters. ChIP analyses of 24,000 promoters confirmed that more than 20% of promoters are bound by E2F1. Including the CLN3 promoter | Bieda, Xiaoqin, Singer, Green, and Farnham (2006) |
| ELAV1 (huR)E2F1 | E2F1_HUMAN | Putative CLN3 promoter has a putative E2F1 binding site. | HuR regulates the stability and translation of numerous mRNAs encoding for stress response and proliferative proteins. HuR was found to bind CLN3 mRNA. The interaction value was higher than the mock controls; albeit very weak. Researchers studied the binding of E2F1 to promoters. ChIP analyses of 24,000 promoters confirmed that more than 20% of promoters are bound by E2F1. Including the CLN3 promoter | Abdelmohsen et al. (2009); Bieda et al. (2006) |
| ETS1ELAV1 (huR) | ETS1 Human | ETS1 binds to the putative CLN3 promoter | The CLN3 promoter contains putative ETS1 binding motifs. HuR regulates the stability and translation of numerous mRNAs encoding for stress responses and proliferative proteins. HuR was found to bind CLN3 mRNA. The interaction value was higher than the mock controls; albeit very weak. | Hollenhorst et al. (2009); Abdelmohsen et al. (2009) |
| ETS1ETS1 | ETS1 MouseETS1 Human | ETS1 binds to the putative CLN3 promoter | CLN3 is downregulated in ETS1‐/‐ mNK cells. The CLN3 promoter contains putative ETS1‐binding motifs. | Ramirez et al. (2012); Hollenhorst et al. (2009) |
| HNF4‐alphaETS1 | HNF4A_HUMANETS1 Mouse | HNF4‐alpha binds to gene CLN3 promoterETS1 | In the referenced paper, CLN3 appears in a list of potential HNF4alpha target genes in differentiated Caco2 colorectal adenocarcinoma cells. CLN3 is downregulated in ETS1‐/‐ mNK cells | Bolotin et al. (2010), Boyd, Bressendorff, Møller, Olsen, and Troelsen (2009) and Ramirez et al. (2012) |
| HSF1HNF4‐alpha | HSF1_HUMANHNF4A_HUMAN | CLN3 harbors an HSF 1 site I at a proximal Alu in an antisense orientationHNF4‐alpha binds to gene CLN3 promoter | CLN3 is downregulated upon heat shock in microarray experiments. In the referenced paper, CLN3 appears in a list of potential HNF4alpha target genes in differentiated Caco2 colorectal adenocarcinoma cells. | Bolotin et al. (2010), Boyd et al. (2009) and Pandey, Mandal, Jha, and Mukerji (2011) |
| TFEB | CLEAR | CLN3 harbors a two CLEAR binding site on its proximal promoter (at −24 nad +6). | TFEB has been shown to bind this (CLEAR) element in the proximal promoter of CLN3 and increase CLN3 transcription. | Palmieri et al. (2011) and Sardiello et al. (2009) |
Additionally, transcription factor binding sites were predicted for the CLN3 promoter using the sequence‐based profiles of known sites. Position‐specific scoring matrices were applied for 202 human transcription factors or factor dimers obtained from the JASPAR database (Portales‐Casamar et al., 2010). The promoter region of CLN3 was obtained from the UCSC Genome Browser using the RefSeq gene boundaries (Pruitt et al., 2014). A 1‐kb region upstream and 500 bases downstream of the transcription start site (TSS) were used for this analysis. The motif search was performed using the MAST tool from the MEME suite (Bailey et al., 2009).
The complete list of predicted transcription factor binding sites is shown in Table 3. Interestingly, the transcription factors SP2, ESRRA, Klf4, and USF2 each have three or more predicted binding sites in the vicinity of the transcription start site of CLN3.
Table 3.
Predicted transcription factor binding sites on the CLN3 gene
| Position Relative to TSS | Transcription factor | p‐value | Length |
|---|---|---|---|
| −329/−123 | E2F3 | 6.9E‐05/5.6E‐07 | 14 |
| −323 | E2F4 | 8.50E‐05 | 10 |
| −734 | EGR2 | 5.90E‐05 | 14 |
| −549 | ELF1 | 3.40E‐06 | 12 |
| −1,396 | ESR1 | 3.50E‐05 | 19 |
| −1,344/−672/−214 | ESRRA | 7.4E‐05/‐9.10E‐05/3.05E‐05 | 10 |
| −919 | FOXA1 | 1.40E‐06 | 14 |
| −915 | Foxa2 | 8.20E‐08 | 11 |
| −131 | FOXC1 | 5.00E‐05 | 7 |
| −1,276 | Foxd3 | 3.20E‐05 | 11 |
| −777 | FOXI1 | 9.80E‐05 | 11 |
| −1,469/−1,271/−807 | FOXP1 | 3.7E‐05/2.2E‐05/3.00E‐05 | 14 |
| −545 | GABPA | 5.50E‐06 | 10 |
| −1,364 | Gata4 | 4.40E‐05 | 10 |
| −603/−407/−388 | Klf4 | 1.4E‐05/3.60E‐05/5.00E‐07 | 9 |
| −1,222 | Meis1 | 9.90E‐05 | 14 |
| −83 | NHLH1 | 7.50E‐05 | 11 |
| −1,349/−677 | NR2F1 | 5.4E‐05/2.00E‐05 | 13 |
| −172 | NRF1 | 9.60E‐05 | 10 |
| −495 | Pax2 | 9.70E‐05 | 7 |
| −1,468 | Pax4 | 2.80E‐05 | 29 |
| −1,493/−1,102 | PAX5 | 2.2E‐06/4.40E‐05 | 18 |
| −1,317 | PBX1 | 4.60E‐05 | 11 |
| −230 | PLAG1 | 1.60E‐06 | 13 |
| −1,285 | POU2F2 | 3.90E‐05 | 12 |
| −1,253 | PRDM1 | 8.20E‐06 | 14 |
| −1,136 | Rfx1 | 7.60E‐05 | 13 |
| −1,167 | RFX5 | 6.30E‐05 | 14 |
| −603/−412/−393/−123 | SP2 | 1.30E‐05/1.90E‐05/6.80E‐05/4.60E‐06 | 14 |
| −715/−305 | Tcfcp2l1 | 8.90E‐05/5.70E‐05 | 13 |
| −1,409/−434 | TFAP2C | 5.70E‐05/1.60E‐05 | 14 |
| −711 | THAP1 | 4.60E‐05 | 8 |
| −308 | TP63 | 2.70E‐05 | 19 |
| −786/−494/−267 | USF2 | 8.00E‐05/7.60E‐06/7.20E‐05 | 10 |
| −242 | ZBTB33 | 8.80E‐05 | 14 |
| −83 | ZEB1 | 4.80E‐05 | 8 |
| −433 | Zfx | 2.60E‐05 | 13 |
Transcription Factors from MetaCore from Clarivate Analytics (Ekins, Nikolsky, Bugrim, Kirillov, & Nikolskaya, 2007).
In addition to regulation by transcription factors, studies have demonstrated that various molecules can indirectly regulate the expression of CLN3. These are listed in Table 4.
Table 4.
Alternative transcriptional regulation
| Alternative Transcription Regulator | Protein details | Methods used to Identify Putative Transcriptional Regulator | PubMed ID | |
|---|---|---|---|---|
| LactoferrinHSF1 | TRFL_HUMAN DeltaLfHSF1_HUMAN | CLN3 harbors an HSF 1 site I at a proximal Alu in an antisense orientation | One form of LF is secreted in body fluids (sLF) whereas an alternative form deltal LF, regulated by a different promoter, is present in normal tissues. Delta LF is downregulated in breast cancer. The CLN3 promoter is downregulated in delta LF expressing HEK cell upon heat shock in microarray experiments. | Kim, Kang, and Kim (2013) and Pandey et al. (2011) |
| MITFLactoferrin | MITF_HUMANTRFL_HUMAN DeltaLf | MITF interacts with the putative CLN3 promoter | Putative CLN3 promoters have been pulled down by gene‐wide chromatin immunoprecipitation of MITF. One form of LF is secreted in body fluids (sLF) whereas an alternative form deltal LF, regulated by a different promoter, is present in normal tissues. Delta LF is downregulated in breast cancer. CLN3 is downregulated in delta LF expressing cell's.promoter. | Bertolotto et al. (2011) and Kim et al. (2013) |
| N‐MyristoylationN‐Myc | N‐myristoyltransferase_(HUMAN)MYCN_MOUSE | N‐Myc regulates transcription of CLN3 | Using SILAC, researchers found that CLN3 is N‐myristoylated. The authors studied changes in gene expression in embryonic stem cells after the induction of various transcription factors. CLN3 was included in the analyses. | Chen et al. (2008) and Nishiyama et al. (2009) |
2.5. Description of the CLN3 protein
The CLN3 gene encodes a highly hydrophobic protein of 438‐amino acids, the three‐dimensional structure of which has not yet been proven through X‐ray crystallography or Nuclear Magnetic Resonance‐spectroscopy. The secondary structure of CLN3 is mainly comprised of transmembrane and low complexity cytosolic or luminal spans (Berman et al., 2000). Computer prediction models suggest CLN3 spans the membrane anywhere from five to ten times (SPEP + Ensemble 1.0, MEMSAT3 + Swissprot, MEMSAT3, TMHMM 2.0, PHOBIUS Constrained, PROFPHD, HMMTOP). However, some experimental studies support a structure where the N‐terminus faces the intraluminal space of organelles with the C‐terminus facing the cytoplasm, the most widely cited peer‐reviewed articles favor a 6‐membrane spanning domain (MSD) model with both the N‐ and C‐termini facing the cytosol. See Figure 2 table below for more detailed information (Ezaki et al., 2003; Mao, Foster, Xia, & Davidson, 2003; Mao, Xia, & Davidson, 2003; Ratajczak, Petcherski, Ramos‐Moreno, & Ruonala, 2014).
Table 5.
Locations of experimentally determined regions/residues
| Region/residue | Location | References |
|---|---|---|
| N‐terminal | Cytoplasmic | Ezaki et al. (2003) and Ratajczak et al. (2014) |
| 1–33 | Cytoplasmic | |
| 2–18 | Lumenal | Mao, Foster, et al. (2003) and Mao, Xia, et al. (2003) |
| N71 | Lumenal | |
| N85 | Lumenal | Storch et al. (2007) |
| *97–121 | MSD | [SPEP + Ensemble 1.0, MEMSAT3, + Swissprot, MEMSAT3 + CLN3, TMHMM 2.0, PHOBIUS Constrained, PROFPHD, HMMTOP] |
| 199 | Lumenal | Mao, Foster, et al. (2003) |
| 210–231 | MSD | [SPEP + Ensemble 1.0, MEMSAT3, + Swissprot, MEMSAT3 + CLN3, TMHMM 2.0, PHOBIUS Constrained, PROFPHD, HMMTOP] |
| 250–264 | Cytoplasmic | Mao, Foster, et al. (2003) and Mao, Xia, et al. (2003) |
| 242–258 | Cytoplasmic | Kyttälä et al. (2004) |
| *276–303 | MSD | [SPEP + Ensemble 1.0, MEMSAT3, + Swissprot, MEMSAT3 + CLN3, TMHMM 2.0, PHOBIUS Constrained, PROFPHD, HMMTOP] |
| N310 | Lumenal | Mao, Foster, et al. (2003) and Storch et al. (2007) |
| 321 | Lumenal | Mao, Foster, et al. (2003) |
| S401 | Cytoplasmic | Kyttälä et al., (2004) and Ratajczak et al. (2014) |
| 406–433 | Cytopasmic | Nugent et al. (2008) |
| Cys435 | Cytoplasmic | Storch et al. (2007) |
| C‐terminal | Cytoplasmic | Mao, Xia, et al. (2003) and Ratajczak et al. (2014) |
Computer Modeling was included for MSDs for which all prediction models support the same conclusion.
2.6. CLN3 protein details
2.6.1. Consensus sequence elements similar to other proteins
The CLN3 protein sequence does not display significant similarities to any protein of known function (Muzaffar & Pearce, 2008). However, some consensus sequence elements within the CLN3 protein allude to its localization, regulation, and function. Early predictions of CLN3 using Position‐Specific Iterative Basic Local Alignment Search Tool (PSI‐BLAST) revealed a distant but significant sequence similarity between CLN3 and members of the SLC29 family of equilibrative nucleoside transporters, of which four members are recognized in mammals (Altschul et al., 1997; Baldwin et al., 2004). More recent algorithms, such as the Structural Classification of Proteins (Andreeva et al., 2008) and Protein families (Pfam) suggest that most of the CLN3 protein (aa 11–433) has a domain structure consistent with members of the major facilitator superfamily (MFS; SCOP superfamily 103473; Pfam clan CL0015). The MFS superfamily is one of the two largest families of membrane transporters and includes small‐solute uniporters, symporters and antiporters (Marger & Saier, 1993). Structural similarities between CLN3 and MFS family member MFSD8, may provide important clues as to its function. Indeed, mutations in MFSD8, which encodes a lysosomal protein with 12‐predicted transmembrane domains and unknown function, result in histological and phenotypical similarities to another form of Batten disease, CLN7, (Siintola et al., 2007). Moreover, sequence alignment and Markov modeling predicted the N‐terminus of CLN3 to be weakly homologous to fatty acid desaturases. Using nervous system and pancreatic tissue samples from a murine homozygous‐knockout model of CLN3, investigators demonstrated that Δ9 desaturase activity was greatly reduced, while heterozygous carriers displayed intermediate desaturase levels (40%) compared to wild‐type animals. Therefore, the loss of CLN3 appears to result in decreased desaturase activity on palmitoyl (C16) moieties of protein substrates (Narayan, Rakheja, Tan, Pastor, & Bennett, 2006; Narayan, Tan, & Bennett, 2008).
Sequence analysis also indicated a multitude of putative trafficking and sorting signals, suggesting CLN3 may populate a variety of organelles. A mitochondrial targeting signal was identified at residue 11 with a cleavage site at residue 19 (Janes et al., 1996). Furthermore, targeting studies demonstrated the existence of two lysosomal sorting signals; (a) a conventional dileucine motif preceded by an acidic patch located in a putative cytosolic loop of the favored 6‐transmembrane structure (Kyttälä et al., 2004) at 242EEE(X)8LI254 and (b) an unconventional motif in the long C‐terminal cytosolic tail consisting of methionine and glycine separated by nine amino acids [M(X)9G] (Kyttälä et al., 2005, 2004; Järvelä et al., 1998; Kida et al., 1999; Storch, Pohl, & Braulke, 2004). Interestingly, green fluorescent protein (GFP)‐tagged CLN3 with a double mutation in the dileucine motif (Leu425Leu426), a putative lysosomal targeting motif, to glycine (Gly425Gly426) still co‐localized with lysosomal associated membrane protein‐1 (LAMP1) in chinese hamster ovary (CHO) cells, suggesting that the dileucine motif is not required for the targeting of CLN3 to the lysosome. Since the dileucine motif is conserved among species, these results suggest that CLN3 contains additional lysosomal targeting sequences or different lysosomal targeting signals altogether (Kida et al., 1999). In contrast truncations of CLN3: GFP‐CLN3(1–322), GFP‐CLN3(138–438), and CLN3(1–138)‐GFP do not localize to lysosomes (Kida et al., 1999) indicating that the missing regions either contain imperative lysosome targeting signals or their absence alters the 3D structure of the protein.
Initial studies suggested that yeast CLN3 homolog Btn1 in S. cerevisiae and S. pombe localizes to yeast vacuoles (Croopnick, Choi, & Mueller, 1998; Gachet et al., 2005; Pearce, Ferea, Nosel, Das, & Sherman, 1999; Wolfe, Padilla‐Lopez, Vitiello, & Pearce, 2011). However, more recent studies suggest that experimental tags may have mislocalized the protein. Indeed, when not tagged at its C‐terminus, Btn1 localizes to the Golgi apparatus (Codlin & Mole, 2009; Dobzinski, Chuartzman, Kama, Schuldiner, & Gerst, 2015; Kama, Kanneganti, Ungermann, & Gerst, 2011; Vitiello, Benedict, Padilla‐Lopez, & Pearce, 2010).
2.6.2. Post‐translational modifications of the CLN3 protein
CLN3 contains several putative post‐translational modification (PTM) motifs which contribute to the targeting and anchoring of CLN3 to distinct biological membranes (Casey, 1995). These motifs include four putative N‐glycosylation sites, two putative O‐glycosylation sites, and consensus sequences for phosphorylation, myristoylation, and farnesylation (Ellgaard & Helenius, 2003; Golabek et al., 1999; Haskell, Carr, Pearce, Bennett, & Davidson, 2000; Järvelä et al., 1998; Kaczmarski et al., 1999; Kida et al., 1999; Mao, Xia, et al., 2003; Michalewski et al., 1998, 1999; Nugent et al., 2008; Pullarkat & Morris, 1997; Sigrist et al., 2013; Storch, Pohl, Quitsch, Falley, & Braulke, 2007; Taschner, de Vos, & Breuning, 1997b).
Glycosylation
Alignment and comparison of the CLN3 amino acid sequences across species (human, canine, murine, and yeast CLN3; Genbank Accession number U32680, L76281.1, U68064, AF058447.1) revealed a number of highly conserved N‐X‐S/T motifs, indicating conservation of putative glycosylation sites. In vitro translation of CLN3 produced a singlet at 43 kilodaltons (kDa) in the absence of microsomal membranes and a doublet at 43 and 45 kDa in the presence of microsomal membranes (using rabbit antibody 385/CLN3 raised against residues 242–258 [EEEAESAARQPLIRTEA], which map to the long cytosolic loop according to the most cited prediction model). Similarly, intracellular synthesis and maturation of CLN3 in COS‐1 and HeLa cells also identified the 43 kDa nonglycosylated and a 45 kDa glycosylated forms of CLN3. In detail, pulse‐chase of transfected COS‐1 cells followed by immunoprecipitation showed a single band of 43 kDa 1 hr following the pulse, while further chase up to 6 hr revealed a characteristic doublet of 43 and 45 kDa. Human N‐glycosylated CLN3 protein is sensitive to endoglycosidase H suggesting a high‐mannose type glycosylation (Järvelä et al., 1998). In contrast, murine CLN3 contains complex‐type N‐linked sugars that differ from the human CLN3 (Ezaki et al., 2003). Moreover, mass spectrometric analyses revealed that CLN3 exhibits tissue‐dependent glycosylation patterns (Ezaki et al., 2003). Thus, the apparent molecular weight of glycosylated CLN3 protein may vary depending on cell type and species (Golabek et al., 1999).
Expression of GFP‐CLN3 fusion protein resulted in a 66 and a 100 kDa bands in neuroblastoma and CHO cells, whereas in COS and HeLa cells only the 66 kDa band is detectable. Expression of GFP alone resulted in a 27kDa band, indicating that CLN3 alone would result in ~39 and ~73 kDa bands, respectively. Pulse‐chase experiments revealed that the 66 kDa form appears first, followed by the 100 kDa band. Both the 66 and 100 kDa forms are digested by complex oligosaccharide amidase Peptide ‐N‐Glycosidase F down to 64 kDa. Whereas glycosidase Endoglycosidase H only digests the 66 kDa form. Thus, the 100 kDa form is a complex oligosaccharide in some cell types (Golabek et al., 1999).
N‐linked glycosylation of integral membrane proteins in the ER and in the early secretory pathways, has been shown to be important for protein folding, oligomerization, quality control, sorting and function (Ellgaard & Helenius, 2003). Human CLN3 possesses four potential N‐glycosylation sites (N49, N71, N85, and N310). Glycosylation of N49 is physically unlikely because this residue is located in the first membrane domain. Mutational analyses demonstrated that N71 and N85, located in the first luminal domain, are N‐linked glycosylated (Storch et al., 2007). It remains unclear whether N310, in the third luminal domain, is N‐linked glycosylated (Mao, Foster, et al., 2003; Storch et al., 2007). N‐glycosylation is not required for the proper trafficking of CLN3, as neither treatment with the N‐glycosylation inhibitor tunicamycin, nor single or double substitution of N71 and N85 affected the stability or the trafficking of CLN3 to lysosomes (Golabek et al., 1999; Kida et al., 1999; Storch et al., 2007).
CLN3 also possesses two putative O‐glycosylation sites at T80 and T256 (Consortium 1995). However, O‐glycosylation sites are poorly defined, not necessarily utilized, and T256 is predicted to be cytoplasmic, which is not compatible with glycosylation.
Phosphorylation
Sequence analyses using the ScanPROSITE tool (Sigrist et al., 2013) suggest that CLN3 contains nine putative phosphorylation sites: six on cytoplasmic loops (Ser12, Ser14, Thr19, Thr232, Ser270, Thr400) and three on luminal loops (Ser69, Ser74, Ser86) (Nugent et al., 2008). Previous computer‐based predictions identified 10 serine and 3 threonine residues that may undergo phosphorylation (Michalewski et al., 1998). GFP‐CLN3 expressed in CHO cells incorporates 32P in both the 66 and 100 kDa forms, when incubated with cAMP‐dependent protein kinase (PKA), cGMP‐dependent protein kinase (PKG) or casein kinase II. The reaction was reversed by alkaline phosphatase, indicating that GFP‐CLN3 is indeed phosphorylated (Michalewski et al., 1998, 1999) by PKA, PKG, and casein kinase II and can be enhanced by inhibition of protein phosphatase 1 or protein phosphatase 2A. However, as these studies relied solely on in vitro assays using kinase activators or phosphatase inhibitors, future studies based on protein knockdown in cellular systems will be useful to better assess the specificity and the biological role of each of these proteins in the regulation of CLN3. However, phosphorylation is important for multiple physiological functions such as membrane targeting, protein–protein interactions and the formation of functional complexes, the phosphorylation states and significance of CLN3 phosphorylation remain elusive. The generation of CLN3 phosphomutants would have merit for fully elucidating the biological relevance of these PTMs.
