

Abstract
Metal triflates have been utilized to catalytically facilitate numerous glycosylation reactions under mild conditions. In some methods, the metal triflate system provides stereocontrol during the glycosylation, rather than the nature of protecting groups on the substrate. Despite these advances, the true activating nature of metal triflates remains unclear. Our findings indicated that the in situ generation of trace amounts of triflic acid from metal triflates can be the active catalyst species in the glycosylation. This fact has been mentioned previously in metal triflate-catalyzed glycosylation reactions; however, a thorough study on the subject and its implications on stereoselectivity has yet to be performed. Experimental evidence from control reactions and 19F NMR spectroscopy have been obtained to confirm and quantify the triflic acid released from nickel triflate, for which it is of paramount importance in achieving a stereoselective 1,2-cis-2-amino glycosidic bond formation via a transient anomeric triflate. A putative intermediate resembling that of a glycosyl triflate has been detected using variable temperature NMR (1H and 13C) experiments. These observations, together with density functional theory calculations and a kinetic study, corroborate a mechanism involving triflic acid-catalyzed stereoselective glycosylation with N-substituted trifluoromethylbenzylideneamino protected electrophiles. Specifically, triflic acid facilitates formation of a glycosyl triflate intermediate which then undergoes isomerization from the stable α-anomer to the more reactive β-anomer. Subsequent SN2-like displacement of the reactive anomer by a nucleophile is highly favorable for the production of 1,2-cis-2-aminoglycosides. Although there is a previously reported work regarding glycosyl triflates, none of these reports have been confirmed to come from the counter ion of the metal center. Our work provides supporting evidence for the induction of a glycosyl triflate through the role of triflic acid in metal triflate-catalyzed glycosylation reactions.
Keywords: Glycosylation Mechanism; Hidden Brønsted Acid; Kinetics; 1,2-Cis-Aminoglycosides; Nickel Triflate
Graphical Abstract
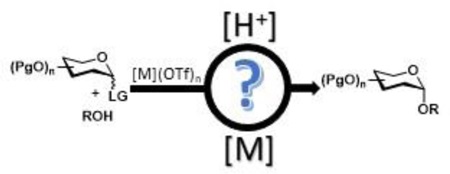
INTRODUCTION
Recent advances in the field of glycoscience have revealed carbohydrates as an essential component of many biologically important molecules in nature.1 Carbohydrate oligosaccharides are often found in low concentrations and heterogeneous forms, greatly complicating their isolation and analysis. In order to attain these carbohydrate targets under mild and operationally simple techniques, many methodologies have begun to utilize catalytic amounts of transition metals.2–4 Of these transition metal catalysts, metal triflates in particular have been found to be highly efficient at facilitating glycosylation reaction and some even provides stereocontrol during the glycoyslation.2–7 Metal triflates have illustrated many advantages over traditional activating reagents for promoting glycosidic bond formation. Since the turnover is high, only substoichiometric amounts of metal triflates have been utilized in the reaction, thus promoting the strategies of “green chemistry”.8 Furthermore, the stereoselectivity of the newly-formed glycosidic bond can be tuned by the nature of the ligand attached to the metal center. In addition, transition metal complexes have been utilized to promote chemoselective activation for orthogonal glycosylation strategies.2–3 However, during many of mechanistic elucidations the question remains for the true role of the metals in the catalytic glycosylations.
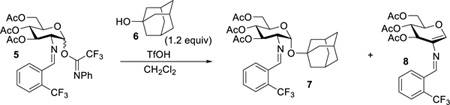
Recently, it has been presented that many organic transformations performed by metal triflate catalysts are truly promoted by triflic acid, which is released from the metal center and has been coined as “hidden Brønsted acid catalysis”.9–14 Although eluted in the literature,15–22 a systematic study on the possible release of triflic acid from a metal triflate to promote a glycosylation has not yet reported. Not only can the release of triflic acid potentially activate the departure of the anomeric leaving group, there is a possibility that it has implications on the stereoselectivity of the reaction, as it has been reported that the triflate anion can play an important role in the diastereoselectivity of the newly-formed glycosidic bond.23 However, the possibility of triflic acid plays a key role in catalytic glycosylations is quickly dismissed because study by our group and others have illustrated that triflic acid was not able to replicate the results obtained by the metal triflate. Typically, the carbohydrate coupling products are obtained in poor yield using triflic acid with an equivalent amount to that of the metal triflate, leading to the impression that triflic acid is less effective and can be prematurely ruled out as the active catalyst.6,11 In order to study the concept of “hidden Brønsted acid catalysis” being the actual promoter for metal triflate-catalyzed glycosylation reactions and its impact on the stereoselectivity, we systematically probed the activation and operative mechanisms of our recently developed method for 1,2-cis-aminoglycoside synthesis via nickel triflate-catalyzed glycosylation with a C(2)-benzylideamino N-phenyl trifluoroacetimidate electrophile (Scheme 1).24
Scheme 1.
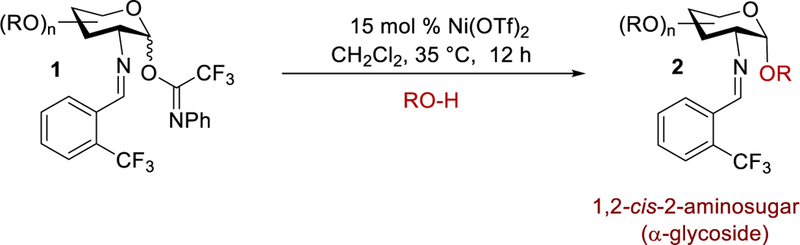
Formation of 1,2-Cis-2-Aminoglycosides
Herein, we report our detailed investigations of 1,2-cis-2-amino glycosylation reaction aimed at understanding the mechanism of α-selective glycosidic bond formation utilizing nickel(II) triflate. Although the hidden Brønsted acid theory has been suggested previously, there are many discrepancies and lapses in the literature pertaining to a detailed study on the subject. Therefore, this work includes: 1) confirming that the actual active catalyst species in the reaction can be triflic acid when utilizing metal triflate catalysts, 2) utilizing computational and experimental studies to detect and analyze a transient glycosyl intermediates after N-phenyl trifluoroacetimidate activation by the “hidden Brønsted acid” released from Ni(OTf)2, and 3) validating the significant role of the metal’s counter ion on selectivity and activation. More importantly, this mechanistic study can have implications on future metal-catalyzed glycosylations and also to previously developed metal-catalyzed mechanisms, of which many contain triflate counter ions as well as a complementary glycosylation reaction can be performed under Brønsted acid conditions.
RESULTS AND DISCUSSION
As part of the effort to develop catalytic stereoselective glycosylations,2,25–27 we recently identified a novel, scalable method to address many of the synthetic limitations previously associated with the construction of 1,2-cis-2-aminoglycosides (Scheme 1).6–7,24,28–32 In our studies, we examined an array of glycosyl electrophiles and metal salts. From this evaluation, we discovered that the combination of C(2)-N-substituted trifluoromethylbenzylideneamino N-phenyl trifluoroacetimidate electrophiles 1 and nickel triflate, Ni(OTf)2, provided high α-selectivity for 1,2-cis-amino linkage formation. Yet some ambiguity about the mechanism and the catalyst still existed. Several reaction pathways have been proposed, including nickel directed glycosylation reaction, hydrogen-bond assisted reaction, glycosyl triflate promoted SN2-like displacement, or imine addition followed by migration (vide infra, Figure 1).
Figure 1.
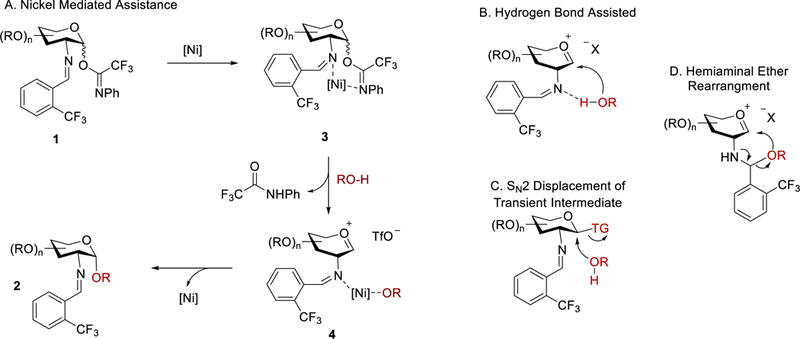
Potential reaction mechanisms for Ni(OTf)2-catalyzed 1,2-cis-2-amino glycosylation.
The 1,2-cis-2-amino glycosylation reaction depicted in Scheme 1 involves a transfer of 15 mol% of Ni(OTf)2, generated in situ from nickel chloride (NiCl2) and silver triflate (AgOTf), to a solution of N-phenyl trifluoroacetimidate 1 (1 equiv.) and a glycosyl nucleophile (RO-H, 1.2 equiv) in methylene chloride at 25 °C. The reaction mixture is then allowed to proceed at 35 °C for 12 h to afford the 1,2-cis-2-amino product 2 in good yield and high diastereoselectivity. Ni(OTf)2 is commercially available from several different sources and air-stable. However, we discovered that discrepancies did arise in the yield when different batches of Ni(OTf)2 were used for the glycosylation.24 In contrast, the in situ generation of Ni(OTf)2, from NiCl2 and AgOTf, resulted reproducible and consistent results.24 From a practical point of view, it is worth noting that both NiCl2 and AgOTf are commercially available and they are not as deliquescent as other metal salts.33 Therefore, in situ generated Ni(OTf)2 became the catalyst of choice for the construction of several highly desired saccharide motifs containing 1,2-cis-2-amino linkages.24,32
Initial Mechanistic Investigations.
