

Abstract
Introduction:
Phosphatidylserine (PS) is a key cell membrane phospholipid normally maintained on the inner cell surface, but externalizes to the outer surface in response to cellular stress. We hypothesized that PS exposure mediates organ dysfunction in hemorrhagic shock.
Our aims were to evaluate PS blockade on: 1) pulmonary, 2) renal, and 3) gut function, as well as 4) serum lysophosphatidic acid (LPA), an inflammatory mediator generated by PS externalization, as a possible mechanism mediating organ dysfunction.
Materials and Methods:
Rats were either: a) monitored for 130 minutes (controls, n=3), b) hemorrhaged then resuscitated (hemorrhage only group, n=3), or c) treated with Diannexin (DA), a PS blocking agent, followed by hemorrhage and resuscitation, (DA + hemorrhage group, n=4). Pulmonary dysfunction was assessed by PaO2, renal dysfunction by serum creatinine, and gut dysfunction by mesenteric endothelial permeability (LP). LPA levels were measured in all groups.
Results:
Pulmonary: There was no difference in PaO2 between groups. Renal: After resuscitation, creatinine levels were lower after PS blockade with Diannexin vs hemorrhage only group (p=0.01). Gut: LP was decreased after PS blockade with Diannexin vs hemorrhage only group (p<0.01). Finally, LPA levels were also lower after PS blockade with Diannexin vs the hemorrhage only group, but higher than the control group (p<0.01).
Conclusion:
PS blockade with Diannexin decreased renal and gut dysfunction associated with hemorrhagic shock and attenuated the magnitude of LPA generation. Our findings suggest potential for therapeutic targets in the future that could prevent organ dysfunction associated with hemorrhagic shock.
Keywords: Diannexin, phosphatidylserine, hemorrhagic shock, organ dysfunction
Introduction
Hemorrhagic shock is responsible for up to 40% of trauma related mortalities with 33-56% of deaths occurring in the prehospital period1. In survivors, hypoperfusion leads to multiple organ failure in nearly a third of cases and is associated with subsequent nosocomial infection and increased mortality2, 3. At the cellular level, hemorrhagic shock results in inadequate oxygen delivery, leading to depletion of ATP stores and cell death through necrosis from membrane rupture, apoptosis or necroptosis3. Signaling molecules, specifically phospholipids, expressed on the cell plasma membrane appear to be key mediators driving the cellular response to tissue hypoxia and may be responsible for the organ dysfunction that follows4–7.
Under normal physiologic conditions, phospholipids are maintained asymmetrically in the plasma membrane by an energy dependent process8. Phosphatidylserine (PS), a negatively charged phospholipid found in eukaryotic cells, is normally secluded inside cells by flippase, an ATP dependent translocase that catalyzes specific cytosol-directed transbilayer movement of phospholipids6–8. PS is externalized to the outer cell surface as a result of flippase inactivation, coupled with activation of scramblase, an ATP independent protein that causes randomization of phospholipid distribution across membranes8. The externalization of PS normally acts as an early indicator for apoptosis during normal homeostasis and allows cells to be recognized and cleared by phagocytes and macrophages without significant inflammation4, 8, 9.
However, in the pathophysiologic setting of severe tissue ischemia with depletion of ATP, such as that occurring during significant hemorrhage, PS appears to be aberrantly upregulated. This results in over activation of the coagulation cascade, platelet activation, and inflammatory response, potentially driving the organ dysfunction seen after hemorrhage10–13. Furthermore, secretory phospholipase A2 binds to externalized PS and subsequently generates lysophosphatidic acid (LPA)14. LPA has been shown to increase vascular permeability, platelet aggregation, and promote macrophage activation14, 15, potentially contributing to the mechanism behind organ dysfunction following hemorrhagic shock.
