

Summary
Medulloblastoma (MB), the most frequent malignant childhood brain tumor, can arise from cellular malfunctions during hindbrain development. Here we generate humanized models for Sonic Hedgehog (SHH)-subgroup MB via MYCN overexpression in primary human hindbrain-derived neuroepithelial stem (hbNES) cells or iPSC-derived NES cells, which display a range of aggressive phenotypes upon xenografting. iPSC-derived NES tumors develop quickly with leptomeningeal dissemination, whereas hbNES-derived cells exhibit delayed tumor formation with less dissemination. Methylation and expression profiling show that tumors from both origins recapitulate hallmarks of infant SHH MB and reveal that mTOR activation, as a result of increased Oct4, promotes aggressiveness of human SHH tumors. Targeting mTOR decreases cell viability and prolongs survival, showing the utility of these varied models for dissecting mechanisms mediating tumor aggression and demonstrating the value of humanized models for a better understanding of pediatric cancers.
Keywords: medulloblastoma, neuroepithelial stem cells, MYCN, metastasis, mTOR, POU5F1, reprogramming
Graphical Abstract
Highlights
-
•
Human iPSC-derived or primary neuroepithelial stem cells can be transformed by MYCN
-
•
MYCN drives infant SHH medulloblastoma with clinically relevant features
-
•
Epigenetically regulated Oct4 promotes mTOR hyperactivation in infant SHH tumors
-
•
mTOR inhibition efficiently targets metastatic SHH medulloblastoma models and PDXs
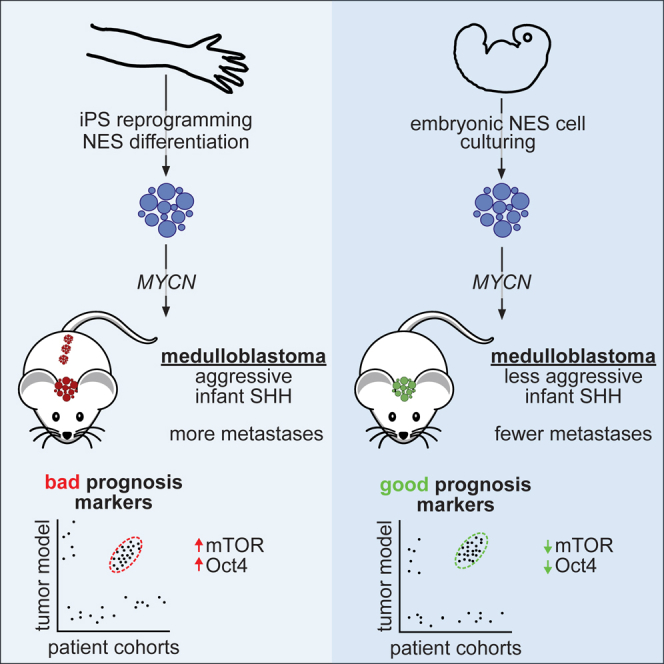
Čančer et al. report multiple models of infant Shh medulloblastoma; human iPSC-derived models display significant malignancy, whereas primary hindbrain neuroepithelial stem cell-derived models are less aggressive. These models were used to identify a role for Oct4 in promoting mTOR activation in metastatic tumors, which were sensitive to mTOR inhibition.
Introduction
Medulloblastoma (MB) is the most common malignant pediatric brain tumor and arises in the developing hindbrain (Markant and Wechsler-Reya, 2012). Although current therapeutic options have been successful in prolonging patients’ survival, survivors usually suffer from severe side effects, including cognitive impairment and endocrine deficiency (Saury and Emanuelson, 2011). Transcriptional, genomic, and epigenomic profiling have revealed four main subgroups of MB—WNT, Sonic Hedgehog (SHH), group 3, and group 4 (Taylor et al., 2012)—with further subtype specification of SHH MB into SHH-α, which mostly contains childhood patients; SHH-β and SHH-γ, which include infants; and SHH-δ, which mostly includes adolescents/adults (Cavalli et al., 2017). Amplification and overexpression of MYC and MYCN genes are frequently found in MB and are associated with poor prognosis (Cavalli et al., 2017). MYCN is expressed in essentially all MBs (Hede et al., 2014, Swartling et al., 2010) but is specifically upregulated in WNT and SHH tumors.
We previously demonstrated that ectopic MYCN expression drives MB from murine neural stem cells and is further required for tumor maintenance (Swartling et al., 2010, Swartling et al., 2012). Animal models of MB have been important tools for understanding of developmental pathways behind tumorigenesis but also for studying therapeutic strategies employed to better target the disease. Although murine SHH models have been primarily developed by either expressing activated SMO or depleting PTCH1, key upstream regulators in the SHH pathway, they face a number of apparent disadvantages in terms of resemblance to human MB (Neumann et al., 2017). Apart from being developed in another species, genetically engineered mouse models (GEMMs) of MB rarely show the histological desmoplastic features often seen in human SHH tumors (Schüller et al., 2004). Most published murine SHH models likely resemble adult SHH tumors and not infant or childhood SHH tumors (Pöschl et al., 2014). Finally, the transcriptional landscapes between the species differ substantially as a result of differential epigenetic regulation (Lin et al., 2014). Thus, it is likely that perturbation in gene expression can result in diverse cellular responses in murine cells compared with human cells.
Here we present several humanized models of SHH MB generated from various types of human neuroepithelial stem (NES) cells by overexpressing MYCN. The models described develop SHH MB with varying malignancy, depending on whether the cells of origin are induced pluripotent stem cell (iPSC)-derived or embryonal hindbrain NES cells.
Results
Ectopic Overexpression of MYCN in Human iPSC-Derived NES and Human Hindbrain Neuroepithelial Stem (hbNES) Cells
To study whether human stem cells can be transformed into brain tumors, we developed a model system in which various types of NES and hbNES cells were genetically engineered by lentiviral transduction of mutationally stabilized MYCNT58A or wild-type MYCNWT protein. We used two types of NES cells: AF22 cells (called NES-1), in which iPSC reprogramming was performed using retroviruses (Falk et al., 2012), and control (CTRL)-3-NES cells (called NES-2), which were derived by integration-free Sendai virus-based reprogramming (Shahsavani et al., 2018) before they were differentiated into long-term self-renewing NES cells. We also studied similarly cultured embryonic hindbrain NES cells isolated at two different time points: Sai2 cells (called hbNES-1) from a gestational age of 36 days and HB930 cells (called hbNES-2) from a gestational age of 46 days. The iPSC-derived NES cells are biologically similar to hbNES cells isolated from human embryos (Tailor et al., 2013). By comparing expression profiles with expression signatures from normal human developing brain, we found that NES cells resembled embryonic stem cells around post-conception weeks 5–7, which also corresponds well with the gestational age of the primary hbNES cells (Figure 1A; Figure S1A). V5-tagged MYCNT58A or MYCNWT was lentivirally overexpressed in iPSC-derived NES-1 and NES-2 cells and primary embryonic hbNES-1 and hbNES-2 cells (Figures 1B and 1C). After selection, MYCN expression was about 15–30 times higher than in parental cells (Figure 1D). MYC overexpression in human neural stem cells is known to cause immortalization (Kim et al., 2006). Similarly, we observed direct activation of TERT, the catalytic subunit of telomerase, following MYCN overexpression in both NES and hbNES cells (Figure S1B).
Figure 1.

Engineering of Cell Lines with Lentiviral Vectors Expressing MYCN
(A) Metagene projection of NES cell lines (AF22, CTRL-3, and CTRL-10) and primary hindbrain hbNES cell lines (Sai2, Sai3, HB901, and HB930) against normal brain profiles (GSE25219), showing that iPSC-derived NES cells display an embryonal expression signature.
(B) Schematic overview. iPSC-derived NES cells and human embryonic hbNES cells were transduced with lentiviruses expressing MYCNT58A and MYCNWT, GFP, and luciferase and transplanted into the cerebella of nude mice.
(C) MYCNT58A or MYCNWT lentiviral vectors contain the tetO-CMV hybrid promoter, used for knockdown of the transgene. The GFP-luciferase vector contains GFP for in vitro visualization and luciferase for in vivo monitoring.
(D) MYCN expression in MYCN-transduced NES1-2 and hbNES1-2 cells was determined by qRT-PCR (n = 3).
See also Figure S1.
NES and hbNES Cells Expressing MYCNT58A or MYCNWT Generate Tumors In Vivo
We transplanted NES and hbNES cells expressing MYCNT58A or MYCNWT into the cerebellum of nude mice. NES-1 and NES-2 cells expressing MYCNT58A generated tumors approximately 2 months post-transplantation (Figure 2A; Table S1), whereas hbNES-1 and hbNES-2 tumors had significantly longer latency (median survival ratio [MSR] NES to hbNES = 0.42; Figure 2A; Table S1). In comparison, transplanted MYCNWT cells generated tumors at a similar latency and with a similar MSR (NES to hbNES = 0.50; Figure 2B). Tumors could be followed with luciferase and were found around the injection site in the cerebellum with occasional spread into the posterior midbrain or the forebrain/olfactory bulb (Figures S2A and S2B).
Figure 2.

Transplanted NES and hbNES Cells Expressing MYCN Give Rise to Highly Proliferative and Metastatic Tumors with MB Histology
(A and B) Tumor-free survival of transplanted NES and hbNES cells expressing MYCNT58A (A) or MYCNWT (B). Dashed lines represent control stem cells. Colored arrows designate the endpoints for the respective tumor model. MSR, median survival ratio.
(C and D) NES tumors expressing MYCNT58A (C) or MYCNWT (D) presented with a significantly higher proportion of leptomeningeal spread compared with hbNES tumors. Metastasis was confirmed by histological analysis of brains and spinal cords of the indicated number of animals examined.
(E) Representative histology of NES and hbNES MYCNT58A MBs. Values indicate the percentage of positive cells (Ki67 and cleaved caspase-3) or relative density (V5-MYCN) measured from three individual tumors.
(F) Representative photos of Reticulin, Synaptophysin, and Ki67 staining of a NES-2 tumor, showing characteristic nodular-desmoplastic MB histology. asterisks, desmoplastic; arrowheads, nodules.
(G) Quantification of the Ki67 staining shown in (F).
Scale bars, 50 μm.
Metastatic spread of transplanted cells was first detected by bioluminescence imaging and later confirmed by histological examination of brains and spinal cords of a significant fraction of tumors. Twelve of 32 NES tumors presented leptomeningeal dissemination, whereas only two of 21 hbNES tumors showed spinal cord metastases (Figures 2C, 2D, and S2A). Detailed histological analysis of tumor tissue showed classical MB histology characterized by small cells with dense nuclei (Figures 2E and S2C). A number of NES-2 tumors presented reticulin-free and synaptophysin-positive pale islands (nodules), characterized by less proliferative cells, surrounded by highly proliferative, reticulin-positive, and synaptophysin-negative desmoplastic zones (Figures 2F and 2G), which is a typical feature of nodular/desmoplastic MB (Schüller et al., 2004). NES and hbNES tumors were further highly proliferative (Figures 2E, S2C, and S2D) and presented a similar rate of apoptotic cells (Figures 2E, S2C, and S2D). All tumors were also characterized by a high level of ectopic MYCNT58A or MYCNWT protein expression (Figures 2E, S2C, and S2D). In contrast, normal NES and hbNES cells injected similarly remained (as silent/non-proliferating cells) in the host brain for months, as seen using the human-specific cell marker Stem121 (Figure S2E), and they never generated any brain tumors.
NES and hbNES Tumors Express Markers of Primitive Embryonal Neuronal Brain Tumors
Because we observed no difference in histology of MYCNT58A and MYCNWT tumors, we continued histological profiling on MYCNT58A tumor models. All tumors showed strong staining of the human tissue markers Stem121 (Figures S3E-S3H) and TUJ1 (Figures 3A–3D) with rare GFAP- and OLIG2-positive cells (Figures 3E–3H and 3I–3L, respectively), indicating a neuronal character of these tumors, with a staining pattern typical for MB (Swartling et al., 2010, Swartling et al., 2012).
Figure 3.

Histological Profiling of NES and hbNES MYCNT58A Tumors
(A–D) Representative photos of tumors stained positive for the neuronal cytoplasmic marker TUJ1 in NES-1 (A), NES-2 (B), hbNES-1 (C), and hbNES-2 (D) tumors.
(E–L) Representative photos of tumors stained for GFAP (E–H) and OLIG2 (I–L), showing weak staining with few positive cells.
Scale bars, 50 μm.
See also Figure S3.
NES expression is essential for the maintenance of neural stem cells and can be found across all MB subtypes (Gilbertson and Ellison, 2008). Many NES and hbNES tumor cells stained positive for Nestin (Figure S3A–S3D), suggesting a primitive origin. Finally, both NES and hbNES tumor cells were able to recruit murine endothelial cells to support neovascularization, as demonstrated by CD34 staining of blood vessels (Figures S3I–3L), where the human tissue marker NuMA was only present in the tumor tissue (Figure S3M).
NES and hbNES Tumors Demonstrate Expression and Methylation Signatures of Infant SHH MB
We next used RNA sequencing to define tumors molecularly. Cross-platform comparison with various brain tumor entities revealed a close alignment of all NES and hbNES tumors with human MB (Figure S4A). We further compared NES and hbNES tumor expression profiles against 737 MB patients (Cavalli et al., 2017) and 20 patient-derived xenografts (PDX) from a number of MB patients similarly transplanted into the cerebellum of immunodeficient mice. Using cross-platform projection of data, we found that NES and hbNES tumors resemble the human SHH subgroup of MB and further cluster closely together with PDXs derived from SHH MB (Figures 4A, 4B, and S4B; data not shown). Next we performed methylation profiling of the tumors and predicted their subgroup affiliation based on a set of hypo- and hypermethylated signature probes of MB subgroups (Cavalli et al., 2017). One hbNES tumor could not be definitively associated with a defined subgroup, but, similar to expression profiling, all other tumors demonstrated methylation signatures typical for SHH MB patients (Figure 4C).