Although a number of reaction pathways can be envisioned for the 1,2-cis-amino glycosylation, it is likely that the mechanism of the transformation proceeds via initial activation of the anomeric N-phenyl trifluoroacetimidate leaving group. In the original proposed mechanism, we hypothesize that Ni(OTf)2 could effectively coordinate to both the benzylidene group at C(2) and the trifluoroacetimidate at C(1) of glycosyl electrophile 1 to form the corresponding nickel-substrate complex 3 (Figure 1a).7 Subsequent ionization of 3 followed by exchange between the trifluoroacetamide group and the glycosyl nucleophile (RO-H) could generate a nickel-alkoxide complex 4, which directs the addition of the alkoxide functionality to the α-face of an oxocarbenium intermediate to afford 1,2-cis-2-aminoglycoside 2. Alternatively, one could invoke several other mechanistic pathways after imidate activation including SN2 displacement of a temporary β-intermediate (Figure 1c). A second possibility is the addition of the acceptor to the N-benzylidene imine to form an unstable hemiaminal followed by migration to the α-face (Figure 1d). Lastly, the C(2)-imine could be utilized as a hydrogen bond acceptor and assist in the delivery to the α-face (Figure 1b). Thus, our goal is to elucidate which mechanism is more likely to operate under Ni(OTf)2-catalyzed glycosylation conditions.
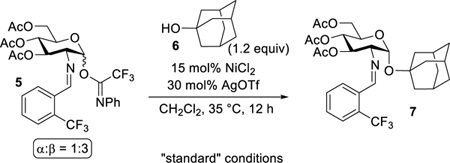
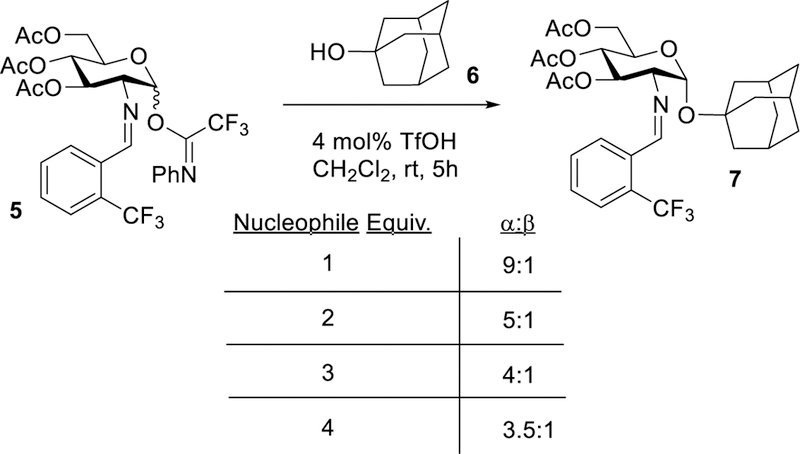
Our first objective is to determine the role of Ni(OTf)2 in the glycosylation or whether “hidden Brønsted acid catalysis” plays a role in the reaction. This is important because many reactions that utilize metal triflates (or a combination of metal halides and silver triflate) as catalysts are indeed activated by a Brønsted acid. In these reactions, traces of triflic acid (TfOH) which are slowly released from metal triflates are sufficient for the reaction to proceed.9–14 As such, we hypothesized that triflic acid released from a combination of nickel chloride (NiCl2) and silver triflate (AgOTf) could be responsible for the observed catalytic glycosylation as triflic acid has been used as a promoter of imidate glycosylation reactions.34 To determine if triflic acid is the source of catalysis, glycosyl electrophile 5 and 1-adamantanol 6 were utilized as model coupling partners in our investigations to simplify the analysis of product mixtures (Table 1). In the experiment, the glycosylation of 6 with 5 was performed in the presence of 15 mol% of in situ generated Ni(OTf)2 and 60 mol% of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) as an acid scavenger (entry 2).9 Consistent with our hypothesis, addition of DTBMP almost completely suppressed the reaction (less than 5% yield of the desired product 7 was detected), indicating that the reaction is activated by triflic acid.21–22 Acid scavenger experiments typically are underutilized when metal triflates are the glycosylation catalyst.35 In the absence of NiCl2 or AgOTf, only minuscule amount of 7 was observed (entries 3 and 4) and the coupling partners 5 and 6 were recovered quantitatively. These results establish that NiCl2 and AgOTf serve as catalyst precursors for the in situ generation of Ni(OTf)2, which then upon formation releases triflic acid.
Table 1.
Effect of Reaction Parameters
 | |||
|---|---|---|---|
| entry | variation from the “standard” condtions | yielda (%) | α:β ratiob |
| 1 | none | 90 | 10:1 |
| 2 | added 60 mol% DTBMP | 5 | n/a |
| 3 | no AgOTf | <1 | n/a |
| 4 | no NiCI2 | 6 | n/a |
| 5 | 1 equiv. AgOTf, instead of in situ generated Ni(OTf)2 | 80 | 5:1 |
| 6 | 15 mol% Zn(OTf)2, instead of in situ generated Ni(OTf)2 | 91 | 10:1 |
| 7 | 15 mol% ln(OTf)3, instead of in situ generated Ni(OTf)2 | 95 | 9:1 |
| 8 | THF, instead of CH2Cl2 | <1 | n/a |
| 9 | EtCN, instead of CH2Cl2 | <1 | n/a |
| 10 | added 3Å MS | <1 | n/a |
Isolated yield.
Determined by 1H NMR analysis
Next, we explored the ability of other metal triflates to promote the 1,2-cis-2-amino glycosylation (Table 1, entries 5 – 7) to further validate whether triflic acid released from these metal salts is the source of catalysis. For metal salts with weakly coordinating anions, the release of Brønsted acids through cation hydrolysis has been documented.36 In addition, there is a correlation between the catalytic activity of metal triflates and their hydrolysis constants (pKh).33 Cationic metals are generally difficult to hydrolyze when their pKh values are greater than 10.1. In contrast, cations with pKh values less than 4.3 are easy to hydrolyze.33 For example, since the pKh value of Ag(I) is too large (pKh=12) for hydrolysis to occur, almost no catalytic activity is observed in the glycosylation reaction, potentially from minimal release of triflic acid (entry 4, only 30 mol% AgOTf was utilized). The reaction proceeds to completion only in the presence of stoichiometric amount of AgOTf (entry 5). In contrast, Ni(II) and Zn(II) have suitable pKh values (9.86 for nickel and 8.96 for zinc), and thus their corresponding Ni(OTf)2 and Zn(OTf)2 sufficiently release triflic acid to catalyze the reaction and provide 1,2-cis glycoside 7 in excellent yield (entries 1 and 6). The easily hydrolysable In(OTf)3, whose pKh value of In(III) is 4.0, was quite effective at catalyzing the reaction (entry 7). To compare, lanthanide triflates which are some of the most commonly used metal triflates for promoting glycosylation reactions have potential to go through a similar hydrolysis reaction as they have pKh = ~7–8.16 Furthermore, we hypothesize that the impact of Brønsted acid catalysis can be attenuated by employing Lewis basic solvents.41 As expected, utilization of tetrahydrofuran (THF) or propionitrile (EtCN) completely suppresses the reaction (Table 1, entries 8 and 9). If Ni(OTf)2 has been producing triflic acid species through hydrolytic interactions with adventitious H2O, then molecular sieves will shut down the coupling event. We have found that no glycosidic bond (<1%) occurs with use of 3Å activated molecular sieves (entry 10).37 This result is consistent with our previous observation that utilization of commercially available Ni(OTf)2 led to inconsistencies in the yield of the product when different batches were used.24 These variances could arise from batch-to-batch variation of moisture content.38
It is well-documented that in situ generated triflic acid from metal triflates has distinct advantages over the direct use of triflic acid for several reasons: (i) triflic acid is not readily soluble in apolar solvents, particularly in a small-scale experiments;14 (ii) the use of triflic acid with equivalent amount to metal triflate in control experiments often can lead to the misleading impression that triflic acid is less effective than metal triflates due to product decomposition and significant amount of side products;7,11 (iii) triflic acid-catalyzed control reactions often show inferior catalytic activity because they cannot receive the optimized amount of triflic acid that metal triflate-catalyzed reactions generally receive. To validate these limitations, we probed the effect of varying amounts of triflic acid on the coupling efficiency (Table 2). These experiments also allowed us to compare qualitatively the rate of the glycosylation in the presence of triflic acid to that in the presence of nickel triflate. As illustrated in Table 2, although the reaction with use of 10 mol% of TfOH proceeded to completion within 1 h at 25 °C (entry 1), a significant amount of elimination product 8 was observed (7:8 = 3:1). Decreasing the triflic acid loading (entries 2 and 3) further suppressed formation of the side product 8, but significantly slowed down the rate of glycosylation. Overall, these observations showed that the rate and the coupling efficiency catalyzed by 1 mol% of TfOH at 35 °C (entry 4) was similar to that catalyzed by 15 mol% of Ni(OTf)2 at 35 °C (Table 1, entry 1).
Table 2.
Effect of the Amount of Triflic Acid
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | TfOH loading (mol%) | Temperature(°C) | reaction time (h) | conversiona (%) | 7:8 ratioa | 7 yield(%)b | 7 α:β ratioa |
| 1 | 10 | 25 | 1 | 100 | 3:1 | 73 | 10:1 |
| 2 | 5 | 25 | 3 | 85 | 11:1 | 78 | 10:1 |
| 3 | 1 | 25 | 24 | 35 | >99:1 | 34 | 10:1 |
| 4 | 1 | 35 | 12 | 95 | >99:1 | 78 | 10:1 |
Determined by 1H NMR analysis.
Isolated yield
At present, the nickel triflate-catalyzed glycosylation protocol is conducted under acidic and elevated temperature conditions. Therefore, there is a possibility that post-coupling anomerization of the 1,2-trans glycosides to the thermodynamically stable 1,2-cis glycosides can occur.39–48 As a result, we subjected an α- and β-mixture of product 7 to the standard glycosylation conditions catalyzed by either nickel triflate or triflic acid. No anomerization was observed after 12 h at 35 °C (Scheme 2). It appears that the α:β ratio of product 7 is kinetically derived and is not reflective of a thermodynamic distribution arising from post-coupling anomeric anomerization.
Scheme 2.