Annexin A5 is an endogenous phospholipid blocking protein with selective affinity for PS16. A homodimer of annexin A5, Diannexin, was synthesized to permit a longer half-life and increased PS binding affinity compared to endogenous annexin A511. Diannexin binds to externalized PS during the early stages of apoptosis, when cellular injury may still be reversible, blocking its downstream effects11, 15. Phosphatidylserine blockade with Diannexin has minimized the detrimental effects of ischemia reperfusion injury in rat models of renal, pancreatic islet cell, lung, and liver transplant as well as in muscle flaps11, 14, 17–21. The underlying pathophysiology of hemorrhagic shock, followed by resuscitation is similar to the insult seen with ischemia reperfusion injury after transplant. Therefore, we hypothesized that PS blockade with Diannexin would provide a protective effect against hemorrhage related organ dysfunction. To test this, we evaluated the outcome of PS blockade on hemorrhage induced pulmonary, renal, and gut dysfunction. As a potential mechanism contributing to organ dysfunction, we also examined LPA levels. We hypothesized PS blockade would reduce organ dysfunction and attenuate the release of systemic LPA after hemorrhage.
Materials and Methods
Animals and Solution Preparation
All studies were approved by an Institutional Committee for the use of animals and complied with institutional animal research protocols. All animals were allowed food and water ad libitum. Animal and Ringer solution preparation have been described previously22.
In brief, the Ringer’s solution was prepared daily in distilled deionized water containing, in mM, 135 NaCl, 4.6 KCl, 2.46 MgSO4, 5.0 NaHCO3, 5.5 dextrose, 9.03 HEPES salt, and 11.04 HEPES acid (Research Organics, Cleveland, OH). A 1% bovine serum albumin (BSA) Ringer’s perfusate was prepared by adding BSA (BSA crystallized, Sigma, St. Louis, MO) to the Ringer’s solution.
Adult female Sprague-Dawley rats (225–300g; Hilltop Lab Animals Inc., Scottsdale, PA), were anesthetized with subcutaneous sodium pentobarbitol (55mg/kg body weight). The femoral vessels were cannulated and venous blood obtained for study serology. The mesentery was exposed via a midline incision and the bowel positioned on an inverted microscope (Diaphot, Nikon; Melville NY). The animal’s mesentery was bathed continuously in Ringer’s solution and body temperature was maintained at 37°C throughout the study period. All rats were allowed a 30-minute stabilization period prior to administration of treatments.
Hemorrhage Protocol
Rats were allocated to one of three experimental groups: control (n=3), hemorrhage only (n=3), and phosphatidylserine blockade with Diannexin followed by hemorrhage (n=4). In brief, after obtaining baseline serology and baseline microvascular leak (Lp) measurements, the rats in the hemorrhage only group were stabilized for 10 minutes. Then, hemorrhage was induced by withdrawal of 30ml/kg into a syringe with 0.5ml heparinized saline (7U/ml) every 10 minutes for a period of 60 minutes. During this time, MAP was maintained at 40 mmHg by withdrawal or reinfusion of blood. Following the 60 minutes of hemorrhage, serology and Lp measurements were again obtained. Resuscitation with shed blood as well as LR (21-32 ml/kg) was then performed for 60 minutes to maintain the original baseline MAP. Serology and Lp were again recorded after the resuscitation period and rats were euthanized.
In the PS blockage group, rats received a continuous infusion of Diannexin blockade at 400μg/kg over the initial 10 minutes. This dose was selected based on a phase II trial using Diannexin in kidney transplantation (NCT00615966). In this phase II trial, administration of Diannexin up to 400μg/kg was not associated with adverse events in recipients and showed reduced dialysis days and incidence of delayed graft function23. Doses of up to 1000μg/kg have been shown to be safe in animal studies18. Diannexin was administered prior to hemorrhage based on our prior studies evaluating Lp in the setting of ischemia reperfusion19. Following Diannexin infusion, hemorrhage was carried out in an identical manner to the hemorrhage only group. Resuscitation was also performed in the same manner.
For control animals, all steps were carried out in the same manner except in lieu of hemorrhage, resuscitation, or infusion with Diannexin, the rats received an infusion with 1% BSA Ringer’s perfusate at 0.65mg/h. To evaluate our study hypothesis, evidence of pulmonary, renal, and gut dysfunction, in addition to LPA levels, were evaluated in each group. Serology and Lp measurements were conducted in the same manner for all groups.