Figure 4.

NES and hbNES Tumors Show Expression Signatures Characteristic of SHH MB
(A and B) Metagene projection of NES-1, NES-2, hbNES-1, and hbNES-2 tumors and PDX models onto human MB samples (GSE85217) followed by hierarchical clustering (A) or PCA (B) indicates that PDX models cluster with their respective human counterparts and that tumor models affiliate with the SHH subgroup.
(C) Tumor models display a methylation signature recapitulating SHH MBs.
(D) Normalized gene expression of lineage markers for NES cells, upper rhombic lip (URL), and granule cells (GC) in NES and hbNES stem cells and tumor models. Values were averaged across samples within each compartment and then gene normalized.
(E) Boxplot comparing MYCN expression levels, normalized by Z score across all samples, between WT and T58A tumors for NES-1, NES-2, hbNES-1, and hbNES-2 models.
(F and G) NES and hbNES tumors display a gene expression profile (F) and a methylation profile (G) recapitulating infant SHH.
See also Figure S4.
In contrast to recent single-cell RNA transcriptomics reports in MB patients (Hovestadt et al., 2019, Vladoiu et al., 2019) suggesting that tumors might be similar to their true cell of origin, our starting cells were indeed more stem-like (NES+, SOX2+), whereas MYCN-driven tumors from this cell type are more similar to granule neuron precursor (GNP) cells (ATOH1+), suggesting that MYCN overexpression is promoting a lineage differentiation toward granule neurons (Figure 4D). Genes involved in SHH signaling were expressed at similar (GLI2, MYCN, SMO, and ATOH1) or slightly lower (GLI1 and PTCH1) levels in NES and hbNES tumors compared with SHH PDX samples (Figure S4C). In comparison, PDXs from group 3 and group 4 MB subgroups showed no or very low expression of these genes (Figure S4C). Further, NES and hbNES tumors showed upregulation of genes involved in oxidative stress/mitochondrial biogenesis in human SHH MBs but rarely in SHH GEMMs (Figure S4D; Łastowska et al., 2019). We treated tumor cells with the SMO agonist SAG but observed no changes in viability or expression of GLI1 (Figures S4E and S4F). The SMO antagonist SANT-1 did not affect GLI1 expression either (Figure S4F). Taken together, our data suggest that the downstream, but not upstream, SHH signaling pathway is activated by MYCN expression.
Interestingly, all MYCNT58A tumors (except for NES-2) have significantly lower expression of MYCN mRNA compared with MYCNWT (Figure 4E), whereas, at the protein level, there was no significant difference in MYCN expression (Figures 2E, S2B, and S2C). The data suggest that MYCN protein levels might be accumulated because of the stabilizing mutation in the MYCNT58A model. SHH-α and SHH-β patients usually express higher levels of MYCN compared with SHH-γ (Figure S4G). Mutations in TP53 are particularly enriched in poor-prognosis childhood (but not infant) SHH-α MB (Cavalli et al., 2017, Zhukova et al., 2013). Still, by using Sanger sequencing, we did not find any insertions or deletions (indels) or point mutations that are associated with loss of function of p53 in NES or hbNES cells (data not shown).
Most SHH MBs occur in either infants or adults (Kool et al., 2012). In contrast to SHH MB models published so far (Pöschl et al., 2014), which resemble adult SHH MB, NES and hbNES tumors demonstrated expression and methylation signatures of infant SHH MBs (Figures 4F and 4G). Among pediatric SHH subtypes, MYCN expression is equally upregulated in SHH-α and SHH-β patients (Figure S4G), which have the worst prognosis in this subgroup. Although MYCN is amplified in young children with SHH-α tumors, it correlates with survival only in infant SHH-β/SHH-γ subtypes (Figures S4H, left, and S4I, left). Differences in terms of aggressiveness found between NES and hbNES tumors (Figures 2A–2D) could be attributed to SHH-β and SHH-γ patients, where metastases further have a prognostic value as compared to SHH-α patients (Figures S4H right and S4I right).
Pediatric cases of SHH MB have a significant rate of chromosome rearrangements (Cavalli et al., 2017). After analyzing our methylation data, we nevertheless found that none of the presented SHH MB models exhibited any broad chromosomal copy number aberrations (Figure S4J; data not shown). This is perhaps not surprising because transformation in our models takes a shortcut by directly activating the strong cancer driver MYCN, whereas tumorigenesis in patients is a slower multi-step process requiring several random events to support tumor progression, including chromosomal rearrangements.
NES Tumors Resemble a More Malignant Subset of SHH MBs and Show Enhanced Stemness and mTOR Signatures
Relative to their stem cells of origin, both NES and hbNES tumors showed increased expression of MYCN but suppressed MYC mRNA levels (Figure 5A). Such mutual exclusivity of MYC and MYCN expression has been reported previously in both MB (Northcott et al., 2017) and neuroblastoma (Westermann et al., 2008). MYCN but not MYC expression limited to infant and childhood SHH MBs (Cavalli et al., 2017) was also associated with worse outcome (Figures 5B and 5C), suggesting that the MYC-to-MYCN switch might contribute to increased malignancy.
Figure 5.

Activation of Stem Cell Signature Genes and PI3K/AKT/mTOR, but Not MYC, Contributes to the Aggressiveness of Infant SHH MB
(A) Bar plot showing the fold change of MYCN and MYC expression of tumor models compared with the respective controls.
(B) Survival plot comparing pediatric (<18 years old) SHH patients (n = 60) with a high MYCN/MYC expression ratio with patients (n = 61) with a low ratio.
(C) Association between normalized MYCN and MYC expression in pediatric SHH patients, indicating samples with high (red) or low (blue) MYCN/MYC expression ratio used in (B).
(D) Key findings of targeted or unbiased GSEA comparing NES and hbNES tumors. Colors indicate significant enrichment in NES (red) or hbNES (blue) tumors, respectively.
(E and F) Heatmap illustrating the gene-normalized expression of stem cell signature genes (E) or genes involved in mTOR signaling (F) across tumors.
(G and H) Protein lysates of NES-2 and hbNES-1 tumor cells overexpressing POU5F1 (G) showing upregulation of phosphorylated 4E-BP1 in Oct4+ cells (H).
(I) POU5F1-transduced NES-2 and hbNES-1 tumor cells developed MB with significantly shorter latency compared with EV-transduced controls. There was no significant difference in median survival for hbNES-1 POU5F1 (70 days) and NES-2 EV (66 days).
(J) POU5F1 promoted an increase in metastatic spread to the spinal cord in POU5F1-transduced hbNES tumors.
(K and L) Transplanted NES-2/POU5F1 and hbNES-1/POU5F1 cells developed Oct4+ tumors (K) that significantly upregulated p-4E-BP1 (L).
(M) Strong staining of p-4E-BP1 in NES-1 tumors and metastatic SHH patient-derived xenografts (PDX), moderate to weak staining in NES-2 and hbNES tumors, and non-metastatic SHH PDXs as shown using IHC.
Scale bars, 50 μm.
See also Figure S5.
We next performed targeted gene set enrichment analyses (GSEAs) on gene sets associated with prognostic markers. Convincingly, the expression profile of hbNES compared with NES tumors showed enrichment of genes associated with good prognosis in MB in general (Pomeroy et al., 2002) and of genes associated with good prognosis in pediatric SHH MB in particular (Figure 5D).
Unbiased GSEAs showed that NES tumors demonstrated enrichment in embryonic stem cell signatures (Figure 5D), including NANOG, SOX2, KLF4, PARP1, and POLR3G (Figure 5E), which all collaborate in transcriptional activation to maintain pluripotency (Boyer et al., 2005, Wong et al., 2011, Roper et al., 2014). The reprogramming factor POU5F1 (encoding Oct4) and stemness markers NANOG, PARP1, NES, and POLR3G were upregulated in NES tumors compared with hbNES tumors (Figure 5E). POU5F1 has been identified as a driver of MB dissemination (da Silva et al., 2017), although it does not correlate significantly to a poor prognosis (data not shown) in any of the four MB subgroups (Cavalli et al., 2017). POU5F1, however, correlates with a poor outcome in patients of SHHβ and SHHγ subtypes but not SHHα (Figure S5A; data not shown). Likewise, NES is a mediator for progression of SHH MB (Li et al., 2016), and SOX2 is a link to a more aggressive SHH MB subset (Vanner et al., 2014). Neither NES or SOX2 nor other reprogramming factors, including NANOG and KLF4, correlate significantly with a poor prognosis in SHHβ and SHHγ MBs in the Cavalli MB dataset (data not shown). Unbiased GSEA also demonstrated upregulation of markers of metastasis and mTOR signaling in NES compared with hbNES tumors (Figure 5D). Here, typical mTOR signaling genes were upregulated only in NES tumors (Figure 5F). Out of these, EIF4EBP1 was found to be significantly associated with a worse survival prognosis in human pediatric SHH tumors (Figure S5B). The gene encodes the 4E-BP1 protein, a master regulator of mRNA translation, and is a direct effector of mTORC1 signaling.
Oct4 Regulates mTOR Hyperactivation to Promote Aggressiveness in Infant SHH Subtypes
Of all tumors, NES tumors, and especially the most aggressive NES-1 tumors, have the highest POU5F1 levels (Figure 5E). When checking the promoter methylation status of POU5F1, we found hypomethylation of the promoter compared with the respective stem cells of origin for NES-1 and NES-2 tumors (Figure S5C). In contrast, in the hbNES-1 model, both tumors and stem cells displayed equally methylated POU5F1 promoters. At the protein level, Oct4 is not expressed, or expressed at very low levels, in both NES and hbNES stem cells (Figure S5D), whereas both NES-1 and NES-2 tumor cells exhibited high Oct4 protein levels upon tumorigenesis compared with hbNES tumor cells (Figure S5D). This phenomenon suggests that hypomethylation of the POU5F1 promoter and subsequent POU5F1 overexpression occur after MYCN transformation and are cell type-specific. Likewise, it has been shown that MYCN amplification in human neuroblastoma cells can increase POU5F1 expression in a positive feedback loop (Kaneko et al., 2015). In comparison, we did not find any changes in methylation in promoter regions of other reprogramming factors, including KLF4, SOX2, MYC, and NANOG (data not shown). Consequently, SOX2 was elevated in most cells but, unlike Oct4, did not significantly change, as shown by comparing its levels in stem cells and corresponding tumor cells (Figure S5E).
We isolated brain tumors from NES- and hbNES-MYCNT58A-transplanted animals and cultured tumor cells (Table S1) under stem cell conditions. Compared with respective NES and hbNES tumors, NES and hbNES tumor-derived cell cultures expressed comparable MYCN levels and closely resembled the methylation signature characteristic of SHH MB patients (data not shown). To functionally test whether POU5F1 promotes aggressiveness of SHH MB, we overexpressed POU5F1 in the more aggressive NES-2 tumor cells and the less aggressive hbNES-1 tumor cells (Figure 5G). These cells exhibited a substantial increase in phosphorylated 4E-BP1 immediately following Oct4 overexpression (Figure 5H), suggesting that Oct4 might be involved in regulating mTOR pathway activity. When orthotopically injected, POU5F1-overexpressing NES-2 and hbNES-1 tumor cells induced tumors at significantly shorter latency compared with empty vector (EV)-transduced counterparts (Figure 5I). POU5F1 overexpression in hbNES-1 cells shortened the latency to be analogous with the latency of NES-2 EV tumors (median survival hbNES-1 POU5F1 = 70 days; median survival NES-2 EV = 66 days) and increased their metastatic potential because clear spinal cord metastases were present (in H&E samples) in half of the tumors screened (2 of 4; Figure 5J). Tumors developed from POU5F1-overexpressing cells showed strong Oct4 staining (Figure 5K and S5F) and were also enriched in p-4E-BP1 (Figure 5L and S5G). We next compared the increase in malignancy because of POU5F1 overexpression with similar experiments in which we inactivated the well-known tumor suppressor p53. Given that our tumor models did not show any mutations in TP53, we lentivirally expressed a dominant-negative p53 mutant (p53DD) to abrogate p53 activity. Following etoposide treatment, p53DD-transduced NES-2 and hbNES-1 tumor cells showed decreased induction of p21 and inhibition of apoptosis (Figure S5H). p53DD overexpression shortened the latency in animals orthotopically transplanted with NES-2 and hbNES-1 tumor cells to a similar extent as when overexpressing POU5F1 in these cell lines (Figures S5I and S5J).
We observed positive staining for p-4E-BP1 in all our models in vivo (Figure 5M), where NES-1 tumors demonstrated significantly higher phosphorylation compared with NES-2, hbNES-1, and hbNES-2. Similarly, p-4E-BP1 was expressed at varying levels in SHH PDX samples, showing particularly high expression in MYCN-amplified and/or metastatic samples (Figure 5M). POU5F1 expression positively correlates with the expression of 4E-BP1 within the SHHβ and SHHγ patient cohorts but not in the SHHα patient cohort (Figure S5K; data not shown), which prompted us to evaluate whether high POU5F1 expression leads to upregulation of phosphorylated 4E-BP1 in NES and hbNES tumors. High POU5F1 mRNA levels in NES-1 tumors positively correlates with phosphorylation of 4E-BP1 (Figure S5L). Collectively, our results suggest that activation of the mTOR pathway as a result of aberrant POU5F1 expression potentiates the malignancy of infant SHH MB.