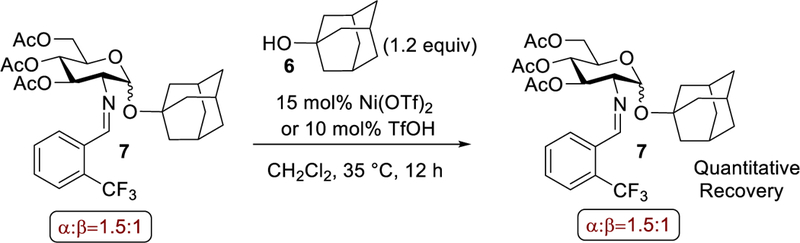
Studies of Post-Coupling Anomerization
To understand how triflic acid is being produced from nickel triflate and acts as the active catalyst to promote the glycosylation, three control 19F NMR experiments were conducted at 35 °C (Figure 2). Triflic acid was not detected from the reagent combination of NiCl2 and AgOTf (Figure 2a) or from the mixture of NiCl2, AgOTf, and an adamantanol nucleophile (Figure 2b). Interestingly, in the presence of glycosyl electrophile 5, a broad singlet 19F resonance at δ −78.99 ppm was detected (Figure 2c), a chemical shift which is consistent with characteristics of triflic acid (δ = −79.4 ± 0.5 ppm) (Figure S1).14 Quantitative analysis of the newly formed broad singlet provides it at a 2 mol% ratio relative to 8 (Figure S1), which is nearly identical to the amount of triflic acid needed to replicate the results of the metal triflate catalyzed glycosylation (Table 2). This result suggests that residual moisture content of the carbohydrate substrate potentially facilitates the initial release of triflic acid from the metal center. Once the reaction initiated, triflic acid can be continuously generated through many other reaction pathways.
Figure 2.
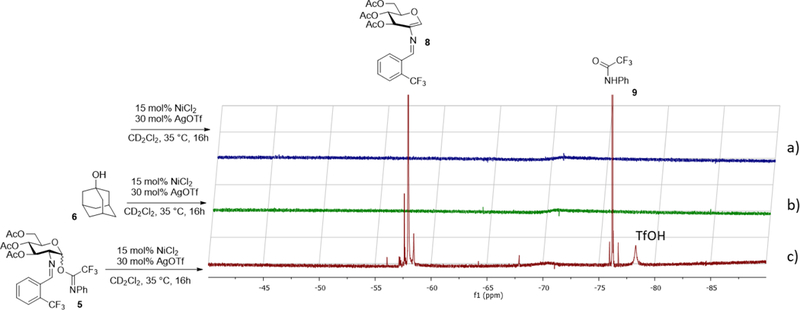
Detection of triflic acid monitored at 35 °C by 19F NMR in CD2Cl2: (a) Ni(OTf)2 generated in situ from NiCl2 and AgOTf; (b) adamantanol 6 and Ni(OTf)2 generated in situ from NiCl2 and AgOTf; (c) electrophile 5 and Ni(OTf)2 generated in situ from NiCl2 and AgOTf. Top portion of resonances corresponding to 8 and 9 has been removed, for full spectrum see Figure S1.
On the basis of the above observations, it is unlikely that nickel-coordinated delivery of an alkoxide nucleophile to generate 1,2-cis-2-amino product is the operative catalytic pathway (Figure 1a). Importantly, these data further support that Brønsted acid catalysis is likely to operate in this reaction. With the understanding that the reaction is catalyzed by “triflic acid” which is slowly released from Ni(OTf)2, it is necessary to compare the benefits of both catalyst conditions. Although utilization of Ni(OTf)2 does requires a two-step procedure, the pre-catalysts (NiCl2 and AgOTf) used in the reaction are moisture stable. In addition, slow release of triflic acid from Ni(OTf)2 makes the method amenable to a wide variety of acid-sensitive protecting groups. On the other hand, use of pure triflic acid can quickly decompose acid-labile substrates; although the procedure is operationally simple, triflic acid is more moisture sensitive leading to unpredictable yields when used at low catalytic loadings.
Next, we examined the role of triflic acid in influencing the stereochemical outcome of the glycosylation. Since the triflic acid released from Ni(OTf)2 is the potential active catalyst, it is likely that a glycosyl triflate could be generated in the subsequent stages of the reaction.23,49 The glycosyl triflate intermediate has been invoked to be involved in the formation of β−1,2-cis-mannoside products.23,50–53 As a result, we hypothesize that substitution of Ni(OTf)2 with nickel triflimide, Ni(NTf2)2 could impact the outcome of the selectivity.51,54–58 Accordingly, the coupling of 6 with 5 was conducted in the presence of 15 mol% of Ni(NTf2)2 (Scheme 3a), generated in situ from NiCl2 and silver triflimide (AgNTf2). Confirming our hypothesis, the reaction proceeded with poor anomeric stereoselectivity (α:β = 1.4:1). Similarly, use of triflimide, Tf2NH (Scheme 3b), also provided a 1.3:1 mixture of α- and β-isomers. Lack of selectivity with use of the triflimide anion implies that the reaction goes through an oxocarbenium ion intermediate in favor of an SN1-type mechanism for the catalytic process. Furthermore, this observation suggests that both the hydrogen bond assisted (Figure 1b) and hemiaminal rearrangement mechanisms (Figure 1d) are not likely to be the operational pathways. For both of the aforementioned pathways to be operative, the selectivity should remain the same regardless whether triflimide or triflate is used in the reaction. Overall, the poor selectivity observed with triflimide illustrates the importance of the triflate anion and further supports the possibility that the reaction goes through a transient glycosyl triflate intermediate.
Scheme 3.
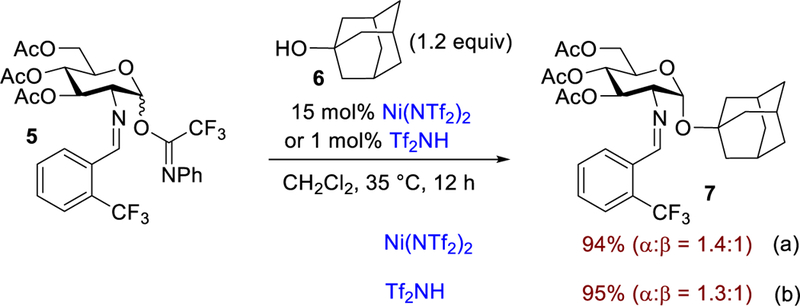
Nickel Triflimide-Catalyzed Glycosylation Reaction
Coinciding with the notion that the glycosyl triflate is the key intermediate in the reaction, we analyzed previously reported data in which we exchanged different substituents (both electron donating and electron withdrawing) on the C(2)-benzylidene group; there was no trend on the selectivity.6 One would hypothesize that if an electron-donating substituent was placed on the aryl ring of the C(2)-benzylidene nitrogen which would become a better hydrogen bond acceptor, higher α-selectivity could be observed (Pathway B, Figure 1). Alternatively, an electron-withdrawing substituent would increase the electrophilicity of the C(2)-benzylidene imine carbon, making nucleophile addition more favorable (Pathway D, Figure 1). Upon analysis, there was no correlation between the electronic nature of the substituent and the stereochemical outcome of the reaction. Thus, the data rules out the proposed pathways B and D (Figure 1) are likely to be the operative pathways in the nickel triflate-catalyzed glycosylation.
Finally, to further confirm that both anomers of N-phenyl trifluoroacetimidate 5 reacted to produce the 1,2-cis-2-amino product with similar selectivity, we separately subjected them to the 15 mol% Ni(OTf)2 reaction conditions. While α-imidate 5 proceeded to completion within 4 h, β-imidate 5 progressed slowly and completed after 16 h. Nevertheless, they produced adamantanol glycoside 7 in α:β = 10:1 and similar yields (α: 95%, β: 88%). This result suggests that both anomers of imidate 5 are likely to proceed through the same reaction pathway.
Overall, our initial observations reveal several key aspects of the 1,2-cis-2-amino glycosylation. First, triflic acid released from nickel triflate is likely to be the active catalyst in effecting stereoselective formation of glycosidic bonds. Second, nickel triflate is not directly involved in the key glycosidic bond-forming process and serves as a precursor to the active triflic acid catalyst. Third, the presence of triflate anion is essential for the observed α-selectivity. Fourth, it is unlikely that the reaction proceeds through a nickel-substrate complex as we originally proposed (Figure 1a). An alternative pathway consistent with the aforementioned observations is illustrated in Figure 3. In the first step, TfOH, released from Ni(OTf)2, can engage in electrophilic activation of the N-phenyl trifluoroacetimidate leaving group of glycosyl electrophile 5 to form the activated donor-complex 10. Subsequent ionization of 10 can lead to an array of reactive glycosyl intermediates. The oxocarbenium ion intermediate 11 can exist in a series of equilibria as it can be either closely associated with the triflate counterion in the form of contact ion pairs (CIPs) or further separated from the triflate counterion in the form of solvent separated ion pairs (SSIPs).50 An intermediate such as 11 is likely as it can be obtained by both anomers of N-phenyl trifluoroacetimidate 5, consistent with previous results that both α- and β- anomers provide the product with similar α-selectivity (α:β = 10:1). Triflic acid promoted stereoselective glycosylation via SN1 pathway would have to bias the addition of a glycosyl nucleophile 6 to either diastereomeric face of this oxocarbenium ion-like intermediate 11 (Figure 3). Alternately, the two covalent glycosyl triflate intermediates 12 and 13 can be formed by nucleophilic attack of a triflate anion and are in equilibrium with oxocarbenium-like intermediate 11.51 Displacement of the covalent triflate species by a nucleophilic partner takes place via an invertive SN2 pathway, in such a way that, the stereochemistry of the 2-aminosugar product would be dictated by the anomeric configuration of the glycosyl triflate intermediates. Although the glycosyl triflate has been reported to exist predominantly with an α-linkage (such as 12) regardless of the electrophilic donor configuration, a dynamic system wherein α-glycosyl triflate is in equilibrium with its less stable but more reactive β-glycosyl triflate (such as 13) has been proposed.23,50,53 The rapid equilibrium between these two triflate anomers can take place at low temperatures (−78 °C). 23,50,53 Therefore, the equilibrium and the relative reactivity of the putative α- and β-covalent triflates could account for the observed selectivity in the formation of the 2-aminoglycoside products. This proposed catalytic glycosylation system, which affords 1,2-cis-2-aminoglycoside product 7 from the more reactive and less stable β-glycosyl triflate, is loosely reminiscent of a mechanism for 1,2-cis glycoside formation for α-glycosyl bromide in the presence of external bromide ion reported by Lemieux.59
Figure 3.