Pulmonary and renal dysfunction:
Arterial and venous blood was obtained after induction of anesthesia to establish baseline creatinine and partial pressure of oxygen (PaO2) parameters. Venous and arterial sampling were repeated after 60 minutes of hemorrhage and then again after 60 minutes of resuscitation (end of study). Cr and PaO2 measurements were obtained at the same three time points in the control group (though hemorrhage and resuscitation were not performed in this group). Laboratory values were measured using Abbot Point of Care i-STAT® System, per manufacture protocol. Serum was centrifuged for 20 minutes at 2000xg and stored at −20°C.
Gut dysfunction (microvascular leak, Lp):
Single vessel Lp (microvascular leak, hydraulic conductivity) was determined using the modified Landis micro-occlusion technique24. The assumptions and limitations of this technique have been previously described25. Mesenteric post capillary venules, 20 to 30 μm in diameter and at least 400μm in length, were identified based on flow patterns. Vessels with no evidence of leukocyte adherence or side branches were chosen. The vessels were cannulated with micropipettes attached to a water manometer to control hydrostatic perfusion pressure. Initial cell velocity (dl/dt) was obtained by recording marker cell position as a function of time. Transmural water flux per unit area (Jv/S) was calculated using the equation Jv/S=(dl/dt)(r/2l), where r is the capillary radius and 1 is the initial distance between the marker cell and the occluded site. Lp was determined using a modified version of Starling’s equation of fluid filtration: Lp=(Jv/S)(1/Pc), where Pc is the capillary hydrostatic pressure. Lp was calculated from the slope of the regression of Jv/S on Pc derived from several occlusions at three different perfusate pressures. Lp values are represented as mean ± standard error of the mean 10−7 cm·s −1·cmH2O −1. Control studies that document the stability of this model over time,and after multiple recannulations of the vessels have been reported22, 26, 27.
Lysophosphatidic Acid (LPA):
LPA was measured using Echelon Biosciences lysophosphatidic acid competitive ELISA kit for rat serum. Concentration of LPA, in μM, was obtained for each group at the same three time points: baseline, after hemorrhage, and after resuscitation.
Statistical analysis:
Data are presented as mean ± standard error of the mean. Statistical analysis was performed with paired t-test and analysis of variance assuming normal distribution. P value of <0.05 was considered statistically significant.
Results
Effect of PS blockade on pulmonary dysfunction:
Pulmonary function, measured by arterial partial pressure of oxygen (PaO2), was similar between the groups at baseline (p=0.63). There was no difference in pulmonary dysfunction between the groups post hemorrhage or post resuscitation (p=0.29 and p=0.59, respectively). (Figure 1).
Figure 1. Phosphatidylserine (PS) blockade with Diannexin on pulmonary dysfunction.
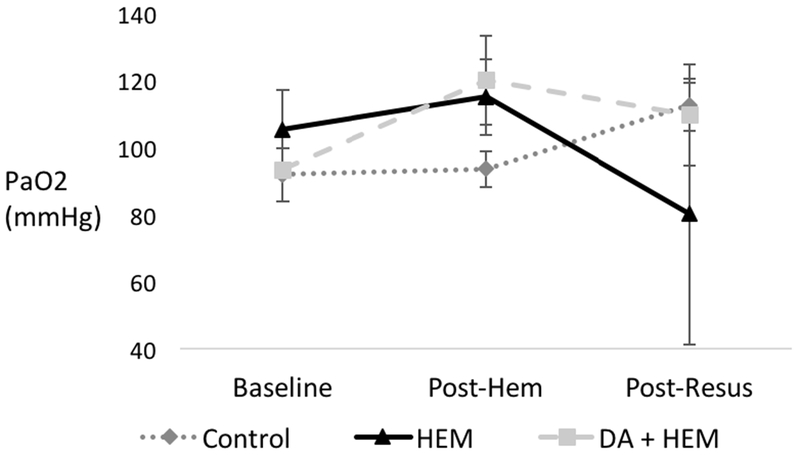
Partial pressure of oxygen (PaO2) in control, hemorrhage only (HEM), and Diannexin followed by hemorrhage (DA + HEM) groups at the following time points: baseline, post-hemorrhage (post-hem), and post-resuscitation (post-resus). * denotes p<0.05 on analysis of variance among the three groups. There was no difference in pulmonary function between the three groups. Units of PaO2 in millimeters of mercury. Error bars indicate SEM.