Tumor-Derived Cell Cultures Demonstrate Clear Differences in Self-Renewal, Differentiation Patterns, and Promoting Metastases
We next studied the behavior of generated tumor cell lines to see whether certain experimental conditions could reflect their variation in overall malignancy. NES- and hbNES-derived tumor cultures showed increased proliferation rates compared with corresponding stem cells (Figures 6A–6D). Withdrawal of growth factors in NES and hbNES stem cell cultures results in differentiation (Falk et al., 2012, Tailor et al., 2013). We analyzed the self-renewal potential and observed that only NES tumor-derived cultures were able to form secondary neurospheres in limiting dilution assays, with NES-1 tumor culture 2 able to form spheres even in media lacking both epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) (Figures 6E and 6F). None of the hbNES tumor cell lines or the NES and hbNES stem cell lines could form secondary spheres under any given condition (data not shown). We then investigated how tumor cell lines respond to differentiation into more mature cell types. As shown in Figure S6A, after 4 weeks of unguided differentiation, normal stem cells showed reduced proliferation (Ki67+), whereas tumor cells sustained proliferation. NES cells preferentially differentiated toward a neuronal fate (TUJ1+ and/or MAP-2+) compared with hbNES cells, which showed a comparably reduced level of neuronal differentiation (Figures S6B–S6E). Of all normal stem cells, hbNES cells from the latest gestational age (hbNES-2) showed a profound capacity for astrocytic differentiation (Figures S6F and S6G). In comparison, tumor cells showed a significantly reduced capacity for any kind of differentiation. Finally, NES tumor cells also showed loss of Oct4 staining following 4 weeks of differentiation, whereas no tumor cells showed any reduction in the number of SOX2-positive cells (Figure S6H).
Figure 6.

Primary Tumor-Derived Cell Cultures Demonstrate Growth Factor Independence, Retain Tumor Regeneration Capacity, and Have a Capacity to Promote Migration
(A–D) NES-1 (A), NES-2 (B), hbNES-1 (C) and hbNES-2 (D) derived tumor cultures (TCs) demonstrated increased proliferation compared with their stem cells of origin.
(E and F) Tumor sphere-forming capacity of two representative NES-1 (E) and NES-2 (F) tumor-derived cultures in the presence or absence of EGF and bFGF.
(G) NES and hbNES tumor lines regenerate tumors when transplanted in mice with a differential latency similar to primary tumors.
(H and I) NES-1 secondary tumors demonstrated distant metastatic spread as shown by in vivo luciferase imaging (H) and histology (I) of spinal cords and spinal cord metastases from NES-1 and metastatic SHH PDX stained positive for phosphorylated 4E-BP1. Scale bars, 50 μm.
(J) Spinal cord metastatic cells and NES cells showed increased migration compared with hbNES cultures in a wound healing assay.
(K) The NES-1 tumor metastasis demonstrated significant enrichment in the mTOR pathway.
See also Figure S6.
When transplanting NES-1 and hbNES-1 MB cultures into the cerebellum of nude mice, they generated secondary tumors with a latency of 1.5–2 and 3–7 months, respectively (Figure 6G), recapitulating the differential median latency observed in primary tumors (MSR = 0.42 for primary tumors and 0.47 for secondary tumors). We again observed increased leptomeningeal spread in NES secondary tumors, a feature rarely shown in hbNES secondary tumors (Figure 6H and 6I). Similar to their parental tumors, spinal cord metastases derived from NES-1 secondary tumors or metastatic MYCN-amplified SHH PDX showed high levels of p-4E-BP1 (Figure 6I). NES tumors presented leptomeningeal spread, and they further demonstrated a significantly higher migration rate and shorter time to wound closure compared with hbNES-derived tumor cells (Figures 6J and S6D). When we specifically dissected out NES-1 spinal cord metastases and cultured them under the same conditions as tumor cell lines, they showed the highest migration rates among all tumor lines (Figures 6J and S6I). Further, when we compared metastases with associated primary tumors, we found significant enrichment in mTOR signaling (Figure 6K).
Specific Knockdown and Inhibition of MYCN Leads to a Decrease in Tumor Cell Viability
To investigate whether tumor-derived cell cultures depend on MYCN expression, we conditionally knocked down MYCN in two representative NES-1-derived tumor cell cultures (Figure S7A). When transducing cells with a Tet repressor vector, MYCN could be partially knocked down in the tumor cells, resulting in a significant decrease in cell viability (Figure S7B). Bromodomain and extraterminal domain (BET) protein inhibition is a promising therapy for MYC/MYCN-amplified tumors in several malignancies, including MB (Bandopadhayay et al., 2014). Although ectopic MYCN DNA does not contain binding sites for BET genes because we use a chimeric CMV promotor, the BET inhibitor JQ1 reduced cell viability by more than 50% in tumor models and metastases (Figure S7C), in line with our previous reports that JQ1 targets the transcriptional output of oncogenic MYCN (Bolin et al., 2018). We also found that, upon BET inhibition, SHH MB tumor cells and metastases initiated apoptosis, as demonstrated by an increase in cleaved caspase-3, but also presented a decrease in Lamin B1 (Figure S7D), which is associated with senescent cells (Freund et al., 2012). Pharmacological inhibition of MYCN signaling had only a modest effect on apoptosis induction in stem cells of origin for tumor models (Figure S7F), whereas the major effect was exercised on inducing cellular senescence (Figure S7F). However, it is important to note that these cells represent embryonic hindbrain neuroepithelial cells (Falk et al., 2012, Tailor et al., 2013) and are not likely to persist in an older individual. In fact, we recently showed that neonatal stem cells were not sensitive to even rather high micromolar concentrations of JQ1 (Bolin et al., 2018).
mTOR Inhibition Suppresses Viability and Migration of Tumor-Derived Cell Cultures
The phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway strongly correlates with increased invasiveness and metastasis of glioblastoma and MB (Crespo et al., 2016). When we exposed NES, hbNES, and NES-1 metastatic tumor cells to AZD2014, a potent mTOR and PI3K inhibitor, we observed a significant reduction in both cell viability and migration (Figures 7A and 7B). AZD2014 induced cell cycle arrest and caused an increase in the apoptotic sub-G1/G0 population in all four SHH MB models and SHH MB metastases (Figure 7C). AZD2014 and two other mTOR inhibitors, GDC-0084 and INK128, all decreased p-4E-BP1 and induced apoptosis and/or senescence in all SHH MB tumor models (Figure 7D). In contrast to JQ1, mTOR inhibition did not induce apoptosis and senescence in normal NES and hbNES stem cells (Figure S7F).
Figure 7.

NES- and hbNES-Derived Tumor Cultures Demonstrate Sensitivity to mTOR Inhibition
(A and B) AZD2014 significantly decreased viability (A) and migration (B) of NES- and hbNES-derived tumor cells and cells derived from NES-1 spinal cord metastases (NES-1 mets).
(C) AZD2014 promoted cell cycle arrest in G1/G0 phase and increased the sub-G1/G0 population of all cells derived from the presented tumor models and spinal cord metastases.
(D) The small-molecule mTOR inhibitors AZD2014, GDC-0084, and INK128 decreased phosphorylation of 4E-BP1 and induced senescence and/or apoptosis in all tumor models and metastases.
(E) Representative bioluminescence images of NES-1-transplanted mice treated for 8 days with vehicle or 1 mg/kg INK128.
(F–H) Quantification of bioluminescence images as a function of total tumor signal (F) or spinal cord metastases signal (G) over time. INK128-treated mice showed a reduction in metastatic spread (H). Orange bars indicate a window when mice received daily i.p. injections of vehicle (n = 6) or 1 mg/kg INK128 (n = 6).
(I and J) INK128-treated mice developed smaller tumors (I) that were significantly less positive for p-4E-BP1 and more apoptotic (I and J). Scale bars, 50 μm.
(K) Daily INK128 treatment (0.5 mg/kg) of NES-1-bearing mice significantly prolonged their survival compared with vehicle control (Wilcoxon test).
(L) Human SHH PDXs treated with 500 μM INK128 show a significant decrease in viability in vitro after 24-h and 48-h inhibition. Metastatic SHH PDX lines with elevated mTOR signaling were particularly sensitive to mTOR inhibition.
(M) Western blot of PDXs treated with INK128, showing a reduction in phosphorylated and total 4E-BP1 and an increase in cleaved caspase-3 in metastatic lines after 24-h and 48-h treatment.
See also Figure S7.
Combination treatment with JQ1 and AZD2014 also had a synergistic effect and decreased the viability of all NES and hbNES tumor cell lines as well as NES-1 metastasis cells (Figure S7E). Although the synergism between the two drugs resulted in increased apoptosis in some of the normal stem cells (Figure S7F), our data suggest that combined BET and mTOR inhibition can be used as potential therapy for these tumors.
mTOR Inhibition Suppresses SHH MB Growth and Prolongs Survival of NES Tumor-Grafted Animals
We next assessed whether mTOR inhibition can suppress tumor growth and metastasis in vivo. Mice grafted with NES-1 tumors were subjected to daily intraperitoneal (i.p.) injections of either vehicle or 1 mg/kg INK128 for 8 days. INK128 inhibition caused primary tumors to dramatically reduce in size after 14 days (Figures 7E, 7F, and 7I) and prevented formation and growth of spinal cord metastases already after 7 days of treatment (Figures 7E, 7G, and 7H). INK128 administration for 8 days resulted in a significant decrease in mTOR activity, as demonstrated by a reduction in p-4E-BP1 (Figures 7I and 7J). At the same time, tumors treated with INK128 underwent apoptosis (Figures 7I and 7J). Some of the animals experienced weight loss when using 1 mg/kg INK128 for more than 8 days, so we had to reduce the dose for long-term treatments. The tolerable concentration of 0.5 mg/kg INK128 still significantly prolonged the survival of mice grafted with NES-1 cells following daily treatments over 30 days relative to mice administered with vehicle (Figure 7K).
In addition, 8-day INK128 treatment of NES-1 tumors, which resulted in dephosphorylation of 4E-BP1 (Figures 7I and 7J), did not affect Oct4 expression (Figure S7G), suggesting that POU5F1 is upstream of mTOR and regulates its activity.
INK128 treatment also suppressed the viability of three representative SHH MB PDX lines (Figure 7L). The two PDX samples that were most sensitive were also highly metastatic tumors with hyperactivation of mTOR signaling (Figure 5M). INK128 treatment not only removed the phosphorylation mark from 4E-BP1 but also resulted in a decrease in total 4E-BP1 (Figure 7M). Further, the treatment led to increased apoptosis in the metastatic lines (Figure 7M). Thus, INK128 inhibition of SHH PDX lines supports our findings regarding humanized SHH MB models and proved mTOR inhibition to also be a valid strategy for SHH MB patients with metastatic tumors.
Discussion
Distinct Reprogramming Factors Are Potentiating MB Malignancy
The use of iPSCs and embryonic stem cells has drawn a lot of attention in recent years in the field of regenerative medicine but also for establishment of various disease models such as cancer (Gingold et al., 2016). It is likely that epigenetic regulation associated with iPSC derivation may promote development of certain types of cancer. The introduction of reprogramming factors into non-tumorigenic mammary epithelial cells can transform them into tumors with cancer stem cell properties (Nishi et al., 2014). In contrast, mesenchymal stem cells (MSCs) derived from iPSCs formed less aggressive tumors than primary tissue-derived MSCs (Zhao et al., 2015). The findings suggest that the contribution of reprogramming in cancer might be context dependent and depends on the nature of the tumor developed. Our data suggest that reprogramming itself is not a link for easier transformation, poor prognosis, and metastasis; instead, specific reprogramming factors, like POU5F1, are important in driving tumor progression in infant SHH MBs. However, there are strikingly few reported mutations in pluripotency factors or stemness genes in tumor biopsies from SHH MB patients (Northcott et al., 2017). This suggests that such events involved in MB progression might be epigenetically controlled, which was also confirmed by our data when analyzing demethylation of the POU5F1 promoter in tumor cells compared with the cells from which they originated.
MYCN-Driven Tumor Initiation Is Spatially Restricted and Gives Rise to Distinct Brain Tumor Types
In a recently published study (Huang et al., 2019), the authors demonstrated malignant transformation of iPSC-derived NES cells into MB following ectopic expression of MYCN. Further, cerebellum-derived neural stem cells transformed by a mix of p53 knockdown (DNp53) and overexpression of MYC, AKT1, and TERT formed aggressive MB when injected into the brains of nude mice (Hanaford et al., 2016). However, previously published work never compared or investigated the difference in tumorigenic potential of transformed iPSC-derived and embryonal NES cells. The T58A mutation will allow MYC to become stabilized by avoiding proteasomal degradation (Hede et al., 2014). This change further promotes anti-apoptotic features and presumably allows NSCs to become immortalized (Hemann et al., 2005). We previously showed that mutationally stabilized MYCN was required to generate MB from primary murine neural stem cells (Swartling et al., 2012), whereas wild-type MYCN instead caused apoptosis and no malignant transformation. Nevertheless, it is likely that wild-type MYC proteins can be expressed at high levels during earlier embryonic time points without causing apoptosis, suggesting that there might be time windows in normal development where cells can malfunction and acquire activation of such oncogenes, allowing specific tumors to arise. Our results indeed show a higher level of apoptosis in MYCNWT tumors but no delay in tumor latency compared with MYCNT58A-induced MBs.
iPSC-Derived and Primary Stem Cells as Resources to Better Model and Treat Human Cancer
The constitution of cells in NES and hbNES cultures is not known, and it is likely a more heterogeneous population of cell types in hbNES culture compared with NES culture. Comparing NES cells and hbNES cells side by side with the same oncogene is a good way to understand differences depending on the tumor cell of origin. We found that, although transformation is driven from NES cells, the generated tumors present markers closely corresponding to GNPs. This suggests that MYCN overexpression does not only cause malignancy but also a glutamatergic neuronal switch in hindbrain developmental fate.