Myristoylation
A putative N‐myristoylation site exists at 2GGCAGS7 in human (Genbank Accession number U32680), canine (Genbank accession number L76281.1) and murine (Genbank Accession number U68064) CLN3. The significance of this lipid modification to CLN3 has not been explored experimentally. However, covalent attachment of myristoyl group by an amide bond to an alpha‐amino group of a N‐terminal glycine has been implicated in protein‐protein and protein‐lipid interactions, membrane targeting, and numerous signal transduction steps. Conservation of N‐myristoylation motifs in human, dog, and mouse as well as isoprenylation motifs in human, dog, mouse and yeast suggest that CLN3 could be a membrane‐attached protein despite the lack of a signaling peptide (Taschner, de Vos, & Breuning, 1997a).
Prenylation/Farnesylation
Prenylation refers to the addition of hydrophobic molecules to a substrate, and involves the transfer of either farnesyl or geranyl‐geranyl moiety to C‐terminal cysteine(s) of the target protein. It is believed that prenyl group modifications facilitate attachment to cell membranes, similar to lipid anchors. Farnesylation is a type of prenylation, where an isoprenyl group is added to a cysteine residue. These modifications are important for protein–protein and protein–membrane interactions. Sequence analyses of CLN3 predict a CAAX motif 435CQLS438 at the C‐terminus that can be prenylated (Taschner et al., 1997a). Coupled translation/prenylation reactions of CLN3 and tetra‐peptides in vitro demonstrate that the CQLS sequence acts as a good acceptor for a farnesylation group (Kaczmarski et al., 1999; Pullarkat & Morris, 1997). Furthermore, glutathione S transferase (GST)‐fusion CLN3 protein, and CLN3 synthesized in a cell‐free environment act as prenylation substrates. Prenylation of GST‐CLN3T greatly enhances its association with membranes. Since prenylation occurs at protein termini, this modification at the C‐terminus of CLN3 may create an additional, terminal loop, which contradicts the assumption that the C‐terminus is free‐floating in the cytosol (Kaczmarski et al., 1999). Substitution of C435 by C435S does not affect CLN3 exit from the endoplasmic reticulum (ER) or transport to lysosomes in COS7 cells but trafficking rate and sorting efficiency are affected (Storch et al., 2007). Incubation with increasing concentrations of farnesyltransferase inhibitor L‐744,832 prevented prenylation of CLN3, which resulted in an increase in the fraction of CLN3 at the plasma membrane, suggesting that C‐terminal lipid modification of CLN3 is important for proper sorting (Storch et al., 2007). It is important to note that while sequence similarities and short‐term in vitro experiments are helpful, no experimental data exists demonstrating the function of PTMs of CLN3 protein in vivo.
2.7. Biosynthesis, trafficking, and intracellular localization of CLN3
In summary, CLN3 contains a farnesylation site at residues that are presumed to anchor the protein to intracellular or plasma membranes. However, mutagenesis of the putative farnesylation motif did not alter lysosomal localization of untagged, overexpressed CLN3. Thus, predicted farnesylation of CLN3 is not required for its lysosomal localization (Haskell et al., 2000; Pullarkat & Morris, 1997) but may have other, yet unidentified, roles.
2.7.1. Biosynthesis, trafficking, and intracellular localization of wild‐type CLN3
Due to its low expression, hydrophobic nature, and lack of suitable antibodies capable of detecting endogenous CLN3, examination of the biosynthesis, PTMs, intracellular trafficking and localization of CLN3 were performed via overexpression in COS‐1, HeLa, baby hamster kidney (BHK), and normal rat kidney epithelia (NRK) cell lines. Most of the results are based on antibodies raised against the N‐terminal domain (h345, aa4‐19) or the large second cytosolic loop of human CLN3 (h385, aa242‐258, Q438, aa251‐265) (Haskell et al., 2000; Järvelä, Lehtovirta, Tikkanen, Kyttälä, & Jalanko, 1999).
Pulse‐chase experiments in transfected COS‐1 cells indicated that CLN3 is synthesized as an N‐glycosylated single‐chain polypeptide and is localized to the lysosomal compartment (Järvelä et al., 1998). Moreover, double immunoflourescence analyses in CLN3 overexpressing HeLa cells revealed strong co‐localization of CLN3 with the lysosomal marker protein Lamp1 (Kyttälä et al., 2004). This study likewise revealed a weak co‐localization of CLN3 with markers of the ER and early endosomes (early endosomal antigen 1; EEA1), whereas no colocalization was detected with the 300 kDa mannose 6 phosphate (M6P) markers of the trans‐Golgi/late endosome network, plasma membrane or mitochondria. Colocalization of CLN3 with lysosomal marker protein Lamp‐1 was also confirmed in transiently transfected BHK cells (Järvelä et al., 1999) and in neuronal cells. In transfected primary hippocampal neurons and glia cells CLN3 co‐localized mainly with the lysosomal marker Lamp‐1 and occasionally with EEA1 (Kyttälä et al., 2004). In transfected mouse primary telencephalic neurons, the distribution of CLN3 overlapped with lysosomal markers and synaptic vesicle marker SV2 (Järvelä et al., 1999). Indeed, the vast majority of studies on CLN3 have been in relation to the lysosome or have assumed that the primary function of CLN3 is lysosomal. However, it is important to note that CLN3 is present in multiple compartments of the cell, the significance of which is unknown. For instance, CLN3 was also detected at the Golgi/trans‐Golgi network and in more peripheral transport vesicles. Some particles were also detected at the plasma membrane (Kyttälä et al., 2004).
To rule out mislocalization of CLN3 due to high overexpression, lysosomal localization of CLN3 was confirmed by double immunoflourescence microscopy in NRK cells stably expressing low levels of CLN3 in an inducible manner (Girotti & Banting, 1996; Kyttälä et al., 2004; Luiro et al., 2004; Reaves & Banting, 1994). Moreover, cryoimmunoelectron microscopy of NRK cells stably transfected with untagged CLN3 demonstrated co‐localization of CLN3 with cathepsin D and LIMPII in lysosomal structures.
Lysosomal sorting motifs of CLN3
Traditionally, M6P‐tagged lysosomal enzymes are transported to late endosomes via vesicular transport. To test whether CLN3 is transported by the same mechanism as lysosomal enzymes, investigators expressed GFP‐CLN3 in CHO cells in presence of I‐M6P. No radioactive signal was incorporated into GFP‐CLN3 suggesting that CLN3 is directed to the lysosomal membrane by an alternative mechanism (Michalewski et al., 1999). Indeed, lysosomal targeting of membrane proteins is mediated by short linear sequences located in their cytosolic domains (Braulke & Bonifacino, 2009). These include tyrosine and acidic cluster dileucine‐based lysosomal sorting motifs which fit the consensus sequences (YXXO) and (D/E)XXXL(L/I), respectively, where X can be any amino acid and O is an amino acid with a large hydrophobic side chain (Bonifacino & Traub, 2003). These sorting motifs interact with cytosolic heterotetrameric adapter proteins AP1‐5 which mediate the packaging of transmembrane cargo into vesicles (Robinson, 2015). Amino acid sequence analysis of the carboxy‐terminal region of CLN3 suggests that the C‐terminal contains one or more tyrosine‐binding motifs (370–374, 378–382, 387–391) which are linked to cytoplasmic adapter complexes involved in sorting of integral membrane proteins to lysosomes (Höning, Sandoval, & von Figura, 1998; Ohno et al., 1998). CLN3 also possess a novel dominant dileucine‐based sorting signal in the predicted second cytoplasmic loop, EEE(X)8LI, and an unconventional M(X)9G sorting motif in the C‐terminal tail (Kyttälä et al., 2004; Storch et al., 2007). These complex sorting motifs are required for the transport of CLN3 to lysosomes. Using binding assays and immunoflourescence techniques, researchers determined that the dileucine motif binds both AP‐1 and AP‐3 in vitro and both adaptor complexes are required for sequential sorting of CLN3 protein (Kyttälä et al., 2005) (Figure 3).
Figure 3.
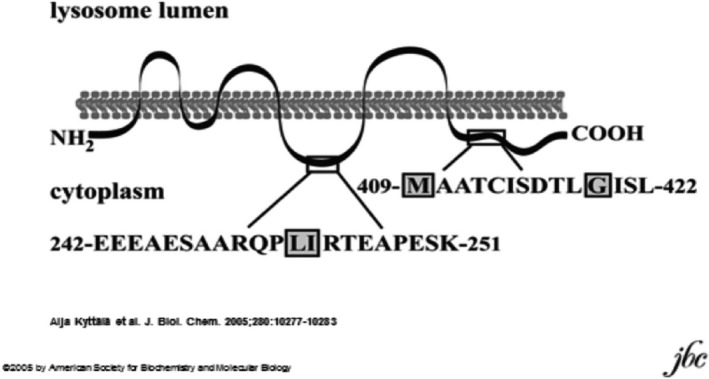
Lysosomal targeting motifs of CLN3
Localization of endogenous CLN3 protein
Ezaki and coworkers demonstrated by subcellular fractionation of mouse livers that endogenous CLN3 is present in lysosomal fractions positive for the lysosomal markers cathepsin B, cathepsin D, Lamp1, Lamp2, and Limp2 (Ezaki et al., 2003). No codistribution of CLN3 with the mitochondrial marker subunit IV of cytochrome oxidase or the Golgi apparatus marker G58K was found in mouse liver. Immunohistochemistry of rat liver showed partial colocalization of endogenous CLN3 with the lysosomal marker acid phosphatase and the late endosomal marker lysobisphosphatidic acid. However, CLN3 did not overlap with EEA1, the cis‐Golgi marker protein GM 130, or the ER marker protein disulfide isomerase (PDI, Ezaki et al., 2003). Lysosomal localization of endogenous CLN3 was also demonstrate in human tissue by a proteomic approach using purified membranes of placental lysosomes (Schröder, Elsässer, Schmidt, & Hasilik, 2007).
Localization of CLN3 mutant protein
More than 60 different mutations in the CLN3 gene have been identified in patients with CLN3 disease (see Table 1 and http://www.ucl.ac.uk/ncl/cln3.shtml). The most common genomic deletion (of 966 bp) results in the biosynthesis of a truncated CLN3 polypeptide composed of 153 canonical amino acids followed by 28 novel amino acids (Batten Disease Consortium, 1995). Based on experimentally determined membrane topology of the Ruonala group the mutant, truncated CLN3 protein is composed of the first two transmembrane domains and a large, C‐terminal cytosolic domain (Ratajczak et al., 2014). Based on an alternative schematic model of Nugent and coworkers the mutant, truncated CLN3 is composed of the first three transmembrane domains followed by 28 novel amino acids located on the luminal side of the membrane (Nugent et al., 2008). In both cases, mutant CLN3 lacks three or four transmembrane domains, the cytosolic loop, and the C‐terminal domain containing the lysosomal sorting motifs and the C‐terminal CQLS farnesylation site (Kyttälä et al., 2004; Storch et al., 2007).
Pulse‐chase analyses of truncated CLN3 expressed in BHK cells revealed a 24 kDa polypeptide which was not further processed in a 6 hr chase period (Järvelä et al., 1999). Double immunoflourescence analyses of truncated CLN3 expressed in BHK cells revealed major co‐localization of CLN3 with the ER marker PDI indicating its retention in the ER (Järvelä et al., 1999). In line with these findings, substitution of the entire C‐terminal domain of CLN3 with cytoplasmic tails of M6P receptors led to retention of chimeric proteins in the ER indicating the importance of the CLN3 C‐terminal for proper ER exit (Storch et al., 2007).
It has long been a matter of debate whether patients that are homozygous for the 966bp deletion in CLN3, produce a biologically active mutant protein. Studies conducted by investigators at the University College London comparing healthy, and affected patient fibroblasts in the presence of RNA interference, support the presence of mutant CLN3 transcripts and postulate the presence of CLN3 protein activity (Kitzmüller et al., 2008). In contrast, researchers at Sanford Children's Health Research Center found a substantial decrease in the transcript level of truncated CLN3 in patient fibroblasts, together with the analysis of transcripts expressed in the Cln3Dex1‐6 mouse and in silico prediction of the expected consequences of truncated protein, support the argument that nonsense‐mediated decay ensures that no functional (mutant) protein is made (Chan, Mitchison, & Pearce, 2008; Miller, Chan, & Pearce, 2013). Moreover, researchers at Massachusetts General Hospital provide further evidence that support the role of nonsense‐mediated decay in the regulation of mutant CLN3 protein. However, Northern blot analyses of liver, kidney and brains of wild‐type and homozygous mutant Cln3Dex7/8 knock‐in mice revealed decreased yet stable levels of mutant RNA consistent with the presence of CLN3 mRNA in patient tissue (Cotman et al., 2002 ; "Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium," 1995 ) . Unfortunately, due to challenges associated with the ability of current reagents (antibodies) to detect endogenous CLN3 protein, no consistent results regarding intracellular localization or quantification of the protein have been possible for either wild‐type, mutant, or variant forms of CLN3 protein.
Expression and localization studies of disease‐associated, CLN3 missense mutations suggest that reduced or complete loss of CLN3 function results in decreased protein half‐life rather than a mislocalization. These studies also indicated that in CLN3 disease, caused by missense mutations, it is the loss of protein and not mislocalization that contributes to pathogenesis. In BHK cells expressing CLN3 E295K, the mutant CLN3 protein co‐localized with Lamp‐1 indicating correct lysosomal localization (Järvelä et al., 1999). Moreover, in transiently transfected human epithelial lung carcinoma cells (A‐549) mutant CLN3 with patient‐derived missense mutations V330F, R334H, L101P, L170P, and E295K colocalized with the lysosomal marker Lamp‐1, further supporting proper lysosomal localization with these mutations (Haskell et al., 2000). Conversely, expression of CLN3 carrying nonsense and frameshift mutations led to a retention of the protein in the ER. In HeLa cells transiently expressing p. Glu399X or p. CLN3 fsG424, mutant CLN3 colocalized with the ER protein PDI in immunoflourescence analyses indicating retention in the ER (Kyttälä et al., 2004). No experimental evidence exists on the consequences of other nonsense mutations (p.Trp35X, p.Glu17X, p.Glu72X, p.Arg89X, p.Ser161X, p.Ser162X, p.Tyr199X, p.Gln211X, p.Lys262X, p.Glu395X, p.Tyr322X, p.Gln327X, p.Gln352X, p.Thr400X, p.Ser423X) or frameshift mutations (p.Thr80Asn fsX12, p.Tyr124Leu fsX36, p.Arg127Pro fsx55, p.Arg127Gly fsX54, p.Gly154Ala fs29, p.Val142Leu fsX39, p.Gly187Asp fsX48, p.Gly190Glu fsX65, p.Ala196Gly fsX40, p.Ser208Phe fsX28, p.Gly264Val fsX29, p.His315Gln fsX67, p.Leu350Cys fsX27, p.Leu379Met fsX11, p.Leu425Ser fsX87) identified in CLN3 patients. For a visual representation of disease‐causing mutations, see Figure 2. For more and continually updated information regarding disease‐causing mutations please see http://www.ucl.ac.uk/ncl/cln3.shtml.
Transient expression of the 966bp deletion, a common JNCL mutation and Q295K, a missense mutation predicted to be in the 5th transmembrane of the 6‐transmembrane model described by Nugent et al (Nugent et al., 2008) demonstrated that CLN3 protein with the common mutation is retained in the ER, whereas, Q295K mutants localize to the expected lysosomal compartment (Järvelä et al., 1999). Q295K is associated with an atypical presentation of juvenile Batten disease. Visual failure initiates and proceeds similar to children with the common deletion. However, normal MRI results have been reported for 2 decades longer than in patients with the common deletion (Järvelä et al., 1997; Wisniewski, Connell, et al., 1998).
NB: There is an error in Jarvela et al 1999 reading the amino acid code. The missense mutation is a change of glutamic acid (not glutamine) to lysine (i.e. E295K, not Q295K)
The 461–677 common deletion mutant localizes to the cell soma whereas wild‐type and Q295K co‐localize to the cell soma and neurites. The authors further report CLN3 co‐localizes with synaptic vesicle marker SV2 (antibody developed by Kathleen Buckley Harvard Medical School, Boston MA). Localization of wild‐type CLN3 protein to synaptic vesicles has not been confirmed by another laboratory. In 2001, Luiro and colleagues reported, using the same polyclonal antibody raised against amino acid residues (242–258, EEEAESAARQPLIRTEA), that CLN3 protein targets to synaptic fractions but not synaptic vesicles (Järvelä et al., 1998; Luiro, Kopra, Lehtovirta, & Jalanko, 2001). Human retinal cells transfected with CLN3 and immunostained with an antibody raised against the CLN3 peptide 242–258 showed a beads‐on‐a‐string pattern in neurites, partial co‐localization with SV2, and no co‐localization with LAMP1 (Luiro et al., 2001).
Localization of epitope‐tagged CLN3 protein
Golabek and colleagues reported results obtained from expressing full‐length CLN3 fused with GFP in COS‐1, HeLa, and human neuroblastoma (SK‐N‐SH) cell lines. Using western blotting, Percoll density gradient fractionation, and Triton X‐114 extraction, the authors demonstrated that the product of the CLN3 gene is a highly glycosylated protein found within membrane‐enriched fractions (Golabek et al., 1999). [The authors state that the results of their experiments indicate that CLN3 protein is lysosomal. However, the fractionation methods used do not separate subcellular and plasma membranes from one another, therefore making it impossible to tease out the precise localization of CLN3].
Kida and colleagues expressed full‐length and truncated CLN3 fused to GFP at its N‐terminus (GFP‐CLN3) in CHO and SK‐N‐SH cell lines (Kida et al., 1999). Using co‐immunoflourescence analyses the authors showed that full‐length GFP‐CLN3 fusion protein colocalizes with lysosomal markers Lamp‐1 and Lamp‐2 and with the late endosomal marker Rab7. GFP‐CLN3 was found in the ER, in a few vesicular structures of the Golgi apparatus, and in COPI‐coated vesicles, most likely due to the presence of newly synthesized CLN3 trafficking from the ER to the Golgi apparatus. GFP‐CLN3 did not colocalize with markers of mitochondria or plasma membrane. In contrast, truncated GFP‐CLN3 that lacked either the C‐terminal domain (GFP‐CLN3 aa 1–322 and GFP‐CLN3 aa 1–138) or the N‐terminal domain (GFP‐CLN3 aa138–438) did not codistribute with lysosomal markers thus, indicating their mislocalization. Most of the truncated fusion proteins either localized to the cytoplasm, nucleus, or ER similar to GFP alone. However, mutant CLN3, with double‐point mutations Leu425Leu426 into Gly425Gly426, at its putative dileucine motif, localized to lysosomes, in a similar fashion to wild‐type, full‐length GFP‐CLN3 (Kida et al., 1999). Studies in CHO cells stably expressing GFP‐CLN3 in the presence of the pharmacological N‐glycosylation inhibitor tunicamycin suggest that N‐glycosylation is not required for correct targeting of CLN3 to lysosomes. However, treatment with Monensin, a Na+ ionophore which blocks glycoprotein secretion, produced retention of GFP‐CLN3 in vesicular structures of the Golgi apparatus in the perinuclear space, suggesting that CLN3 fusion protein is transported to the lysosomal compartments through the trans‐Golgi cisternae (Kida et al., 1999).
In contrast to the data described above, Haskell and coworkers showed a nonvesicular distribution of N‐terminal tagged GFP‐CLN3 which overlapped with the Golgi marker beta‐COP in transfected A549 cells, indicating its localization to the Golgi apparatus (Haskell, Derksen, & Davidson, 1999). Haskell et al also found no colocalization of GFP‐CLN3 with lysosomal marker Lamp‐1 or the mitochondrial marker mtHSP60. When disrupted in the presence of brefeldin A, ER‐like staining was noted. The authors postulated that, if wild‐type CLN3 protein localizes to lysosomes and mitochondria under normal conditions, their N‐terminal tag disrupts such localization.
CLN3 fused to GFP at its C terminus (CLN3‐GFP) mainly colocalized with Golgi markers as determined by immunofluorescence analysis (Kremmidiotis et al., 1999). In transiently transfected fibroblasts, HeLa and COS‐7 cells and stably transfected HeLa cells CLN3‐GFP fluorescence codistributed with wheat germ agglutinin coupled to Texas red. Stable expression of CLN3‐GFP in HeLa cells showed perinuclear, asymmetric localization with the Golgi apparatus, minor localization to the ER and lysosomes, and no apparent localization to the nucleus, mitochondria, or cell surface membrane. A juxtanuclear, asymmetric Golgi‐like localization pattern was also observed in transiently transfected HeLa, COS‐7, and fibroblast cells (Kremmidiotis et al., 1999).
2.7.2. Overexpressed, mutant CLN3 protein
More than 80% of the GFP–CLN3 fusion protein can be extracted by phase separation in a solution of Triton X‐114 indicating that CLN3 is a highly hydrophobic membrane protein (Michalewski et al., 1999).
Using immunoelectron microscopy, investigators analyzed the intracellular processing and localization of two CLN3 protein mutants, 461–677 deletion, or the “1 kb” deletion present in 85% of CLN3 alleles (73% of affected patients) and E295K [corrected], a rare missense mutation. Pulse‐chase labeling and immunoprecipitation of the 461–677 deletion and E295K mutation indicated that 461–677 deletion protein is synthesized as a ~24 kDa polypeptide, whereas the maturation of E295K mutant [corrected] resembles wild‐type CLN3 protein. Transient expression of the two mutants in BHK cells showed that 461–677 deletion protein is retained in the ER, whereas E295K [corrected] mutant was capable of reaching the lysosomal compartment. Using mouse primary neurons, investigators showed that wild‐type and E295K [corrected] mutant CLN3 proteins localize to the soma and neurites, whereas the 461–677 deletion protein is not found in neurites (Järvelä et al., 1999).