Effect of PS blockade on renal dysfunction: Creatinine levels were similar at baseline and after hemorrhage between the groups (p=0.33 and p=0.07, respectively). Creatinine levels were different between groups after resuscitation. After resuscitation, PS blockade decreased Cr by 46% compared to the hemorrhage only group, from 0.93 to 0.5 (p=0.01) (Figure 2).
Figure 2. Phosphatidylserine (PS) blockade with Diannexin on renal dysfunction.
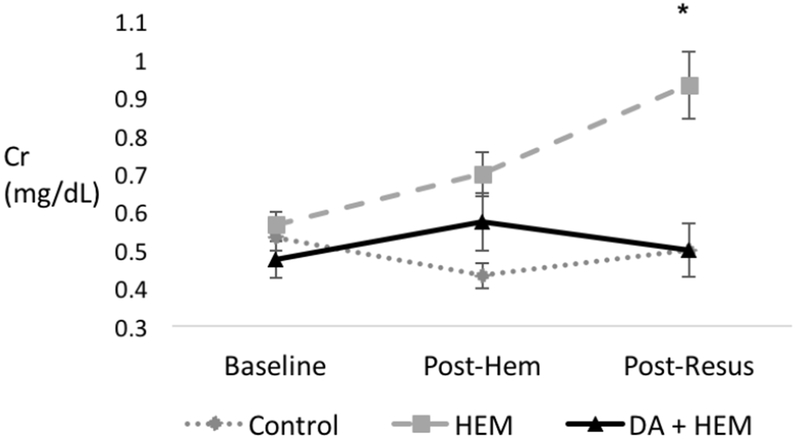
Renal dysfunction, measured by serum creatinine, among the three groups: control, hemorrhage only (HEM), and Diannexin followed by hemorrhage (DA + HEM). Data were collected at the following time points: baseline, post-hemorrhage (post-hem), and post-resuscitation (post-resus). PS blockade improved renal dysfunction after resuscitation. * denotes p<0.05 on analysis of variance among the three groups. Units of Cr in mg/dL. Error bars indicate SEM.
Effect of PS blockade on gut dysfunction:
At baseline, there was a small difference in mesenteric microvascular leak rate (Lp) between the three groups. However, all three groups had low Lp rates at baseline consistent with historic baseline measurements. At the second time point (post hemorrhage), there was no difference in Lp between the three groups (p=0.12). However, at the final time point (post resuscitation), Lp increased by 45% from baseline in the hemorrhage only group compared to a 7% decrease in controls (p<0.05) and PS blockade effectively reduced Lp compared to the hemorrhage only group by 27% (p<0.001) (Figure 3).
Figure 3. Phosphatidylserine (PS) blockade with Diannexin on gut dysfunction (Microvascular Leak Rate, Lp).
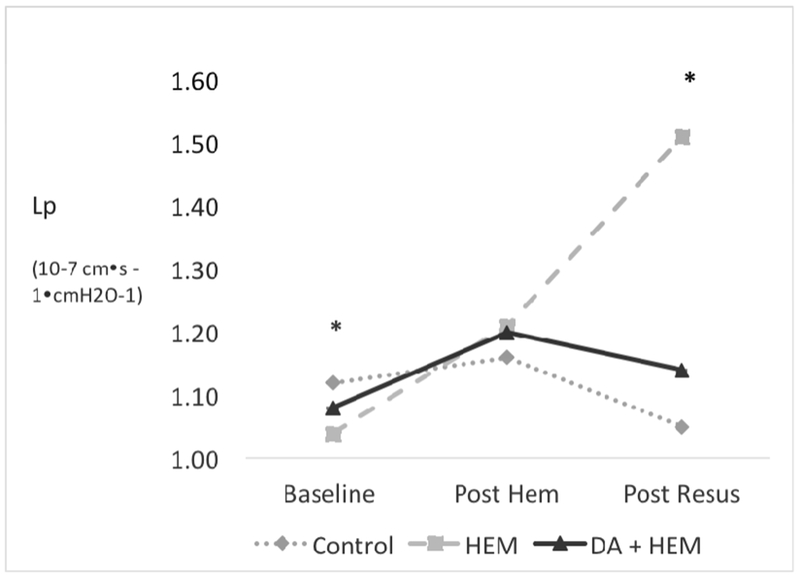
Gut dysfunction as measured by microvascular leak rate, Lp in the control, hemorrhage only (HEM), and Diannexin followed by hemorrhage (DA + HEM) groups. Data were collected at the following time points: baseline, post-hemorrhage (post-hem), and post-resuscitation (post-resus). PS blockade improved gut dysfunction after resuscitation. * denotes p<0.05 on analysis of variance among the three groups. Lp units = 10−7 cm·s −1·cmH2O−1. Error bars indicate SEM.