Although SMO inhibition has shown some promise in adult SHH tumors, it few effects in pediatric SHH tumors (Kieran et al., 2017, Robinson et al., 2015) and further causes severe bone defects after long-term treatment (Robinson et al., 2017). In our tumor models, we have shown that the mTOR pathway is downstream of both MYCN and POU5F1, rendering mTOR inhibition efficient when the expression of these genes is upregulated. A previous chromatin immunoprecipitation sequencing (ChIP-seq) study in human embryonic stem cells (ESCs) has further revealed putative Oct4 binding sites in the vicinity of the promoter region of EIF4EBP1 (Gifford et al., 2013), although the transcriptional effect of such direct binding remains to be elucidated.
Infant patients are spared from radiotherapy because of severe side effects on the developing brain and spinal cord and are thus in great need of better drug treatments. mTOR inhibitors have few severe long-term side effects in pediatric patients, as suggested from recent reports using everolimus in children under the age of 3 with epilepsy (Kotulska et al., 2013), which is in line with the non-toxic effect seen by using the blood-brain barrier-permeable mTOR inhibitor INK128 during early cerebellar development in mice (Wu et al., 2017). This fact, along with our significant results regarding suppressed viability, tumor migration, and spread in vivo following mTOR inhibition, suggests that effective mTOR inhibitors should be a preferential choice to prevent metastatic dissemination and impede recurrence of these fatal childhood malignancies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Ki67 antibody [SP6] - Proliferation Marker | Abcam | Cat# ab16667; RRID: AB_302459 |
| Rabbit CD34 antibody [EP373Y] | Abcam | Cat# ab81289; RRID: AB_1640331 |
| Rabbit V5 tag antibody - ChIP Grade | Abcam | Cat# ab9116; RRID: AB_307024 |
| Rabbit Anti-Lamin B1 Polyclonal Antibody | Abcam | Cat# ab16048; RRID:AB_443298 |
| Rabbit Anti-AU1 tag polyclonal antibody | Abcam | Cat# ab3401; RRID: AB_303770 |
| Rabbit Anti-p21 [EPR3993] monoclonal antibody | Abcam | Cat# ab109199; RRID: AB_10861551 |
| Rabbit Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit 4E-BP1 (53H11) mAb | Cell Signaling Technology | Cat# 9644; RRID: AB_2097841 |
| Rabbit Anti-4E-BP1, phospho (Thr37 / Thr46) Monoclonal Antibody | Cell Signaling Technology | Cat# 2855; RRID: AB_560835 |
| GLI1 (V812) Antibody | Cell Signaling Technology | Cat# 2534S; RRID: AB_2294745 |
| Rabbit Oct-4 Antibody | Cell Signaling Technology | Cat# 2750; RRID:AB_823583 |
| Rabbit Anti-PARP monoclonal antibody, clone 46D11 | Cell Signaling Technology | Cat# 9532; RRID: AB_659884 |
| Mouse Neuronal Class III beta-Tubulin (TUJ1) Monoclonal Antibody, Purified | Covance Research Products Inc | Cat# MMS-435P; RRID: AB_2313773 |
| Sheep ECL Mouse IgG, HRP-linked whole Ab | GE Healthcare Life Sciences | Cat# NA931V; RRID: AB_772193 |
| Donkey ECL Rabbit IgG, HRP-linked whole Ab | GE Healthcare Life Sciences | Cat# NA934V; RRID: AB_772206 |
| Synaptophysin, clone SY38 | Milipore | Cat# MAB5258; RRID: AB_2313839 |
| Mouse Anti-Nestin, clone 10C2 antibody | Millipore | Cat# MAB5326; RRID: AB_2251134 |
| Mouse Anti-Glial Fibrillary Acidic Protein, clone GA5 antibody | Millipore | Cat# MAB3402; RRID: AB_94844 |
| Rabbit Anti-Olig-2 antibody | Millipore | Cat# AB9610; RRID: AB_10141047 |
| Mouse Anti-Actin, beta Monoclonal antibody, Unconjugated, Clone c4 | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_626632 |
| Monoclonal Anti-MAP2 antibody produced in mouse, clone HM-2 | Sigma-Aldrich | Cat# M4403; RRID: AB_477193 |
| Mouse STEM121 Monoclonal Antibody Specific for Human Cytoplasmic Marker | TaKaRa | Cat# Y40410; RRID: AB_2632385 |
| Donkey Anti-Rabbit IgG (H+L) Polyclonal Antibody, Alexa Fluor 555 Conjugated | Thermo Fisher Scientific | Cat# A-31572; RRID:AB_162543 |
| Donkey anti-Mouse IgG (H+L) Polyclonal Antibody, Alexa Fluor 647 Conjugated | Thermo Fisher Scientific | Cat# A-31571; RRID:AB_162542 |
| Donkey Anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A31570; RRID: AB_2536180 |
| Bacterial and Virus Strains | ||
| Oct4-GFP lentivirus | Cellomics Technology | Cat# PLV-10050-50 |
| Biological Samples | N/A | |
| Patient-derived xenografts (PDX) | Robert J. Wechsler-Reya lab | Gift |
| Chemicals, Peptides, and Recombinant Proteins | ||
| INK128 | Apexbio | Cat#A8551 |
| Blotting-Grade Blocker | Bio-Rad | Cat#1706404 |
| Cell Lysis Buffer (10X) | Cell Signaling Technology | Cat#9803 |
| JQ1 | From James Bradner Lab | Gift |
| pLP1, pLP2, pLP/VSVG | From Magnus Essand Lab | Gift |
| Mayer HTX | HistoLab | Cat#01820 |
| Eosin | HistoLab | Cat#01650 |
| Modified decalcification solution (Formic acid) | Histolab | Cat#HL24150.1000 |
| Primocin | Invivogen | Cat#ant-pm-1 |
| Recombinant Human EGF | Peprotech | Cat#AF-100-15 |
| Recombinant Human Basic FGF | Peprotech | Cat#AF-100-18B |
| XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate | PerkinElmer | Cat#122799 |
| Polyethyleneimine Linear MW 25.000 | Polysciences | Cat#23966-2 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#04693159001 |
| PhosSTOP | Roche | Cat#PHOSS-RO |
| AZD2014 | Selleckchem | Cat#S2783 |
| INK128 | Selleckchem | Cat#S2811 |
| GDC-0084 | Selleckchem | Cat#S8163 |
| SAG | Selleckchem | Cat#S7779 |
| SANT-1 | Selleckchem | Cat#S7092 |
| Laminin | Sigma-Aldrich | Cat#L2020-1MG |
| Poly-L-Ornithine | Sigma-Aldrich | Cat#P3655-500MG |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat#P0781 |
| L-Glutamine | Sigma-Aldrich | Cat#G7513 |
| Puromycin | Sigma-Aldrich | Cat#P8833 |
| Resazurin | Sigma-Aldrich | Cat#R7017 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891-5G |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A8531-1VL |
| Etoposide | Sigma-Aldrich | Cat#E1383 |
| Kolliphor® EL | Sigma-Aldrich | Cat#C5135 |
| Sodium chloride | Sigma-Aldrich | Cat#S7653 |
| Sodium dodecyl sulfate | Sigma-Aldrich | Cat#L3771 |
| L-Cysteine | Sigma-Aldrich | Cat#168149 |
| DMEM | Thermo Fisher Scientific | Cat#31966047 |
| DMEM/F12 | Thermo Fisher Scientific | Cat#21331020 |
| OptiMEM | Thermo Fisher Scientific | Cat#31985070 |
| FBS | Thermo Fisher Scientific | Cat#16000044 |
| D-PBS | Thermo Fisher Scientific | Cat#14190250 |
| N2 supplement | Thermo Fisher Scientific | Cat#17502001 |
| B27 minus vitamin A supplement | Thermo Fisher Scientific | Cat#12587010 |
| NEAA | Thermo Fisher Scientific | Cat#11140050 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat#11360070 |
| Geneticin | Thermo Fisher Scientific | Cat#10131027 |
| Blasticidin | Thermo Fisher Scientific | Cat#R21001 |
| Trizol | Thermo Fisher Scientific | Cat#15596018 |
| TrypLE Express | Thermo Fisher Scientific | Cat#12604013 |
| NuPAGE MES SDS Running Buffer (20X) | Thermo Fisher Scientific | Cat#NP0002 |
| 4% formaldehyde | VWR | Cat#AAJ60401-AP |
| Ethanol, absolute | VWR | Cat#20821-365 |
| Papain | Worthington | Cat#LS003126 |
| ISOFLO vet100% isofluoran | Zoetis Finland OY | Cat#002185 |
| Critical Commercial Assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat#200521 |
| Reticulin Stain Kit | Histolab | Cat#AS-RRSK105-100 |
| GoTaq® DNA Polymerase | Promega | Cat#M3001 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat#69504 |
| Nuclear Fast Red solution | Sigma-Aldrich | Cat#N3020 |
| NeuroCult NS-A Differentiation Kit (Human) | STEMCELL Technologies | Cat#05752 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34080 |
| Qubit RNA BR Assay Kit | Thermo Fisher Scientific | Cat#Q10210 |
| Qubit dsDNA BR Assay Kit | Thermo Fisher Scientific | Cat#Q32853 |
| SuperScript VILO cDNA Synthesis Kit | Thermo Fisher Scientific | Cat#11754050 |
| SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat#4368706 |
| Phusion High-Fidelity DNA Polymerase | Thermo Fisher Scientific | Cat#F530 |
| FxCycle PI/RNase Staining Solution | Thermo Fisher Scientific | Cat#F10797 |
| Horse ImmPRESS HRP Anti-Mouse IgG (Peroxidase) Polymer Detection Kit | Vector Laboratories | Cat#MP-7402; RRID: AB_2336528 |
| Horse ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit | Vector Laboratories | Cat#MP-7401; RRID: AB_2336529 |
| ImmPACT DAB Peroxidase (HRP) Substrate Kit | Vector Laboratories | Cat#SK-4105; RRID: AB_2336520 |
| Antigen Unmasking Solution, Citric Acid Based | Vector Laboratories | Cat# H-3300 |
| Deposited Data | ||
| Expression profiling by high throughput sequencing Methylation profiling by genome tiling array | This paper | GEO: GSE106728 |
| Experimental Models: Cell Lines | ||
| Human: AF22 | From Anna Falk lab | Gift |
| Human: CTRL-3-NES | From Anna Falk lab | Gift |
| Human: CTRL-10-NES | From Anna Falk lab | Gift |
| Human: Sai2 | From Anna Falk lab | Gift |
| Human: Sai3 | From Anna Falk lab | Gift |
| Human: HB901 | From Austin Smith lab | Gift |
| Human: HB930 | From Austin Smith lab | Gift |
| Human: HEK293T | From Magnus Essand lab | Gift |
| Primary tumor-derived cell lines See Table S1 | This paper | N/A |
| PDX: ICb-984MB (SHH) aka. PDX MYCNamp M0 | Shu et al., 2008 | N/A |
| PDX: ICb-1572MB (Group 3) | Shu et al., 2008 | N/A |
| PDX: Med-211-FH (Group 3) | Girard et al., 2015 | N/A |
| PDX: Med-411-FH (Group 3) | Morfouace et al., 2014 | N/A |
| PDX: BT-084 (SHH) aka. PDX MYCNamp M+ | From Till Milde lab | Gift |
| PDX: DMB006 (Group 4) | From Rob Wechsler-Reya lab | Gift |
| PDX: DMB012 (SHH) aka. PDX PTCHmut M0 | From Rob Wechsler-Reya lab | Gift |
| PDX: RCMB18 (SHH) aka. PDX MYCNamp M0 | From Rob Wechsler-Reya lab | Gift |
| PDX: RCMB20 (Group 3) | From Rob Wechsler-Reya lab | Gift |
| PDX: RCMB32 (SHH) aka. PDX PTCHmut M+ | From Rob Wechsler-Reya lab | Gift |
| PDX: RCMB40 (Group 3) | From Rob Wechsler-Reya lab | Gift |
| Experimental Models: Organisms/Strains | ||
| Athymic Nude Mouse - Hsd:Athymic Nude-Foxn1nu | Envigo | Cat#069 |
| NSG, NOD scid gamma | The Jackson Laboratory | Cat#005557 |
| Oligonucleotides | ||
| RT-qPCR primers; see Table S3 | This paper | N/A |
| Primers for p53 sequencing; see Table S2 | Pfaff et al., 2010 | N/A |
| Recombinant DNA | ||
| pBMN(CMV-G2L2P) | From Magnus Essand lab | Gift |
| MYCNT58A cDNA | From William A. Weiss lab | Gift |
| MYCNWT cDNA | From William A. Weiss lab | Gift |
| p53DD cDNA | From Moshe Oren lab | Gift |
| Gateway pLenti6.3/TO/V5-DEST | Thermo Fisher Scientific | Not available anymore from vendor |
| Gateway pLenti3.3/TR | Thermo Fisher Scientific | Not available anymore from vendor |
| Gateway pDONR221 Vector | Thermo Fisher Scientific | Cat#12536017 |
| pLenti6.3/TO/V5-MYCN-WT | This paper | N/A |
| pLenti6.3/TO/V5-MYCN-T58A | This paper | N/A |
| pLenti6.3/p53DD-AU1 | This paper | N/A |
| Software and Algorithms | ||
| Fiji ImageJ | Schindelin et al., 2012 | https://fiji.sc/; RRID: SCR_002285 |
| Excel | Microsoft | https://products.office.com/en-us/excel |
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| IndiGO | Berthold Technologies | https://www.berthold.com/de/bio/in_vivo_Imaging_System_NightOWL_LB983 |
| IncuCyte® Scratch Wound Cell Migration Software Module | Essen Bioscience | Cat#9600-0012 |
| Metagene projection script | Tamayo et al., 2007 | https://doi.org/10.1073/pnas.0701068104 |
| Maxstat 0.7.25 | Hothorn and Lausen 2003 | https://cran.r-project.org/web/packages/maxstat/index.html |
| Limma 3.32.5 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Survminer 0.4.0 | Alboukadel Kassambara and Marcin Kosinski | https://cran.r-project.org/web/packages/survminer/index.html |
| pamr 1.55 | T. Hastie, R. Tibshirani, Balasubramanian Narasimhan, and Gil Chu | https://cran.r-project.org/web/packages/pamr/index.html |
| minfi 1.22.1 | Aryee et al., 2014 | http://bioconductor.org/packages/release/bioc/html/minfi.html |
| IlluminaHumanMethylationEPICmanifest 0.3.0 | Kasper Daniel Hansen | https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylationEPICmanifest.html |
| IlluminaHumanMethylation450kmanifest 0.4.0 | Kasper Daniel Hansen and Martin Aryee | http://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylation450kmanifest.html |
| ZEN Black | Zeiss | https://www.zeiss.com/microscopy/int/downloads.html?vaURL=www.zeiss.com/microscopy/int/downloads/zen.html |
| CytExpert 2.0 | Beckman Coulter | http://www.beckman.com/ |
| ChAMP 2.9.10 | Tian et al., 2017 | http://bioconductor.org/packages/release/bioc/html/ChAMP.html |
| Survival 2.41.3 | Therneau and Grambsch, 2000 | https://cran.r-project.org/web/packages/survival/index.html |
| Other | ||
| Synergy HTX multi-mode reader | BioTek | N/A |
| NuPAGE Novex 4-12% Bis-Tris Protein Gels, 1.0 mm, 10-well | Thermo Fisher Scientific | Cat#NP0321BOX |
| iBlot gel transfer stack nitrocellulose, mini | Thermo Fisher Scientific | Cat#IB301032 |
| StepOne Plus Real Time PCR System | Thermo Fisher Scientific | Cat#4376600 |
| ImageQuant LAS4000 | GE Healthcare Life Sciences | N/A |
| CFX96 Touch Real-Time PCR Detection System | BioRad | Cat#1855195 |
| IncuCyte® Cell Migration Kit | Essen Bioscience | Cat#4493 |
| CytoFlex | Beckman Coulter | Cat#B96620 |
| Leica DMi8 Microscope | Leica | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Fredrik J. Swartling (fredrik.swartling@igp.uu.se).