3. TISSUE DISTRIBUTION
CLN3 is universally expressed in multiple human tissues. Immunoblot, immunohistochemistry, Northern blot, and PCR analyses reveal CLN3 protein and mRNA expression in the nervous system, glandular/secretory system, skeletal muscle, gastrointestinal tract, and cancer tissues (Chattopadhyay & Pearce, 2000; Margraf et al., 1999; Persaud‐Sawin, McNamara, Rylova, Vandongen, & Boustany, 2004; Rylova et al., 2002).
In the brain, reactivity for CLN3 is present in astrocytes and neurons, and is more pronounced in the cells of the gray matter, where a larger percentage of astrocytic cells were stained. Capillary endothelium also showed cytoplasmic CLN3 expression. Overall, the expression is similar in intensity and distribution in all of the areas of brain examined, including frontal and temporal cerebral lobes, hippocampus, basal ganglia, and pons (Chattopadhyay & Pearce, 2000; Margraf et al., 1999). Peripheral nerves also express CLN3 (Margraf et al., 1999; Persaud‐Sawin et al., 2004).
In the glandular/ secretory system CLN3 is present in the pancreas (islet somatostatin‐secreting delta cells) (Boriack & Bennett, 2001; Margraf et al., 1999), kidney, testis (in the Sertoli and maturing germ cells), lungs, lymph nodes, placenta, uterus, prostate, ovary, liver, adrenal gland, thyroid, salivary gland, and mammary gland (Chattopadhyay & Pearce, 2000; Margraf et al., 1999).
In the gastrointestinal tract, CLN3 expression is found in stomach, duodenum, jejunam, ileum, ileocecum, appendix, colon and rectum (Chattopadhyay & Pearce, 2000; Rylova et al., 2002). CLN3 is also expressed in fibroblasts (Persaud‐Sawin et al., 2004), heart, and skeletal muscle (Chattopadhyay & Pearce, 2000).
In cancer tissues, CLN3 mRNA and protein are overexpressed in glioblastoma (U‐373G and T98g), neuroblastoma (IMR‐32, SH‐SY5Y, and SK‐N‐MC), prostate (Du145, PC‐3, and LNCaP), ovarian (SK‐OV‐3, SW626, and PA‐1), breast (BT‐20, BT‐549, and BT‐474), and colon (SW1116, SW480, and HCT 116) cancer cell lines, but not in pancreatic (CAPAN and As‐PC‐1) or lung (A‐549 and NCI‐H520) cancer cell lines. Indeed, CLN3 expression is 22%–330% higher in 8 of 10 solid colon tumors when compared with the corresponding normal colon tissue control (anHaack et al., 2011; Rylova et al., 2002; Zhu et al., 2014).
3.1. Gene expression data analysis for CLN3
Tissue expression of CLN3 was retrieved from several sets of expression data collected across the ArrayExpress database (Rustici et al., 2013) and NCBI GEO (GSE1133, GSE2361, GSE7307, GSE30611) (Barrett et al., 2013) (Table 6).
Table 6.
NCBI GEO data sets used for tissue specific expression analysis
| Figure (below) | Dataset | Species | Platform | Description |
|---|---|---|---|---|
| Figure 4 | E‐MTAB‐62 | Human | Affymetrix HG‐U133A | Human gene expression atlas of 5,372 samples representing 369 different cell and tissue types, disease states and cell lines. |
| Figure 5 | GSE10246 | Mouse | Affymetrix Mouse Genome 430 2.0 Array | Multiple tissues were taken from 182 naïve male C57BL6 mice and hybridized to mouse genome arrays to profile a range of gene expressions in normal tissues. |
| Figure 6 | GSE53960 | Rat | Illumina HiSeq 2000 | As part of the SEQC consortium efforts, a comprehensive rat transcriptomic BodyMap created by performing RNA Seq on 320 samples from 11 organs of both sexes of juvenile, adolescent, adult and aged Fischer 344 rats. |
| Figure 7 | GSE1133 | Human | Affymetrix HG‐U133A, GNF1M (non‐commercial), GNF1H (non‐commercial) | Custom arrays that interrogate the expression of the vast majority of protein‐encoding human genes were developed and used to profile a panel of 79 human tissues. The resulting data set provides the expression pattern for thousands of predicted genes, as well as known and poorly characterized genes. |
| Figure 8 | GSE7307 | Human | Affymetrix HG‐U133 Plus 2 | 677 samples representing 90 distinct tissues from normal and diseased human tissues were profiled for gene expression using the Affymetrix U133 plus 2.0 array |
Briefly, each Affymetrix microarray data set was downloaded and preprocessed by MAS5.0 algorithm followed by quantile normalization. Updated Entrez‐centric BrainArray CDF files were used for normalization. RNA‐Seq datasets were downloaded in the already pre‐processed formats from the ReCount and SEQC ("Rat Body Map") websites, respectively. In each dataset, gene expression levels were transformed into Z‐scores by gene centering on zero and dividing the centered profiles by their standard deviation. The box plot for each tissue reflects the distribution of probe set expression signals (log2‐scaled) across the samples of this tissue. The median signal is depicted as black line in the middle of the box; box borders represent the 25th and 75th percentiles of signal distribution. Empty dots represent the “outliers” – samples with unusually high or low expression signal.
The expression of human CLN3 across the E‐MTAB‐62 dataset is plotted in Figure 4, while the mouse GSE10246 and rat GSE53960 data sets are presented in Figure 5 and Figure 6 respectively. We see the highest CLN3 expression in human placenta and leukocytes. Similarly, CLN3 expression is highest in mouse placenta and leukocyte‐associated tissues (bone marrow, spleen, and bone marrow). The tissue distribution available in the rat dataset is limited but the CLN3 expression profile differs noticeably, in that expression is highest in rat testis and then spleen, while testis expression in human is relatively low (~ −1.5 relative to median tissue expression). Additional human tissue distribution plots of each dataset are available in Figures 7 and 8.
Figure 4.
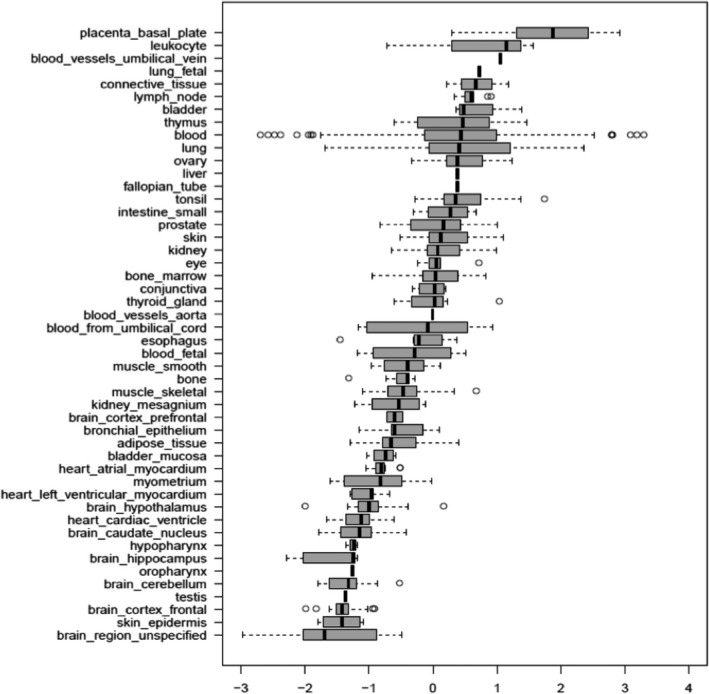
Tissue expression profile of CLN3 across dataset E‐MTAB‐62 (human)NCBI GEO GSE2361
Figure 5.
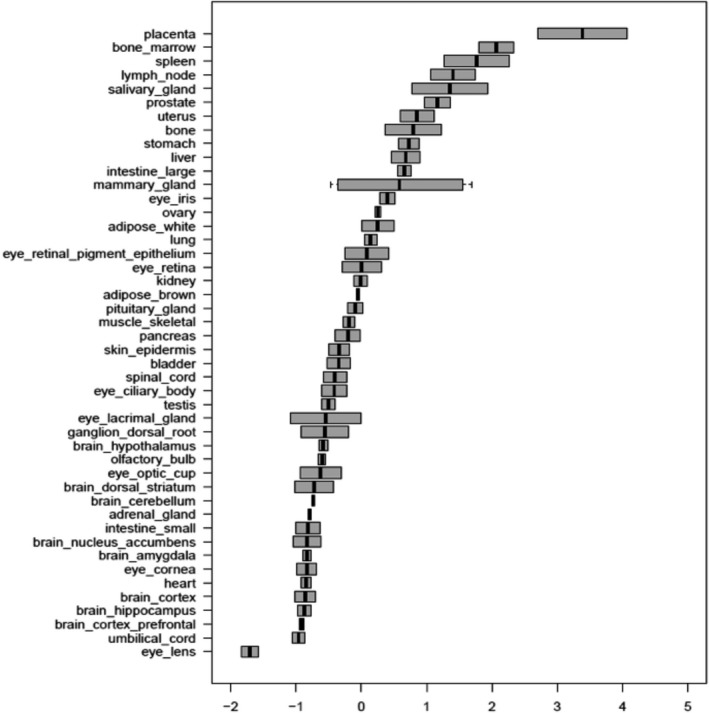
Tissue expression of CLN3 across NCBI GEO data set GSE10246 (mouse)
Figure 6.
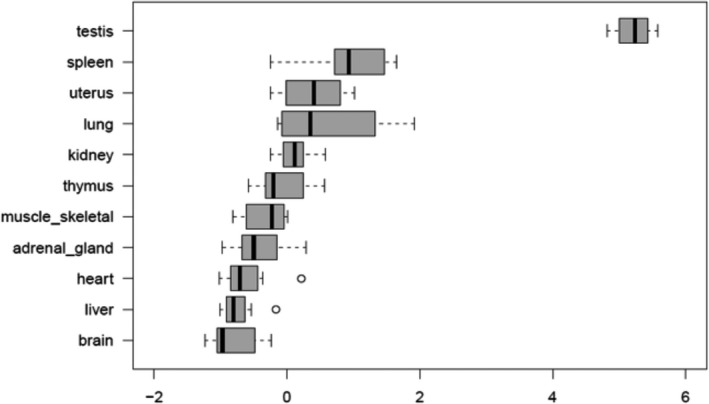
Tissue expression of CLN3 across NCBI GEO data set GSE5396 (rat)
Figure 7.
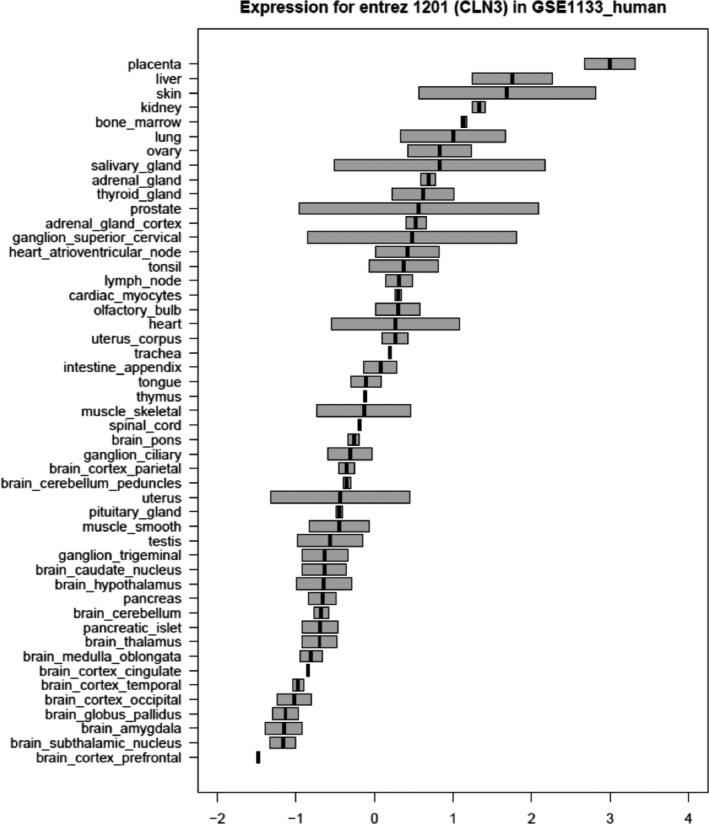
Expression of CLN3 in human tissues according to the NCBI GEO GSE1133 data set
Figure 8.
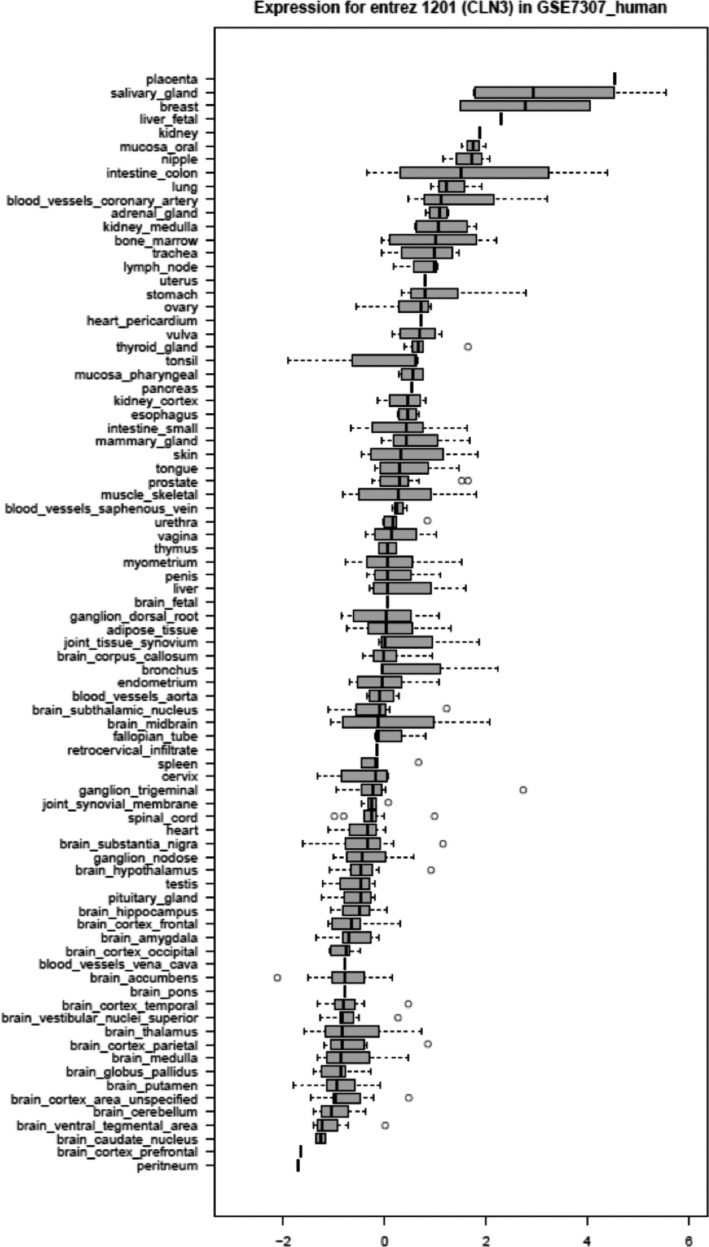
Expression of CLN3 in human tissues according to the NCBI GEO GSE7307 data set
Expression of CLN3 across human tissues and cell types was independently assessed using large‐scale data: such as public microarray data sets. The most widely used data set for whole‐genome human tissue expression is GenAtlas; with 79 human tissues examined by Affymetrix HG U133A microarrays normalized using the GCRMA algorithm. Most tissue samples have only 2 replicates, so the conclusions about expression specificity must be drawn with caution (Figure 7).
Expression of CLN3 is uniform across the majority of tissues profiled in the GeneAtlas data set (Figure 9). The first (dashed) line indicates mean signal of 9.5 RFU. A few tissues have elevated signal of 3X mean (BDCA4 + Dendritic cells, CD4 + helper T‐Cells) and maximum expression is seen in placenta.
Figure 9.
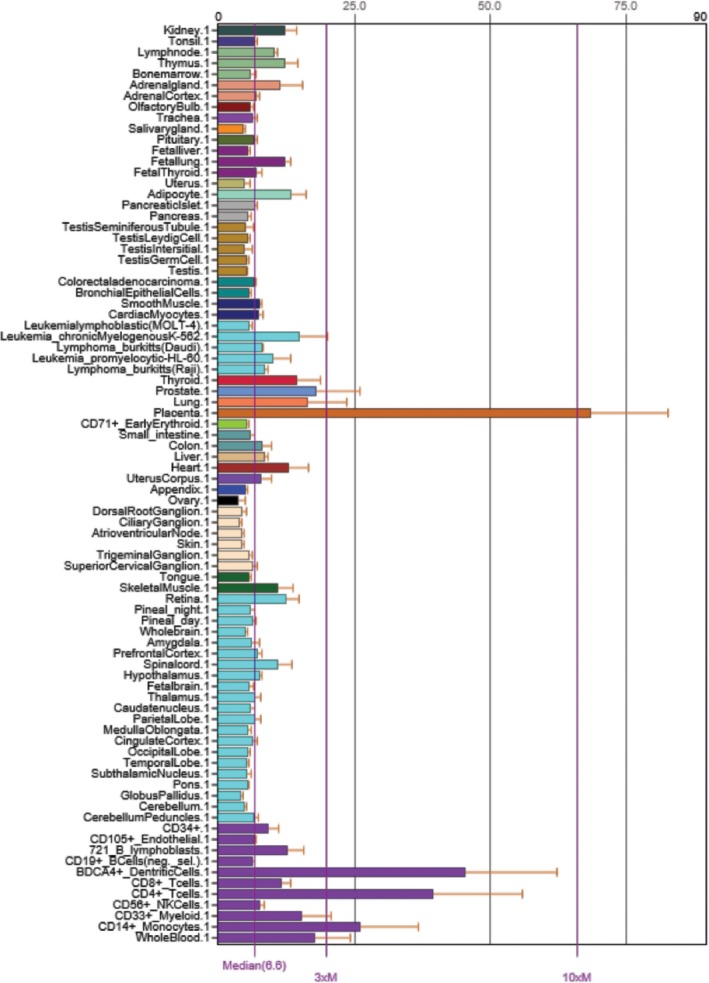
Expression of CLN3 in human tissues according to the Gene Atlas data set (Su et al., 2004; Wu et al., 2009)
When compared across species, the shared tissue samples show a clear trend towards high expression in placenta (Figure 10).
Figure 10.
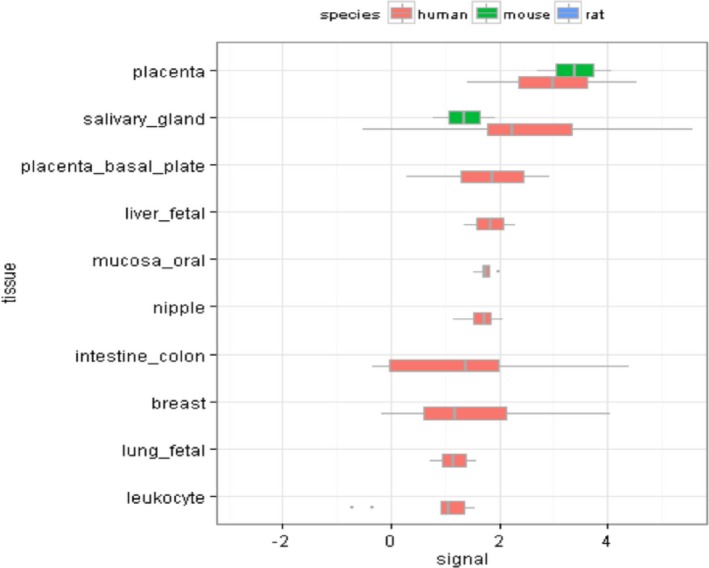
Cross species tissue expression for top ten tissues by expression level in human
3.1.1. CLN3 RNA expression data using GTex Portal
Quantification of gene expression in this data set is mainly done using RNA‐seq. Tissue‐specific gene expression data can be found at http://www.gtexportal.org/home/gene/CLN3 (Lonsdale et al., 2013). CLN3 seems to be highly expressed in colon as compared to other tissues in this dataset. This data does not include placenta and hence, cannot be compared to the above microarray datasets.
3.1.2. Protein expression data analysis for CLN3
The largest proteomics database on tissue expression, Protein Atlas (Uhlen et al., 2010), also confirms widespread expression of CLN3 throughout the organism. Unfortunately, no immunohistochemistry or western blot analysis are available for CLN3 to determine the range or intensity of protein distribution across tissues. This is most likely due to the lack of high‐quality antibodies to CLN3 protein. To date, there are over 30 antibodies in the academic and pharmaceutical sectors. All exhibit either limited CLN3 reactivity, high nonspecific binding or both.
3.1.3. Tissue distribution of CLN3 protein in mice
Using two CLN3‐ specific antibodies, one directed at the N‐terminal (5–19) and the other at the mid‐region (225–280), CLN3 was detected in various mouse tissues (Ezaki et al., 2003). Brain, liver, pancreas, and spleen were tested using both the antibodies. Mouse CLN3 protein was detected as a smear between 45 and 66 kDa in liver, kidney, pancreas, and spleen. In the brain, a CLN3 band is observed closer to 55 kDa. With both antibodies CLN3 signal in brain tissue was weaker than in the other tissues mentioned above.
4. PROTEIN‐PROTEIN INTERACTIONS
This section presents protein‐protein and nucleic acid‐protein interactions of CLN3. The interactions contain directionality, effect (activation, inhibition, etc.) and mechanism (binding, phosphorylation, transcriptional regulation, etc.).