Effect of PS blockade on LPA levels:
There was no difference in LPA levels between the groups at baseline (p=0.2). After hemorrhage, there was no change in LPA levels between the PS blockade group compared to the hemorrhage only group (p=0.16). After hemorrhage, LPA levels in the PS blockade group remained 77% higher than the levels in the control group (p<0.001). After resuscitation, there was a difference in LPA levels among the three groups (p<0.001). After resuscitation, PS blockade decreased the level of LPA by 31% compared to the hemorrhage only group, from 571 μM to 395 μM (p<0.001). LPA level in the control group remained stable over time. (Figure 4).
Figure 4. Phosphatidylserine (PS) blockade with Diannexin attenuates lysophosphatidic acid (LPA) response after hemorrhage.
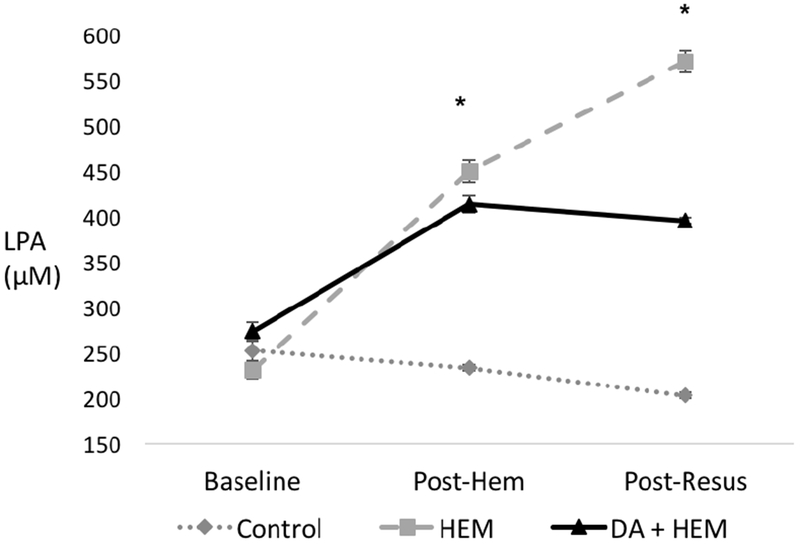
Lysophosphatidic acid (LPA) levels in the control, hemorrhage only (HEM), and Diannexin followed by hemorrhage (DA + HEM) groups. Data were collected at the following time points: baseline, post-hemorrhage (post-hem), and post-resuscitation (post-resus). PS blockade reduced LPA levels compared to the HEM group. * denotes p<0.05 on analysis of variance among the three groups. LPA units are in μM. Error bars indicate SEM.
Discussion
Hemorrhage induced ischemia followed by resuscitation results in a complex pathophysiologic host response, marked by severe inflammation, dysregulated immune response, tissue injury, and ultimately, organ dysfunction3,28,29. Severe ischemia results in cellular stress and cell death via cell membrane rupture, apoptosis, or necroptosis3. Regardless of the exact means of cell death, early and aberrant over-externalization of phosphatidylserine to the exterior membrane of the cell surface appears to be a key signaling molecule early in the cascade of events associated with cell death and the ensuing host response10–13. Diannexin, a homodimer of annexin A5, is known to bind with high affinity to externalized PS, blocking its signaling11, 15. Therefore, our hypothesis was that PS blockade with Diannexin would reduce organ dysfunction and attenuate the generation of systemic LPA after hemorrhage. We found that Diannexin infusion prior to hemorrhage decreased renal and gut dysfunction, but did not decrease pulmonary dysfunction. PS blockade also decreased LPA levels after hemorrhage.