All unique/stable reagents and biological material generated in this study are available from the Lead Contact, Dr. Fredrik J. Swartling, with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Animals
All experiments were performed on 6-7-week-old female mice, housed in individually ventilated cages (3-5 animals per cage) in accordance with Uppsala University regulations on mice with appropriate organic bedding, paper house enrichments, food and water ad libitum and 12/12-hour light/dark cycle. All animals were determined to be healthy according to guidelines set up at Uppsala University. Prior to experiments performed within the scope of this study, animals were not used in any other procedure nor subject to another test or drug analysis. Patient-derived xenografts (see Key Resources Table) were propagated in immunodeficient NSG animals as previously described (Pei et al., 2016). Athymic Nude and NSG mice were maintained in the animal facilities at Uppsala University and at SBP Medical Discovery Institute and the Sanford Consortium for Regenerative Medicine. Animals were euthanized 4 weeks post transplantation (exploratory procedure), upon development of tumor symptoms (hunched back, paralysis, reduction of fur quality, weight loss and tilted head) or one year post transplantation.
All experiments were performed in accordance with national guidelines and regulations, and with the approval of the animal care and use committees at Uppsala University, SBP Medical Discovery Institute and at the University of California San Diego (UCSD).
Cell Cultures and Primary Tumor Cell Cultures
All cell cultures were maintained in humidified incubators set to 37°C in a 5% CO2/95% air atmosphere. Human embryonic neuroepithelial stem (hbNES) cell lines Sai2 and Sai3, human embryonic neural stem (hbNES) cell lines HB901 and HB930, and human induced pluripotent NES-like (NES) cell lines AF22, CTRL-3 and CTRL-10 were cultured on poly-L-ornithine (PO)/laminin coated dishes in feeder-free stem cell conditions (DMEM/F12 media supplemented with 1 × Glutamax, 100 units of Penicillin, 0.1 mg/ml Streptomycin, 1 × N2, 0.05 × B27-vitamin A, 10ng/ml EGF and 10ng/ml bFGF) as previously described (Falk et al., 2012, Tailor et al., 2013). Cells were passaged when confluent in 1:3 ratio using TrypLE Express. Primary tumor cultures were propagated for 7-14 days on PO/laminin coated dishes in PTC media (DMEM/F12 media supplemented with 1 × Glutamax, 100μg/ml Primocin, 1 × N2, 0.5 × B27-vitamin A, 10ng/ml EGF and 10ng/ml bFGF). Established tumor cultures (from day 15 onward) were propagated in TC media (PTC media where Primocin is replaced with 100 units of Penicillin and 0.1 mg/ml Streptomycin). HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units of Penicillin and 0.1 mg/ml Streptomycin, 6mM L-Glutamine, 1 × NEAA, 1mM Sodium Pyruvate.
Patient-derived xenograft cells, a MYCN amplified non-metastatic line (RCMB18), a PTCH1-mutated metastatic line (RCMB32) and a MYCN amplified metastatic line (BT084) (Kool et al., 2014, Pei et al., 2016, Purzner et al., 2018), were cultured in ultra-low attachment plates in PTC media supplemented with 1% fetal bovine serum instead of growth factors. Cells were cultured and examined within 48 hours (as described in respective publications) as it is known that SHH MB cells quickly lose their SHH pathway activity upon cell culturing (Sasai et al., 2006).
Cell lines and PDXs used in experiments were negative for Mycoplasma as determined at btrl.org or by using MycoAlert kits (Lonza)
Method Details
Viral Constructs and Cell Transductions
MYCNT58A or MYCNWT cDNA was cloned into pLenti6.3/TO/V5-DEST vector using Gateway cloning system, hence the name of the vector pLenti6.3/TO/V5-MYCN-T58A or pLenti6.3/TO/V5-MYCN-WT. pLenti6.3/p53DD-AU1 was generated by replacing the SpeI/NheI fragment of the pLenti6.3/TO/V5-DEST vector with the cDNA of the AU1-tagged p53DD (Bowman et al., 1996, Gottlieb et al., 1994). Errors in the open reading frame of p53DD were beforehand corrected using site-directed mutagenesis. pLenti6.3/TO/V5-MYCN-T58A, pLenti6.3/TO/V5-MYCN-WT, pBMN(CMV-G2L2P) (Figure 1B) and pLenti6.3/p53DD-AU1 were transfected into HEK293T cells to produce infectious lentiviral particles as described (Hillerdal et al., 2012). Briefly, cells were transfected with lentiviral vectors and the pLP1, pLP2 and pLP/VSVG packaging plasmids at a 1:1:1:2 ratio using polyethyleneimine. Viral supernatant was collected 48 and 72 h post transfection, filtered (0.45 μm), and concentrated by ultracentrifugation at 75,000 × g for 90 min at 4°C. AF22 (NES-1), CTRL-3 (NES-2), Sai2 (hbNES-1) and HB930 (hbNES-2) cells were transduced with pLenti6.3/TO/V5-MYCN-T58A or pLenti6.3/TO/V5-MYCN-WT virus and then selected with Blasticidin 72h post-transduction for at least 72h. NES-1, NES-2, hbNES-1 and hbNES-2 expressing MYCNT58A or MYCNWT were addressed as NES-1 MYCNT58A or WT, NES-2 MYCNT58A or WT, hbNES-1 MYCNT58A or WT and hbNES-2 MYCNT58A or WT respectively. As a control, NES-1, NES-2, hbNES-1 and hbNES-2 cells were transduced with pBMN(CMV-G2L2P) virus and selected with Puromycin. Prior to transplantation, all MYCNT58A or WT expressing cells were transduced with pBMN(CMV-G2L2P). NES-2 or hbNES-1 tumor cells were transduced with lentiviral particles coding for POU5F1 or p53DD-AU1 as described above. Cells were propagated for 3 passages before they were transplanted into nude mice.
Model Establishment
In vivo propagation of NES-1 MYCNT58A or WT, NES-2 MYCNT58A or WT, hbNES-1 MYCNT58A or WT, hbNES-2 MYCNT58A or WT primary tumor cell cultures and corresponding normal control stem cells, as well as propagation of NES-2 and hbNES-1 cells expressing POU5F1 was commenced in Athymic Nude Mouse. Cells were transplanted into cerebellum by stereo-tactical injections of 100,000 cells at a 30° angle over skull bone, 1mm lateral of sagittal suture, 1mm posterior of lambda suture and 2mm into the cerebellum. Tumor development was monitored by in vivo luciferase imaging as previously described (Swartling et al., 2010). Briefly, mice were anesthetized by isoflurane inhalation and injected with 150mg/kg luciferin. Detection of bioluminescent signal commenced 5 min post injection and was detected at the exposure time of 5 min with binning set to 4x4. Same bioluminescent settings were used from first imaging until experimental endpoint (tumor development or one-year post implantation of cells). From a symptomatic, euthanized animal, a small piece of tumor tissue was collected in PTC medium or snap-frozen, while the rest of the brain was fixed in 4% formaldehyde and embedded in paraffin.
Primary Tumor Cell Culture Establishment
Primary tumor cell cultures were established by mechanical disruption of a tumor biopsy in PTC media (gentle pipetting up and down) and the cells were spun down at 300g for 5 minutes. Media was aspirated and cells were seeded on poly-L-ornithine (PO, 1:500 dilution)/laminin (1:1000 dilution) coated dishes in DMEM/F12 media supplemented with 1 × Glutamax, 100μg/ml Primocin, 1 × N2, 0.5 × B27-vitamin A, 10ng/ml EGF and 10ng/ml bFGF (PTC media). Metastasis-derived primary cultures were established by dissecting tumor metastases from spinal cord under fluorescent microscope by selecting GFP positive cells, followed by the same procedure described above for establishing primary tumor cell cultures from tumor biopsies. All primary tumor cell cultures were passaged as described above and maintained in TC media (PTC media where Primocin is replaced with 100 units of Penicillin and 0.1 mg/ml Streptomycin).
Cell Proliferation, Migration and Inhibition Assays
Cell proliferation was determined as a function of cell metabolism over time. 5,000 cells were seeded in triplicates in TC media in PO (1:500) and laminin (1:1000) coated 96-well plates. Cell metabolism was assessed at 0, 1 and 3 days by adding 10% resazurin reagent and incubating for 4 hours at 37°C. Fluorescence was detected by excitation at 530nm and emission at 590nm using Synergy HTX multi-mode reader. Data points were normalized against day 0 measurements. Cell proliferation assays were performed in two biological replicates.
To analyze cell viability upon exposure to cytotoxic drugs and small molecule inhibitors, 10,000 cells were seeded in triplicates in 96-well plates coated with PO (1:500)/laminin (1:1000). Cells were incubated in TC media supplemented with SAG (500nM), JQ1 (500nM), INK128 (500nM), GDC-0084 (500nM) or AZD2014 (500nM) for 72h, when the viability was assessed using resazurin reagent, as a function of cell metabolism compared to cells exposed to DMSO. To assess the synergy of combined JQ1 and AZD2014 inhibition, 10,000 cells were seeded in triplicates on PO (1:500) and laminin (1:1000) coated 96-well plates and exposed to 125, 250, 500, 1000 or 2000nM of either single inhibitor or a combination of both JQ1 and AZD2014 (where the concentration of e.g., JQ1 = AZD2014 = 125nM etc.) for 72h, after which the viability was assessed using resazurin reagent, as a function of cell metabolism compared to cells exposed to DMSO.
Cell migration was assessed in scratch-wound healing assay. 1.6 × 105 cells/cm2 were seeded in 6 replicates in TC media on PO (1:500) and laminin (1:1000) coated 96-well plates 24h prior the wound (700-800 μm in size) was made using IncuCyte Cell Migration Kit. Cells were than washed and incubated in TC media alone or supplemented with AZD2014 (500nM) for 24h. Images were taken every 1h and migration rate was calculated as a function of wound size over time using IncuCyte Scratch Wound Cell Migration Software Module according to the manual provided by the manufacturer.
Patient-derived xenograft cell lines RCMB32, RCMB18 and BT084 were maintained in Athymic Nude Mice as described above. Frozen vials of the cell lines were thawed and cells were plated at a density of 50,000 cells per well in triplicate for each condition on two 96 well Ultra-Low attachment plates using primary tumor cell media (see above) supplemented with 1% fetal bovine serum instead of growth factors. Cells were allowed to recover for 4 hours after which cell viability was measured, 10% resazurin agent was added 2 hours prior detection, cells from the same condition were then pooled and isolated for protein extraction. INK128 (500nM) or DMSO control was added to the remaining wells 4 hours after plating, then incubated for 24h or 48h. Cell viability was measured at each end point and cells were isolated for protein extraction.
To induce apoptosis, 500,000 NES-2 or hbNES-1 tumor cells were treated with 20 μM etoposide in 2ml of TC media for 8 hours and cell lysates were subsequently analyzed by western blotting.
In Vivo and In Vitro Propagation of PDX Models
PDX cells of various medulloblastoma subgroups were obtained from btrl.org or from Dr. Robert Wechsler-Reyás lab. Cells were quickly thawed and spun down in PTC media to remove DMSO. Cells were counted and 100,000 cells were stereotactically injected at 30° over the skull bone, 1mm posterior and lateral to lambda and 2mm inside the cerebellum. Mice with tumors were euthanized and cells were prepared by dissecting the tumor tissue followed by Papain digestion (10U/ml) supplemented with 0,2mg/ml L-cysteine and 25U/ml DNase for 30min at 37°C. Papain reaction was stopped by 1% FBS solution (v/v in PBS), single cells were strained through 0,7 μm strainer, spun down at 300g and resuspended in PTC media, alternatively in FBS supplemented with 10% DMSO (v/v) for freezing.