As stated previously, the function of the CLN3 has not been completely elucidated. This is not surprising as transmembrane proteins, like CLN3, are highly hydrophobic and difficult to stabilize in solutions for study. This section is designed to answer specific questions regarding binding of CLN3 to other proteins. Some statements mention putative functions for CLN3. For a full exploration of the proposed functions, the reader should consult the primary literature.
4.1. Interactions with Kinases and Phosphatases
As mentioned in the phosphorylation section above, CLN3 has been demonstrated to interact with, and be phosphorylated by, PKA, PKG, and casein kinase II. Moreover, it is dephosphorylated by protein phosphatase 1 or protein phosphatase 2A (Michalewski et al., 1998, 1999) see phosphorylation section for more information. The phosphorylation of CLN3 may play a role in its interaction with membrane compartments, regulation of protein interactions and formation of functional complexes.
4.2. C2‐Ceramide
The genes that are transcriptionally regulated during ceramide‐mediated cell death are poorly understood. Puranam et al. found that CLN3 does not inhibit C2‐ceramide‐induced apoptosis but modulates endogenous ceramide synthesis and suppresses apoptosis by preventing the generation of ceramide (Puranam, Guo, Qian, Nikbakht, & Boustany, 1999). Accordingly, overexpression of CLN3 protects cells from vincristine‐, staurosporine‐, and topside‐induced apoptosis, but not from ceramide‐induced cell death (Puranam et al., 1999). In addition, C2‐ceramide has been found to induce the expression of CLN3 in PC12 cells, which could represent a negative feedback mechanism regulating endogenous ceramide generation for cellular protection from cell death (Decraene et al., 2002).
4.3. Interactions with Endosomal‐Lysosomal proteins
Although the function of CLN3 protein remains unknown, several findings support the conclusion that lysosomes and endosomes are prominent sites of CLN3 activity. CLN3 contains multiple lysosomal targeting signals including a non‐conventional signal in the C‐terminus, and although any of these are sufficient for transport of CLN3 to lysosomes, all are required for optimal transport efficiency (Kyttälä et al., 2004). CLN3 binds directly to active, guanosine triphosphate (GTP)‐bound Rab7 and Rab‐interacting lysosomal protein as confirmed by mammalian two‐hybrid experiments with a peptide corresponding to amino acids 1–40 of CLN3. These experiments suggest that CLN3 binds to Rab7 via its N‐terminus and that this interaction occurs most favorably with the GTP‐bound form of Rab7 (Kyttälä et al., 2004; Uusi‐Rauva et al., 2012). Rab7 facilitates vesicular transport and delivery from early to late endosomes and late endosomes to lysosomes. The role of Rab7 in vesicular transport is dependent on its interactions with effector proteins, among them RILP, which aids in the recruitment of active Rab7 (GTP‐bound) onto dynein‐dynactin motor complexes to facilitate late endosomal transport on the cytoskeleton (Agola et al., 2015).
To examine putative interactions between CLN3 and microtubule‐binding protein, Hook1, investigators conducted in vitro binding assays with cytoplasmic Hook1 and two putative cytoplasmic domains of CLN3 (1–33 and 232–280). When compared with the GST vector alone, putative CLN3 cytoplasmic domains (1–33 and 232–280) bound with low affinity to Hook1 (Luiro et al., 2004). When Hook1 and CLN3 proteins were co‐expressed with individual GFP‐tagged Rab 7, 9, and 11, Hook1 was found to specifically interact with Rab7, Rab9, and Rab11. In contrast no direct interactions between CLN3 and the Rab proteins were found (Luiro et al., 2004). These findings implicate CLN3 in a complex cellular machinery connecting cytoskeletal dynamics to endocytic membrane trafficking. It is proposed that this interaction may be disturbed in the absence of CLN3, thus leading to endocytic dysfunction in CLN3 deficient cells (Luiro et al., 2004). Indeed wild‐type CLN3 protein (CLN3p, 48–52 kD) traffics from Golgi to lipid rafts at the plasma membrane via Rab4‐ and Rab11‐positive endosomes (Persaud‐Sawin et al., 2004). However, mutant CLN3 protein does not appear to localize to the plasma membrane in JNCL fibroblasts. Instead, mutant CLN3p was retained within the Golgi and partially mis‐localized to lysosomes, failing to reach recycling endosomes, plasma membrane, or lipid rafts (Persaud‐Sawin et al., 2004). Moreover, the yeast homolog of CLN3, BTN1, has also been shown to play a role in endosome‐Golgi retrograde transport by regulating SNARE protein function (Kama et al., 2011). Although BTN1 does not directly interact with SNAREs, it was shown to modulate Sed5 phosphorylation by regulating Yck3, a palmitoylated endosomal kinase. This may involve modification of the Yck3 lipid anchor, as substitution with a transmembrane domain suppresses the deletion of BTN1 and restores trafficking (Kama et al., 2011).
4.4. Interactions with Calsenilin/dream/KChIP3
Yeast two hybrid (Y2H) and immunoprecipitation assays show that Calsenilin (also known as downstream regulatory element antagonist modulator (DREAM) and K+ channel interacting protein 3 (KChIP3)), a neuronal Ca2+‐binding protein, interacts with the C‐terminal region of CLN3 (385–438) and that an increase in Ca2+ concentration promotes the dissociation of CLN3 from calsenilin. Calsenilin has been found to act as a transcriptional repressor, and its activity has been linked with neuronal excitation and repolarization of K+ channel (An et al., 2000). Calsenilin binds DNA in a Ca2+ dependent manner and increases the expression of wild‐type or C‐terminal CLN3 and suppresses thapsigargin mediated cell death. Thapsigargin is a sarco/endoplasmic reticulum Ca2+‐ATPase pump inhibitor, a research tool used to raise cytosolic Ca2+ and induce Ca2+‐mediated cell death. In the absence of CLN3, such as in the case of CLN3 knockout mice or human SH‐SY5Y cells deficient in CLN3, cells are more sensitive to thapsigargin (Chang et al., 2007). The CLN3‐Calsenilin interaction was not confirmed by Tandem Affinity Purification coupled to Mass Spectrometry (TAM‐MS) combined with Significance Analysis of Interactome (SAINT) in human SH‐SY5Y cells (SH‐SY5Y‐NTAP‐CLN3, (Scifo et al., 2013)). More work is needed to understand the physical properties and functional role(s) of CLN3 and Calsenilin interactions.
In 2015, investigators utilized an autophagy assay, a process previously shown to be disrupted in the CbCln3 Δ ex7/8/Δex7/8 mouse model of the disease (Cao et al., 2006), which used green fluorescent protein‐tagged LC3 transgene to label autophagosomes in mouse cerebellar CbCln3 Δ ex7/8/ Δ ex7/8 cell lines. Using these cell lines, investigators screened small molecule modifiers of autophagy to discover the sensitivity of disease cell models to alterations in autophagy which impact Ca2+ regulation. In these experiments, thapsigargin reproducibly displayed significantly more activity in mouse knock‐in cerebellar neurons as well as in induced pluripotent stem cells derived from patients with the common deletion. The mechanism of thapsigargin sensitivity was Ca2+‐mediated, and autophagosome accumulation in JNCL cells could be reversed by cytosolic Ca2+ chelation. Interrogation of intracellular Ca2+ handling highlighted alterations in ER, mitochondrial, and lysosomal Ca2+ pools and in store‐operated Ca2+ uptake (Chandrachud et al., 2015).
4.5. Interactions with other NCL proteins
It has long been presumed, due to the similarity of clinical features and pathological hallmarks between various NCLs, that NCL proteins are part of the same or similar cellular pathways and that there is some degree of interaction between NCL proteins. Some studies support this conclusion. The 13 proteins encoded by NCL genes do not all localize to endosomal/lysosomal pathways, some are situated in compartments of the secretory system such as the ER; these can be found in Table 7.
Table 7.
CLN3 interactions with other NCLs
| Gene | Gene Product (protein) | Protein description | Primary and Neuronal Location(s) | References |
|---|---|---|---|---|
| CLN1 | Palmitoyl protein thioesterase 1, PPT1 | soluble enzyme | Lysosomal | Persaud‐Sawin et al. (2007) |
| CLN2 | Tripeptidyl peptidase 1, TPP1 | soluble enzyme | Lysosomal | Vesa et al. (2002) and Persaud‐Sawin et al. (2007) |
| CLN3 | CLN3 transmembrane protein | transmembrane protein | Late endosomal/Lysosomal, synaptosomes, axons | Storch et al. (2007) |
| CLN4/DNAJC5 | Cysteine string proteinα | secretory vesicle protein | ||
| CLN5 | Ceroid‐lipofuscinosis neuronal protein 5 | soluble (non) enzyme glycoprotein | ER, Lysosomal, neurites | Lyly et al. (2009) |
| CLN6 | Ceroid‐lipofuscinosis neuronal protein 6 | transmembrane protein | Endoplasmic Reticulum | Persaud‐Sawin et al. (2007) |
| CLN7 | Major facilitator superfamily domain‐containing protein 8 | transmembrane protein, endolysosomal transporter | Late endosomal/Lysosomal | |
| CLN8 | unknown transmembrane protein, ER, ER‐Golgi intermediate complex | transmembrane protein | Endoplasmic Reticulum, ER‐Golgi intermediate complex | Persaud‐Sawin et al. (2007) |
| CLN10/CTSD | Cathepsin D | soluble lysosomal enzyme | Lysosomal | |
| CLN11/GRN | Progranulin | non enzyme; poorly understood | ||
| CLN12/ATP13A2 | P‐type ATPase | non enzyme; poorly understood | ||
| CLN13 | Cathepsin F | soluble lysosomal enzyme | ||
| CLN14/KCTD7 | Potassium channel tetramerization domain‐containing protein 7 | probable transmembrane protein voltage‐gated potassium channel complex |
4.5.1. Interaction with CLN1
Palmitoyl protein thioesterase 1 (PPT‐1), encoded by ceroid‐lipofuscinosis, neuronal 1 (CLN1), is a small soluble glycoprotein involved in the catabolism of lipid‐modified proteins during lysosomal degradation. The encoded enzyme removes thioester‐linked fatty acyl groups such as palmitate from cysteine residues on multiple protein targets (Cho & Dawson, 1998; Cho, Dawson, & Dawson, 2000). Defects in this gene are linked to a rapidly progressing lysosomal disease by the same name, CLN1 disease. CLN1 disease may also be referred to as infantile neuronal ceroid lipofuscinosis (INCL) as the vast majority of reported cases of the disease present around 18 months of age. Like most pediatric forms of NCL, patients experience progressive vision loss, cognitive and motor deficits, seizures and early death. With the advent of increased genetic testing, cases of late infantile, juvenile and adult‐onset CLN1 have been reported suggesting that less severe mutations of the gene produce some or modified versions of the PPT‐1 protein (Van Diggelen et al., 2001). The possibility that CLN1 and CLN3 gene products interact with one another was raised by studies demonstrating that PPT‐1 localizes to synaptic vesicles and CLN3 to synaptosomes (Ahtiainen, Diggelen, Jalanko, & Kopra, 2003; Hellsten, Vesa, Olkkonen, Jalanko, & Peltonen, 1996; Lehtovirta et al., 2001; Luiro et al., 2001; Sleat et al., 1997). However, direct interaction between PPT1 and CLN3 protein was not demonstrated by co‐immunoprecipitation, while parallel studies with CLN1 and CLN2 demonstrated interaction between the two. In the same study no benefit to cellular growth or apoptosis was observed in CLN3 deficient cells transfected with CLN1, whereas benefits were observed with CLN2 and CLN6 expression (Persaud‐Sawin et al., 2007). More recent studies using TAM‐MS combined with bioinformatics SAINT demonstrated an interaction between CLN3 and PPT1; however, it is not clear whether this interaction is direct (Scifo et al., 2013).
4.5.2. Interaction with CLN2
The ceroid‐lipofuscinosis, neuronal 2 (CLN2) gene encodes tripeptidyl peptidase I (TPP‐1), a serine protease which cleaves N‐terminal tripeptides from the free N‐termini of small polypeptides and also shows minor endoprotease activity (Golabek et al., 2003, 2004). Mutations in CLN2 result in late‐infantile neuronal ceroid lipofuscinosis previously referred to as LINCL, but now more commonly known as CLN2. Normal human lymphoblasts and COS‐7 cell lysates immunoprecipitated with an anti‐CLN3 antibody and probed with an anti‐CLN2 antibody, detected a 48–50 kD band. This suggested that CLN2 and CLN3 physically interact with one another, which was further supported by co‐localization experiments performed in the same study (Persaud‐Sawin et al., 2007). In addition, C57BL/6 mice homozygous for targeted disruption of the CLN3 gene exhibit elevated CLN2/TPP1 protease activity in the brain, implying a biochemical connection between the gene products of CLN3 and CLN2 (Mitchison et al., 1999). More recent studies using TAM‐MS combined with bioinformatics SAINT also demonstrated an interaction between CLN3 and TPP‐1; however, it is not clear whether this interaction is direct (Scifo et al., 2013).
4.5.3. Dimerization of CLN3
A transmembrane topology where CLN3 contains 6 transmembrane domains with both the N‐ and C‐terminal domains facing the cytosol is currently favored and is supported by both computer modeling and experimental evidence (Kyttälä et al., 2004; Nugent et al., 2008; Ratajczak et al., 2014; Storch et al., 2007). An alternative model which predicts a 5‐transmembrane topology of CLN3 also exists (Mao, Foster, et al., 2003). However, neither the function of CLN3 nor its functional tertiary structure have been solved yet. When COS7 cells overexpressing N‐terminally Myc‐tagged CLN3 are permeabilized and incubated in the absence or presence of chemical cross‐linkers BS3 and DMS, tagged CLN3 forms SDS‐stable 88‐kDa proteins, presumed to correspond to a CLN3 homodimer (Storch et al., 2007). However, experimental artifacts may result in the formation of dimers or oligomers simply via hydrophobic interactions therefore, whether CLN3 forms a functional dimer remains to be confirmed.
4.5.4. Interaction with CLN5
CLN5 protein (CLN5p) is a highly glycosylated protein of unknown function which, similar to CLN3, localizes to lysosomes and neurites (Holmberg et al., 2004; Isosomppi, Vesa, Jalanko, & Peltonen, 2002; Jalanko, Patrakka, Tryggvason, & Holmberg, 2001). Pathogenic mutations lead to its retention in ER/ Golgi and the Finnish variant late infantile form of NCL (vLINCLFin, (Holmberg et al., 2000; Savukoski et al., 1998). Late infantile, juvenile, and adult‐onset forms of CLN5 disease have been reported. Co‐immunoprecipitation and in vitro binding assays revealed that CLN3 protein interacts directly with wild‐type CLN5 synthesized as 47‐, 44‐, 41‐, and 39‐kDa polypeptides, as well as CLN5 mutants FINM, EUR, and SWE. In this study, both CLN3 and CLN5 were transfected into COS cells as this cell line did not have sufficient endogenous levels of the proteins for investigation (Vesa et al., 2002, Figure 1a).
All forms of CLN5 retained their localization to lysosomes and their ability to interact with CLN3 protein (synaptosome fraction not tested, Vesa & Peltonen, 2002). Pull‐down experiments by Lyly and colleagues in 2009 with GST‐mCLN5 also captured CLN3 protein, supporting the conclusions of Vesa and colleagues (Lyly et al., 2009). When CLN5 protein, mutated to restrict its localization to the ER, is expressed in healthy cells, it is shown to colocalize with CLN3 in the ER (Lebrun et al., 2009), the significance of this is unknown. CLN5 protein has molecular connections to CLN3 and at least to four other NCL proteins; CLN1/PPT1, CLN2/TPP1, CLN6 and CLN8 (Lyly et al., 2009). Studies using TAM‐MS combined with bioinformatics SAINT found that 18 of 31 CLN5 interactors also interacted with CLN3, further supporting the functional overlap between the two (Scifo et al., 2013).
4.5.5. Interaction with CLN6
The CLN6 gene encodes a polytopic transmembrane protein, which localizes to the ER (Heine et al., 2007, 2004; Mole et al., 2004). Mutations in the CLN6 gene have been linked to autosomal dominant, adult‐onset known as Kuf's Type A disease. Patients with Kuf's Type A disease present with progressive myoclonic epilepsy in adulthood followed by dementia. Dissimilar to early‐onset CLN6 disease and most forms of NCL, Kuf's Type A disease does not exhibit a retinal phenotype. To test whether CLN3 and CLN6 proteins interact with one another, lymphoblast lysates were immunoprecipitated with an anti‐CLN6/CLN8 antibody and probed with a CLN3 targeted antibody. Indeed, a CLN3 band was detected, suggesting CLN6 and CLN3 physically interact. These results were confirmed by reciprocal experiments using transfected COS‐7 cells. Expression of CLN6 cDNA led to some correction of growth defects in CLN3‐deficient cells, and CLN6 was also shown to colocalize with CLN3 in fibroblasts. Together, these results suggest that CLN3 and CLN6 interact with one another (Persaud‐Sawin et al., 2007). Moreover, TAM‐MS combined with bioinformatics SAINT analysis further supports an interaction between CLN3 and CLN6; however, it is not clear whether this interaction is direct (Scifo et al., 2013).
4.5.6. Interaction with CLN8
The CLN8 gene encodes a transmembrane protein of unknown function whose ER‐Golgi intracellular location is inferred from confocal immunofluorescence microscopy of transiently transfected BHK cells (Lonka, Kyttälä, Ranta, Jalanko, & Lehesjoki, 2000). Two distinct mutations in the CLN8 gene have been shown to result in mutation‐specific phenotypes – juvenile‐onset progressive epilepsy with mental retardation (EPMR, (Hirvasniemi, Herrala, & Leisti, 1995; Hirvasniemi & Karumo, 1994; Hirvasniemi, Lang, Lehesjoki, & Leisti, 1994) and a more severe late variant NCL with pathological similarities to CLN5‐, CLN6‐, and CLN7‐disease (Cannelli et al., 2006; Haltia, Herva, Suopanki, Baumann, & Tyynelä, 2001; Herva, Tyynelä, Hirvasniemi, Syrjäkallio‐Ylitalo, & Haltia, 2000; Ranta, Hirvasniemi, Herva, Haltia, & Lehesjoki, 2002; Ranta, Savukoski, Santavuori, & Haltia, 2001; Ranta et al., 2004; Vantaggiato et al., 2009). Expression of CLN3 cDNA in CLN8‐deficient mouse fibroblasts reduced the aberrant cellular growth of these cells. Interaction of the two proteins was observed by western blot analysis of lymphoblast lysates immunoprecipitated with an anti‐CLN8 antibody and probed with an antibody targeted at CLN3. These results were confirmed by reciprocal experiments using transfected COS‐7 cells suggesting that CLN3 and CLN8 proteins interact with one another (Persaud‐Sawin et al., 2007). Investigation of the cellular localization of CLN8 showed co‐localization of CLN3 and CLN8, which contradicts earlier studies (Persaud‐Sawin et al., 2007). However, studies using TAM‐MS combined with bioinformatics SAINT also demonstrate an interaction between CLN3 and CLN8; however, it is not clear whether this interaction is direct (Scifo et al., 2013).
4.5.7. Lack of interaction with CLN4/DNAJC5, CLN7, CLN10/CTSD, CLN11/GRN, CLN12/ATP13A2/PARK9, CLN13/Cathepsin F, CLN14/KCTD7
No direct interactions between CLN3 and CLN4, CLN7, and CLN10‐14 gene products have been reported, although not all potential interactions have been explored and single‐experiment negative results may not be definitive. Moreover, it must also be considered that experimental approaches that solely interrogate direct interaction do not preclude CLN3 and the other gene products from contributing to the same cellular pathways or loosely associating in a complex.
4.5.8. Lack of interaction with subunit c of mitochondrial ATP synthase; the major component of characteristic intracellular storage material build‐up
The accumulation of subunit c in CLN3 disease (Johnson et al., 1995; Westlake, Jolly, Bayliss, & Palmer, 1995) raises the possibility that CLN3 may be involved in processing or degrading subunit c, which could potentially be mediated by direct physical interaction. Using the Y2H, investigators screened fragments of the CLN3 peptide against a human fetal brain library yet failed to demonstrate a direct interaction between CLN3 and subunit c of mitochondrial ATP synthase (Leung, Greene, Munroe, & Mole, 2001).
4.6. Non‐bona‐fide CLN3 interactors
In addition to bona‐fide interactors, several other proteins have been found to bind CLN3 in vitro using the Cytotrap Y2H system. The original Y2H systems required fusion proteins to be expressed in the nucleus and were thus were not suitable for transmembrane proteins like CLN3. With Cytotrap, instead, the interaction occurs in the cytoplasm with the reporter system associated with the plasma membrane. While this technology is very useful, its use of overexpressed fusion proteins may create artificial conditions. Therefore, subsequent experiments are needed to validate findings where the only reported interaction between CLN3 and another protein of interest was as result of the use of this method.
4.6.1. Interaction with myosin IIB
The C‐terminal region of CLN3 has been found to interact with myosin IIB (Getty, Benedict, & Pearce, 2011). This interaction was found by Y2H and confirmed by co‐immunoprecipitation of overexpressed CLN3 and endogenous myosin‐IIB. Non‐muscle myosin IIB interacts with adenosine triphosphate and F‐actin to promote cytoskeletal integrity and force generation for multiple cellular processes such as cell migration, shape changes, adhesion dynamics, endocytosis, exocytosis and autophagy (Heissler & Manstein, 2013). In addition to the cellular functions listed, myosin IIB has been shown to be important for numerous neuron‐specific cell functions such as polarization, dendritic spine morphology, growth‐cone motility and presynaptic vehicle trafficking. Even though the significance of the interaction between CLN3 and myosin IIB has not been fully elucidated, it stands to reason that CLN3 may act in concert with myosin IIB to regulate cytoskeletal dynamics and that the loss of CLN3 function could disrupt myosin IIB activity.