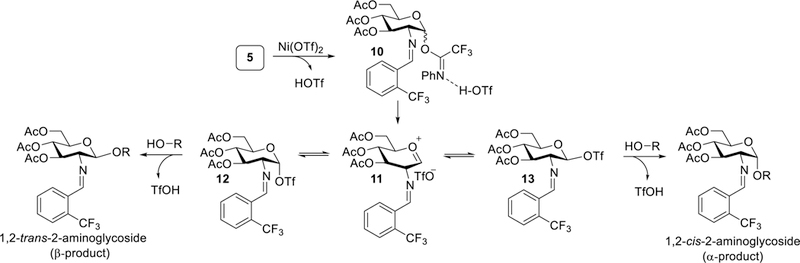
Proposed mechanism of triflic acid-catalyzed glycosylation.
Computational Investigation.
To provide further insight into the reaction mechanism that triflic acid play a key role in controlling the 1,2-cis selectivity through rapid isomerization of transient glycosyl triflate intermediates followed by nucleophilic attack to the more reactive triflate species, we utilized density functional theory (DFT) calculations performed by the Gaussian program.60 All geometries were optimized using the SMD61 implicit solvent model and the B3LYP62 functional with the 6–31G(d) basis set and corrected for basis set superposition error. Vibrational frequency calculations were used to confirm that the optimized structures are minima or transition state structures on the potential energy surface. To test the reliability of the B3LYP results, the single point energies for the B3LYP optimized geometries were also computed at the wB97XD/6–31+G(d) level of theory (Figure S22).63 The potential energy surface computed at the wB97XD/6–31+G(d) level is consistent with the calculations at B3LYP/6–31G(d).
To benchmark our calculations, the proper orientation for the substituents in our computational model were determined from the crystal structure of the β-N-phenyl trifluoroacetimidate 5 (Figures 4 and S21) which was obtained by a CH2Cl2/hexanes slow diffusion at low temperature. We were able to confirm that the saccharide ring of the electrophile exists as a 4C1 conformer and the N-benzylidene prefers the E-configuration with the trifluoromethyl group positioned upwards and in line with the imine group. From the derived structure, the initial computational study found the reaction free energies for the overall glycosylation involving protonated imidate 14 and methanol to be −4.6 and −3.6 kcal/mol, respectively, for the α- and β-methyl glycoside products (15 and 16, Figure 4). The formation of α- and β-methyl glycoside products is similar in reaction free energy, potentially rationalizing why a low α:β selectivity was obtained when the triflimide or nickel triflimide was used as the catalyst experimentally (Scheme 3). This observation also implies: (a) there is the formation of an intermediate that plays a major role in controlling the product distribution and (b) the selectivity does not come from a major inherent difference in the product energies.
Figure 4.
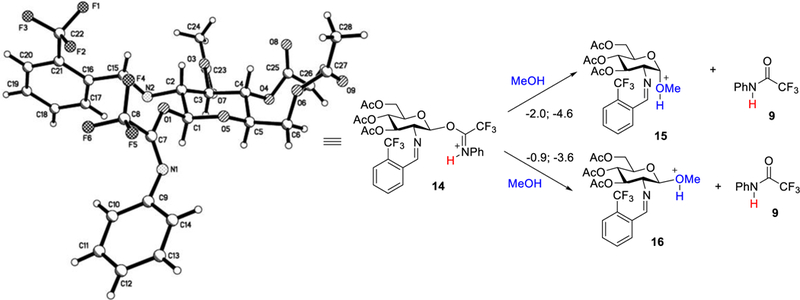
Overall reaction free energy for the formation of methyl glycosides. The initial computational model was formed from the crystal structure of the β-N-phenyl trifluoroacetimidate 5 (CCDC 1854007). Thermal ellipsoidal figure is included in the Supporting Information (Figure S21). Relative enthalpies (first number) and free energies (second number) are in kcal/mol (at 298 K) calculated using B3LYP/6–31G(d) with a SMD implicit solvent model.
To investigate the role of the glycosyl intermediate that enhances the 1,2-cis selectivity of the methyl glycoside product, a possible reaction pathway for the formation of α- and β-triflates (12 and 13, Figure 3) was examined. Upon protonation, the departure of the N-phenyl trifluoroacetimidate leads to formation of the contact oxocarbenium/triflate ion pair intermediate 18 (Figure 5) in the 4H3 half-chair conformation by an exothermic process at −5.1 kcal/mol for the protonated α-imidate 17 and −5.5 kcal/mol for the protonated β-imidate 14 (for unprotonated data, see Figure S2). After the contact ion pair is formed, the oxocarbenium ion intermediate can produce either the α- and β-glycosyl triflates (12 and 13), both in an exothermic fashion (−20.0 and −17.2 kcal/mol). The large release in energy upon formation of the covalent triflates can be attributed to the electron-withdrawing nature of the N-benzylidene group destabilizing the oxocarbenium ion.49,64 This observation is consistent with what has been observed with other short-lived transient intermediates (such as sulfonium ions) that direct stereoselectivity of the glycosylation.65 Furthermore, electron-withdrawing protecting groups (such as acetyl groups) also provide stabilization of a covalent triflate avoiding the glycosylation to proceed via an oxocarbenium ion intermediate, but rather proceeding through an SN2 displacement of a triflate.65 We have previously observed that the SN1-SN2 glycosylation paradigm can be slightly shifted for C(2)-N-benzylidene imidates by replacement of the electron-withdrawing acetyl groups with the electron-donating benzyl groups (α only→10:1). 29,31,54
Figure 5.
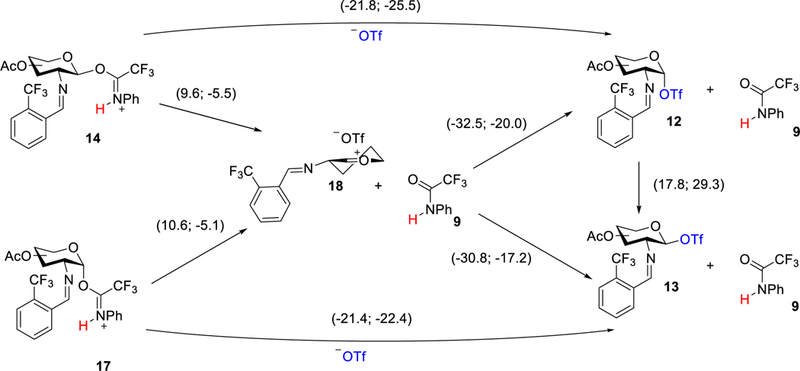
Energy of formation of glycosyl triflate intermediates 12 and 13. Relative enthalpies (first number) and free energies (second number) are in kcal/mol (at 298 K).
Calculations indicate that the α-glycosyl triflate is 2.8 kcal/mol more stable than β-glycosyl triflate (Figure 5).46–47 Knowing that these anomers need to isomerize, we studied the conversion of α-triflate 12 to β-triflate 13 via a direct SN2-like displacement with the triflate anion (TS19, Figure 6). However, the activation barrier for this direct substitution was calculated as 29.3 kcal/mol (Figure 5), suggesting that the preferred pathway for formation of β-glycosyl triflate 13 is the addition of the triflate anion to an oxocarbenium intermediate 18 in a CIP type mechanism.
Figure 6.
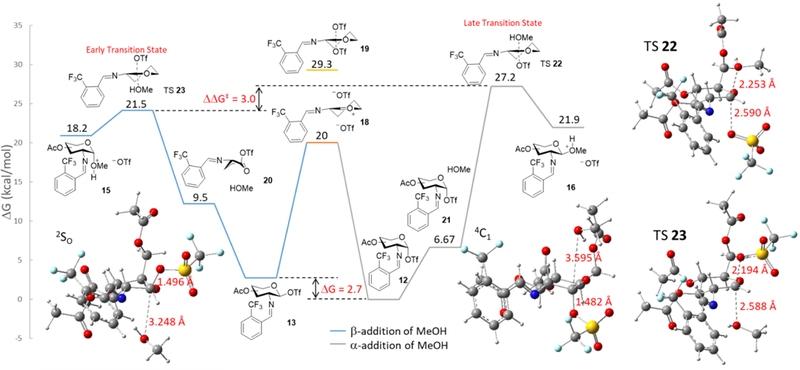
Energetic profile for the comparison of the isomerization of α-glycosyl triflate 12 to β-glycosyl triflate 13 versus their respective SN2 displacements. Energies on the blue line are in respect to 13 and energies on the gray line are in respect to 12 (Units = kcal/mol).
Following the calculations of the glycosyl triflate intermediates, the energy profile was calculated for the triflic acid-mediated formation of 1,2-cis-2-aminoglycoside and is shown in Figure 6. When a methanol nucleophile is approaching the α-face of β-glycosyl triflate 13, the methanol-β-triflate complex intermediate begins to pucker into a 2SO conformation 20 (dC(1)-MeOH = 3.248 Å and dC(1)-OTf = 1.496 Å) with a change of 9.5 kcal/mol with respect to 13. On the other hand, when a methanol nucleophile is approaching the opposite face of an α-glycosyl triflate 12, the methanol-α-triflate complex intermediate maintains a 4C1 conformation 21 (dC(1)-MeOH = 3.595 Å and dC(1)-OTf = 1.482 Å) with a barrier of 6.67 kcal/mol with respect to 12. From 21, nucleophilic attack of methanol to the β-face of α-triflate 12, via transition state (TS22), requires an activation energy of 27.2 kcal/mol to form the β-protonated methyl glycoside 16. Conversely, methanol attack to the α-face of β-triflate 13, via TS 23, requires an activation barrier of only 21.5 kcal/mol to form the α-protonated methyl glycoside 15. Once the protonated glycoside product is generated, it will be quickly deprotonated to complete the catalytic cycle. Comparing two transition states (TS22 and TS23), the energy different (ΔΔG‡) is 3.0 kcal/mol in favor of the α-nucleophilic attack of methanol to the more reactive β-glycosyl triflate species. The Curtin-Hammett principle states that the product distribution can be influenced if anomerization of the α-triflate to the corresponding β-triflate intermediate is rapid and more favorable than the subsequent nucleophilic attack.66 Accordingly, the calculations for the product determining transition states have presented a higher activation barrier for nucleophilic attack to the opposite face of α-triflate (27.2 kcal/mol) in comparison to the anomerization of an α-triflate via an oxocarbenium ion to the β-triflate (20.0 kcal/mol) which is then followed by a quick subsequent nucleophilic attack to the opposite face (21.5 kcal/mol). These calculations are reflected in the experimental product distribution (vide infra, Figure 10) and supports the proposed mechanism in Figure 3.