Authentication of PDXs from btrl.org were performed as previously described (Brabetz et al., 2018) and was based on single tandem repeat analysis (STR) and SNPs on Infinium HumanMethylation450 BeadChip. The identity and subgroup of each PDX line obtained from the Wechsler-Reya lab was validated by gene expression and/or methylation analysis as previously described (Pei et al., 2016).
In Vivo Inhibition of mTOR Signaling
For the in vivo mTOR inhibition experiments, 100,000 NES-1 tumor cells were orthotopically transplanted in mice cerebella by stereotactic injection device 1mm posterior to lambda suture, lateral to sagittal suture and 2mm in the cerebellum 30° over the skull bone and propagated for 20 days before commencing drug treatment. Tumor burden was estimated by bioluminescent imaging (as described in “Model Establishment” section) and mice were randomized into two groups consisting of six mice with similar bioluminescent signals. Mice received daily i.p. injections of either vehicle (13.3% Kolliphor EL and 6.7% ethanol in 0.9% NaCl) or 1mg/kg INK128 (in vehicle), for eight days. Tumor development was followed up for 14 days by bioluminescent imaging every seven days.
To assess INK128 effects on animal survival, 100,000 NES-1 tumor cells were injected as described above and propagated for 14 days before commencing drug treatment. Tumor burden was estimated by bioluminescent imaging (as described in “Model Establishment” section) and mice were randomized into two groups consisting of five mice with similar bioluminescent signals. Mice received daily i.p. injections of either vehicle (13.3% Kolliphor EL and 6.7% ethanol in 0.9% NaCl) or 0.5mg/kg INK128 (in vehicle), for 30 days. Mice were followed up for tumor development until appearance of the tumor symptoms (hunched back, head tilt, paralysis, impaired fur quality), upon which brains and spinal cords were harvested as described above.
Cell Cycle Analysis
For the cell cycle analysis, 500,000 cells were seeded on PO (1:500) and laminin (1:1000) coated dishes, and incubated in TC media supplemented with 500nM AZD2014 for 72h. Cells were fixed in 70% ice-cold ethanol on ice for 30 minutes, washed twice with ice-cold PBS and stained using FxCycle staining solution according to manufacturer’s protocol. Cell cycle was assessed on CytoFlex flow cytometry instrument and data was analyzed using CytExpert software. 10,000 events were recorder for analysis of cell cycle phase distribution.
Histology, Immunohistochemistry and Immunofluorescence
Tumor histology was analyzed on 6μm thick brain sections stained with hematoxylin-eosin as previously described (Swartling et al., 2010). Briefly, histological sections were deparaffinized in xylene, rehydrated in a series of ethanol solutions (100, 95, 90, 70 and 60% v/v in deionized water), followed by staining in eosin and counter-staining in hematoxylin. Sections were dehydrated in ethanol solutions (70, 90, 95 and 100% v/v in deionized water) and in xylene, and embedded in mounting media. Whole brain section microphotographs were created as composite images of single images stitched together using ZEN Black software. White background image was subtracted from an individual image to account for uneven illumination of the specimen. Specific protein and marker expression was analyzed by immunostaining using antibodies against CD34 (1:100), Cleaved Caspase 3 (1:500), GFAP (1:500), Ki67 (1:1500), Nestin (1:200), Olig2 (1:500), phospho-4E-BP1 (1:1600), Stem121 (1:1000), Synaptophysin (1:500), Tuj1 (1:500) and V5 tag (1:1000) as previously described (Swartling et al., 2010). Briefly, sections were deparaffinized in Xylene, rehydrated in serial ethanol solutions as above, followed by antigen unmasking according to manufacturer’s protocol. Unspecific activity of tissue peroxidases was blocked with 0.3% solution of H2O2 for 30min. Sections were then blocked for 1h with horse serum blocking solution (ImmPRESS kit). Primary antibodies against above-mentioned targets were diluted in blocking solution (ImmPRESS kit) and incubated in humidified chambers overnight at 4°C. ImmPRESS and ImmPACT DAB Peroxidase (HRP) Substrate Kit was used to develop the signal. Sections were counter-stained in hematoxylin, dehydrated in ethanol solutions as above and in xylene, and embedded in mounting media. Reticulin staining was performed according to manufacturer’s protocol.
For immunofluorescence, cells were then fixed in 4% formaldehyde for 15 minutes, permeabilized using PBS with 0.2% Triton X, washed once with PBS and then blocked with 0.5% bovine serum albumin in PBS for 30 minutes at room temperature (RT). Primary antibodies were diluted in blocking solution and incubated with the cells for two hours at RT directly after the blocking step. Using antibodies against OCT4 (1:400), SOX2 (1:1000), TUJ1 (1:1000), MAP2 (1:200), GFAP (1:1000) and Ki67 (1:1000). Cells were then washed with 0.1% TBST 3 × 10 minutes and incubated with secondary antibodies (Alexa Fluor 555 anti-rabbit and anti-mouse) and DAPI diluted in blocking solution for 1 hour at RT. Cells were again washed 3 × 10 minutes with 0.1% TBST and mounted using Fluoromount. All immunofluorescence images were created as composite images of a single fluorescence channel. Individual channels were merged into a single composite image using Fiji ImageJ software.
Detection and Analysis of Metastatic Dissemination
Mice bearing tumors were imaged as as described in “Model Establishment” section. Bioluminescent imaging was subsequently used to detect both primary intracranial tumor and metastases in spinal cords. Upon development of brain tumor symptoms, mice were euthanized and in a set of animals both brains and spinal cords were harvested and immediately fixed in 4% formalin for at least 24 hours. Spinal cords were decalcified overnight in Modified Decalcification Solution, washed in 70% ethanol, embedded in paraffin and subsequently stained with Hematoxylin-Eosin or IHC against p-4E-BP1, as described above.
MYCNT58A Knock-down
Exogenous MYCNT58A expressed in primary tumor cultures was knocked-down by transducing them with pLenti3.3/TR viral vector. Cells were transduced with virus (without estimation of concentration and IU) and cultured for 72h before selecting the positive cells using TC media supplemented with 500μg/ml Geneticin. Positive selection was maintained for at least 7 days before continuing expression analysis. MYCNT58A expression was maintained by incubating cells in TC media supplemented with 1μg/ml doxycycline and suppression was established by removing the doxycycline. MYCN RNA and protein expression was detected by RT-qPCR and western blot, as described below.
Sphere Forming Assay and Differentiation
Self-renewability of stem cells and primary tumor cultures, in conditions with or without growth factors, was assayed as previously described (Swartling et al., 2012). Both primary and secondary spheres were allowed to form from 10 cells/well in ultra-low attachment 96-well plates in complete TC media or in TC media where EGF, bFGF, or both were removed. Seeded cells were allowed to form primary spheres for seven days, after which spheres were dissociated as described in tumor cell culture establishment section. These cells were then re-seeded (10 cells/well in ultra-low attachment plates) and allowed to grow for another seven days to form secondary spheres. Secondary spheres having diameter larger than 10, 20, 40 and 80μm were counted and statistical analysis was performed on three replicates. Sphere forming assays were done in two biological replicates.
Differentiation experiments were done by culturing stem cells and primary tumor cultures for four weeks using STEMCELL Technologies NeuroCult NS-A Differentiation Kit (Human), given new media weekly or split when needed, after which they were seeded on PO (1:500)/laminin (1:1000) coated chamber slides and cultured for an additional four days then fixed and processed for immunofluorescence. Undifferentiated cells were seeded on coated chamber slides two days before fixation.
p53 Sequencing
The entire coding sequence of the TP53 gene was amplified using the GoTaq DNA Polymerase and primers listed below. Amplicons were purified using the QIAquick PCR purification kit and sequenced by GATC Biotech (Cologne, Germany). Primers used: exon 2 + 3 forward (CTGTCTCAGACACTGGCATGG), exon 2 + 3 reverse (GGCAAGGGGGACTGTA), exon 4 forward (GGACTGACTTTCTGCTCTTGTCTTT), exon 4 reverse (CAGAGATCACACATTAAGTGGGTAA), exon 5 forward (CTCTCTAGCTCGCTAGTGGGT), exon 5 reverse (CGAAAAGTGTTTCTGTCATCCAAAT), exon 6 forward (GCCATGGCCATCTACAAGCA), exon 6 reverse (TGGGGTTATAGGGAGGTCAAA), exon 7 forward (ACAGGTCTCCCCAAGG), exon 7 reverse (AAACTGAGTGGGAGCAGTAAGGAGA), exon 8 + 9 forward (GGACAAGGGTGGTTGGGAGTAGA), exon 8 + 9 reverse (CCCAATTGCAGGTAAAACAGTCAAG), exon 10 forward (CAGTTTCTACTAAATGCATGTTGCT), exon 10 reverse (ATACACTGAGGCAAGAATGTGGTTA), exon 11 forward (CATCTTGATTTGAATTCCCGTTGT), exon 11 reverse (CACCAGTGCAGGCCAACTTGTTCAG).
Protein Biochemical Analysis
500,000 cells were seeded in 6-well PO (1:500) and laminin (1:1000) coated plates 24h before commencing treatment. Cells were incubated in TC media supplemented with SAG (500nM), SANT-1 (500nM), JQ1 (500nM), INK128 (500nM), GDC-0084 (500nM) or AZD2014 (500nM) for 72h and lysed with 1x Cell Lysis Buffer supplemented with 1% SDS, 1:10 PhosSTOP and 1:100 Protease Inhibitor Cocktail. To analyze protein expression after lentiviral gene transfer, 500,000 cells were harvested and lysed as described above. A total 10-20μg of protein lysate was loaded onto 4%–12% Bis Tris gel, run for 55 min at 180V and 400mA in 1X MES running buffer and transferred to iBlot Gel Transfer Stacks. Membranes were blocked with 5% bovine serum albumin or 5% milk in 0.5% TBST (0.5% Tween 20 in Tris-Buffered Saline) and probed against beta-Actin (1:1000), Cleaved Caspase-3 (1:500), V5 tag (1:1000), AU1 tag (1:1000), p21 (1:1000), PARP (1:1000), Lamin B1 (1:1000), Oct4 (1:1000), 4E-BP1 (1:1000) and p-4E-BP1 (Thr37/46) (1:1000) overnight at 4 degrees centigrade. Signal was developed by probing the membranes with secondary HRP-conjugated anti-mouse or anti-rabbit antibodies diluted in blocking buffer (1:10,000) and incubating them with SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Femto Chemiluminescent Substrate. Signal was detected on ImageQuant LAS 4000 or an Amersham Imager 680.
Real-Time Quantitative PCR Analysis
cDNA was synthesized from 200ng of total RNA using SuperScript VILO cDNA Synthesis Kit according to manufacturer’s protocol in following conditions: 25°C for 10min, 42°C for 60min, 85°C for 5min. For qPCR reaction, each cDNA sample was diluted 1:100 in MQ water. Expression of MYCN and TERT was determined using SYBR Green PCR Master Mix chemistry and following primers: GAPDH (CCGACCCCTTCATTGACCTCAACT and ATATTTCTCGTGGTTCACACCCAT), MYCN (AAGAACCCAGACCTCGAGTTTGAC and GCAGCAGCTCAAACTTCTTCCAGA) and TERT (TGGCTGCGTGGTGAACTTG and GCGGTTGAAGGTGAGACTGG). Thermal cycler was set to following parameters: hold at 95°C for 10min, 95°C for 15 s, 60°C for 1min (40 cycles). GAPDH expression was used as an endogenous control. All qPCR experiments were run in triplicates on StepOne Plus Real Time PCR System and CFX96 Touch Real-Time PCR Detection System. The data was analyzed by comparative ΔΔCt method.
RNA and DNA Extraction and Quantification
RNA from fresh frozen tumor samples (∼10mg) and primary tumor cell cultures (300,000 cells) was extracted using Trizol reagent and RNeasy Mini Kit according to manufacturer’s protocol. RNA concentration was determined with Qubit RNA BR Assay Kit according to manufacturer’s protocol. DNA from fresh frozen (∼10mg) and primary tumor cell cultures (300,000 cells) was prepared using DNeasy Blood and Tissue Kit according to manufacturer’s protocol. DNA concentration was determined using Qubit dsDNA BR Assay Kit according to manufacturer’s protocol.
RNA Expression and DNA Methylation Profiling
Targeted RNA-Seq was performed at Uppsala Genome Center, SciLifeLab, Uppsala University on Ion-Proton platform using Ion AmpliSeq Transcriptome Human Gene Expression panel. Methylation profiling was performed by the SNP&SEQ Technology Platform in Uppsala (http://snpseq.medsci.uu.se/). The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory on Illumina Platform using Illumina MethylationEPIC_v-1-0 DNA analysis BeadChip.
External RNA expression (GSE85217, Cavalli et al., 2017; GSE50161, Griesinger et al., 2013; GSE25219, Kang et al., 2011) and DNA methylation datasets (GSE85212, Cavalli et al., 2017) were downloaded as raw CEL or IDAT files from the Gene Expression Omnibus (GEO), respectively.
Microarray expression CEL files were processed in the Affymetrix Expression Console (AEC) using the Robust Multichip Average (RMA) method for background correction, quantile normalization and probe summarization. Raw AmpliSeq counts were normalized differently depending on downstream use. Specifically, for classification analyses, we performed within-sample normalization by dividing raw read counts within each sample by the total number of reads in the sample. For between-sample comparisons of expression values, we performed TMM normalization followed by a conversion to logCPM units. Finally, for differential analysis, including pre-ranking of genes prior to GSEApreranked, we conducted TMM normalization followed by VOOM processing. Prior to TMM normalization, genes were filtered by requiring that a certain number of samples had a CPM value above 1, i.e., sum(cpm > 1) > 10 for obtaining a normalized expression array and sum(cpm > 1) > 3 for normalization prior to between-condition limma analyses.