4.6.2. Interaction with Shwachman‐Bodian Diamond Syndrome (SBDS) protein
To determine which proteins interact with CLN3, investigators screened fragments of the CLN3 peptide against a human fetal brain library using a cytotrap Y2H system (Vitiello et al., 2010). The C‐terminal fragment of CLN3, predicted to be cytosolic, was found to interact with the N‐terminus of Schwachman‐Bodian‐Diamond‐syndrome (SBDS) protein. These results were confirmed by co‐immunoprecipitation and co‐localization studies in NIH/3T3 cells with C‐terminal c‐myc and V5 tags (Vitiello et al., 2010). The interaction between CLN3 and SBDS is evolutionarily conserved since Sdop1 and Btn1p, the yeast homologs of SBDS and CLN3, respectively, have been found to interact with one another. Loss of SBDS protein results in Shwachman‐Bodian Diamond syndrome, an autosomal recessively‐inherited neutropenia syndrome characterized by bone marrow dysfunction and associated cumulative risk of aplastic anemia progressing to myelodysplastic syndrome and acute myeloid leukemia (Donadieu et al., 2005). Previous studies on Sdo1p revealed that this protein is involved in ribosomal biogenesis and RNA processing (Luz, Georg, Gomes, Machado‐Santelli, & Oliveira, 2009). More recently, SBDS has been found to regulate the expression of C/EBPalpha and C/EBPbeta, which are critical transcription factors for myelomonocytic lineage commitment. In particular, SBDS patients have reduced C/EBPbeta‐LIP levels (In et al., 2016). Defective expression of these factors may affect myeloid cell proliferation and differentiation, driving neutropenia which is the most prominent hallmark in almost all SBDS patients.
4.6.3. Interaction with ß‐fodrin and Na+‐K+‐ATPase complex
To determine which proteins interact with CLN3 and could therefore, provide clues as to its function, investigators screened fragments of the CLN3 peptide (N1‐40 and N232‐280), both predicted to be cytoplasmic, with a LacZ/beta‐galactosidase Y2H system. Interactions were subsequently confirmed by co‐immunoprecipitation by overexpressing the full‐length CLN3 protein in COS1 cells and by using CLN3 antibodies. The results showed that full‐length CLN3 interacts with cytoskeletal protein β‐fodrin (β‐II‐spectrin) and its plasma/endosomal interaction partner Na+, K+ ATPase, a heteromeric protein with varying α‐β isoform combinations (Uusi‐Rauva et al., 2008). Beta‐fodrin, known also as non‐erythroid spectrin, is concentrated in the synaptosome fraction and is associated with synaptic membranes (Sobue, Kanda, & Kakiuchi, 1982). In erythrocyte membrane skeleton, beta fodrin has been found in heterotetrameric complexes with alpha fodrin (two alpha and two beta chains), which drive the formation of polygonal network linked to actin filaments. This network is located at the plasma membrane bilayer by interaction with ankyrin protein and the cytoplasmic domain of the Na+, K+, ATPase and it is involved in protein stability and polarization (Bennett & Baines, 2001). Na+, K+, ATPase is an ubiquitous heterodimeric transmembrane enzyme composed of varying alpha and beta isoform combinations that transport Na+ and K+ across the plasma membrane by hydrolysis of ATP (Lingrel et al., 1994). Follow‐up studies revealed that the ion pumping activity of Na+, K+ ATPase is unchanged in CLN3 disease mouse models created by a homozygous deletion of exons 1–6 of CLN3 bred onto a C57/BL genetic background. However, the immunostaining pattern of fodrin appeared abnormal in CLN3 patient fibroblasts and Cln3‐/‐ mouse brains suggesting disturbances in the fodrin cytoskeleton. Furthermore, the basal subcellular distribution as well as ouabain‐induced endocytosis of neuron‐specific Na+, K+ ATPase were markedly affected in Cln3‐/‐ mouse primary neurons. Studies using TAM‐MS combined with bioinformatics SAINT confirm a direct interaction between CLN3, ß‐fodrin and the Na+, K+ ATPase complex (Scifo et al., 2013; Uusi‐Rauva et al., 2008). However, the interaction between fodrin and Na+, K+, ATPase in CLN3‐/‐ mouse models has not been evaluated. Further studies are needed to confirm the role of CLN3 protein in the regulation of plasma membrane‐fordin cytoskeleton and, consequently, the plasma membrane association of Na+, K+ ATPase.
4.6.4. Human Autophagy Interacting Network (AIN)
However, specific aspects of the autophagy pathway have been studied extensively, less is known about the overall architecture and associated regulation of the autophagy interaction network (AIN). To generate a framework for the human AIN that could be followed up by direct mechanistic and other functional studies, Behrends and colleagues performed a systematic proteomic analysis by retrovirally expressing CLN3 and 31 additional proteins as Flag‐HA‐fusion proteins in 293T cells, isolating α‐HA immune complexes by mass spectrometry and processing the complexes using Comparative Proteomics Analysis Software Suite (CompASS©) to identify high‐confidence candidate interaction proteins (HCIPs). TAM‐MS and combined with bioinformatics SAINT yielded 58 CLN3 interacting partners including IMMT, GCN1L1, PRKDC, XPO1, CPT1A, HSD17B12, RPN2, PHGDH, COX15, SLC25A11, DDOST, AUP1, KIAA0368, SLC25A22, SLC25A10, and NUP205 (Scifo et al., 2013). The significance of computer‐generated results is unknown and needs to be followed up with direct mechanistic and functional studies (Figure 11).
Figure 11.
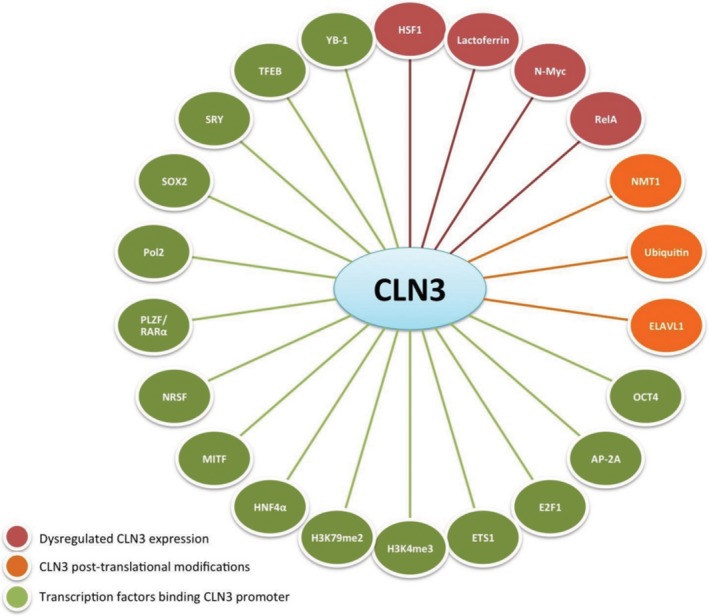
Network diagram of CLN3 interactors. These interactions have been found by ChIP‐chip and Chip‐Seq analysis (Transcription factors binding CLN3 promoter), Microarray analysis (Dysregulated CLN3 expression) and Mass spectrometry analysis (CLN3 PTMs). The majority of these interactions require further validation
5. CONCLUSION
The discovery of juvenile Batten disease occurred more than 100 years ago (Stengel, 1826). The responsible genetic dysfunction was discovered more than 20 years ago (International Batten Disease Consortium, 1995). Since that time, over 500 unique discoveries have been made relevant to the CLN3 gene, its protein, regulation or dysfunction in its absence. Despite all of this, the function of CLN3 protein remains elusive. The authors hope that the historical research findings presented here will contribute to determining the function of the CLN3 protein and exactly why, CLN3 gene defects adversely affect endosomal‐lysosomal homeostasis, proteome, lipidome, trafficking, maturation, and recycling activities (anHaack et al., 2011; Appu et al., 2019; Burkovetskaya et al., 2014; Cao et al., 2006; Chandrachud et al., 2015; Codlin et al., 2008; Codlin & Mole, 2009; Gachet et al., 2005; Golabek et al., 2000; Holopainen, Saarikoski, Kinnunen, & Jarvela, 2001; Fossale et al., 2004; Kama et al., 2011; Luiro et al., 2004; Metcalf, Calvi, Seaman, Mitchison, & Cutler, 2008; Schmidtke et al., 2019; Stein et al., 2010; Tecedor et al., 2013; Vidal‐Donet, Cárcel‐Trullols, Casanova, Aguado, & Knecht, 2013). Why does CLN3 protein deficiency impact protein palmitoylation? How then does palmitoylated reversible trafficking and localization of proteins to cell membranes contribute to cell death (Narayan et al., 2006, 2008)? Are the defects found in the endoplasmic reticulum, trans‐golgi network, and mitochondria of CLN3‐deficient cells primary or secondary to disease pathogenesis (Fossale et al., 2004; Luiro et al., 2006; Metcalf et al., 2008)? If the breakdown in intracellular communication between membranous compartments of the endosomal‐lysosomal system and other organelles were resolved in glia, would neuronal survivability improve (Parviainen et al, 2017)? Which defects lead to the selective vulnerability of neurons in disease? Overexpression of CLN3 protein has been reported in several primary cancerous tissues and cell lines (Dhar et al., 2002; Makoukji et al., 2015; Mao et al., 2015; Narayan et al., 2006; Oltulu et al., 2019; Ren et al., 2016; Rylova et al., 2002; Xu et al., 2019; Zhang et al., 2008; Zhu et al., 2014). Are the anti‐apoptotic properties of CLN3 protein responsible for promising preclinical CLN3 gene therapy reports or does gene therapy affect one or more of the deficient processes listed above (Bosch et al., 2016; Lane, Jolly, Schmechel, Alroy, & Boustany, 1996).
CONFLICT OF INTEREST
The authors certify that they have NO affiliations or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership; employment; consultancies; stock ownership, or other equity interest; and expert testimony or patent licensing arrangements), or nonfinancial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Supporting information
APPENDIX 1.
ABBREVIATIONS
aa: amino acid
AIN: (Human) Autophagy Interacting Network
ATP: Adenosine triphosphate
BHK: baby hamster kidney
bp: base pair
CLN1: ceroid‐lipofuscinosis, neuronal 1
CLN2: ceroid‐lipofuscinosis, neuronal 2
CLN3: ceroid‐lipofuscinosis, neuronal 3
CLN4: ceroid‐lipofuscinosis, neuronal 4
CLN5: ceroid‐lipofuscinosis, neuronal 5
CLN6: ceroid‐lipofuscinosis, neuronal 6
CLN7: ceroid‐lipofuscinosis, neuronal 7
CLN8: ceroid‐lipofuscinosis, neuronal 8
DREAM: downstream regulatory element antagonist modulator
EEA1: early endosome antigen 1
EPMR: Progressive Epilepsy with Mental Retardation
GFP: Green Flourescent Protein
GST: Glutathione S Transferase
GTP: guanosine triphosphate
hp: haptoglobin
INCL: infantile neuronal ceroid lipofuscinosis
JNCL: juvenile Neuronal Ceroid Lipofuscinosis
kb: kilobase
kDa: kilodalton
LINCL: late infantile Neuronal Ceroid Lipofuscinosis
M6P: mannose 6 phosphate
MFS: Major Facilitator Superfamily
MSD: Membrane Spanning Domain
NCBI: National Center for Biotechnology Information
NLM: National Library of Medicine
NRK: Normal Rat Kidney
PDI: protein disulfide isomerase
PKA: protein kinase A
PKG: protein kinase G
PPT1: palmitoyl‐protein thioesterase 1
PTM: post‐translational modification
RFU: relative fluorescent units
S. cerevisiae: schizosaccharomyces cerevisae
S. pombe: schizosaccharomyces pombe
SBDS: Shwachman‐Bodian Diamond Syndrome
SNP: Single Nucleotide Polymorphism
TAM‐MS: Tandem Affinity Purification coupled with Mass Spectrometry
TPP1: Tripeptidyl Peptidase 1
TSS: Transcription Start Site
UTR: Untranslated Region
vLINCL fin: Finnish variant late infantile form of Neuronal Ceroid Lipofuscinosis
Y2H: yeast‐2‐hybrid
Mirza M, Vainshtein A, DiRonza A, et al. The CLN3 gene and protein: What we know. Mol Genet Genomic Med. 2019;7:e859 10.1002/mgg3.859
REFERENCES
- Abdelmohsen, K. , Srikantan, S. , Yang, X. , Lal, A. , Kim, H. H. , Kuwano, Y. , … Gorospe, M. (2009). Ubiquitin‐Mediated proteolysis of HuR by heat shock. The EMBO Journal, 28(9), 1271–1282. 10.1038/emboj.2009.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agola, J. O. , Sivalingam, D. , Cimino, D. F. , Simons, P. C. , Buranda, T. , Sklar, L. A. , & Wandinger‐Ness, A. (2015). Quantitative bead‐based flow cytometry for assaying Rab7 GTPase interaction with the Rab‐Interacting Lysosomal Protein (RILP) effector protein. Methods in Molecular Biology (Clifton, N.J.) 1298: 331–354. 10.1007/978-1-4939-2569-8_28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahtiainen, L. , Van Diggelen, O. P. , Jalanko, A. , & Kopra, O. (2003). Palmitoyl protein thioesterase 1 is targeted to the axons in neurons. The Journal of Comparative Neurology, 455(3), 368–377. 10.1002/cne.10492 [DOI] [PubMed] [Google Scholar]
- Altschul, S. F. , Madden, T. L. , Schäffer, A. A. , Zhang, J. , Zhang, Z. , Miller, W. , & Lipman, D. J. (1997). Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Research, 25(17), 3389–3402. http://www.ncbi.nlm.nih.gov/pubmed/9254694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, W. F. , Bowlby, M. R. , Betty, M. , Cao, J. , Ling, H.‐P. , Mendoza, G. , … Rhodes, K. J. (2000). Modulation of A‐Type potassium channels by a family of calcium sensors. Nature, 403(6769), 553–556. 10.1038/35000592 [DOI] [PubMed] [Google Scholar]
- Andreeva, A. , Howorth, D. , Chandonia, J.‐M. , Brenner, S. E. , Hubbard, T. J. P. , Chothia, C. , & Murzin, A. G. (2008). Data growth and its impact on the SCOP database: New developments. Nucleic Acids Research, 36 (Database issue): D419–D425. 10.1093/nar/gkm993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- anHaack, K. , Narayan, S. B. , Li, H. , Warnock, A. , Tan, L. , & Bennett, M. J. (2011). Screening for calcium channel modulators in CLN3 SiRNA knock down SH‐SY5Y neuroblastoma cells reveals a significant decrease of intracellular calcium levels by selected L‐Type calcium channel blockers. Biochimica et Biophysica Acta (BBA) ‐ General Subjects, 1810(2), 186–191. 10.1016/j.bbagen.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appu, A. P. , Bagh, M. B. , Sadhukkan, T. , Mondal, A. , Casey, S. , & Mukherjee, A. B. (2019). CLN3‐mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of Palmitoyl‐protein thioesterases‐1 (Ppt1)‐protein and Ppt‐enzyme activity in the lysosome. Journal of Inherited Metabolic Disease. 1–11. 10.1002/jimd.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, T. L. , Boden, M. , Buske, F. A. , Frith, M. , Grant, C. E. , Clementi, L. , … Noble, W. S. (2009). MEME SUITE: Tools for Motif discovery and searching. Nucleic Acids Research, 37(Web Server issue), W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, S. A. , Beal, P. R. , Yao, S. Y. M. , King, A. E. , Cass, C. E. , & Young, J. D. (2004). The equilibrative nucleoside transporter family, SLC29. Pflugers Archiv: European Journal of Physiology, 447(5), 735–743. 10.1007/s00424-003-1103-2 [DOI] [PubMed] [Google Scholar]
- Barnett, T. R. , Pickle, W. , & Elting, J. J. (1990). Characterization of two new members of the pregnancy‐specific Beta 1‐Glycoprotein family from the Myeloid Cell Line KG‐1 and suggestion of two distinct classes of transcription unit. Biochemistry, 29(44), 10213–10218. http://www.ncbi.nlm.nih.gov/pubmed/2271648 [DOI] [PubMed] [Google Scholar]
- Barrett, T. , Wilhite, S. E. , Ledoux, P. , Evangelista, C. , Kim, I. F. , Tomashevsky, M. , … Soboleva, A. (2013). NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Research, 41(D1), D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, V. , & Baines, A. J. (2001). Spectrin and Ankyrin‐based pathways: metazoan inventions for integrating cells into tissues. Physiological Reviews, 81(3), 1353–1392. http://www.ncbi.nlm.nih.gov/pubmed/11427698 [DOI] [PubMed] [Google Scholar]
- Bensaoula, T. , Shibuya, H. , Katz, M. L. , Smith, J. E. , Johnson, G. S. , John, S. K. , & Milam, A. H. (2000). Histopathologic and immunocytochemical analysis of the retina and ocular tissues in batten disease. Ophthalmology, 107(9), 1746–1753. http://www.ncbi.nlm.nih.gov/pubmed/10964839 [DOI] [PubMed] [Google Scholar]
- Berman, H. M. , Westbrook, J. , Feng, Z. , Gilliland, G. , Bhat, T. N. , Weissig, H. , … Bourne, P. E. (2000). The protein Data Bank. Nucleic Acids Research, 28(1), 235–242. http://www.ncbi.nlm.nih.gov/pubmed/10592235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto, C. , Lesueur, F. , Giuliano, S. , Strub, T. , de Lichy, M. , Bille, K. , … Bressac‐de Paillerets, B. (2011). A SUMOylation‐defective MITF Germline mutation predisposes to melanoma and renal carcinoma. Nature, 480(7375), 94–98. 10.1038/nature10539 [DOI] [PubMed] [Google Scholar]
- Bieda, M. , Xiaoqin, X. U. , Singer, M. A. , Green, R. , & Farnham, P. J. (2006). Unbiased location analysis of E2F1‐binding sites suggests a widespread role for E2F1 in the human genome. Genome Research, 16(5), 595–605. 10.1101/gr.4887606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin, E. , Liao, H. , Ta, T. C. , Yang, C. , Hwang‐Verslues, W. , Evans, J. R. , … Sladek, F. M. (2010). Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology (Baltimore, MD), 51 (2): 642–653. 10.1002/hep.23357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J. S. , & Traub, L. M. (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annual Review of Biochemistry, 72(1), 395–447. 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- Boriack, R. L. , & Bennett, M. J. (2001). CLN‐3 protein is expressed in the pancreatic somatostatin‐secreting delta cells. European Journal of Paediatric Neurology, 5 (Suppl A), 99–102. 10.1053/ejpn.2001.0478 [DOI] [PubMed] [Google Scholar]
- Boyd, M. , Bressendorff, S. , Møller, J. , Olsen, J. , & Troelsen, J. T. (2009). Mapping of HNF4alpha target genes in intestinal epithelial cells. BMC Gastroenterology, 9(1), 68 10.1186/1471-230X-9-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke, T. , & Bonifacino, J. S. (2009). Sorting of lysosomal proteins. Biochimica et Biophysica Acta, 1793(4), 605–614. 10.1016/j.bbamcr.2008.10.016 [DOI] [PubMed] [Google Scholar]
- Bosch, M. E. , Aldrich, A. , Fallet, R. , Odvody, J. , Burkovetskaya, M. , Schuberth, K. , … Kielian, T. (2016). Self‐complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis (CLN3). Journal of Neuroscience, 36(37), 9669–9682. 10.1523/JNEUROSCI.1635-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkovetskaya, M. , Karpuk, N. , Xiong, J. , Bosch, M. , Boska, M. D. , Takeuchi, H. , … Kielian, T. (2014). Evidence for aberrant astrocyte hemichannel activity in juvenile neuronal ceroid lipofuscinosis (JNCL). PLoS ONE, 9(4), e95023 10.1371/journal.pone.0095023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen, D. F. , Baker, E. , Lane, S. , Nancarrow, J. , Thompson, A. , Whitmore, S. A. , … Järvelä, I. (1991). Regional mapping of the Batten Disease Locus (CLN3) to human chromosome 16p12. American Journal of Human Genetics, 49(6), 1372–1377. http://www.ncbi.nlm.nih.gov/pubmed/1746562 [PMC free article] [PubMed] [Google Scholar]
- Callen, D. F. , Doggett, N. A. , Stallings, R. L. , Chen, L. Z. , Whitmore, S. A. , Lane, S. A. , … Sutherland, G. R. (1992). High‐Resolution cytogenetic‐based physical map of human chromosome 16. Genomics, 13(4), 1178–1185. http://www.ncbi.nlm.nih.gov/pubmed/1505951 [DOI] [PubMed] [Google Scholar]
- Cannelli, N. , Cassandrini, D. , Bertini, E. , Striano, P. , Fusco, L. , Gaggero, R. , … Santorelli, F. M. (2006). Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: Another genetic hit in the mediterranean. Neurogenetics, 7(2), 111–117. 10.1007/s10048-005-0024-y [DOI] [PubMed] [Google Scholar]
- Cao, Y. , Espinola, J. A. , Fossale, E. , Massey, A. C. , Cuervo, A. M. , MacDonald, M. E. , & Cotman, S. L. (2006). Autophagy Is disrupted in a knock‐in mouse model of juvenile neuronal ceroid lipofuscinosis. Journal of Biological Chemistry, 281(29), 20483–20493. 10.1074/jbc.M602180200 [DOI] [PubMed] [Google Scholar]