Figure 10.
Relationship of reactivity of nucleophile and α:β selectivity of product under nickel triflate catalyzed conditions.
We also performed a computational analysis to account for the reactivity differences between the two transition states, TS22 and TS23 (Figure S3). Looking at the optimized transition state structure of TS23 for the α-nucleophile attack by methanol, the C-O bond length between the departing β-triflate group and the anomeric carbon is shorter than that between the incoming methanol and the anomeric carbon (2.194 Å vs 2.588 Å, Figure 6). A non-covalent interaction (NCI) plot was calculated for TS23 (Figure S4) and no hydrogen bonding interactions were found between the C(2)-imine nitrogen and the methanol molecule, further supporting that the stereoselectivity is not being derived from hydrogen bonding (see Figure 1b). The six-membered ring structure in TS23 for the α-nucleophilic attack by methanol closely resembles the 2SO conformation of intermediate 20, suggesting that the reaction proceeds through early transition state. The puckered conformation of 20 is achieved in order to relieve potential steric interactions between methanol and the N-benzylidene in the transition state structure (TS23). According to the Hammond postulate, an early transition state (TS23) is preferred over a late transition state (TS22) as illustrated with the β-nucleophilic attack, wherein a six-membered ring remains in a product-like 4C1 conformation as the nucleophile approaches.67
NMR Studies.
With the possibility that the reaction goes through a transient triflate intermediate which is generated from the reaction of triflic acid with the glycosyl electrophile, we next attempted to detect this putative species spectroscopically. Although different types of glycosyl triflates have been observed by low-temperature NMR spectroscopy, there are few reports of α-triflate intermediates (12, Figure 3) generated from N-phenyl trifluoroacetimidate electrophiles.49,68 As previously described by Yu, N-phenyl trifluoroacetimidate electrophiles present a unique challenge when monitoring by NMR.69 These electrophiles are known to undergo interconversion via syn-anti isomerization around the carbon-nitrogen bond, resulting in a 1H NMR spectrum with broad resonances.69 As expected, both α- and β-imidates 5 were found at room temperature to have the broad anomeric proton resonances at δ 6.22 and 6.50, respectively (Figures 7a and h). However, at −60 °C interconversion is slow and these anomeric proton resonances are no longer broad (Figures 7b and g). As a result, we could be able to confirm the anomeric configuration of both α- and β-imidates 5 (α−5: 3JH1H2=3.3 Hz, β−5 3JH1H2=8.3 Hz) at −60°C. The imine proton of the trifluromethylbenzylidene group for both α- and β-imidates 5 appeared as a singlet resonance (δ 8.75) at room temperature (Figures 7a and h). Interestingly, we detected two sets of proton resonances (β−5: δ 8.75 and 8.65; α−5: δ 8.75 and 8.45) for these imine protons at −60 °C (Figures 7b and g), indicating that the N-phenyl group becomes locked in either the E- and Z-isomers at low temperature; however, the chemical environment around the imine proton is changed. To validate this proposal, a control study was performed in which each anomer in CD2Cl2 was cooled to −60 °C and gradually warmed to room temperature. Coalescence of these resonances into a single resonance δ 8.75 while taking the 1H NMR spectra at different temperature intervals confirmed them as isomers (see Figures S5 and S6 in SI).
Figure 7.
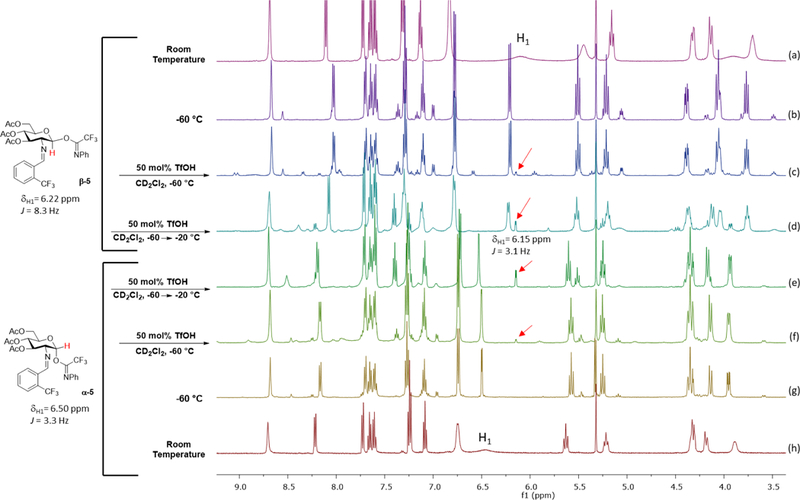
Addition of triflic acid to α- and β-imidates 5 was monitored by 1H NMR in CD2Cl2: (a) and (h) α- and β-imidates 5 at room temperature; (b) and (g) α- and β-imidates 5 at −60 °C; (c) and (f) −60 °C, 20 min after addition of TfOH; (d) and (e) −60 → −20 °C, 20 min.
Next, triflic acid (50 mol%) was added to the NMR tube containing β-imidate 5 at −60 °C (Figure 7c). Upon addition of TfOH, several new sets of proton resonances were detected and there was a distinct doublet resonance resided at δ 9.0 ppm with a coupling constant value of 16.4 Hz. This downfield doublet resonance had corresponding integrations to two other resonances in the anomeric region, δ 6.60 (d, J = 8.4 Hz), δ 5.96 (t, J = 9.9 Hz). We established the identity of the proton resonance at δ 9.0 as a protonated iminium ion of β-imidate 5 by the addition of 50 mol% TfOH β-glycosyl acetate S1 at −60 °C (Figure S7). The 1H NMR spectrum of the corresponding protonated glycosyl acetate S2 is illustrated in Figure S7, wherein the iminium proton resonance has a large trans coupling value of 15.4 Hz and resides at δ 9.03. The data is consistent with that of protonation of β-imidate 5. The same findings were found for α-imidate 5.
After identifying several proton resonances of the protonated α- and β-imidates 5, our next goal is to determine the identity of the doublet resonance with a coupling constant value of 3.1 Hz at δ 6.15 (Figure 7). Consistent with our hypothesis that the reaction proceeds through the same intermediate, the resonance at δ 6.15 was detected in both α- and β-imidate 5 upon addition of TfOH (Figures 7c and f) at −60 °C. This also rules out anomerization of the imidate leaving group, as the anomeric resonance β-imidate was not detected in the spectrum of the α-counterpart and vice versa.30 Since the chemical shift and coupling constant of this proton corresponds to the range of the previously reported anomeric proton resonance of α-glycosyl triflates (δ 6.0 – 6.5),49 it was tentatively assigned as α-triflate 12 (Figure 3). A COSY 2D-NMR experiment of this putative glycosyl triflate intermediate indicated the resonance at δ 6.15 to be at the C(1) position (Figure S8a). Additionally, a HSQC 2D-NMR correlated the C(1) proton resonance at δ 6.15 to a 13C resonance of δ 105.62, which is analogous to the 13C resonance for the C(1) carbon of previously reported glycosyl triflates (Figure S8b).49 Next, the reaction was allowed to gradually warm at 10 °C increments. At −20 °C, 1H NMR spectral data contained only two major carbohydrate species which were identified as the unreacted imidate substrate 5 and the putative α-glycosyl triflate 12 (Figures 7d and e). Unfortunately, imidate 5 was not completely converted to α-triflate 12 even in the presence of excess TfOH. Any further temperature increase above −20 °C led to decomposition into the highly conjugated glycal 8 and the undesired α-glycosyl acetate 26 (vide infra, Figure 10. Also see Figures S9 and S10 for VT-NMR decomposition experiments for α- and β-imidates 5).x (Here, you can add in the reference to discuss the outcome of the 19F NMR experiment for Figure S11). To further verify the identity of the principal carbohydrate species as a glycosyl triflate, we conducted a control experiment using Tf2NH in lieu of TfOH. We reasoned that if this species is the principal intermediate in the reaction, it would not be formed upon addition of Tf2NH to α- and β-imidates 5. Confirming our hypothesis, the proton resonance at δ 6.15 ppm was not observed the reaction (see Figures S12 and S13 in SI).
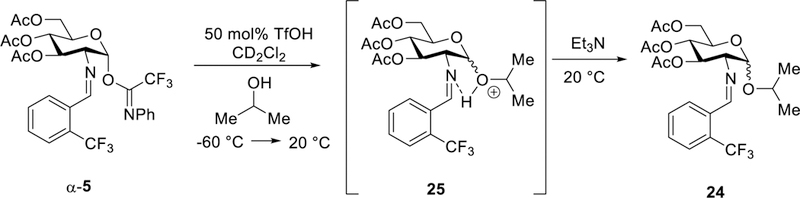
The subsequent stage in the glycosylation procedure involved the introduction of a nucleophile. We chose to use 2-propanol as a nucleophilic partner for ease of characterization of the carbohydrate intermediates. In the first scenario, 2-propanol (IPA) was introduced at the beginning of the reaction (Figure 8). A principal carbohydrate species having the proton resonance at δ 6.15 ppm was not detected. Upon warming of the reaction to 20 °C followed by quenching the mixture with triethylamine, formation of the isopropyl glycoside 24 was, however, observed with two doublet proton resonances (one at δ 4.96 ppm and other at δ 4.85 ppm) whose coupling constant values were 3.5 Hz and 7.8 Hz, respectively. This data suggests that the desired product 24 exists as a mixture of α- and β-isomers (Figure S14). As expected, 24 was isolated in 85% yield and 6:1 α:β selectivity and no side products were detected. In comparison, when the glycosylation reaction was conducted in a Schlenk flask under nickel triflate conditions, a similar outcome was observed (78%, α:β = 8:1). It is worth noting that the 1H NMR spectrum at 20 °C after full conversion did not look like that of the isolated isopropyl glycoside 24. However, upon addition of triethylamine, the 1H NMR spectrum became identical to the isolated product 24, indicating that the protonated isopropyl glycoside 25 (Figure S14) was initially formed under acidic conditions. To validate that the principal carbohydrate species prior to triethylamine addition is simply the protonated glycoside 25, the isolated product 24 was treated with 1 equivalent of TfOH (see Figure S15) wherein the 1H NMR spectrum matched that for the monitored NMR experiment. After quenching the reaction with triethylamine, the protonated glycoside product 25 was reverted to the isopropyl glycoside 24 with no change in α:β selectivity (Figure S15).