Gene symbols of both AmpliSeq and microarray expression datasets were translated to official gene symbols using the HUGO Gene Nomenclature Committee (HGNC). Multiple rows mapping to the same HGNC gene symbol were either collapsed using the mean value (microarrays) or removed from the expression matrix (AmpliSeq).
DNA methylation IDAT files were processed in R using the minfi and IlluminaHumanMethylation450kmanifest or IlluminaHumanMethylationEPICmanifest packages. Probes were removed, if their p value p > 0.01 in more than 5% of samples. The data was normalized using the preprocessNoob scheme on red-green channels before extraction of beta values.
Differential Expression/Methylation Analyses
The discovery of differentially expressed genes in AmpliSeq or microarray expression data was conducted in R using the limma package and the VOOM functionalities (only AmpliSeq). P values were corrected for multiple testing using Benjamini-Hochberg’s False Discovery Rate (FDR), and a differential expression was concluded significant if the adjusted p value q < 0.05.
Differentially methylated genomic positions were estimated in R using the dmpFinder function in the minfi package. Positions were regarded as differentially methylated if the adjusted p value q < 0.05. Row-normalized RNA expression data was used to analyze MYC and MYCN expression in tumor biopsies. The gene expression was analyzed by pooling together samples from the same experimental group (NES-1, NES-2, hbNES-1 and hbNES-2) and normalized to the expression levels of the respective stem cell of origin. Normalized log fold change values > 1 indicate overexpression, while values < −1 indicate suppression. Error bars indicate SEM.
Classification Analyses
Cross-platform classification of AmpliSeq RNA expression profiles against cerebellar samples of different developmental stages (GSE25219), against different classes of brain tumors (GSE50161) and against MB subgroups (GSE85217) was performed in R using the Metagene algorithm for cross-platform, cross-species projection of transcription profiles (Tamayo et al., 2007). For classification analyses, the GSE85217 dataset was filtered to exclude samples with transcription profiles that were arguably less clearly affiliated with a distinct MB subgroup (Weishaupt et al., 2019), resulting in a final dataset comprising 737 samples. The results of principal component analyses (PCA) and hierarchical clustering (HC) analyses generated by the Metagene algorithm were used to visualize the projection, i.e., PCA plots were either shown as produced by the metagene package or redrawn from the produced PCA results, while HC data was redrawn from the projected data in R using the heatmap.3 script. The plots were used in conjunction with the support vector machine (SVM) predictions from the metagene script in order to infer class affiliations.
A comparison of AmpliSeq expression profiles against SHH infant and SHH adult profiles was conducted in R using the pamr classification package. Specifically, reference infant (≤3 years, n = 70) and adult (> = 18 years, n = 69) cases were obtained from the GSE85217 dataset. The reference and AmpliSeq data were matched based on common HGNC gene symbols. Subsequently, 60 classifier genes were extracted as the 30 top significantly upregulated genes in infant cases (FDR < 0.05, average sample z-score in infant group > 0.25, average sample z-score in adult group < −0.25) and 30 top significantly downregulated genes in infant cases (FDR < 0.05, average sample z-score in infant group < −0.25, average sample z-score in adult group > 0.25). The classifier was trained using the pamr.train function applied on the classifier genes and the reference samples. The age group of the AmpliSeq samples was subsequently predicted using the pamr.predict function on the same set of genes in the AmpliSeq data.
The classification of methylation samples against MB subgroups and age groups was also performed using the pamr package and utilizing training samples extracted from the GSE85212 dataset. Specifically, for the classification of MB subgroups, 2000 signature probes were extracted for each subgroup as the 1000 top significantly hyper-methylated and 1000 top significantly hypo-methylated probes in that subgroup as compared to samples of all other subgroups. For the classification against infant or adult age groups, a total of 2000 classifier probes were extracted as the 1000 top significantly hypermethylated or 1000 top significantly hypomethylated genes in the infant group as compared to the adult group. Training and prediction was performed using the pamr package as described above for the RNA expression data.
Gene Set Enrichment Analyses
Gene Set Enrichment Analyses (GSEAs) were conducted using the GSEApreranked module on the GenePattern web platform (Reich et al., 2006). Specifically, genes were preranked by assigning scores according to the logFC and p values results from respective limma analyses, where individual scores were set as score = -sign(logFC)∗log10(p value). Statistics were calculated using gene set permutations and the classic scoring scheme was utilized. Unbiased GSEA screens were performed against 7 databases of gene sets (Hallmark, Kegg, Reactome, chemical & genetic pertubations (CGP), Transcription factor targets (TFT), Gene Ontology (C5), and Oncogenic Signatures (C6)).
To directly test, whether transcriptional differences between NES and hbNES correlated with clinical prognostic signatures, we performed targeted GSEA analyses against 4 gene sets. Of those, 2 gene sets had been established previously and reflected genes upregulated in patients with good prognosis (POMEROY_MEDULLOBLASTOMA_PROGNOSIS_UP) or bad prognosis (POMEROY_MEDULLOBLASTOMA_PROGNOSIS_DN), respectively. Two additional gene sets were established by identifying genes in pediatric SHH patients (see details of survival analyses below), high expression of which conferred either a significantly improved survival probability (n = 61; GOOD PROGNOSIS) or decreased survival probability (n = 28; BAD PROGNOSIS).
Patient Survival Analyses
Survival analyses for MB patients were performed in R using the survival package utilizing MB samples and survival/clinical/molecular annotations from the GSE85217 dataset. For analyses of individual target genes, survival differences were investigated between patients with high gene expression and patients with low gene expression, where gene expression cutoffs were based either on the mean or median expression and the corresponding p value was obtained using the score test (corresponding to a log-rank test) from the coxph-function.
In order to determine genes associated with good or bad prognosis in pediatric SHH patients (age < 18 years, n = 121), we utilized only the 3000 genes with highest standard deviation. For a single gene, the survival difference was investigated between patients with high gene expression and patients with low gene expression, where gene expression cutoffs and corresponding p values for the survival difference were estimated in R using the maxstat package. P values were corrected for multiple testing using Benjamini-Hochberg’s False Discovery Rate (FDR), and a survival difference was concluded significant if the adjusted p value q < 0.05.
Copy Number Analyses
Plots of broad copy number status in tumor samples were established via analysis of methylation data through the ChAMP (Tian et al., 2017) package in R. Specifically, a CNA profile for each tumor was estimated via the champ.CNA function by utilizing the respective normal cell as controls.
Quantification and Statistical Analysis
Immunohistochemistry and Immunofluorescence Quantification
Quantification of immunohistochemistry staining was performed in Fiji ImageJ on three representative microphotographs from three tumors and expressed as % of positively stained nuclei or relative density of DAB signal. Quantification of Immunofluorescence staining was performed in ImageJ on three representative microphotographs and expressed as % of DAPI positive nuclei. Data values represent mean and standard error of mean. Statistical significance of Ki67 measurements in nodular-desmoplastic, Oct4 and p-4E-BP1 levels in NES-2 and hbNES-1 EV/POU5F1 tumor staining, levels of Cleaved Caspase 3 and p-4E-BP1, and levels of Ki67, TUJ1, MAP-2 and GFAP in stem and tumor cells under normal and differentiating conditions, was estimated using Student’s t test. Differentiation experiments were repeated once.
Quantification of Metastatic Dissemination
Quantification and statistical analysis of metastatic spread was performed on samples where primary and metastatic tumors were analyzed both by bioluminescence and histological examination of H&E-stained coronal spinal cord sections (at least 5 sections from various regions/mouse per slide). Only samples with clear aggregates of more than ten tumor cells in any of the spinal cord sections were considered metastatic. Numbers of analyzed animals are shown in respective figures. Statistical analysis was performed using Chi-square test on primary tumors, where the observed outcome of NES-2, hbNES-1 and hbNES-2 was compared to observed outcome of NES-1. Student’s t test was applied to compare metastatic spread of tumors in mice treated with INK128 or vehicle.
Statistical Analysis
qPCR, cell proliferation, migration, inhibition, cell cycle, sphere forming assays and western blot were performed in two biological replicates. Student’s t test was applied to determine significant differences in a single gene expression (qPCR and RNA-Seq), cell proliferation and inhibition, while ANOVA with post hoc Tukey multiple comparison test was applied to assess significance of migration, cell cycle and sphere forming assays. Inhibition of PDX lines and accompanied western blot analysis was repeated once. Combination index (CI) was computed using CompuSyn software (Chou and Martin, 2005) according to developers guidelines for computing CI at fixed drug-to-drug ratio. CI < 0.8 indicated synergy, 0.8 < CI < 1.2 indicated addition, CI > 1.2 indicated antagonism. All data values represent mean of replicates and standard error of mean.
Animal Survival Statistics
All animal experiments (primary tumor penetrance), POU5F1 overexpression, p53 suppression and INK128 treatments were done once. Animal survival was shown as Kaplan-Meier curve and analyzed using GraphPad Prism 6 software, where survival difference between the two groups was assessed with Log-rank (all survival analyses except Figure 7K) or Gehan-Breslow-Wilcoxon test (Figure 7K).
Reporting of p Values, Sample Size and Statistical Significance
Significant p values are reported for each figure either in the figure description or at the respective figure panel. Samples size (n) for each analysis can be found in figure panels or figure descriptions. Error bars represent the standard error of mean. ∗, ∗∗, ∗∗∗ and ∗∗∗∗ indicate p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively.
Randomization and Inclusion/Exclusion Criteria
Animals included in INK128 in vivo drug evaluation were stratified into INK128 or vehicle group based on the bioluminescent signal to achieve a similar distribution of tumor sizes among the treatment groups. Animals that were found dead without obvious signs of tumor (both part of primary tumor penetrance studies, and POU5F1, p53 and INK128 treatment experiments) were marked as censored. No other data was excluded from the analysis and reporting in this study. No blinding was done at any stage of the study.
Data and Code Availability
Raw data of samples and cell lines from expression and methylation analysis is provided in GEO accession number GEO: GSE106728. A complete list of deposited data and software for data analysis can be found in the Key Resources Table.
Acknowledgments
This work was generously sponsored by the European Research Council under Horizon 2020 (project 640275, Medulloblastoma), the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, the Swedish Research Council, and the Ragnar Söderberg’s Foundation. We would like to thank Dr. Austin Smith (Wellcome Trust/MRC Stem Cell Institute, University of Cambridge) for providing us with human embryonic stem cells. We further acknowledge Drs. William A. Weiss and Miller Huang (UCSF, San Francisco) for providing us with MYCN constructs and Dr. Moshe Oren (Weizmann Institute, Rehovot) for the p53DD construct. We thank Dr. Till Milde (DKFZ, Heidelberg), Dr. Xiao-Nan Li (Texas Children’s Hospital, Houston), the Brain Tumor Resource Laboratory (https://research.fhcrc.org/olson/en/btrl.html), and Dr. Jim Olson’s lab (Fred Hutchinson Cancer Research Center, Seattle) for sharing their PDX models. We further acknowledge the BioVis facility, the National Genomics Infrastructure (NGI)/Uppsala Genome Center, and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure funded by RFI/VR and SciLifeLab, Sweden. Methylation profiling was performed by the SNP&SEQ Technology Platform in Uppsala, which is part of the NGI Sweden and SciLifeLab. The SNP& SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Author Contributions
Conceptualization, F.J.S. and M.Č.; Methodology, F.J.S. and M.Č.; Investigation, M.Č., S.H., K.O.H., T.B., and G.R.; Formal Analysis, M.Č., H.W., A.S., K.O.H., and S.H.; Resources, F.J.S., M.Č., M.E., J.T., A.G., R.J.W.-R., and A.F.; Visualization, M.Č., H.W., K.O.H., A.S., and S.H.; Writing – Original Draft, M.Č., H.W., and F.J.S.; Funding Acquisition, F.J.S; Supervision, F.J.S.
Declaration of Interest
The authors declare no competing interests.
Published: November 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.10.005.