- Carss, K. J. , Arno, G. , Erwood, M. , Stephens, J. , Sanchis‐Juan, A. , Hull, S. , … Yu, P. (2017). Comprehensive rare variant analysis via whole‐genome sequencing to determine the molecular pathology of inherited retinal disease. American Journal of Human Genetics, 100(1), 75–90. 10.1016/j.ajhg.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey, P. J. (1995). Protein lipidation in cell signaling. Science (New York, N.Y.), 268 (5208), 221–225. http://www.ncbi.nlm.nih.gov/pubmed/7716512 [DOI] [PubMed] [Google Scholar]
- Chan, C.‐H. , Mitchison, H. M. , & Pearce, D. A. (2008). Transcript and in silico analysis of CLN3 in Juvenile neuronal ceroid lipofuscinosis and associated mouse models. Human Molecular Genetics, 17(21), 3332–3339. 10.1093/hmg/ddn228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrachud, U. , Walker, M. W. , Simas, A. M. , Heetveld, S. , Petcherski, A. , Klein, M. , … Cotman, S. L. (2015). Unbiased cell‐based screening in a neuronal cell model of batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. The Journal of Biological Chemistry, 290(23), 14361–14380. 10.1074/jbc.M114.621706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, J.‐W. , Choi, H. , Kim, H.‐J. , Jo, D.‐G. , Jeon, Y.‐J. , Noh, J.‐Y. , … Jung, Y.‐K. (2007). Neuronal vulnerability of CLN3 deletion to calcium‐induced cytotoxicity is mediated by calsenilin. Human Molecular Genetics, 16(3), 317–326. 10.1093/hmg/ddl466 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, S. , & Pearce, D. A. (2000). Neural and extraneural expression of the neuronal ceroid lipofuscinoses genes CLN1, CLN2, and CLN3: Functional implications for CLN3. Molecular Genetics and Metabolism, 71(1–2), 207–211. 10.1006/mgme.2000.3056 [DOI] [PubMed] [Google Scholar]
- Mao, D. , Che, J. , Han, S. , Zhao, H. , Zhu, Y. , & Zhu, H. (2015). RNAi‐mediated knockdown of the CLN3 gene inhibits proliferation and promotes apoptosis in drug‐resistant ovarian cancer cells. Molecular Medicine Reports, 12(5), 6635–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. I. , Xu, H. , Yuan, P. , Fang, F. , Huss, M. , Vega, V. B. , … Ng, H.‐H. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell, 133(6), 1106–1117. 10.1016/j.cell.2008.04.043 [DOI] [PubMed] [Google Scholar]
- Cho, S. , & Dawson, G. (1998). Enzymatic and molecular biological analysis of palmitoyl protein thioesterase deficiency in infantile neuronal ceroid lipofuscinosis. Journal of Neurochemistry, 71(1), 323–329. http://www.ncbi.nlm.nih.gov/pubmed/9648881 [DOI] [PubMed] [Google Scholar]
- Cho, S. , Dawson, P. E. , & Dawson, G. (2000). In Vitro Depalmitoylation of neurospecific peptides: Implication for infantile neuronal ceroid lipofuscinosis. Journal of Neuroscience Research, 59(1), 32–38. http://www.ncbi.nlm.nih.gov/pubmed/10658183 [PubMed] [Google Scholar]
- Codlin, S. , Haines, R. L. , & Mole, S. E. (2008). btn1 affects endocytosis, polarization of sterol‐rich membrane domains and polarized growth in Schizosaccharomyces pombe . Traffic, 9(6), 936–950. 10.1111/j.1600-0854.2008.00735.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codlin, S. , & Mole, S. E. (2009). S. Pombe Btn1, the orthologue of the batten disease gene CLN3, is required for vacuole protein sorting of Cpy1p and Golgi Exit of Vps10p. Journal of Cell Science, 122(8), 1163–1173. 10.1242/jcs.038323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters, F. , Van Schil, K. , Bauwens, M. , Verdin, H. , De Jaegher, A. , Syx, D. , … De Baere, E. (2014). Identity‐by‐Descent–guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genetics in Medicine, 16(9), 671–680. 10.1038/gim.2014.24 [DOI] [PubMed] [Google Scholar]
- Cortese, A. , Tucci, A. , Piccolo, G. , Galimberti, C. A. , Fratta, P. , Marchioni, E. , … Moggio, M. (2014). Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology, 82(23), 2072–2076. 10.1212/WNL.0000000000000490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman, S. L. , & Staropoli, J. F. (2012). The Juvenile batten disease protein, CLN3, and its role in regulating anterograde and retrograde Post‐Golgi Trafficking. Clinical Lipidology, 7(1), 79–91. 10.2217/clp.11.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman, S. L. , Vrbanac, V. , Lebel, L.‐A. , Lee, R. L. , Johnson, K. A. , Donahue, L.‐R. , … MacDonald, M. E. (2002). Cln3(Deltaex7/8) knock‐in Mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Human Molecular Genetics, 11(22), 2709–2721. http://www.ncbi.nlm.nih.gov/pubmed/12374761 [DOI] [PubMed] [Google Scholar]
- Croopnick, J. B. , Choi, H. C. , & Mueller, D. M. (1998). The subcellular location of the yeast saccharomyces cerevisiae homologue of the protein defective in the juvenile form of batten disease. Biochemical and Biophysical Research Communications, 250(2), 335–341. 10.1006/bbrc.1998.9272 [DOI] [PubMed] [Google Scholar]
- de los Reyes, E. , Dyken, P. R. , Phillips, P. , Brodsky, M. , Bates, S. , Glasier, C. , & Mrak, R. E. (2004). Profound infantile neuroretinal dysfunction in a heterozygote for the CLN3 genetic defect. Journal of Child Neurology, 19(1), 42–46. 10.1177/08830738040190010703 [DOI] [PubMed] [Google Scholar]
- Decraene, C. , Brugg, B. , Ruberg, M. , Eveno, E. , Matingou, C. , Tahi, F. , … Pietu, G. (2002). Identification of genes involved in ceramide‐dependent neuronal apoptosis using CDNA arrays. Genome Biology, 3 (8), RESEARCH0042 http://www.ncbi.nlm.nih.gov/pubmed/12186649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, S.‐L. , Tecedor, L. , Stein, C. S. , & Davidson, B. L. (2011). A Knock‐in reporter mouse model for batten disease reveals predominant expression of Cln3 in visual, limbic and subcortical motor structures. Neurobiology of Disease, 41(2), 237–248. 10.1016/j.nbd.2010.09.011 [DOI] [PubMed] [Google Scholar]
- Dhar, S. , Bitting, R. L. , Rylova, S. N. , Jansen, P. J. , Lockhart, E. , Koeberl, D. D. , … Boustany, R. M. (2002). Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3‐ and CLN2‐deficient neurons. Annals of Neurology, 51(4), 448–466. [DOI] [PubMed] [Google Scholar]
- Dobzinski, N. , Chuartzman, S. G. , Kama, R. , Schuldiner, M. , & Gerst, J. E. (2015). Starvation‐dependent regulation of golgi quality control links the TOR signaling and vacuolar protein sorting pathways. Cell Reports, 12(11), 1876–1886. 10.1016/j.celrep.2015.08.026 [DOI] [PubMed] [Google Scholar]
- Donadieu, J. , Michel, G. , Merlin, E. , Bordigoni, P. , Monteux, B. , Beaupain, B. , … Stephan, J. L. (2005). Hematopoietic stem cell transplantation for shwachman‐diamond syndrome: Experience of the French neutropenia registry. Bone Marrow Transplantation, 36(9), 787–792. 10.1038/sj.bmt.1705141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drack, A. V. , Miller, J. N. , & Pearce, D. A. (2013). A Novel c.1135_1138delCTGT mutation in CLN3 leads to juvenile neuronal ceroid lipofuscinosis. Journal of Child Neurology, 28(9), 1112–1116. 10.1177/0883073813494812 [DOI] [PubMed] [Google Scholar]
- Eiberg, H. , Gardiner, R. M. , & Mohr, J. (1989). Batten disease (Spielmeyer‐Sjøgren Disease) and Haptoglobins (HP): Indication of linkage and assignment to Chr. 16. Clinical Genetics, 36(4), 217–218. http://www.ncbi.nlm.nih.gov/pubmed/2805379 [DOI] [PubMed] [Google Scholar]
- Ekins, S. , Nikolsky, Y. , Bugrim, A. , Kirillov, E. , & Nikolskaya, T. (2007). Pathway mapping tools for analysis of high content data. Methods in Molecular Biology (Clifton, N.J.), 356, 319–350 http://www.ncbi.nlm.nih.gov/pubmed/16988414 [DOI] [PubMed] [Google Scholar]
- Eksandh, L. B. , Ponjavic, V. B. , Munroe, P. B. , Eiberg, H. E. , Uvebrant, P. E. , Ehinger, B. E. , … Andréasson, S. (2000). Full‐Field ERG in patients with Batten/Spielmeyer‐Vogt disease caused by mutations in the CLN3 gene. Ophthalmic Genetics, 21(2), 69–77. http://www.ncbi.nlm.nih.gov/pubmed/10916181 [PubMed] [Google Scholar]
- Eliason, S. L. , Stein, C. S. , Mao, Q. , Tecedor, L. , Song‐Lin Ding, D. , Gaines, M. , & Davidson, B. L. (2007). A knock‐in reporter model of batten disease. The Journal of Neuroscience, 27(37), 9826–9834. 10.1523/JNEUROSCI.1710-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard, L. , & Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nature Reviews Molecular Cell Biology, 4(3), 181–191. 10.1038/nrm1052 [DOI] [PubMed] [Google Scholar]
- Ezaki, J. , Takeda‐Ezaki, M. , Koike, M. , Ohsawa, Y. , Taka, H. , Mineki, R. , … Kominami, E. (2003). Characterization of Cln3p, the gene product responsible for juvenile neuronal ceroid lipofuscinosis, as a lysosomal integral membrane glycoprotein. Journal of Neurochemistry, 87(5), 1296–1308. http://www.ncbi.nlm.nih.gov/pubmed/14622109 [DOI] [PubMed] [Google Scholar]
- Fossale, E. , Wolf, P. , Espinola, J. A. , Lubicz‐Nawrocka, T. , Teed, A. M. , Gao, H. , … Cotman, S. L. (2004). Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BioMed Central Neuroscience, 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet, Y. , Codlin, S. , Hyams, J. S. , & Mole, S. E. (2005). Btn1, the Schizosaccharomyces Pombe Homologue of the human batten disease gene CLN3, regulates vacuole homeostasis. Journal of Cell Science, 118(Pt 23), 5525–5536. 10.1242/jcs.02656 [DOI] [PubMed] [Google Scholar]
- Gardiner, M. , Sandford, A. , Deadman, M. , Poulton, J. , Cookson, W. , Reeders, S. , … Julier, C. (1990). Batten disease (Spielmeyer‐Vogt Disease, Juvenile Onset Neuronal Ceroid‐Lipofuscinosis) gene (CLN3) maps to human chromosome 16. Genomics, 8(2), 387–390. http://www.ncbi.nlm.nih.gov/pubmed/2249854 [DOI] [PubMed] [Google Scholar]
- Getty, A. L. , Benedict, J. W. , & Pearce, D. A. (2011). A novel interaction of CLN3 with nonmuscle Myosin‐IIB and defects in cell motility of Cln3(‐/‐) cells. Experimental Cell Research, 317(1), 51–69. 10.1016/j.yexcr.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti, M. , & Banting, G. (1996, December). TGN38‐Green fluorescent protein hybrid proteins expressed in stably transfected eukaryotic cells provide a tool for the real‐time, in vivo study of membrane traffic pathways and suggest a possible role for RatTGN38. Journal of Cell Science, 109, 2915–2926. http://www.ncbi.nlm.nih.gov/pubmed/9013339 [DOI] [PubMed] [Google Scholar]
- Golabek, A. A. , Kaczmarski, W. , Kida, E. , Kaczmarski, A. , Michalewski, M. P. , & Wisniewski, K. E. (1999). Expression studies of CLN3 Protein (Battenin) in fusion with the green fluorescent protein in mammalian cellsin Vitro. Molecular Genetics and Metabolism, 66(4), 277–282. 10.1006/mgme.1999.2836 [DOI] [PubMed] [Google Scholar]
- Golabek, A. A. , Kida, E. , Walus, M. , Kaczmarski, W. , Michalewski, M. , & Wisniewski, K. E. (2000). CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid‐beta protein precursor and cathepsin D in human cells. Molecular Genetics and Metabolism, 70(3), 203–213. [DOI] [PubMed] [Google Scholar]
- Golabek, A. A. , Kida, E. , Walus, M. , Wujek, P. , Mehta, P. , & Wisniewski, K. E. (2003). Biosynthesis, glycosylation, and enzymatic processing in vivo of human Tripeptidyl‐Peptidase I. The Journal of Biological Chemistry, 278(9), 7135–7145. 10.1074/jbc.M211872200 [DOI] [PubMed] [Google Scholar]
- Golabek, A. A. , Wujek, P. , Walus, M. , Bieler, S. , Soto, C. , Wisniewski, K. E. , & Kida, E. (2004). Maturation of human Tripeptidyl‐Peptidase I in vitro. The Journal of Biological Chemistry, 279(30), 31058–31067. 10.1074/jbc.M400700200 [DOI] [PubMed] [Google Scholar]
- Goyal, S. , Jäger, M. , Robinson, P. N. , & Vanita, V. (2016). Confirmation of TTC8 as a disease gene for nonsyndromic autosomal Recessive Retinitis Pigmentosa (RP51). Clinical Genetics, 89(4), 454–460. 10.1111/cge.12644 [DOI] [PubMed] [Google Scholar]
- Haeussler, M. , Zweig, A. S. , Tyner, C. , Speir, M. L. , Rosenbloom, K. R. , Raney, B. J. , … Kent, W. J. (2019). The UCSC genome browser database: 2019 update. Nucleic Acids Research, 47(D1), D853–D858. 10.1093/nar/gky1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia, M. , Herva, R. , Suopanki, J. , Baumann, M. , & Tyynelä, J. (2001). Hippocampal lesions in the neuronal ceroid lipofuscinoses. European Journal of Paediatric Neurology, 5 (Suppl A), 209–211. http://www.ncbi.nlm.nih.gov/pubmed/11588999 [DOI] [PubMed] [Google Scholar]
- Haskell, R. E. , Carr, C. J. , Pearce, D. A. , Bennett, M. J. , & Davidson, B. L. (2000). Batten disease: Evaluation of CLN3 mutations on protein localization and function. Human Molecular Genetics, 9(5), 735–744. http://www.ncbi.nlm.nih.gov/pubmed/10749980 [DOI] [PubMed] [Google Scholar]
- Haskell, R. E. , Derksen, T. A. , & Davidson, B. L. (1999). Intracellular trafficking of the JNCL protein CLN3. Molecular Genetics and Metabolism, 66(4), 253–260. 10.1006/mgme.1999.2802 [DOI] [PubMed] [Google Scholar]
- Heine, C. , Heine, C. , Quitsch, A. , Storch, S. , Martin, Y. , Lonka, L. , … Braulke, T. (2007). Topology and endoplasmic reticulum retention signals of the lysosomal storage disease‐related membrane protein CLN6. Molecular Membrane Biology, 24(1), 74–87. 10.1080/09687860600967317 [DOI] [PubMed] [Google Scholar]
- Heine, C. , Koch, B. , Storch, S. , Kohlschütter, A. , Palmer, D. N. , & Braulke, T. (2004). Defective endoplasmic reticulum‐resident membrane protein CLN6 affects lysosomal degradation of endocytosed Arylsulfatase A. The Journal of Biological Chemistry, 279(21), 22347–22352. 10.1074/jbc.M400643200 [DOI] [PubMed] [Google Scholar]
- Heissler, S. M. , & Manstein, D. J. (2013). Nonmuscle Myosin‐2: Mix and match. Cellular and Molecular Life Sciences, 70(1), 1–21. 10.1007/s00018-012-1002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten, E. , Vesa, J. , Olkkonen, V. M. , Jalanko, A. , & Peltonen, L. (1996). Human palmitoyl protein thioesterase: Evidence for lysosomal targeting of the enzyme and disturbed cellular routing in infantile neuronal ceroid lipofuscinosis. The EMBO Journal, 15(19), 5240–5245. http://www.ncbi.nlm.nih.gov/pubmed/8895569 [PMC free article] [PubMed] [Google Scholar]
- Herva, R. , Tyynelä, J. , Hirvasniemi, A. , Syrjäkallio‐Ylitalo, M. , & Haltia, M. (2000). Northern epilepsy: A novel form of neuronal ceroid‐lipofuscinosis. Brain Pathology (Zurich, Switzerland), 10(2), 215–222. http://www.ncbi.nlm.nih.gov/pubmed/10764041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvasniemi, A. , Herrala, P. , & Leisti, J. (1995). Northern epilepsy syndrome: Clinical course and the effect of medication on seizures. Epilepsia, 36(8), 792–797. http://www.ncbi.nlm.nih.gov/pubmed/7635097 [DOI] [PubMed] [Google Scholar]
- Hirvasniemi, A. , & Karumo, J. (1994). Neuroradiological findings in the northern epilepsy syndrome. Acta Neurologica Scandinavica, 90(6), 388–393. http://www.ncbi.nlm.nih.gov/pubmed/7892756 [DOI] [PubMed] [Google Scholar]
- Hirvasniemi, A. , Lang, H. , Lehesjoki, A. E. , & Leisti, J. (1994). Northern epilepsy syndrome: An inherited childhood onset epilepsy with associated mental deterioration. Journal of Medical Genetics, 31(3), 177–182. http://www.ncbi.nlm.nih.gov/pubmed/8014963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst, P. C. , Chandler, K. J. , Poulsen, R. L. , Evan Johnson, W. , Speck, N. A. , & Graves, B. J. (2009). DNA specificity determinants associate with distinct transcription factor functions. Edited by Michael Snyder. Plos Genetics, 5(12), e1000778 10.1371/journal.pgen.1000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg, V. , Jalanko, A. , Isosomppi, J. , Fabritius, A.‐L. , Peltonen, L. , & Kopra, O. (2004). The mouse ortholog of the neuronal ceroid lipofuscinosis CLN5 gene encodes a soluble lysosomal glycoprotein expressed in the developing brain. Neurobiology of Disease, 16(1), 29–40. 10.1016/j.nbd.2003.12.019 [DOI] [PubMed] [Google Scholar]
- Holmberg, V. , Lauronen, L. , Autti, T. , Santavuori, P. , Savukoski, M. , Uvebrant, P. , … Järvelä, I. (2000). Phenotype‐genotype correlation in eight patients with finnish variant late infantile NCL (CLN5). Neurology, 55(4), 579–581. http://www.ncbi.nlm.nih.gov/pubmed/10953198 [DOI] [PubMed] [Google Scholar]
- Holopainen, J. M. , Saarikoski, J. , Kinnunen, P. K. , & Järvelä, I. (2001). Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs). European Journal of Biochemistry, 268(22), 5851–5856. [DOI] [PubMed] [Google Scholar]
- Höning, S. , Sandoval, I. V. , & von Figura, K. (1998). A Di‐Leucine‐based motif in the cytoplasmic tail of LIMP‐II and tyrosinase mediates selective binding of AP‐3. The EMBO Journal, 17(5), 1304–1314. 10.1093/emboj/17.5.1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- In, K. , Zaini, M. A. , Müller, C. , Warren, A. J. , von Lindern, M. , & Calkhoven, C. F. (2016). Shwachman‐Bodian‐Diamond Syndrome (SBDS) protein deficiency impairs translation re‐initiation from C/EBPα and C/EBPβ MRNAs. Nucleic Acids Research, 44(9), 4134–4146. 10.1093/nar/gkw005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Batten Disease Consortium . (1995). Isolation of a novel gene underlying batten disease, CLN3. The international batten disease consortium. Cell, 82(6), 949–957. http://www.ncbi.nlm.nih.gov/pubmed/7553855 [DOI] [PubMed] [Google Scholar]
- Isosomppi, J. , Vesa, J. , Jalanko, A. , & Peltonen, L. (2002). Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Human Molecular Genetics, 11(8), 885–891. http://www.ncbi.nlm.nih.gov/pubmed/11971870 [DOI] [PubMed] [Google Scholar]
- Jalanko, H. , Patrakka, J. , Tryggvason, K. , & Holmberg, C. (2001). Genetic kidney diseases disclose the pathogenesis of proteinuria. Annals of Medicine, 33(8), 526–533. http://www.ncbi.nlm.nih.gov/pubmed/11730159 [DOI] [PubMed] [Google Scholar]
- Janes, R. W. , Munroe, P. B. , Mitchison, H. M. , Gardiner, R. M. , Mole, S. E. , & Wallace, B. A. (1996). A model for batten disease protein CLN3: Functional implications from homology and mutations. FEBS Letters, 399(1–2), 75–77. http://www.ncbi.nlm.nih.gov/pubmed/8980123 [DOI] [PubMed] [Google Scholar]
- Järvelä, I. , Autti, T. , Lamminranta, S. , Aberg, L. , Raininko, R. , & Santavuori, P. (1997). Clinical and magnetic resonance imaging findings in batten disease: Analysis of the major mutation (1.02‐Kb Deletion). Annals of Neurology, 42(5), 799–802. 10.1002/ana.410420517 [DOI] [PubMed] [Google Scholar]
- Järvelä, I. , Lehtovirta, M. , Tikkanen, R. , Kyttälä, A. , & Jalanko, A. (1999). Defective intracellular transport of CLN3 is the molecular basis of batten disease (JNCL). Human Molecular Genetics, 8(6), 1091–1098. http://www.ncbi.nlm.nih.gov/pubmed/10332042 [DOI] [PubMed] [Google Scholar]
- Järvelä, I. E. , Mitchison, H. M. , Callen, D. F. , Lerner, T. J. , Doggett, N. A. , Taschner, P. E. , … Mole, S. E. (1995). Physical map of the region containing the gene for batten disease (CLN3). American Journal of Medical Genetics, 57(2), 316–319. 10.1002/ajmg.1320570242 [DOI] [PubMed] [Google Scholar]