Figure 8.
Glycosylation monitored by 1H NMR in CD2Cl2.
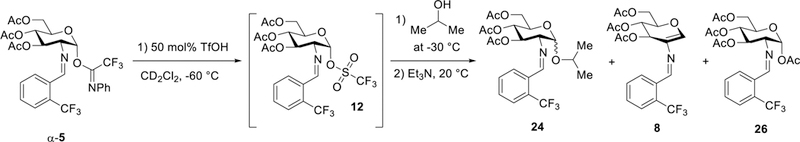
The complementary experiments were also performed by adding 2-propanol nucleophile after the putative glycosyl triflate with the proton resonance at δ 6.15 ppm was detected at −30 °C (12, Figure 9 and Figure S16). Upon gradually warming of the reaction at 10 °C increments, the desired glycoside 24 was detected along with other significant side products 8 and 26 (Figure 9). At the temperature range between −20 and −10 °C, an unexpected α-glycosyl acetate 26 (H1 = δ 6.24, d, 3JH1H2 = 3.2 Hz, Figure S16) was detected presumably via intermolecular acetyl migration. Formation of glycal 8 was also detected between −20 and −10 °C (Figure S16). At low temperatures, we hypothesize that the thermodynamically favored α-triflate has an increased population and could not rapidly undergo anomerization to the more reactive β-triflate. As a result of the increased pools of glycosyl triflate 12, the elimination pathway to produce the conjugated glycal 8 is competing with the nucleophilic attack of 2-propanol to form the desired product 24. On the other hand, under standard conditions at elevated temperature (35 °C), isomerization of the transient glycosyl triflate followed by nucleophilic attack occurs faster than the competing elimination pathway. As a result, glycal 8 was not detected when the reaction was conducted at 35 °C (Table 1, entry 1) or when a nucleophile was introduced at the beginning of the reaction (Figure 8).
Figure 9.
Glycosylation monitored by 1H NMR in CD2Cl2.
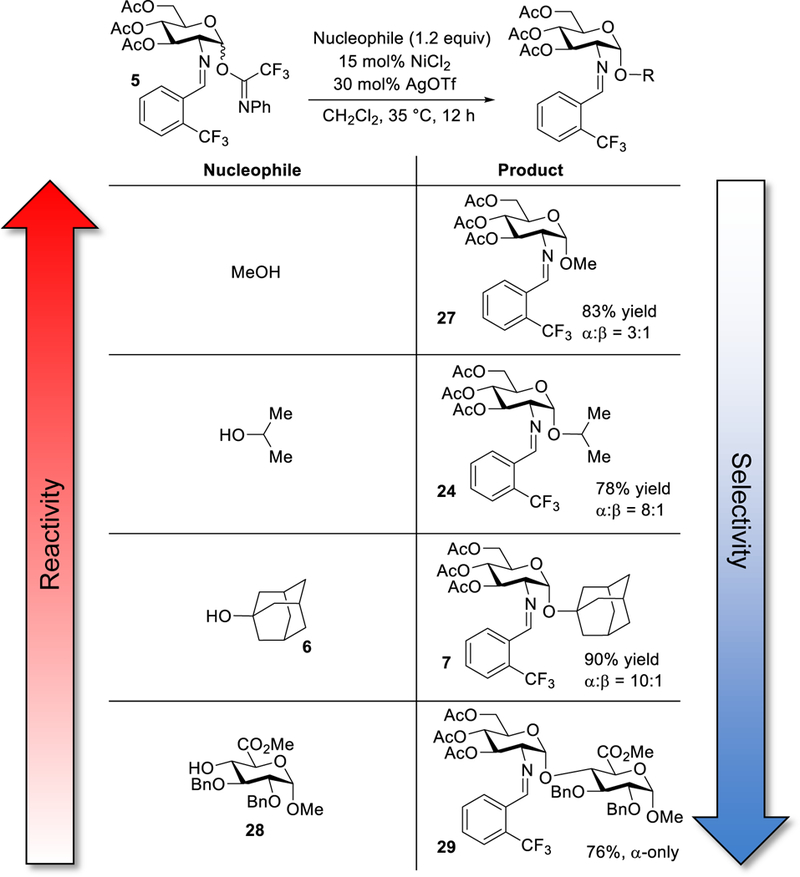
The need for interconversion from the α- to the β-glycosyl triflate intermediate (12 and 13, Figure 3) explains why we have observed selectivity changes with different glycosyl nucleophiles (Figure 10).70 By using bulky or deactivated nucleophiles such as glucuronic acid 28 (α:β = 1:0)24 and adamantanol 6 (α:β = 10:1), the reaction become very α-selective because more time is allowed for anomerization of the α-triflate to the more reactive β-triflate. An alternative explanation is that these acceptors are not reactive enough to displace the stable α-glycosyl triflate.52,54,70–72 On the other hand, when a more reactive nucleophile is used in the reaction, the α-selectivity decreases significantly. 52,54,70–72 For instance, use of the highly reactive methanol provided the product 27 as a 3:1 α:β mixture, albeit in favor of α-anomer (for additional examples of acceptors, see ref 24).
It was also observed in a kinetics experiment that varying the equivalents of adamantanol 6 (Figure 11) had a significant impact on the selectivity of the reaction; however, there was no observed pattern for change in rate (Figures S17).72 In contrast, there was a first order correlation when the amount of triflic acid catalyst was varied (Figure S18). For each different nucleophile concentration, the α:β selectivity were individually taken at multiple different time points. At a specific time point (5 h), increasing adamantanol 6 to 4 equivalents dropped the α:β selectivity to 3.5:1, while only using 1 equivalent of 6 produced the desired coupling product 7 in α:β = 9:1. We hypothesize that increasing the nucleophile concentration allows the nucleophilic attack to compete with the isomerization of α-glycosyl triflate to the corresponding β-glycosyl triflate, thus increasing the amount of β-product. Over the duration of the experiment, each nucleophile concentration showed an increase in α:β selectivity as time progressed because there is more time for the isomerization of the glycoysl triflate (Figure S17). These rate orders and changes in selectivity based on nucleophile concentration support of a mechanism in which the rate is limited by the formation and isomerization of a glycosyl triflate, that can then be facilely displaced by a nucleophile. Formation of the β-glycosyl triflate induces a bottleneck, making the nucleophile appear zeroth order.72
Figure 11.
Kinetic study with different equivalents of nucleophile.
To further support the triflate isomerization hypothesis and to probe the influence of the C(2)-N-benzylidene on the formation of a glycosyl triflate intermediate, a C(2)-azido- and C(2)-OBn-derived imidates were employed (30 and 31, Figure 12).73–75 Upon subjection of 50 mol% of TfOH at −50 °C, the previously reported C(2)-azido glycosyl α-triflate 32 was formed with a new anomeric resonance at δ 6.13 ppm (Figure S19 in SI).76 Upon warming the NMR probe, the C(2)-azido glycosyl triflate had a decomposition temperature of 0 °C, which is greater than that of the C(2)-N-benzylidene glycosyl triflate 12 (−20 °C). Alternatively, glycosyl triflate 33, which was formed from subjecting C(2)-OBn imidate 31 to 50 mol% of TfOH at −50 °C, decomposed at a slightly lower temperature of −30 °C (Figure S20). According to Crich and Codèe, a more stable α-glycosyl triflate does not allow for isomerization to the β-glycosyl triflate and will result in higher β-selectivity of the glycosylation product.49,64,70 As it is expected, when the more electron-withdrawing C(2)-azido donor was subjected to a nickel triflate mediated coupling reaction with adamantanol 6, the selectivity of the coupling product dropped to α:β = 4:1.70 In contrast, the N-benzylidene and C(2)-benzyl ether modulate the electronics suitably allowing for the triflate to isomerize, thus resulting in higher α-selectivity (Figure 12).
Figure 12.
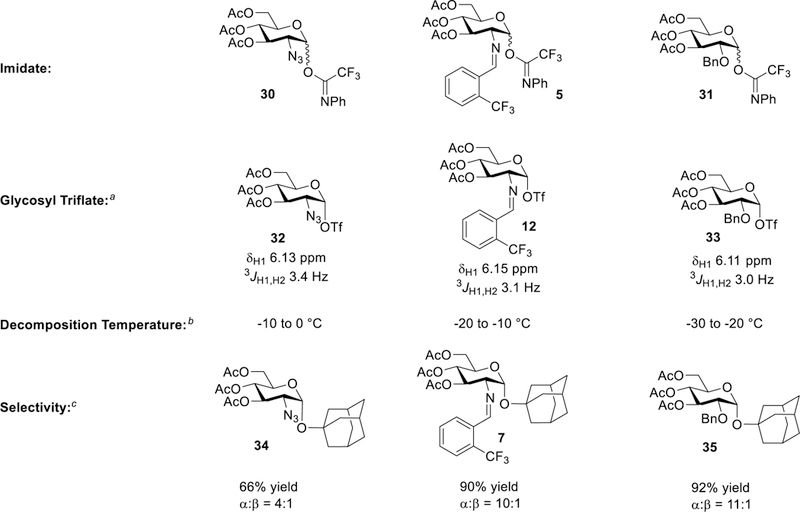
Relating the glycosyl triflate decomposition temperature to α:β selectivity of C(2)-protected glycosyl donors under Ni(OTf)2-catalyzed conditions. a Upon subjection of 50 mol% of TfOH at −50 °C in CD2Cl2. b Glycosyl triflate allowed to warm from −50 °C to room temperature while 1H NMR was taken at 10 °C intervals. c 15 mol% Ni(OTf)2 in CH2Cl2.