Supplemental Information
References
- Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandopadhayay P., Bergthold G., Nguyen B., Schubert S., Gholamin S., Tang Y., Bolin S., Schumacher S.E., Zeid R., Masoud S. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014;20:912–925. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin S., Borgenvik A., Persson C.U., Sundström A., Qi J., Bradner J.E., Weiss W.A., Cho Y.-J., Weishaupt H., Swartling F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene. 2018;37:2850–2862. doi: 10.1038/s41388-018-0135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman T., Symonds H., Gu L., Yin C., Oren M., Van Dyke T. Tissue-specific inactivation of p53 tumor suppression in the mouse. Genes Dev. 1996;10:826–835. doi: 10.1101/gad.10.7.826. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabetz S., Leary S.E.S., Gröbner S.N., Nakamoto M.W., Şeker-Cin H., Girard E.J., Cole B., Strand A.D., Bloom K.L., Hovestadt V. A biobank of patient-derived pediatric brain tumor models. Nat. Med. 2018;24:1752–1761. doi: 10.1038/s41591-018-0207-3. [DOI] [PubMed] [Google Scholar]
- Cavalli F.M.G., Remke M., Rampasek L., Peacock J., Shih D.J.H., Luu B., Garzia L., Torchia J., Nor C., Morrissy A.S. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.C., Martin N. ComboSyn Inc.; 2005. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values. [Google Scholar]
- Crespo S., Kind M., Arcaro A. The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis. J. Cancer Metastasis Treat. 2016;2:80–89. [Google Scholar]
- da Silva P.B.G., Teixeira Dos Santos M.C., Rodini C.O., Kaid C., Pereira M.C.L., Furukawa G., da Cruz D.S.G., Goldfeder M.B., Rocha C.R.R., Rosenberg C., Okamoto O.K. High OCT4A levels drive tumorigenicity and metastatic potential of medulloblastoma cells. Oncotarget. 2017;8:19192–19204. doi: 10.18632/oncotarget.15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A., Koch P., Kesavan J., Takashima Y., Ladewig J., Alexander M., Wiskow O., Tailor J., Trotter M., Pollard S. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS ONE. 2012;7:e29597. doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A., Laberge R.-M., Demaria M., Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford C.A., Ziller M.J., Gu H., Trapnell C., Donaghey J., Tsankov A., Shalek A.K., Kelley D.R., Shishkin A.A., Issner R. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson R.J., Ellison D.W. The origins of medulloblastoma subtypes. Annu. Rev. Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- Gingold J., Zhou R., Lemischka I.R., Lee D.-F. Modeling Cancer with Pluripotent Stem Cells. Trends Cancer. 2016;2:485–494. doi: 10.1016/j.trecan.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard E., Ditzler S., Lee D., Richards A., Yagle K., Park J., Eslamy H., Bobilev D., Vrignaud P., Olson J. Efficacy of cabazitaxel in mouse models of pediatric brain tumors. Neuro-oncol. 2015;17:107–115. doi: 10.1093/neuonc/nou163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E., Haffner R., von Rüden T., Wagner E.F., Oren M. Down-regulation of wild-type p53 activity interferes with apoptosis of IL-3-dependent hematopoietic cells following IL-3 withdrawal. EMBO J. 1994;13:1368–1374. doi: 10.1002/j.1460-2075.1994.tb06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger A.M., Birks D.K., Donson A.M., Amani V., Hoffman L.M., Waziri A., Wang M., Handler M.H., Foreman N.K. Characterization of distinct immunophenotypes across pediatric brain tumor types. J. Immunol. 2013;191:4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaford A.R., Archer T.C., Price A., Kahlert U.D., Maciaczyk J., Nikkhah G., Kim J.W., Ehrenberger T., Clemons P.A., Dančík V. DiSCoVERing Innovative Therapies for Rare Tumors: Combining Genetically Accurate Disease Models with In Silico Analysis to Identify Novel Therapeutic Targets. Clin. Cancer Res. 2016;22:3903–3914. doi: 10.1158/1078-0432.CCR-15-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hede S.M., Savov V., Weishaupt H., Sangfelt O., Swartling F.J. Oncoprotein stabilization in brain tumors. Oncogene. 2014;33:4709–4721. doi: 10.1038/onc.2013.445. [DOI] [PubMed] [Google Scholar]
- Hemann M.T., Bric A., Teruya-Feldstein J., Herbst A., Nilsson J.A., Cordon-Cardo C., Cleveland J.L., Tansey W.P., Lowe S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillerdal V., Nilsson B., Carlsson B., Eriksson F., Essand M. T cells engineered with a T cell receptor against the prostate antigen TARP specifically kill HLA-A2+ prostate and breast cancer cells. Proc. Natl. Acad. Sci. USA. 2012;109:15877–15881. doi: 10.1073/pnas.1209042109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T., Lausen B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003;43:121–137. [Google Scholar]
- Hovestadt V., Smith K.S., Bihannic L., Filbin M.G., Shaw M.L., Baumgartner A., DeWitt J.C., Groves A., Mayr L., Weisman H.R. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Tailor J., Zhen Q., Gillmor A.H., Miller M.L., Weishaupt H., Chen J., Zheng T., Nash E.K., McHenry L.K. Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell. 2019;25:433–446.e7. doi: 10.1016/j.stem.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y., Suenaga Y., Islam S.M.R., Matsumoto D., Nakamura Y., Ohira M., Yokoi S., Nakagawara A. Functional interplay between MYCN, NCYM, and OCT4 promotes aggressiveness of human neuroblastomas. Cancer Sci. 2015;106:840–847. doi: 10.1111/cas.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran M.W., Chisholm J., Casanova M., Brandes A.A., Aerts I., Bouffet E., Bailey S., Leary S., MacDonald T.J., Mechinaud F. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-oncol. 2017;19:1542–1552. doi: 10.1093/neuonc/nox109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.U., Park I.H., Kim T.H., Kim K.S., Choi H.B., Hong S.H., Bang J.H., Lee M.A., Joo I.S., Lee C.S., Kim Y.S. Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease. Neuropathology. 2006;26:129–140. doi: 10.1111/j.1440-1789.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- Kool M., Korshunov A., Remke M., Jones D.T.W., Schlanstein M., Northcott P.A., Cho Y.-J., Koster J., Schouten-van Meeteren A., van Vuurden D. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M., Jones D.T.W., Jäger N., Northcott P.A., Pugh T.J., Hovestadt V., Piro R.M., Esparza L.A., Markant S.L., Remke M., ICGC PedBrain Tumor Project Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotulska K., Chmielewski D., Borkowska J., Jurkiewicz E., Kuczyński D., Kmieć T., Łojszczyk B., Dunin-Wąsowicz D., Jóźwiak S. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2013;17:479–485. doi: 10.1016/j.ejpn.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Łastowska M., Karkucińska-Więckowska A., Waschek J.A., Niewiadomski P. Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma. Cells. 2019;8:E216. doi: 10.3390/cells8030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Lee E.H., Du F., Gordon R.E., Yuelling L.W., Liu Y., Ng J.M.Y., Zhang H., Wu J., Korshunov A. Nestin Mediates Hedgehog Pathway Tumorigenesis. Cancer Res. 2016;76:5573–5583. doi: 10.1158/0008-5472.CAN-16-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Lin Y., Nery J.R., Urich M.A., Breschi A., Davis C.A., Dobin A., Zaleski C., Beer M.A., Chapman W.C. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA. 2014;111:17224–17229. doi: 10.1073/pnas.1413624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markant S.L., Wechsler-Reya R.J. Review: personalized mice: modelling the molecular heterogeneity of medulloblastoma. Neuropathol. Appl. Neurobiol. 2012;38:228–240. doi: 10.1111/j.1365-2990.2011.01235.x. [DOI] [PubMed] [Google Scholar]
- Morfouace M., Shelat A., Jacus M., Freeman B.B., 3rd, Turner D., Robinson S., Zindy F., Wang Y.-D., Finkelstein D., Ayrault O. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell. 2014;25:516–529. doi: 10.1016/j.ccr.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J.E., Swartling F.J., Schüller U. Medulloblastoma: experimental models and reality. Acta Neuropathol. 2017;134:679–689. doi: 10.1007/s00401-017-1753-3. [DOI] [PubMed] [Google Scholar]
- Nishi M., Sakai Y., Akutsu H., Nagashima Y., Quinn G., Masui S., Kimura H., Perrem K., Umezawa A., Yamamoto N. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2014;33:643–652. doi: 10.1038/onc.2012.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott P.A., Buchhalter I., Morrissy A.S., Hovestadt V., Weischenfeldt J., Ehrenberger T., Gröbner S., Segura-Wang M., Zichner T., Rudneva V.A. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y., Liu K.-W., Wang J., Garancher A., Tao R., Esparza L.A., Maier D.L., Udaka Y.T., Murad N., Morrissy S. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell. 2016;29:311–323. doi: 10.1016/j.ccell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff E., Remke M., Sturm D., Benner A., Witt H., Milde T., von Bueren A.O., Wittmann A., Schöttler A., Jorch N. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J. Clin. Oncol. 2010;28:5188–5196. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- Pomeroy S.L., Tamayo P., Gaasenbeek M., Sturla L.M., Angelo M., McLaughlin M.E., Kim J.Y.H., Goumnerova L.C., Black P.M., Lau C. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Pöschl J., Stark S., Neumann P., Gröbner S., Kawauchi D., Jones D.T.W., Northcott P.A., Lichter P., Pfister S.M., Kool M., Schüller U. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014;128:123–136. doi: 10.1007/s00401-014-1297-8. [DOI] [PubMed] [Google Scholar]
- Purzner T., Purzner J., Buckstaff T., Cozza G., Gholamin S., Rusert J.M., Hartl T.A., Sanders J., Conley N., Ge X. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci. Signal. 2018;11:eaau5147. doi: 10.1126/scisignal.aau5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J.P. GenePattern 2.0. Nat. Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson G.W., Orr B.A., Wu G., Gururangan S., Lin T., Qaddoumi I., Packer R.J., Goldman S., Prados M.D., Desjardins A. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015;33:2646–2654. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson G.W., Kaste S.C., Chemaitilly W., Bowers D.C., Laughton S., Smith A., Gottardo N.G., Partap S., Bendel A., Wright K.D. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017;8:69295–69302. doi: 10.18632/oncotarget.20619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper S.J., Chrysanthou S., Senner C.E., Sienerth A., Gnan S., Murray A., Masutani M., Latos P., Hemberger M. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014;42:8914–8927. doi: 10.1093/nar/gku591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai K., Romer J.T., Lee Y., Finkelstein D., Fuller C., McKinnon P.J., Curran T. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006;66:4215–4222. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- Saury J.-M.G., Emanuelson I. Cognitive consequences of the treatment of medulloblastoma among children. Pediatr. Neurol. 2011;44:21–30. doi: 10.1016/j.pediatrneurol.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller U., Schober F., Kretzschmar H.A., Herms J. Bcl-2 expression inversely correlates with tumour cell differentiation in medulloblastoma. Neuropathol. Appl. Neurobiol. 2004;30:513–521. doi: 10.1111/j.1365-2990.2004.00553.x. [DOI] [PubMed] [Google Scholar]
- Shahsavani M., Pronk R.J., Falk R., Lam M., Moslem M., Linker S.B., Salma J., Day K., Schuster J., Anderlid B.M. An in vitro model of lissencephaly: expanding the role of DCX during neurogenesis. Mol. Psychiatry. 2018;23:1674–1684. doi: 10.1038/mp.2017.175. [DOI] [PubMed] [Google Scholar]
- Shu Q., Wong K.K., Su J.M., Adesina A.M., Yu L.T., Tsang Y.T.M., Antalffy B.C., Baxter P., Perlaky L., Yang J. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells. 2008;26:1414–1424. doi: 10.1634/stemcells.2007-1009. [DOI] [PubMed] [Google Scholar]
- Swartling F.J., Grimmer M.R., Hackett C.S., Northcott P.A., Fan Q.-W., Goldenberg D.D., Lau J., Masic S., Nguyen K., Yakovenko S. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling F.J., Savov V., Persson A.I., Chen J., Hackett C.S., Northcott P.A., Grimmer M.R., Lau J., Chesler L., Perry A. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor J., Kittappa R., Leto K., Gates M., Borel M., Paulsen O., Spitzer S., Karadottir R.T., Rossi F., Falk A., Smith A. Stem cells expanded from the human embryonic hindbrain stably retain regional specification and high neurogenic potency. J. Neurosci. 2013;33:12407–12422. doi: 10.1523/JNEUROSCI.0130-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P., Scanfeld D., Ebert B.L., Gillette M.A., Roberts C.W., Mesirov J.P. Metagene projection for cross-platform, cross-species characterization of global transcriptional states. Proc. Natl. Acad. Sci. USA. 2007;104:5959–5964. doi: 10.1073/pnas.0701068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M.D., Northcott P.A., Korshunov A., Remke M., Cho Y.-J., Clifford S.C., Eberhart C.G., Parsons D.W., Rutkowski S., Gajjar A. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau T.M., Grambsch P.M. Modeling Survival Data: Extending the Cox Model. Springer; New York: 2000. [Google Scholar]
- Tian Y., Morris T.J., Webster A.P., Yang Z., Beck S., Feber A., Teschendorff A.E. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–3984. doi: 10.1093/bioinformatics/btx513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanner R.J., Remke M., Gallo M., Selvadurai H.J., Coutinho F., Lee L., Kushida M., Head R., Morrissy S., Zhu X. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014;26:33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladoiu M.C., El-Hamamy I., Donovan L.K., Farooq H., Holgado B.L., Sundaravadanam Y., Ramaswamy V., Hendrikse L.D., Kumar S., Mack S.C. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572:67–73. doi: 10.1038/s41586-019-1158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupt H., Johansson P., Sundström A., Lubovac-Pilav Z., Olsson B., Nelander S., Swartling F.J. Batch-normalization of cerebellar and medulloblastoma gene expression datasets utilizing empirically defined negative control genes. Bioinformatics. 2019;35:3357–3364. doi: 10.1093/bioinformatics/btz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann F., Muth D., Benner A., Bauer T., Henrich K.O., Oberthuer A., Brors B., Beissbarth T., Vandesompele J., Pattyn F. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008;9:R150. doi: 10.1186/gb-2008-9-10-r150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R.C., Pollan S., Fong H., Ibrahim A., Smith E.L., Ho M., Laslett A.L., Donovan P.J. A novel role for an RNA polymerase III subunit POLR3G in regulating pluripotency in human embryonic stem cells. Stem Cells. 2011;29:1517–1527. doi: 10.1002/stem.714. [DOI] [PubMed] [Google Scholar]
- Wu C.-C., Hou S., Orr B.A., Kuo B.R., Youn Y.H., Ong T., Roth F., Eberhart C.G., Robinson G.W., Solecki D.J. mTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell. 2017;43:673–688.e5. doi: 10.1016/j.devcel.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Gregory C.A., Lee R.H., Reger R.L., Qin L., Hai B., Park M.S., Yoon N., Clough B., McNeill E. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc. Natl. Acad. Sci. USA. 2015;112:530–535. doi: 10.1073/pnas.1423008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukova N., Ramaswamy V., Remke M., Pfaff E., Shih D.J.H., Martin D.C., Castelo-Branco P., Baskin B., Ray P.N., Bouffet E. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013;31:2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data of samples and cell lines from expression and methylation analysis is provided in GEO accession number GEO: GSE106728. A complete list of deposited data and software for data analysis can be found in the Key Resources Table.