- Järvelä, I. E. , Mitchison, H. M. , O'rawe, A. M. , Munroe, P. B. , Taschner, P. E. , Devos, N. , … Mole, S. E. (1995). YAC and cosmid contigs spanning the batten disease (CLN3) Region at 16p12.1‐P11.2. Genomics, 29(2), 478–489. http://www.ncbi.nlm.nih.gov/pubmed/8666398 [DOI] [PubMed] [Google Scholar]
- Järvelä, I. , Sainio, M. , Rantamäki, T. , Olkkonen, V. M. , Carpén, O. , Peltonen, L. , & Jalanko, A. (1998). Biosynthesis and intracellular targeting of the CLN3 protein defective in batten disease. Human Molecular Genetics, 7(1), 85–90. http://www.ncbi.nlm.nih.gov/pubmed/9384607 [DOI] [PubMed] [Google Scholar]
- Jin, V. X. , Rabinovich, A. , Squazzo, S. L. , Green, R. , & Farnham, P. J. (2006). A computational genomics approach to identify Cis‐regulatory modules from chromatin immunoprecipitation microarray Data–a case study using E2F1. Genome Research, 16(12), 1585–1595. 10.1101/gr.5520206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D. W. , Speier, S. , Qian, W. H. , Lane, S. , Cook, A. , Suzuki, K. , … Boustany, R. M. (1995). Role of Subunit‐9 of mitochondrial ATP synthase in batten disease. American Journal of Medical Genetics, 57(2), 350–360. 10.1002/ajmg.1320570250 [DOI] [PubMed] [Google Scholar]
- Kaczmarski, W. , Wisniewski, K. E. , Golabek, A. , Kaczmarski, A. , Kida, E. , & Michalewski, M. (1999). Studies of membrane association of CLN3 protein. Molecular Genetics and Metabolism, 66(4), 261–264. 10.1006/mgme.1999.2833 [DOI] [PubMed] [Google Scholar]
- Kama, R. , Kanneganti, V. , Ungermann, C. , & Gerst, J. E. (2011). The yeast batten disease orthologue Btn1 controls Endosome‐Golgi retrograde transport via SNARE assembly. The Journal of Cell Biology, 195(2), 203–215. 10.1083/jcb.201102115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, M. L. , Shibuya, H. , Liu, P. C. , Kaur, S. , Gao, C. L. , & Johnson, G. S. (1999). A mouse gene knockout model for Juvenile Ceroid‐Lipofuscinosis (Batten Disease). Journal of Neuroscience Research, 57(4), 551–556. http://www.ncbi.nlm.nih.gov/pubmed/10440905 [PubMed] [Google Scholar]
- Kent, W. J. , Sugnet, C. W. , Furey, T. S. , Roskin, K. M. , Pringle, T. H. , Zahler, A. M. , & Haussler, D. (2002). The Human Genome Browser at UCSC. Genome Research, 12(6), 996–1006. 10.1101/gr.229102. Article published online before print in May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida, E. , Kaczmarski, W. , Golabek, A. A. , Kaczmarski, A. , Michalewski, M. , & Wisniewski, K. E. (1999). Analysis of intracellular distribution and trafficking of the CLN3 protein in fusion with the green fluorescent proteinin vitro. Molecular Genetics and Metabolism, 66(4), 265–271. 10.1006/mgme.1999.2837 [DOI] [PubMed] [Google Scholar]
- Kim, B. , Kang, S. , & Kim, S. J. (2013). Genome‐wide pathway analysis reveals different signaling pathways between secreted lactoferrin and intracellular delta‐lactoferrin. Edited by Francisco Jos? Esteban. PLoS ONE, 8(1), e55338 10.1371/journal.pone.0055338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzmüller, C. , Haines, R. L. , Codlin, S. , Cutler, D. F. , & Mole, S. E. (2008). A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Human Molecular Genetics, 17(2), 303–312. 10.1093/hmg/ddm306 [DOI] [PubMed] [Google Scholar]
- Kousi, M. , Lehesjoki, A.‐E. , & Mole, S. E. (2012). Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Human Mutation, 33(1), 42–63. 10.1002/humu.21624 [DOI] [PubMed] [Google Scholar]
- Kremmidiotis, G. , Lensink, I. L. , Bilton, R. L. , Woollatt, E. , Chataway, T. K. , Sutherland, G. R. , & Callen, D. F. (1999). The Batten Disease Gene Product (CLN3p) is a golgi integral membrane protein. Human Molecular Genetics, 8(3), 523–531. http://www.ncbi.nlm.nih.gov/pubmed/9949212 [DOI] [PubMed] [Google Scholar]
- Ku, C. A. , Hull, S. , Arno, G. , Vincent, A. , Carss, K. , Kayton, R. , … Pennesi, M. E. (2017). Detailed clinical phenotype and molecular genetic findings in CLN3‐associated isolated retinal degeneration. JAMA Ophthalmology, 135(7), 749–760. 10.1001/jamaophthalmol.2017.1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, J. M. , Rothberg, P. G. , Leman, A. R. , Weimer, J. M. , Mink, J. W. , & Pearce, D. A. (2005). Novel CLN3 mutation predicted to cause complete loss of protein function does not modify the classical JNCL phenotype. Neuroscience Letters, 387(2), 111–114. 10.1016/j.neulet.2005.07.023 [DOI] [PubMed] [Google Scholar]
- Kyttälä, A. , Ihrke, G. , Vesa, J. , Schell, M. J. , Paul, J. , & Luzio., (2004). Two motifs target batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Molecular Biology of the Cell, 15(3), 1313–1323. 10.1091/mbc.E03-02-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttälä, A. , Yliannala, K. , Schu, P. , Jalanko, A. , Paul, J. , & Luzio., (2005). AP‐1 and AP‐3 facilitate lysosomal targeting of batten disease protein CLN3 via its Dileucine Motif. The Journal of Biological Chemistry, 280(11), 10277–10283. 10.1074/jbc.M411862200 [DOI] [PubMed] [Google Scholar]
- Lane, S. C. , Jolly, R. D. , Schmechel, D. E. , Alroy, J. , & Boustany, R. M. (1996). Apoptosis as the mechanism of degeneration in Batten’s disease. Journal of Neurochemistry, 67(2), 677–683. [DOI] [PubMed] [Google Scholar]
- Lauronen, L. , Munroe, P. B. , Jarvela, I. , Autti, T. , Mitchison, H. M. , O'Rawe, A. M. , … Santavuori, P. (1999). Delayed classic and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis. Neurology, 52(2), 360–365. http://www.ncbi.nlm.nih.gov/pubmed/9932957 [DOI] [PubMed] [Google Scholar]
- Lebrun, A.‐H. , Storch, S. , Rüschendorf, F. , Schmiedt, M.‐L. , Kyttälä, A. , Mole, S. E. , … Schulz, A. (2009). Retention of lysosomal protein CLN5 in the endoplasmic reticulum causes neuronal ceroid lipofuscinosis in Asian sibship. Human Mutation, 30(5), E651–E661. 10.1002/humu.21010 [DOI] [PubMed] [Google Scholar]
- Lehtovirta, M. , Kyttälä, A. , Eskelinen, E. L. , Hess, M. , Heinonen, O. , & Jalanko, A. (2001). Palmitoyl Protein Thioesterase (PPT) localizes into synaptosomes and synaptic vesicles in neurons: Implications for Infantile Neuronal Ceroid Lipofuscinosis (INCL). Human Molecular Genetics, 10(1), 69–75. http://www.ncbi.nlm.nih.gov/pubmed/11136716 [DOI] [PubMed] [Google Scholar]
- Leman, A. R. , Pearce, D. A. , & Rothberg, P. G. (2005). Gene symbol: CLN3. Disease: Juvenile neuronal ceroid lipofuscinosis (Batten Disease). Human Genetics, 116(6), 544 http://www.ncbi.nlm.nih.gov/pubmed/15991331 [PubMed] [Google Scholar]
- Lerner, T. J. , Boustany, R. M. , MacCormack, K. , Gleitsman, J. , Schlumpf, K. , Breakefield, X. O. , … Haines, J. L. (1994). Linkage disequilibrium between the juvenile neuronal ceroid lipofuscinosis gene and marker Loci on chromosome 16p 12.1. American Journal of Human Genetics, 54(1), 88–94. http://www.ncbi.nlm.nih.gov/pubmed/8279474 [PMC free article] [PubMed] [Google Scholar]
- Leung, K. Y. , Greene, N. D. , Munroe, P. B. , & Mole, S. E. (2001). Analysis of CLN3‐Protein interactions using the yeast two‐hybrid system. European Journal of Paediatric Neurology, 5 Suppl A (2), 89–93. 10.1053/ejpn.2001.0472 [DOI] [PubMed] [Google Scholar]
- Licchetta, L. , Bisulli, F. , Fietz, M. , Valentino, M. L. , Morbin, M. , Mostacci, B. , … Tinuper, P. (2015). A novel mutation Af Cln3 associated with delayed‐classic juvenile ceroid lipofuscinois and autophagic vacuolar myopathy. European Journal of Medical Genetics, 58(10), 540–544. 10.1016/j.ejmg.2015.09.002 [DOI] [PubMed] [Google Scholar]
- Lingrel, J. B. , Van Huysse, J. , O’Brien, W. , Jewell‐Motz, E. , Askew, R. , & Schultheis, P. (1994). Structure‐function studies of the Na, K‐ATPase. Kidney International. Supplement, 44(January), S32–S39. http://www.ncbi.nlm.nih.gov/pubmed/8127032 [PubMed] [Google Scholar]
- Lonka, L. , Kyttälä, A. , Ranta, S. , Jalanko, A. , & Lehesjoki, A. E. (2000). The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Human Molecular Genetics, 9(11), 1691–1697. http://www.ncbi.nlm.nih.gov/pubmed/10861296 [DOI] [PubMed] [Google Scholar]
- Lonsdale, J. , Thomas, J. , Salvatore, M. , Phillips, R. , Lo, E. , Shad, S. , … Moore, H. F. (2013). The Genotype‐tissue expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiro, K. , Kopra, O. , Blom, T. , Gentile, M. , Mitchison, H. M. , Hovatta, I. , … Jalanko, A. (2006). Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments. Journal of Neuroscience Research, 84(5), 1124–1138. [DOI] [PubMed] [Google Scholar]
- Luiro, K. , Kopra, O. , Lehtovirta, M. , & Jalanko, A. (2001). CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: new clues to batten disease. Human Molecular Genetics, 10(19), 2123–2131. http://www.ncbi.nlm.nih.gov/pubmed/11590129 [DOI] [PubMed] [Google Scholar]
- Luiro, K. , Yliannala, K. , Ahtiainen, L. , Maunu, H. , Järvelä, I. , Kyttälä, A. , & Jalanko, A. (2004). Interconnections of CLN3, Hook1 and Rab proteins link batten disease to defects in the endocytic pathway. Human Molecular Genetics, 13(23), 3017–3027. 10.1093/hmg/ddh321 [DOI] [PubMed] [Google Scholar]
- Luz, J. S. , Georg, R. C. , Gomes, C. H. , Machado‐Santelli, G. M. , & Oliveira, C. C. (2009). Sdo1p, the yeast orthologue of shwachman‐bodian‐diamond syndrome protein, binds RNA and interacts with nuclear RRNA‐processing factors. Yeast (Chichester, England), 26(5), 287–298. 10.1002/yea.1668 [DOI] [PubMed] [Google Scholar]
- Lyly, A. , von Schantz, C. , Heine, C. , Schmiedt, M.‐L. , Sipilä, T. , Jalanko, A. , & Kyttälä, A. (2009). Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biology, 10(1), 83 10.1186/1471-2121-10-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makoukji, J. , Raad, M. , Genadry, K. , El‐Sitt, S. , Makhoul, N. J. , Saad Aldin E., … Boustany, R. M. (2015). Association between CLN3 (Neuronal Ceroid Lipofuscinosis, CLN3 Type) Gene expression and clinical characteristics of breast cancer patients. Frontiers in Oncology, 5, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Q. , Foster, B. J. , Xia, H. , & Davidson, B. L. (2003). Membrane Topology of CLN3, the protein underlying batten disease. FEBS Letters, 541(1–3), 40–46. http://www.ncbi.nlm.nih.gov/pubmed/12706816 [DOI] [PubMed] [Google Scholar]
- Mao, Q. , Xia, H. , & Davidson, B. L. (2003). Intracellular trafficking of CLN3, the protein underlying the childhood neurodegenerative disease, batten disease. FEBS Letters, 555(2), 351–357. http://www.ncbi.nlm.nih.gov/pubmed/14644441 [DOI] [PubMed] [Google Scholar]
- Marger, M. D. , & Saier, M. H. (1993). a major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends in Biochemical Sciences, 18(1), 13–20. http://www.ncbi.nlm.nih.gov/pubmed/8438231 [DOI] [PubMed] [Google Scholar]
- Margraf, L. R. , Boriack, R. L. , Routheut, A. A. J. , Cuppen, I. , Alhilali, L. , Bennett, C. J. , & Bennett, M. J. (1999). Tissue expression and subcellular localization of CLN3, the batten disease protein. Molecular Genetics and Metabolism, 66(4), 283–289. 10.1006/mgme.1999.2830 [DOI] [PubMed] [Google Scholar]
- Metcalf, D. J. , Calvi, A. A. , Seaman, M. N. J. , Mitchison, H. M. , & Cutler, D. F. (2008). Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannos 6‐phosphate receptor. Traffic. 9(11), 1905–1914. 10.1111/j.1600-0854.2008.00807.x [DOI] [PubMed] [Google Scholar]
- Michalewski, M. P. , Kaczmarski, W. , Golabek, A. A. , Kida, E. , Kaczmarski, A. , & Wisniewski, K. E. (1998). Evidence for phosphorylation of CLN3 protein associated with batten disease. Biochemical and Biophysical Research Communications, 253(2), 458–462. 10.1006/bbrc.1998.9210 [DOI] [PubMed] [Google Scholar]
- Michalewski, M. P. , Kaczmarski, W. , Golabek, A. A. , Kida, E. , Kaczmarski, A. , & Wisniewski, K. E. (1999). Posttranslational modification of CLN3 protein and its possible functional implication. Molecular Genetics and Metabolism, 66(4), 272–276. 10.1006/mgme.1999.2818 [DOI] [PubMed] [Google Scholar]
- Miller, J. N. , Chan, C.‐H. , & Pearce, D. A. (2013). The role of nonsense‐mediated decay in neuronal ceroid lipofuscinosis. Human Molecular Genetics, 22(13), 2723–2734. 10.1093/hmg/ddt120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, W. A. , Porter, M. , Kuwabara, P. , & Mole, S. E. (2001). Genomic structure of three CLN3‐like genes in caenorhabditis elegans. European Journal of Paediatric Neurology, 5 (Suppl A), 121–125, http://www.ncbi.nlm.nih.gov/pubmed/11588982 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , Bernard, D. J. , Greene, N. D. E. , Cooper, J. D. , Junaid, M. A. , Pullarkat, R. K. , … Nussbaum, R. L. (1999). Targeted disruption of the Cln3 gene provides a mouse model for batten disease. The Batten Mouse Model Consortium [Corrected]. Neurobiology of Disease, 6(5), 321–334. 10.1006/nbdi.1999.0267 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , Munroe, P. B. , O'Rawe, A. M. , Taschner, P. E. M. , de Vos, N. , Kremmidiotis, G. , … Mole, S. E. (1997). Genomic structure and complete nucleotide sequence of the batten disease gene, CLN3. Genomics, 40(2), 346–350. 10.1006/geno.1996.4576 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , O’Rawe, A. M. , Taschner, P. E. , Sandkuijl, L. A. , Santavuori, P. , de Vos, N. , … Järvelä, I. E. (1995). Batten disease gene, CLN3: Linkage disequilibrium mapping in the finnish population, and analysis of European Haplotypes. American Journal of Human Genetics, 56(3), 654–662. http://www.ncbi.nlm.nih.gov/pubmed/7887419 [PMC free article] [PubMed] [Google Scholar]
- Mitchison, H. M. , O'Rawe, A. M. , Lerner, T. J. , Taschner, P. E. M. , Schlumpf, K. , D'Arigo, K. , … Mole, S. E. (1995). Refined localization of the batten disease gene (CLN3) by haplotype and linkage disequilibrium mapping to D16S288‐D16S383 and exclusion from this region of a variant form of batten disease with granular osmiophilic deposits. American Journal of Medical Genetics, 57(2), 312–315. 10.1002/ajmg.1320570241 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , Taschner, P. E. M. , O'Rawe, A. M. , de Vos, N. , Phillips, H. A. , Thompson, A. D. , … Lerner, T. J. (1994). Genetic mapping of the batten disease locus (CLN3) to the interval D16S288‐D16S383 by analysis of haplotypes and allelic association. Genomics, 22(2), 465–468. http://www.ncbi.nlm.nih.gov/pubmed/7806237 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , Thompson, A. D. , Mulley, J. C. , Kozman, H. M. , Richards, R. I. , Callen, D. F. , … Gardiner, R. M. (1993). Fine genetic mapping of the batten disease locus (CLN3) by haplotype analysis and demonstration of allelic association with chromosome 16p microsatellite loci. Genomics, 16(2), 455–460. http://www.ncbi.nlm.nih.gov/pubmed/8314582 [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , Williams, R. E. , McKay, T. R. , Callen, D. F. , Thompson, A. D. , Mulley, J. C. , … Gardiner, R. M. (1993). Refined genetic mapping of juvenile‐onset neuronal ceroid‐lipofuscinosis on chromosome 16. Journal of Inherited Metabolic Disease, 16(2), 339–341. http://www.ncbi.nlm.nih.gov/pubmed/8105142 [DOI] [PubMed] [Google Scholar]
- Mole, S. E. , & Gardiner, M. (1991). Molecular genetic analysis of neuronal ceroid lipofuscinosis. International Journal of Neurology, 25–26, 52–59. http://www.ncbi.nlm.nih.gov/pubmed/11980063 [PubMed] [Google Scholar]
- Mole, S. E. , Michaux, G. , Codlin, S. , Wheeler, R. B. , Sharp, J. D. , & Cutler, D. F. (2004). CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Experimental Cell Research, 298(2), 399–406. 10.1016/j.yexcr.2004.04.042 [DOI] [PubMed] [Google Scholar]
- Mole, S. E. , Zhong, N. A. , Sarpong, A. , Logan, W. P. , Hofmann, S. , Yi, W. , … Taschner, P. E. M. (2001). New Mutations in the Neuronal Ceroid Lipofuscinosis Genes. European Journal of Paediatric Neurology 5 (Suppl A), 7–10. http://www.ncbi.nlm.nih.gov/pubmed/11589012 [DOI] [PubMed] [Google Scholar]
- Moore, S. J. J. , Buckley, D. J. J. , MacMillan, A. , Marshall, H. D. D. , Steele, L. , Ray, P. N. N. , …. Parfrey, P. S. (2008). The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clinical Genetics, 74 (3), 213–222. 10.1111/j.1399-0004.2008.01054.x [DOI] [PubMed] [Google Scholar]
- Munroe, P. B. , Mitchison, H. M. , O'Rawe, A. M. , Anderson, J. W. , Boustany, R.‐M. , Lerner, T. J. , … Mole, S. E. (1997). Spectrum of mutations in the batten disease gene, CLN3. American Journal of Human Genetics, 61(2), 310–316. http://www.ncbi.nlm.nih.gov/pubmed/9311735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzaffar, N. E. , & Pearce, D. A. (2008). Analysis of NCL proteins from an evolutionary standpoint. Current Genomics, 9(2), 115–136. 10.2174/138920208784139573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan, S. B. , Rakheja, D. , Tan, L. , Pastor, J. V. , & Bennett, M. J. (2006). CLN3P, the batten’s disease protein, is a novel palmitoyl‐protein delta‐9 desaturase. Annals of Neurology, 60(5), 570–577. 10.1002/ana.20975 [DOI] [PubMed] [Google Scholar]
- Narayan, S. B. , Rakheja, D. , Pastor, J. V. , Rosenblatt, K. , Greene, S. R. , Yang, J. , Bennett, M. J. (2006). Over‐expression of CLN3P, the Batten disease protein, inhibits PANDER‐induced apoptosis in neuroblastoma cells: further evidence that CLN3P has anti‐apoptotic properties. Molecular Genetics and Metabolism, 88(2), 178–183. [DOI] [PubMed] [Google Scholar]
- Narayan, S. B. , Tan, L. U. , & Bennett, M. J. (2008). Intermediate levels of neuronal palmitoyl‐protein delta‐9 desaturase in heterozygotes for murine batten disease. Molecular Genetics and Metabolism, 93(1), 89–91. 10.1016/j.ymgme.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Nishiyama, A. , Xin, L. I. , Sharov, A. A. , Thomas, M. , Mowrer, G. , Meyers, E. , … Ko, M. S. H. (2009). Uncovering early response of gene regulatory networks in ESCs by systematic induction of transcription factors. Cell Stem Cell, 5(4), 420–433. 10.1016/j.stem.2009.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent, T. , Mole, S. E. , & Jones, D. T. (2008). The transmembrane topology of batten disease protein CLN3 determined by consensus computational prediction constrained by experimental data. FEBS Letters, 582(7), 1019–1024. 10.1016/j.febslet.2008.02.049 [DOI] [PubMed] [Google Scholar]
- Ohno, H. , Aguilar, R. C. , Yeh, D. , Taura, D. , Saito, T. , & Bonifacino, J. S. (1998). The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine‐based sorting signals. The Journal of Biological Chemistry, 273(40), 25915–25921. http://www.ncbi.nlm.nih.gov/pubmed/9748267 [DOI] [PubMed] [Google Scholar]
- Oltulu, F. , Kocatürk, D.Ç. , Adali, Y. , Özdil, B. , Aciköz, E. , Gürel, Ç. , … Aktuğ, H. (2019). Autophagy and mTOR pathways in mouse embryonic stem cell, lung cancer and somatic fibroblast cell lines. Journal of Cellular Biochemistry. 1–11. 10.1002/jcb.29110 [DOI] [PubMed] [Google Scholar]