An additional rationalization for the difference of these three imidates for high α-selectivity comes from the induction of an early-transition state as the α-face attacking nucleophile interacts with the steric bulk of the N-benzylidene. On this basis, it has been reported that the N-benzylidene and O-benzyl protecting groups have similar size and can induce similar reactions because of their steric A value of 1.39. 77–78 Whereas, the azido group is smaller (steric A value = 0.45–0.62) and can perhaps not as induced the early transition state as readily. 77–78
CONCLUSIONS
In summary, we have presented the first systematic investigation of the release of triflic acid by a metal triflate for activation and its impact on the stereochemical outcome of metal triflate-catalyzed glycosylation. As metal triflates are one of the most commonly utilized catalysts for glycosylations and provide many benefits over traditional activation methods, we have laid the foundation for future studies involving these catalysts as elucidations of their operative mechanism can be complex. We have shown that these catalysts are capable of producing triflic acid and that their hydrolysis constants should be taken into consideration. Once released, as systematically studied in our nickel triflate-catalyzed glycosylation mechanism, triflic acid can influence the anomeric selectivity through a generation of a transient glycosyl triflate. However, a glycosyl triflate mechanism is not universal in effecting the selectivity of the newly formed glycosidic bond; a multitude of other factors need to be taken into consideration when trying to rationalize selectivity, including protecting groups and carbohydrate coupling partners. Under our nickel triflate-catalyzed conditions, the release of triflic acid in combination with the unique stereoelectronic nature of the C(2)-N-benzylidene group on glycosyl N-phenyl trifluoroacetimidate provided the formation of 1,2-cis-aminoglycosides to be highly favorable.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (R01 GM098285) and Wayne State University. E.T.S. acknowledges the University of Iowa for a graduate fellowship. The authors also thank Professor David Crich at Wayne State University for helpful discussions, Dr. Dale Swenson at the University of Iowa for assistance in X-ray crystallography analysis, and the Wayne State University Chemistry Lumigen Center for instrument assistance.
Footnotes
Notes: The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Full experimental procedures and characterization data for all new compounds (PDF).
X-ray data for compound β-imidate 5 (CIF).
REFERENCES
- (1).Varki A Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McKay MJ; Nguyen HM Recent Advances in Transition Metal-Catalyzed Glycosylation. ACS Catal 2012, 2, 1563–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Li X; Zhu J Glycosylation via Transition-Metal Catalysis: Challenges and Opportunities. Eur. J. Org. Chem 2016, 2016, 4724–4767. [Google Scholar]
- (4).Nielsen MM; Pedersen CM Catalytic Glycosylations in Oligosaccharide Synthesis. Chem. Rev 2018, 118, 8285–8358. [DOI] [PubMed] [Google Scholar]
- (5).Zhu Y; Yu B Highly Stereoselective β-Mannopyranosylation via the 1-α-Glycosyloxy-isochromenylium-4-gold(I) Intermediates. Chem. Eur. J 2015, 21, 8771–8780. [DOI] [PubMed] [Google Scholar]
- (6).Mensah EA; Yu F; Nguyen HM Nickel-Catalyzed Stereoselective Glycosylation with C(2)-N-Substituted Benzylidene D-Glucosamine and Galactosamine Trichloroacetimidates for the Formation of 1,2-Cis-2-Amino Glycosides. Applications to the Synthesis of Heparin Disaccharides, GPI Anchor Pseudodisaccharides, and Alpha-GalNAc. J. Am. Chem. Soc 2010, 132, 14288–14302. [DOI] [PubMed] [Google Scholar]
- (7).Mensah EA; Nguyen HM Nickel-Catalyzed Stereoselective Formation of Alpha-2-Deoxy-2-Amino Glycosides. J. Am. Chem. Soc 2009, 131, 8778–8780. [DOI] [PubMed] [Google Scholar]
- (8).Zhu X; Schmidt RR New Principles for Glycoside-Bond Formation. Angew. Chem. Int. Ed 2009, 48, 1900–1934. [DOI] [PubMed] [Google Scholar]
- (9).Wabnitz TC; Yu J-Q; Spencer JB Evidence That Protons Can Be the Active Catalysts in Lewis Acid Mediated Hetero-Michael Addition Reactions. Chem. Eur. J 2004, 10, 484–493. [DOI] [PubMed] [Google Scholar]
- (10).Li Z; Zhang J; Brouwer C; Yang C-G; Reich NW; He C Brønsted Acid Catalyzed Addition of Phenols, Carboxylic Acids, and Tosylamides to Simple Olefins. Org. Lett 2006, 8, 4175–4178. [DOI] [PubMed] [Google Scholar]
- (11).Rosenfeld DC; Shekhar S; Takemiya A; Utsunomiya M; Hartwig JF Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett 2006, 8, 4179–4182. [DOI] [PubMed] [Google Scholar]
- (12).Taylor JG; Adrio LA; Hii KK Hydroamination Reactions by Metal Triflates: Brønsted Acid vs. Metal Catalysis? Dalton Trans 2010, 39, 1171–1175. [DOI] [PubMed] [Google Scholar]
- (13).McKinney Brooner RE; Widenhoefer RA Stereochemistry and Mechanism of the Brønsted Acid Catalyzed Intramolecular Hydrofunctionalization of an Unactivated Cyclic Alkene. Chem. Eur. J 2011, 17, 6170–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dang TT; Boeck F; Hintermann L Hidden Brønsted Acid Catalysis: Pathways of Accidental or Deliberate Generation of Triflic Acid from Metal Triflates. J. Org. Chem 2011, 76, 9353–9361. [DOI] [PubMed] [Google Scholar]
- (15).Crasto CF; Jones GB A Practical method for preparation of β-glycosides of N-acetylglucosamine. Tetrahedron Lett 2004, 45, 4891–4894. [Google Scholar]
- (16).Sanders WJ; Kiessling LL Stereoselective, Lewis Acid-Catalyzed Glycosylation of Alcohols by Glucose 1,2-Cyclic Sulfites. Tetrahedron Lett 1994, 35, 7335–7338. [Google Scholar]
- (17).Kristensen SK; Salamone S; Rasmussen MR; Marqvorsen MHS; Jensen HH Glycosyl ortho-Methoxybenzoates: Catalytically Activated Glycosyl Donors with an Easily Removable and Recyclable Leaving Group. Eur. J. Org. Chem 2016, 2016, 5365–5376. [Google Scholar]
- (18).Adinolfi M; Barone G; Iadonisi A; Schiattarella M Efficient Activation of Glycosyl N-(Phenyl)trifluoroacetimidate Donors with Ytterbium(III) Triflate in the Glycosylation Reaction. Tetrahedron Lett 2002, 43, 5573–5577. [Google Scholar]
- (19).Christensen H; Christiansen MS; Petersen J; Jensen HH Direct Formation of β-Glycosides of N-Acetyl Glycosamines Mediated by Rare Earth Metal Triflates. Org. Biomol. Chem 2008, 6, 3276–3283. [DOI] [PubMed] [Google Scholar]
- (20).Rasmussen MR; Marqvorsen MHS; Kristensen SK; Jensen HH A Protocol for Metal Triflate Catalyzed Direct Glycosylations with GalNAc 1-OPiv Donors. J. Org. Chem 2014, 79, 11011–11019. [DOI] [PubMed] [Google Scholar]
- (21).Dumeunier R; Markó IE On the Role of Triflic Acid in the Metal Triflate-Catalysed Acylation of Alcohols. Tetrahedron Lett 2004, 45, 825–829. [Google Scholar]
- (22).Bizier NP; Atkins SR; Helland LC; Colvin SF; Twitchell JR; Cloninger MJ Indium Triflate Catalyzed Peracetylation of Carbohydrates. Carbohydr. Res 2008, 343, 1814–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Crich D; Sun S Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1H, 13C, and 19F NMR Spectroscopic Investigation. J. Am. Chem. Soc 1997, 119, 11217–11223. [Google Scholar]
- (24).Sletten ET; Ramadugu SK; Nguyen HM Utilization of Bench-Stable and Readily Available Nickel(II) Triflate for Access to 1,2-Cis-2-Aminoglycosides. Carbohydr. Res 2016, 435, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yang J; Cooper-Vanosdell C; Mensah EA; Nguyen HM Cationic Palladium-Catalyzed Stereoselective Glycosylation with Glycosyl Trichloroacetimidates. J. Org. Chem 2008, 73, 794–800. [DOI] [PubMed] [Google Scholar]
- (26).Mensah EA; Azzarelli JM; Nguyen HM Palladium-Controlled Beta-Selective Glycosylation in the Absence of the C(2)-Ester Participatory Group. J. Org. Chem 2009, 74, 1650–1657. [DOI] [PubMed] [Google Scholar]
- (27).McKay MJ; Naab BD; Mercer GJ; Nguyen HM Selective Formation of Beta-O-Aryl Glycosides in the Absence of the C(2)-Ester Neighboring Group. J. Org. Chem 2009, 74, 4705–4711. [DOI] [PubMed] [Google Scholar]
- (28).Yu F; Nguyen HM Studies on the Selectivity Between Nickel-Catalyzed 1,2-Cis-2-Amino Glycosylation of Hydroxyl Groups of Thioglycoside Acceptors with C2-Substituted Benzylidene N-Phenyl Trifluoroacetimidates and Intermolecular Aglycon Transfer of the Sulfide Group. J. Org. Chem 2012, 77, 7330–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McConnell MS; Yu F; Nguyen HM Nickel-Catalyzed Alpha-Glycosylation of C(1)-Hydroxyl D-Myo-Inositol: A Formal Synthesis of Mycothiol. Chem. Commun 2013, 49, 4313–4315. [DOI] [PubMed] [Google Scholar]
- (30).McConnell MS; Mensah EA; Nguyen HM Stereoselective Alpha-Glycosylation of C(6)-Hydroxyl Myo-Inositols via Nickel Catalysis-Application to the Synthesis of GPI Anchor Pseudo-Oligosaccharides. Carbohydr. Res 2013, 381, 146–152. [DOI] [PubMed] [Google Scholar]
- (31).Yu F; McConnell MS; Nguyen HM Scalable Synthesis of Fmoc-Protected GalNAc-Threonine Amino Acid and Tn Antigen via Nickel Catalysis. Org. Lett 2015, 17, 2018–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bennett CS, Selective Glycosylations Synthetic Methods and Catalysts Wiley-VCH: Weinheim, Germany, 2017; p 1–378. [Google Scholar]
- (33).Kobayashi S; Nagayama S; Busujima T Lewis Acid Catalysts Stable in Water. Correlation between Catalytic Activity in Water and Hydrolysis Constants and Exchange Rate Constants for Substitution of Inner-Sphere Water Ligands. J. Am. Chem. Soc 1998, 120, 8287–8288. [Google Scholar]
- (34).Glibstrup E; Pedersen CM Scalable Synthesis of Anomerically Pure Orthogonal-Protected GlcN3 and GalN3 from d-Glucosamine. Org. Lett 2016, 18, 4424–4427. [DOI] [PubMed] [Google Scholar]
- (35).There are several examples in the literature in which the reaction still proceeding when an acid scavenger was used in the presence of a metal triflate. Chem. Commun 2015, 51, 8939–8941. [Google Scholar]
- (36).Baes CF; Mesmer RE, The Hydrolysis of Cations Wiley: New York a.o., 1976; p XXI, 489 p. [Google Scholar]
- (37).Similar triflic acid promoted reactions have been known to proceed in the presence of molecular sieves: Chem. Commun, 2014, 50, 1067. [DOI] [PubMed] [Google Scholar]
- (38).Godeau J; Fontaine-Vive F; Antoniotti S; Duñach E Experimental and Theoretical Studies on the Bismuth-Triflate-Catalysed Cycloisomerisation of 1,6,10-Trienes and Aryl Polyenes. Chem. Eur. J 2012, 18, 16815–16822. [DOI] [PubMed] [Google Scholar]
- (39).Lemieux RU; Shyluk WP; Huber G The Acetolyses of the Alpha and Beta-Methyl D-Glucopyranoside Tetraacetates. Can. J. Chem 1955, 33, 148–162. [Google Scholar]
- (40).Normant JF; Alexakis A; Ghribi A; Mangeney P Boron Fluoride Promoted Cleavage of Acetals by Organocopper Reagents Application to Asymmetric Synthesis. Tetrahedron 1989, 45, 507–516. [Google Scholar]
- (41).Ikemoto N; Kim OK; Lo L-C; Satyanarayana V; Chang M; Nakanishi K Ferric Chloride, an Anomerization Aatalyst for the Preparation of Alkyl α-Glycopyranosides. Tetrahedron Lett 1992, 33, 4295–4298. [Google Scholar]
- (42).Olsson R; Rundström P; Persson B; Frejd T Organotitanium-Induced Stereoselective Alkylative Endo-Cleavage of Benzyl Pentopyranosides. Carbohydr. Res 1998, 307, 13–18. [Google Scholar]
- (43).O’Brien C; Poláková M; Pitt N; Tosin M; Murphy PV Glycosidation–Anomerisation Reactions of 6,1-Anhydroglucopyranuronic Acid and Anomerisation of β-D-Glucopyranosiduronic Acids Promoted by SnCl4. Chem. Eur. J 2007, 13, 902–909. [DOI] [PubMed] [Google Scholar]
- (44).Wang Y; Cheon H-S; Kishi Y Unique Reactivity of the Mukaiyama Glycosidation Catalyst (SnCl3ClO4) Toward β-Mannopyranosides. Chem. Asian J 2008, 3, 319–326. [DOI] [PubMed] [Google Scholar]
- (45).Pilgrim W; Murphy PV SnCl4- and TiCl4-Catalyzed Anomerization of Acylated O- and S-Glycosides: Analysis of Factors That Lead to Higher α:β Anomer Ratios and Reaction Rates. J. Org. Chem 2010, 75, 6747–6755. [DOI] [PubMed] [Google Scholar]
- (46).Manabe S; Satoh H; Hutter J; Lüthi HP; Laino T; Ito Y Significant Substituent Effect on the Anomerization of Pyranosides: Mechanism of Anomerization and Synthesis of a 1,2-cis Glucosamine Oligomer from the 1,2-trans Anomer. Chem. Eur. J 2013, 20, 124–132. [DOI] [PubMed] [Google Scholar]
- (47).Satoh H; Manabe S; Ito Y; Lüthi HP; Laino T; Hutter J Endocyclic Cleavage in Glycosides with 2,3-trans Cyclic Protecting Groups. J. Am. Chem. Soc 2011, 133, 5610–5619. [DOI] [PubMed] [Google Scholar]
- (48).Vidadala SR; Pimpalpalle TM; Linker T; Hotha S Gold-Catalyzed Reactions of 2-C-Branched Carbohydrates: Mild Glycosidations and Selective Anomerizations. Eur. J. Org. Chem 2011, 2011, 2426–2430. [Google Scholar]
- (49).Frihed TG; Bols M; Pedersen CM Mechanisms of Glycosylation Reactions Studied by Low-Temperature Nuclear Magnetic Resonance. Chem. Rev 2015, 115, 4963–5013. [DOI] [PubMed] [Google Scholar]
- (50).Huang M; Garrett GE; Birlirakis N; Bohe L; Pratt DA; Crich D Dissecting the Mechanisms of a Class of Chemical Glycosylation using Primary 13C Kinetic Isotope Effects. Nat. Chem 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dhakal B; Bohe L; Crich D Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J. Org. Chem 2017, 82, 9263–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).van der Vorm S; Hansen T; Overkleeft HS; van der Marel GA; Codée JDC The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem. Sci 2017, 8, 1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Crich D; Cai W Chemistry of 4,6-O-Benzylidene-D-glycopyranosyl Triflates: Contrasting Behavior between the Gluco and Manno Series. J. Org. Chem 1999, 64, 4926–4930. [DOI] [PubMed] [Google Scholar]
- (54).Adero PO; Amarasekara H; Wen P; Bohe L; Crich D The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kowalska K; Pedersen CM Catalytic Stereospecific O-Glycosylation. Chem. Commun 2017, 53, 2040–2043. [DOI] [PubMed] [Google Scholar]
- (56).Qiao Y; Ge W; Jia L; Hou X; Wang Y; Pedersen CM Glycosylation intermediates studied using low temperature 1H- and 19F-DOSY NMR: new insight into the activation of trichloroacetimidates. Chem. Commun 2016, 52, 11418–11421. [DOI] [PubMed] [Google Scholar]
- (57).Zandanel C; Dehuyser L; Wagner A; Baati R Highly Chemo- and Stereoselective Glycosidation of Permethacrylated O-Glycosyl Trichloroacetimidate Reagents Promoted by TMSNTf2. Tetrahedron 2010, 66, 3365–3369. [Google Scholar]
- (58).Elferink H; Pedersen CM l-Rhamnosylation: The Solvent is the Solution. Eur. J. Org. Chem 2016, 2017, 53–59. [Google Scholar]
- (59).Lemieux RU; Hendriks KB; Stick RV; James K Halide Ion Catalyzed Glycosidation Reactions. Syntheses of alpha-Linked Disaccharides. J. Am. Chem. Soc 1975, 97, 4056–4062. [Google Scholar]
- (60).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- (61).Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (62).Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (63).Grimme S Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem 2006, 27, 1787–1799. [DOI] [PubMed] [Google Scholar]
- (64).Crich D; Vinogradova O Synthesis and Glycosylation of a Series of 6-Mono-, Di-, and Trifluoro S-Phenyl 2,3,4-Tri-O-Benzyl-Thiorhamnopyranosides. Effect of the Fluorine Substituents on Glycosylation Stereoselectivity. J. Am. Chem. Soc 2007, 129, 11756–11765. [DOI] [PubMed] [Google Scholar]
- (65).Fang T; Gu Y; Huang W; Boons G-J Mechanism of Glycosylation of Anomeric Sulfonium Ions. J. Am. Chem. Soc 2016, 138, 3002–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Seeman JI Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett Winstein-Holness Kinetics. Chem. Rev 1983, 83, 83–134. [Google Scholar]
- (67).Hammond GS A Correlation of Reaction Rates. J. Am. Chem. Soc 1955, 77, 334–338. [Google Scholar]
- (68).Walvoort MTC; Lodder G; Mazurek J; Overkleeft HS; Codée JDC; van der Marel GA Equatorial Anomeric Triflates from Mannuronic Acid Esters. J. Am. Chem. Soc 2009, 131, 12080–12081. [DOI] [PubMed] [Google Scholar]
- (69).Yu B; Tao H Glycosyl Trifluoroacetimidates. 2. Synthesis of Dioscin and Xiebai Saponin I. J. Org. Chem 2002, 67, 9099–9102. [DOI] [PubMed] [Google Scholar]
- (70).van der Vorm S; Overkleeft HS; van der Marel GA; Codée JDC Stereoselectivity of Conformationally Restricted Glucosazide Donors. J. Org. Chem 2017, 82, 4793–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schumann B; Parameswarappa SG; Lisboa MP; Kottari N; Guidetti F; Pereira CL; Seeberger PH Nucleophile-Directed Stereocontrol Over Glycosylations Using Geminal-Difluorinated Nucleophiles. Angew. Chem. Int. Ed 2016, 55, 14431–14434. [DOI] [PubMed] [Google Scholar]
- (72).D’Angelo KA; Taylor MS Borinic Acid Catalyzed Stereo- and Regioselective Couplings of Glycosyl Methanesulfonates. J. Am. Chem. Soc 2016, 138, 11058–11066. [DOI] [PubMed] [Google Scholar]
- (73).Paulsen H; Stenzel W Bausteine von Oligosacchariden, X: Synthese α−1→4- und α−1→3-Verknüpfter Disaccharide der 2-Amino-2-Desoxy-D-Glucopyranose nach der Azid-Methode. Chem. Ber 1978, 111, 2348–2357. [Google Scholar]
- (74).Paulsen H; Stenzel W Bausteine von Oligosacchariden, IX: Stereoselektive Synthese α-Glycosidisch Verknüpfter Di- und Oligosaccharide der 2-Amino-2-Desoxy-D-Glucopyranose. Chem. Ber 1978, 111, 2334–2347. [Google Scholar]
- (75).Lemieux RU; Ratcliffe RM The Azidonitration of Tri-O-Acetyl-D-Galactal. Can. J. Chem 1979, 57, 1244–1251. [Google Scholar]
- (76).Nokami T; Shibuya A; Manabe S; Ito Y; Yoshida J -i. α- and β-Glycosyl Sulfonium Ions: Generation and Reactivity. Chem. Eur. J 2009, 15, 2252–2255. [DOI] [PubMed] [Google Scholar]
- (77).Crich D; Xu H Direct Stereocontrolled Synthesis of 3-Amino-3-Deoxy-Beta-Mannopyranosides: Importance of the Nitrogen Protecting Group on Stereoselectivity. J. Org. Chem 2007, 72, 5183–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Crich D Mechanism of a Chemical Glycosylation Reaction. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.