Renal dysfunction was prevented in the PS blockade group after resuscitation, with similar levels of Cr found between the PS blockade group and the control group, suggesting a renal protective effect of PS blockade in the setting of hemorrhage. In ischemic conditions, PS is externalized on the cell membrane of endothelial cells, hematologic cells, and microvesicles are shed from injured cells30, 31. This then initiates generation of thrombin, complement, and inflammatory lipid mediators leading to microvascular thrombotic occlusion, endothelial injury, and edema32, 33. These events are known to contribute to renal injury and renal dysfunction. PS blockade with Diannexin resulted in decreased renal ischemic injury in prior studies examining renal perfusion injury and renal transplantation21, 23. Specifically, Diannexin decreased proximal tubule damage and leukocyte influx in a mouse ischemia reperfusion injury model21, and reduced rates of delayed graft function and hemodialysis days in marginal donor renal transplant recipients23. Our study results are in agreement with prior studies that show PS blockade protects against renal dysfunction, and extend applicability to the setting of hemorrhage in addition to transplantation. There was no improvement in pulmonary function after hemorrhage or after resuscitation in the PS blockade group. We suspect this was due to the limited sensitivity of PaO2 as a marker of pulmonary dysfunction.
In our model of gut dysfunction, we found that PS blockade effectively decreased hemorrhage induced mesenteric microvascular leak after resuscitation. In addition to a direct effect of PS blockade and prevention of downstream effects of PS signaling, the findings may in part be accounted for by the relatively early diversion of splanchnic blood flow to more critical organs during hemorrhagic shock. The gastrointestinal endothelial cells would therefore experience more profound ischemia compared to other organs. This may lead to greater expression of externalized PS since its exposure has been shown to correlate with a decline in intracellular ATP concentrations10. In our study, blockade of PS with Diannexin attenuated the elevated Lp during hemorrhage and resuscitation, suggesting a protective effect against gut dysfunction associated with hemorrhage.
We next sought a potential mechanism of how PS exposure may lead to organ dysfunction. Externalized PS is bound by secretory phosphoplipase A2 (sPLA2), thereby allowing sPLA2 to generate LPA14. LPA is known to induce platelet aggregation15, 20 and loss of vascular integrity34, 35. This is due in part to rounding of normally flat endothelial cells, resulting in passage of fluids and proteins between them and subsequent tissue edema35, 36. Selective blockage of LPA receptors was shown to decrease ischemia reperfusion injury in a murine model37. Given its direct relationship with PS exposure and role in subsequent downstream detrimental effects, LPA levels were evaluated in our hemorrhage model as a potential mechanism driving organ dysfunction. We found that LPA levels were reduced in the PS blockade group compared to the hemorrhage only group. However, the PS blockade group had higher levels of LPA than controls, with the hemorrhage only group having the highest LPA levels. This suggests that PS blockade prior to hemorrhage attenuates the response of LPA to cellular stress, but does not completely prevent its response.
In our study, PS blockade was performed prior to the onset hemorrhagic shock. This timing has potential for therapeutic benefit by means of prophylactically blocking PS prior to an additional systemic insult or “second hit,” such as damage control laparotomy19. Prior studies, including a phase II trial, have shown benefits of Diannexin when administered after an ischemic insult19, 23. Additionally, in murine models of ischemia reperfusion with Diannexin, reduction in renal tubule damage was apparent up to 8 days after ischemic insult, suggesting the benefits of PS blockade are durable21.
There are several limitations of our study. First, Diannexin was administered prior to hemorrhage, which is less clinically relevant when compared to administration after hemorrhage or during resuscitation. We had selected this timing based on our prior studies, however it would be useful to re-evaluate the potential benefits of Diannexin administration after hemorrhage or during resuscitation in future studies. Next, only laboratory markers of end organ function were evaluated. Histologic specimens would provide additional evidence for end organ damage, especially in the evaluation of pulmonary dysfunction. Lastly, this study used a rat model and given the much smaller blood volume and different physiology, the results may not be transferable to humans.
Conclusions
Hemorrhagic shock results in tissue ischemia and gradual ATP depletion leading to cell death. Intracellular ATP levels are inversely correlated with PS externalization and therefore microvascular thrombosis, ischemic conditions, endothelial leak, edema, and tissue injury10,33,38. We have shown that PS blockade with Diannexin decreases the renal and gut dysfunction associated with hemorrhagic shock and attenuates the magnitude of LPA generation. Our findings suggest the potential for therapeutic targets in the future that could prevent organ dysfunction associated with hemorrhagic shock.
ACKNOWLEDGEMENTS:
Diannexin was given as a gift by Anthony Allison, Ph.D., Alavita Pharmaceuticals, Mountain View, CA.
Funding: This work was supported by the National Institutes of Health (NIH/NIGMS) #K08GM081361
Financial Support: NIH/NIGMS #K08GM081361
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
References
- 1.Kauvar DS, Lefering R, Wade CE. Impact of Hemorrhage on Trauma Outcome: An Overview of Epidemiology, Clinical Presentations, and Therapeutic Considerations. The Journal of Trauma: Injury, Infection, and Critical Care. 2006;60:S3–S11. [DOI] [PubMed] [Google Scholar]
- 2.Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, et al. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Critical Care Medicine. 2012;40:1129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon JW. Hemorrhagic Shock. N Engl J Med. 2018;378:370–9. [DOI] [PubMed] [Google Scholar]
- 4.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. 2001;276:1071–7. [DOI] [PubMed] [Google Scholar]
- 5.Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016;23:1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagata S, Suzuki J, Segawa K, Fujii T. Exposure of phosphatidylserine on the cell surface. Cell Death & Differentiation. 2016;23:952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segawa K, Nagata S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–50. [DOI] [PubMed] [Google Scholar]
- 8.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27. [DOI] [PubMed] [Google Scholar]
- 9.Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y. Silent Cleanup of Very Early Apoptotic Cells by Macrophages. The Journal of Immunology. 2003;171:4672–9. [DOI] [PubMed] [Google Scholar]
- 10.Gleiss BGV, Orrenius S, Fadeel B. Fas-triggered phosphatidylserine exposure is modulated by intracellular ATP. Federation of European Biochemical Societies. 2002;519:153–8. [DOI] [PubMed] [Google Scholar]
- 11.Shen XD, Ke B, Zhai Y, Tsuchihashi SI, Gao F, Duarte S, et al. Diannexin, a novel annexin V homodimer, protects rat liver transplants against cold ischemia-reperfusion injury. Am J Transplant. 2007;7:2463–71. [DOI] [PubMed] [Google Scholar]
- 12.Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Progress in Lipid Research. 2003;42:423–38. [DOI] [PubMed] [Google Scholar]
- 13.Mold C, Morris CA. Complement activation by apoptoic endothelial cells following hypoxia/reoxygenation. Immunology. 2001;102:359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molski M, Groth A, Allison AC, Hendrickson M, Siemionow M. Diannexin treatment decreases ischemia-reperfusion injury at the endothelial cell level of the microvascular bed in muscle flaps. Ann Plast Surg. 2009;63:564–71. [DOI] [PubMed] [Google Scholar]
- 15.Kuypers FA, Larkin SK, Emeis JJ, Allison A. Interaction of an annexin V homodimer (Diannexin) with phosphatidylserine on cell surfaces and consequent antithrombotic activity. Thrombosis and Haemostasis. 2017;97:478–86. [PubMed] [Google Scholar]
- 16.Reutelingsperger CPM, van Heerde, WL. Annexin V, the regulator of phosphatidylserine-catalyzed inflammation and coagulation during apoptosis. Cellular and Molecular Life Sciences. 1997;53:527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng EY, Sharma VK, Chang C, Ding R, Allison AC, Leeser DB, et al. Diannexin decreases inflammatory cell infiltration into the islet graft, reduces beta-cell apoptosis, and improves early graft function. Transplantation. 2010;90:709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto K, Kim H, Oishi H, Chen M, Iskender I, Sakamoto J, et al. Annexin V homodimer protects against ischemia reperfusion-induced acute lung injury in lung transplantation. J Thorac Cardiovasc Surg. 2016;151:861–9. [DOI] [PubMed] [Google Scholar]
- 19.Strumwasser A, Bhargava A, Victorino GP. Attenuation of endothelial phosphatidylserine exposure decreases ischemia-reperfusion induced changes in microvascular permeability. J Trauma Acute Care Surg. 2018;84:838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teoh NC, Ito Y, Field J, Bethea NW, Amr D, McCuskey MK, et al. Diannexin, a novel annexin V homodimer, provides prolonged protection against hepatic ischemia-reperfusion injury in mice. Gastroenterology. 2007;133:632–46. [DOI] [PubMed] [Google Scholar]
- 21.Wever KE, Wagener FA, Frielink C, Boerman OC, Scheffer GJ, Allison A, et al. Diannexin protects against renal ischemia reperfusion injury and targets phosphatidylserines in ischemic tissue. PLoS One. 2011;6:e24276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Victorino GP, Newton CR, Curran B. Effect of angiotensin II on microvascular permeability. J Surg Res. 2002;104:77–81. [DOI] [PubMed] [Google Scholar]
- 23.Cooper MKS, Stratta R, D’Alessandro A, Malgaonkar S, Allison A, et al. Diannexin, a novel ischemia/reperfusion therapeutic agent, reduces delayed graft function (DGF) in renal transplant recipients from marginal donors. American Journal of Transplant. 2010;10. [Google Scholar]
- 24.Landis EM. Micro-Injection Studies of Capillary Permeability. The American Journal of Physiology. 1927;82. [Google Scholar]
- 25.Curry FHV, Sarelius I. Cardiovascular Physiology: Techniques in the Life Sciences. New York: Elsevier; 1983. [Google Scholar]
- 26.Curry FE, Sarelius, I. Measurement of Permeability, Pressure, and Flow. New York: Elsevier; 1983. [Google Scholar]
- 27.Victorino GP, Chong TJ, Curran B. Albumin impacts the effects of tonicity on microvascular hydraulic permeability. J Surg Res. 2004;122:167–72. [DOI] [PubMed] [Google Scholar]
- 28.Lord JM, Midwinter MJ, Chen Y-F, Belli A, Brohi K, Kovacs EJ, et al. The systemic immune response to trauma: an overview of pathophysiology and treatment. The Lancet. 2014;384:1455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierce A, Pittet JF. Inflammatory response to trauma: implications for coagulation and resuscitation. Curr Opin Anaesthesiol. 2014;27:246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ran SDA, Thorpe P. Increased Exposure of Anionic Phospholipids on the Surface of Tumor Blood Vessels. Cancer Research. 2002;62:6132–40. [PubMed] [Google Scholar]
- 31.Rongen GA, Oyen WJ, Ramakers BP, Riksen NP, Boerman OC, Steinmetz N, et al. Annexin A5 scintigraphy of forearm as a novel in vivo model of skeletal muscle preconditioning in humans. Circulation. 2005;111:173–8. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Zhang S, Luo L, Norstrom E, Braun OO, Morgelin M, et al. Platelet-derived microparticles regulates thrombin generation via phophatidylserine in abdominal sepsis. J Cell Physiol. 2018;233:1051–60. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Jiang R, Liu L, Watkins T, Zhang F, Dong JF. Traumatic brain injury-associated coagulopathy. J Neurotrauma. 2012;29:2597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fourcade OSM, Viode C, Rugani N, Leballe F, Ragab A, et al. Secretory Phospholipase A2 Generates the Novel Lipid Mediator Lysophosphatidic Acid in Membrane Microvesicles Shed from Activated Cells. Cell. 1995;80:919–27. [DOI] [PubMed] [Google Scholar]
- 35.Neidlinger NA, Larkin SK, Bhagat A, Victorino GP, Kuypers FA. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006;281:775–81. [DOI] [PubMed] [Google Scholar]
- 36.van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VWM. Role of RhoA and Rho Kinase in Lysophosphatidic Acid-Induced Endothelial Barrier Dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20. [DOI] [PubMed] [Google Scholar]
- 37.Okusa MDYH, Huang L, Sigismund L, Macdonald T, Lynch KR. Selective blockade of lysophosphatidic acid LPA3 receptors reduces murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2003;285:F565–F74. [DOI] [PubMed] [Google Scholar]
- 38.Lee WL, Slutsky AS. Sepsis and Endothelial Permeability The New England Journal of Medicine. 2010:689–91. [DOI] [PubMed] [Google Scholar]