- Palmieri, M. , Impey, S. , Kang, H. , di Ronza, A. , Pelz, C. , Sardiello, M. , & Ballabio, A. (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Human Molecular Genetics, 20(19), 3852–3866. 10.1093/hmg/ddr306 [DOI] [PubMed] [Google Scholar]
- Pandey, R. , Mandal, A. K. , Jha, V. , & Mukerji, M. (2011). Heat shock factor binding in Alu repeats expands its involvement in stress through an antisense mechanism. Genome Biology, 12(11), R117 10.1186/gb-2011-12-11-r117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvainen, L. , Dihanich, S. , Anderson, G. W. , Wong, A. M. , Brooks, H. R. , Abeti, R. , … Cooper, J. D. (2017). Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathological Communications, 5(1), 74 10.1186/s40478-017-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce, D. A. , Ferea, T. , Nosel, S. A. , Das, B. , & Sherman, F. (1999). Action of BTN1, the yeast orthologue of the gene mutated in batten disease. Nature Genetics, 22(1), 55–58. 10.1038/8861 [DOI] [PubMed] [Google Scholar]
- Pearce, D. A. , & Sherman, F. (1997). BTN1, a yeast gene corresponding to the human gene responsible for batten’s disease, is not essential for viability, mitochondrial function, or degradation of mitochondrial ATP synthase. Yeast (Chichester, England), 13(8), 691–697. 10.1002/(SICI)1097-0061(19970630)13:8<691:AID-YEA123>3.0.CO;2-D [DOI] [PubMed] [Google Scholar]
- Pérez‐Poyato, M.‐S. , Milà Recansens, M. , Ferrer Abizanda, I. , Montero Sánchez, R. , Rodríguez‐Revenga, L. , Cusí Sánchez, V. , … Pineda Marfà, M. (2011). Juvenile neuronal ceroid lipofuscinosis: Clinical course and genetic studies in spanish patients. Journal of Inherited Metabolic Disease, 34(5), 1083–1093. 10.1007/s10545-011-9323-7 [DOI] [PubMed] [Google Scholar]
- Persaud‐Sawin, D.‐A. , McNamara, J. O. , Rylova, S. , Vandongen, A. , & Boustany, R.‐M. (2004). A Galactosylceramide binding domain is involved in trafficking of CLN3 from Golgi to Rafts via recycling endosomes. Pediatric Research, 56(3), 449–463. 10.1203/01.PDR.0000136152.54638.95 [DOI] [PubMed] [Google Scholar]
- Persaud‐Sawin, D.‐A. , Mousallem, T. , Wang, C. , Zucker, A. , Kominami, E. , & Boustany, R.‐M. (2007). Neuronal ceroid lipofuscinosis: A common pathway? Pediatric Research, 61(2), 146–152. 10.1203/pdr.0b013e31802d8a4a [DOI] [PubMed] [Google Scholar]
- Portales‐Casamar, E. , Thongjuea, S. , Kwon, A. T. , Arenillas, D. , Zhao, X. , Valen, E. , … Sandelin, A. (2010). JASPAR 2010: The greatly expanded open‐access database of transcription factor binding profiles. Nucleic Acids Research, 38 (Database issue): D105–D110. 10.1093/nar/gkp950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K. D. , Brown, G. R. , Hiatt, S. M. , Thibaud‐Nissen, F. , Astashyn, A. , Ermolaeva, O. , … Ostell, J. M. (2014). RefSeq: An update on mammalian reference sequences. Nucleic Acids Research, 42(Database issue), D756–D763. 10.1093/nar/gkt1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullarkat, R. K. , & Morris, G. N. (1997). Farnesylation of batten disease CLN3 protein. Neuropediatrics, 28(1), 42–44. 10.1055/s-2007-973665 [DOI] [PubMed] [Google Scholar]
- Puranam, K. L. , Guo, W. X. , Qian, W. H. , Nikbakht, K. , & Boustany, R. M. (1999). CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Molecular Genetics and Metabolism, 66(4), 294–308. 10.1006/mgme.1999.2834 [DOI] [PubMed] [Google Scholar]
- Ramirez, K. , Chandler, K. J. , Spaulding, C. , Zandi, S. , Sigvardsson, M. , Graves, B. J. , & Kee, B. L. (2012). Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity, 36(6), 921–932. 10.1016/j.immuni.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranta, S. , Hirvasniemi, A. , Herva, R. , Haltia, M. , & Lehesjoki, A.‐E. (2002). Northern epilepsy and the gene error behind It. Duodecim; Laaketieteellinen Aikakauskirja, 118(15), 1551–1558. http://www.ncbi.nlm.nih.gov/pubmed/12244629 [PubMed] [Google Scholar]
- Ranta, S. , Savukoski, M. , Santavuori, P. , & Haltia, M. (2001). Studies of homogenous populations: CLN5 and CLN8. Advances in Genetics, 45, 123–140. http://www.ncbi.nlm.nih.gov/pubmed/11332769 [DOI] [PubMed] [Google Scholar]
- Ranta, S. , Topcu, M. , Tegelberg, S. , Tan, H. , Üstübütün, A. , Saatci, I. , … Lehesjoki, A.‐E. (2004). Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to northern epilepsy. Human Mutation, 23(4), 300–305. 10.1002/humu.20018 [DOI] [PubMed] [Google Scholar]
- Ratajczak, E. , Petcherski, A. , Ramos‐Moreno, J. , & Ruonala, M. O. (2014). FRET‐assisted determination of CLN3 membrane topology. Edited by Pedro Fernandez‐Funez. PLoS ONE, 9(7), e102593 10.1371/journal.pone.0102593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves, B. , & Banting, G. (1994). Overexpression of TGN38/41 leads to mislocalisation of Gamma‐Adaptin. FEBS Letters, 351(3), 448–456. http://www.ncbi.nlm.nih.gov/pubmed/8082813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, L. R. , Zhang, L. P. , Huang, S. Y. , Zhu, Y. F. , Li, W. J. , Fang, S. Y. , … Gao, Y. L. (2016). Effects of sialidase NEU1 siRNA on proliferation, apoptosis, and invasion in human ovarian cancer. Molecular and Cellular Biochemistry, 411(1‐2), 213–219. 10.1007/s11010-015-2583-z [DOI] [PubMed] [Google Scholar]
- Rivolta, C. , Sweklo, E. A. , Berson, E. L. , & Dryja, T. P. (2000). Missense mutation in the USH2A Gene: Association with recessive retinitis pigmentosa without hearing loss. American Journal of Human Genetics, 66(6), 1975–1978. 10.1086/302926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M. S. (2015). Forty years of clathrin‐coated vesicles. Traffic, 16(12), 1210–1238. 10.1111/tra.12335 [DOI] [PubMed] [Google Scholar]
- Rustici, G. , Kolesnikov, N. , Brandizi, M. , Burdett, T. , Dylag, M. , Emam, I. , … Sarkans, U. . (2013). ArrayExpress update‐Trends in database growth and links to data analysis tools. Nucleic Acids Research, 41(D1), D987–D990. 10.1093/nar/gks1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylova, S. N. , Amalfitano, A. , Persaud-Sawin, D. A. , Guo, W. X. , Chang, J. Jansen, P. J. , … Boustany, R. M. (2002). The CLN3 gene is a novel molecular target for cancer drug discovery. Cancer Research, 62(3), 801–808. http://www.ncbi.nlm.nih.gov/pubmed/11830536 [PubMed] [Google Scholar]
- Sardiello, M. , Palmieri, M. , di Ronza, A. , Medina, D. L. , Valenza, M. , Gennarino, V. A. , … Ballabio, A. (2009). A gene network regulating lysosomal biogenesis and function. Science (New York, N.Y.), 325 (5939), 473–477. 10.1126/science.1174447 [DOI] [PubMed] [Google Scholar]
- Sarpong, A. , Schottmann, G. , Rüther, K. , Stoltenburg, G. , Kohlschütter, A. , Hübner, C. , & Schuelke, M. (2009). Protracted course of juvenile ceroid lipofuscinosis associated with a novel CLN3 mutation (p. Y199X). Clinical Genetics, 76(1), 38–45. 10.1111/j.1399-0004.2009.01179.x [DOI] [PubMed] [Google Scholar]
- Savukoski, M. , Klockars, T. , Holmberg, V. , Santavuori, P. , Lander, E. S. , & Peltonen, L. (1998). CLN5, a novel gene encoding a putative transmembrane protein mutated in finnish variant late infantile neuronal ceroid lipofuscinosis. Nature Genetics, 19(3), 286–288. 10.1038/975 [DOI] [PubMed] [Google Scholar]
- Schmidtke, C. , Tiede, S. , Thelen, M. , Kakela, R. , Jabs, S. , Makrypidi, G. , … Braulke, T. (2019). Lysosomal proteome analysis reveals that CLN3‐defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. Journal of Biological Chemistry, 294(24), 9592–9604. 10.1074/jbc.RA119.008852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, B. , Elsässer, H.‐P. , Schmidt, B. , & Hasilik, A. (2007). Characterisation of lipofuscin‐like lysosomal inclusion bodies from human placenta. FEBS Letters, 581(1), 102–108. 10.1016/j.febslet.2006.12.005 [DOI] [PubMed] [Google Scholar]
- Schultz, M. L. , Tecedor, L. , Stein, C. S. , Stamnes, M. A. , & Davidson, B. L. (2014). CLN3 deficient cells display defects in the ARF1‐Cdc42 pathway and actin‐dependent events. PLoS ONE, 9(5), e96647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scifo, E. , Szwajda, A. , Dębski, J. , Uusi‐Rauva, K. , Kesti, T. , Dadlez, M. , … Lalowski, M. (2013). Drafting the CLN3 Protein interactome in SH‐SY5Y human neuroblastoma cells: A label‐free quantitative proteomics approach. Journal of Proteome Research, 12(5), 2101–2115. 10.1021/pr301125k [DOI] [PubMed] [Google Scholar]
- Shevach, E. , Ali, M. , Mizrahi‐Meissonnier, L. , McKibbin, M. , El‐Asrag, M. , Watson, C. M. , … Sharon, D. (2015). Association between missense mutations in the BBS2 gene and nonsyndromic retinitis pigmentosa. JAMA Ophthalmology, 133(3), 312–318. 10.1001/jamaophthalmol.2014.5251 [DOI] [PubMed] [Google Scholar]
- Sigrist, C. J. A. , de Castro, E. , Cerutti, L. , Cuche, B. A. , Hulo, N. , Bridge, A. , … Xenarios, I. (2013). New and continuing developments at PROSITE. Nucleic Acids Research, 41(Database issue), D344–D347. 10.1093/nar/gks1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siintola, E. , Topcu, M. , Aula, N. , Lohi, H. , Minassian, B. A. , Paterson, A. D. , … Lehesjoki, A.‐E. (2007). The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. American Journal of Human Genetics, 81(1), 136–146. 10.1086/518902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat, D. E. , Donnelly, R. J. , Lackland, H. , Liu, C. G. , Sohar, I. , Pullarkat, R. K. , & Lobel, P. (1997). Association of mutations in a lysosomal protein with classical late‐infantile neuronal ceroid lipofuscinosis. Science (New York, N.Y.), 277 (5333): 1802–1805. http://www.ncbi.nlm.nih.gov/pubmed/9295267 [DOI] [PubMed] [Google Scholar]
- Sobue, K. , Kanda, K. , & Kakiuchi, S. (1982). Solubilization and partial purification of protein kinase systems from brain membranes that phosphorylate calspectin. A spectrin‐like calmodulin‐binding protein (Fodrin). FEBS Letters, 150(1), 185–190. http://www.ncbi.nlm.nih.gov/pubmed/7160470 [DOI] [PubMed] [Google Scholar]
- Stein, C. S. , Yancey, P. H. , Martins, I. , Sigmund, R. D. , Stokes, J. B. , & Davidson, B. L. (2010). Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. American Journal of Physiology‐Cell Physiology, 298(6), C1388–C1400. 10.1152/ajpcell.00272.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel, C. (1826). Beretning om et maerkeligt Sygdomstilfaelde hos fire Soedskende I Naerheden af Roeraas, Eyr et medicinsk Tidskrift, 1, 347–352. [Google Scholar]
- Storch, S. , Pohl, S. , & Braulke, T. (2004). A dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of the batten disease‐related CLN3 protein are required for efficient lysosomal targeting. The Journal of Biological Chemistry, 279(51), 53625–53634. 10.1074/jbc.M410930200 [DOI] [PubMed] [Google Scholar]
- Storch, S. , Pohl, S. , Quitsch, A. , Falley, K. , & Braulke, T. (2007). C‐Terminal prenylation of the CLN3 membrane glycoprotein is required for efficient endosomal sorting to lysosomes. Traffic (Copenhagen, Denmark), 8(4), 431–444. 10.1111/j.1600-0854.2007.00537.x [DOI] [PubMed] [Google Scholar]
- Su, A. I. , Wiltshire, T. , Batalov, S. , Lapp, H. , Ching, K. A. , Block, D. , … Hogenesch, J. B. (2004). A gene atlas of the mouse and human protein‐encoding transcriptomes. Proceedings of the National Academy of Sciences, 101(16), 6062–6067. 10.1073/pnas.0400782101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner, P. E. , de Vos, N. , & Breuning, M. H. (1997a). Cross‐species homology of the CLN3 Gene. Neuropediatrics, 28(1), 18–20. 10.1055/s-2007-973658 [DOI] [PubMed] [Google Scholar]
- Taschner, P. E. , de Vos, N. , & Breuning, M. H. (1997b). Rapid detection of the major deletion in the batten disease gene CLN3 by Allele specific PCR. Journal of Medical Genetics, 34(11), 955–956. http://www.ncbi.nlm.nih.gov/pubmed/9391897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecedor, L. , Stein, C. S. , Schultz, M. L. , Farwanah, H. , Sandhoff, K. , & Davidson, B. L. (2013). CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. Journal of Neuroscience, 33(46), 18065–18079. 10.1523/JNEUROSCI.0498-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, C. , Guimarães, A. , Bessa, C. , Ferreira, M. J. , Lopes, L. , Pinto, E. , … Ribeiro, M. G. (2003). Clinicopathological and molecular characterization of neuronal ceroid lipofuscinosis in the portuguese population. Journal of Neurology, 250(6), 661–667. 10.1007/s00415-003-1050-z [DOI] [PubMed] [Google Scholar]
- Tuxworth, R. I. , Chen, H. , Vivancos, V. , Carvajal, N. , Huang, X. , & Tear, G. (2011). The batten disease gene CLN3 is required for the response to oxidative stress. Human Molecular Genetics, 20(10), 2037–2047. 10.1093/hmg/ddr088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen, M. , Oksvold, P. , Fagerberg, L. , Lundberg, E. , Jonasson, K. , Forsberg, M. , … Ponten, F. (2010). Towards a knowledge‐based human protein atlas. Nature Biotechnology, 28(12), 1248–1250. 10.1038/nbt1210-1248 [DOI] [PubMed] [Google Scholar]
- Uusi‐Rauva, K. , Kyttälä, A. , van der Kant, R. , Vesa, J. , Tanhuanpää, K. , Neefjes, J. , … Jalanko, A. (2012). Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cellular and Molecular Life Sciences, 69(12), 2075–2089. 10.1007/s00018-011-0913-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusi‐Rauva, K. , Luiro, K. , Tanhuanpää, K. , Kopra, O. , Martín‐Vasallo, P. , Kyttälä, A. , & Jalanko, A. (2008). Novel interactions of CLN3 protein link batten disease to dysregulation of Fodrin–Na+, K+ ATPase complex. Experimental Cell Research, 314(15), 2895–2905. 10.1016/j.yexcr.2008.06.016 [DOI] [PubMed] [Google Scholar]
- Van Diggelen, O. P. , Keulemans, J. L. , Kleijer, W. J. , Thobois, S. , Tilikete, C. , & Voznyi, Y. V. (2001). Pre‐ and postnatal enzyme analysis for infantile, late infantile and adult neuronal ceroid lipofuscinosis (CLN1 and CLN2). European Journal of Paediatric Neurology, 5 Suppl A (4), 189–192. 10.1053/ejpn.2001.0509 [DOI] [PubMed] [Google Scholar]
- Vantaggiato, C. , Redaelli, F. , Falcone, S. , Perrotta, C. , Tonelli, A. , Bondioni, S. , … Bassi, M. T. (2009). A novel CLN8 mutation in late‐infantile‐onset neuronal ceroid lipofuscinosis (LINCL) reveals aspects of CLN8 neurobiological function. Human Mutation, 30(7), 1104–1116. 10.1002/humu.21012 [DOI] [PubMed] [Google Scholar]
- Vesa, J. , Chin, M. H. , Oelgeschläger, K. , Isosomppi, J. , DellAngelica, E. C. , Jalanko, A. , & Peltonen, L. (2002). Neuronal ceroid lipofuscinoses are connected at molecular level: Interaction of CLN5 protein with CLN2 and CLN3. Molecular Biology of the Cell, 13(7), 2410–2420. 10.1091/mbc.E02-01-0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesa, J. , & Peltonen, L. (2002). Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinoses encode lysosomal proteins. Current Molecular Medicine, 2(5), 439–444. http://www.ncbi.nlm.nih.gov/pubmed/12125809 [DOI] [PubMed] [Google Scholar]
- Vidal‐Donet, J. M. , Carcel-Trullois, J. , Casanova, B. , Aguado, C. , & Knecht, E. (2013). Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS ONE, 8(2), e55526 10.1371/journal.pone.0055526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello, S. P. , Benedict, J. W. , Padilla‐Lopez, S. , & Pearce, D. A. (2010). Interaction between Sdo1p and Btn1p in the saccharomyces cerevisiae model for batten disease. Human Molecular Genetics, 19(5), 931–942. 10.1093/hmg/ddp560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , Wang, H. , Tuan, H.‐F. , Nguyen, D. H. , Sun, V. , Keser, V. , … Chen, R. (2014). Next generation sequencing‐based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype‐phenotype correlation and clinical refinements. Human Genetics, 133(3), 331–345. 10.1007/s00439-013-1381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisschuh, N. , Mayer, A. K. , Strom, T. M. , Kohl, S. , Glöckle, N. , Schubach, M. , … Wissinger, B. (2016). Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. Edited by Andreas R. Janecke. PLoS ONE, 11(1), e0145951 10.1371/journal.pone.0145951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake, V. J. , Jolly, R. D. , Bayliss, S. L. , & Palmer, D. N. (1995). Immunocytochemical studies in the ceroid‐lipofuscinoses (Batten Disease) using antibodies to subunit c of mitochondrial ATP synthase. American Journal of Medical Genetics, 57(2), 177–181. 10.1002/ajmg.1320570214 [DOI] [PubMed] [Google Scholar]
- Wisniewski, K. E. , Connell, F. , Kaczmarski, W. , Kaczmarski, A. , Siakotos, A. , Becerra, C. R. , & Hofmann, S. L. (1998). Palmitoyl‐protein thioesterase deficiency in a novel granular variant of LINCL. Pediatric Neurology, 18(2), 119–123. http://www.ncbi.nlm.nih.gov/pubmed/9535296 [DOI] [PubMed] [Google Scholar]
- Wisniewski, K. E. , Zhong, N. , Kaczmarski, W. , Kaczmarski, A. , Kida, E. , Brown, W. T. , … Wisniewski, T. M. (1998). Compound heterozygous genotype is associated with protracted juvenile neuronal ceroid lipofuscinosis. Annals of Neurology, 43(1), 106–110. 10.1002/ana.410430118 [DOI] [PubMed] [Google Scholar]
- Wolfe, D. M. , Padilla‐Lopez, S. , Vitiello, S. P. , & Pearce, D. A. (2011). PH‐dependent localization of Btn1p in the yeast model for batten disease. Disease Models & Mechanisms, 4(1), 120–125. 10.1242/dmm.006114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. , Orozco, C. , Boyer, J. , Leglise, M. , Goodale, J. , Batalov, S. , … Su, A. I. (2009). BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biology, 10(11), R130 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Wang, H. , Zeng, Y. , Tian, Y. , Shen, Z. , Xie, Z. , … Luo, H. (2019). Overexpression of CLN3 contributes to tumour progression and predicts poor prognosis in hepatocellular carcinoma. Surgical Oncology, 28, 180–189. [DOI] [PubMed] [Google Scholar]
- Zerbino, D. R. , Achuthan, P. , Akanni, W. , Amode, M. R. , Barrell, D. , Bhai, J. , … Flicek, P. (2018). Ensembl 2018. Nucleic Acids Research, 46(D1), D754–D761. 10.1093/nar/gkx1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Wu, J. , Nguyen, A. , Wang, B. D. , He, P. , Laurent, G. S. , … Su, Y. A. (2008). Molecular mechanism underlying differential apoptosis between human melanoma cell lines UACC903 and UACC903(+6) revealed by mitochondria-focused cDNA microarrays. Apoptosis, 13(8), 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, N. , Wisniewski, K. E. , Kaczmarski, A. L. , Ju, W. , Xu, W. M. , Xu, W. W. , … Brown, W. T. (1998). Molecular screening of batten disease: Identification of a missense mutation (E295K) in the CLN3 gene. Human Genetics, 102(1), 57–62. http://www.ncbi.nlm.nih.gov/pubmed/9490299 [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Huang, Z. , Chen, Y. , Zhou, J. , Hu, S. , Zhi, Q. , … He, S. (2014). Effect of CLN3 silencing by RNA interference on the proliferation and apoptosis of human colorectal cancer cells. Biomedicine & Pharmacotherapy, 68(3), 253–258. 10.1016/j.biopha.2013.12.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials