

Abstract
In 2018, we reported a hybrid pipeline that predicts protein structures with I-TASSER and function with COFACTOR. I-TASSER/COFACTOR achieved Gene Ontology (GO) high prediction accuracies of Fmax = 0.69 and 0.57 for molecular function (MF) and biological process (BP), respectively, on 100 comprehensively annotated proteins. Now we report blinded analyses of newly annotated proteins in the critical assessment of function annotation (CAFA) three function prediction challenge and in neXtProt. For CAFA3 results released in May 2019, our predictions on 267 and 912 human proteins with newly annotated MF and BP terms achieved Fmax = 0.50 and 0.42, respectively, on “No Knowledge” proteins, and 0.51 and 0.74, respectively, on “Limited Knowledge” proteins. While COFACTOR consistently outperforms simple homology-based analysis, its accuracy still depends on template availability. Meanwhile, in neXtProt 2019–01, 25 proteins acquired new function annotation through literature curation at UniProt/Swiss-Prot. Before the release of these curated results, we submitted to neXtProt blinded predictions of free-text function annotation based on predicted GO terms. For 10 of the 25, a good match of free-text or GO term annotation was obtained. These blind tests represent rigorous assessments of I-TASSER/COFACTOR. neXtProt now provides links to precomputed I-TASSER/COFACTOR predictions for proteins without function annotation to facilitate experimental planning on “dark proteins”.
Keywords: uncharacterized human proteins validated at protein level (uPE1), CP50 challenge, structure-based function annotation, I-TASSER, COFACTOR, critical assessment of function annotation (CAFA) 3, neXtProt
Graphical Abstract
INTRODUCTION
For systematic structure and function annotations for uncharacterized human proteins validated at protein level (uPE1), we proposed in 2018 a computational pipeline1 combining the I-TASSER2 protein structure prediction algorithm and the COFACTOR3 structure-based function annotation method. On a benchmark test of 100 randomly selected very well-annotated proteins from chromosome 17 (with at least 3 Gold GO terms each for all three aspects of MF, BP, and CC), I-TASSER/COFACTOR achieved a high Gene Ontology (GO) term4 annotation accuracy of 0.69 and 0.57, as measured by Fmax (see Materials and Methods for mathematical explanation), for molecular function (MF) and biological process (BP), respectively, and consistently outperformed two state-of-the-art methods (GoFDR5 and GOtcha6). Cellular component (CC) is not considered as “function” in neXtProt. Using this pipeline (with C-score ≥0.5, see Materials and Methods), we then confidently assigned MF and BP for 13 and 33 proteins, respectively, of all the 66 uPE1 proteins from chromosome 17 in neXtProt as of 2017–07–01.
Three shortcomings emerged as a result of our discussions with other experts in the proteomics community,7 particularly at neXtProt. First, when benchmarking our algorithm, we had removed experimentally studied close homologues to the query protein if sharing >30% sequence identity. While this is a close approximation of real-life challenges for distant- or non-homologous protein function prediction, such benchmarks could have been biased in the following aspects: (a) different from community-wide protein structure and function challenges such as CASP8 and CAFA,9 this benchmark was not a blind test; thus, the prediction result could potentially be enhanced by prior knowledge of known protein functions; and (b) the benchmark is akin to a cross-validation test in machine learning, where the same version of a database is partitioned into the training set (template database after close homologue removal) and the validation set (the benchmark set of 100 query protein), and the labels (GO terms) of the validation set are predicted from the training set. This is different from a real-lifetime-elapsed prediction, where only older functional annotations in the template database are used to predict functions of new proteins to be annotated. Cross-validation tests have been reported to be less challenging than time-elapsed tests.10 That is partly because a single high-throughput experimental paper could lead to a dramatic increase of annotation for a few specific GO terms, and such a change in GO term distribution over time cannot realistically be modeled in a cross-validation setting.11
Second, our benchmark test focuses mainly on evaluation of predicted GO terms, which are standard vocabulary for function annotation that are amenable for programmatic parsing. However, experimental biologists are usually more accustomed to free-text function annotation instead of computationally friendly GO terms, and almost all experimental literature of function characterization presents the findings in free-text form, until processed into GO terms separately by database curators later.12
Third, the interpretation of prediction confidence scores from our pipeline for human proteins is not straightforward because the confidence scores were originally calibrated on the E. coli proteome3 instead of on the human proteome.
This manuscript addresses the three issues by recalibrating the confidence scores of our pipeline and performing two rigorous time-elapsed blind tests of protein function prediction. The recalibration of the correspondence between the confidence score of our predictions and the precision of function prediction was performed on a large set of 1995 well-annotated human PE1 proteins among the 17 694 PE1 proteins.
In the first blind test, we evaluated the performance of COFACTOR in the CAFA3 GO term prediction challenge. On the 267 and 912 CAFA3 human targets used for MF and BP evaluations, respectively, we found a clear advantage of COFACTOR compared to simple sequence homology search or background probability modeling, though its performance is still dependent on the availability of high scoring templates.
In the second blind test, an independent assessor (coauthor L.L.) identified a set of 44 neXtProt13 proteins undergoing function curation based on manually gathered publications during 2018.14 Meanwhile, predictors (coauthors C.Z., G.S.O., and Y.Z.) performed protein structure and function predictions using the same automated pipeline as in our 2018 study. On the basis of the automatically predicted GO terms, these three predictors assigned a free-text function interpretation for each query protein. Both the automated GO predictions and the manual free-text interpretations were performed blind to the pending curation of the proteins and were submitted to the assessor before the neXtProt 2019–01 release. For both predicted GO terms and the respective free-text interpretation, consistency of the predicted functions with neXtProt curation was assessed upon publication of the neXtProt 2019–01 release.
These analyses should serve as an incremental step toward completion of structure and function modeling of all remaining uPE1 and even PE2,3,4 proteins in the human proteome.15 All data discussed in this study are available at https://zhanglab.ccmb.med.umich.edu/COFACTOR2/nx2019addition/GOterm.html. Additionally, all modeling results for each of the 44 blindly tested neXtProt proteins in this study are provided as a link on neXtProt (https://www.nextprot.org/entry/NX_P0C870/gh/zhanglabs/COFACTOR, where “NX_P0C870” can be replaced by the neXtProt ID for each other target of interest). These capabilities have now been made available to all investigators through the “Community” link in neXtProt.16
MATERIALS AND METHODS
Our computational pipeline for structure and function prediction of human uPE1 proteins is largely identical to our previous report,1 except for the use of specific versions of the protein function databases including UniProt, STRING, and BioLiP (see further). For the sake of completeness, we briefly summarize the computational methods as follows.
I-TASSER Protein Structure Prediction
The input of the I-TASSER structure prediction pipeline is the query protein sequence. From the query sequence, residue position-specific local sequence and structure features such as sequence profiles, secondary structures, and backbone torsion angles are derived. These local features are used by the LOMETS17 meta-threading server to search the query protein against a custom PDB library (https://zhanglab.ccmb.med.umich.edu/library/); templates in this PDB library are nonredundant, that is, any two templates in the library share <70% sequence identity with each other. From the structure templates identified by LOMETS threading, continuous fragments are collected and assembled into full length structures by replica-exchange Monte Carlo (REMC) simulation in I-TASSER. This REMC simulation is guided by a composite energy function combining knowledge-based potential derived from PDB statistics, together with residue—residue distance restraints calculated from threading templates. The REMC simulation trajectory produces tens of thousands of conformations, also known as “decoys”, which are clustered by SPICKER18 according to structure similarity. The largest cluster, which corresponds to the set of conformations with lowest free energy, and the cluster centroid are selected to undergo atomic-level structure refinement by FG-MD19 to derive the final structure model. While I-TASSER can generate up to five structure models corresponding to the five largest clusters, this study used only the first model for subsequent function modeling. This model is usually closest to the native structure among all models and can reliably estimate the global quality (estimated TM-score) according to LOMETS threading alignment scores and simulation convergency.20
COFACTOR Automatic Structure-Based Function Annotation
To functionally annotate the query structure model obtained above, the COFACTOR algorithm uses a modified TM-align21 program to search structure analogs of the query structure in the BioLiP22 structure—function database. BioLiP functions from template structures with significant global structure similarity are rescored by a combination of global structure and local binding site structure alignment to measure the global structure similarity, active site local similarity, and matching of sequence profiles.23 Functions from templates are then transferred to the query according to the composite globallocal structure alignment score.
While structure alignment provides a sensitive approach for function annotations, its power is limited when the protein in question is unstructured (i.e., intrinsically disordered) or proteins with relevant function annotation lack an experimental structure in the BioLiP database. To mitigate this shortcoming of structure-based function annotation, COFACTOR also extracts GO term annotations by a sequence-based approach and a protein—protein interaction (PPI)-based approach. The sequence-based approach derives functions from BLAST and PSI-BLAST24 hits in the UniProt database search. The PPI-based approach maps the query to a PPI network defined by the STRING25 database to find the PPI partners of the query. In the earlier version of COFACTOR,3 including the version participated in the CAFA3 predictions in 2017, the functions were inferred from GO terms annotated to the PPI partners. When we later developed MetaGO,23 an extension for the GO term prediction component of COFACTOR, we found that functions inferred from UniProt26 sequences homologous to the PPI partners were more accurate than functions directly inferred from PPI partners. Therefore, the current COFACTOR program uses this improved PPI-based method originally developed for MetaGO, where functions are predicted from UniProt sequences homologous to PPI partners of the query protein.
For a given GO term q, the confidence of the final consensus prediction ranges between 0 and 1 and is a weighted average of the three approaches (structure, PPI, and sequence):
| (1) |
Here, wm is the weighting score for method m. Cscorem(q) is the confidence score of the mth method for GO term q and takes the following form:
| (2) |
Nm is the total number of templates detected by method m. Sim is the weighting score of the ith template detected by method m. The template weighting score could be (PSI-)BLAST sequence identity for sequence-based method, and interaction score assigned by STRING database for PPI based method. Nm(q) and Sim(q) are the template number and weighting score of ith template, respectively, in method m for the subset of templates associated with GO term q. We note that, instead of using the most confident template for each GO term, eq 2 represents a weighted k-nearest-neighbor approach where all N templates are considered in the consensus voting for each predicted GO term. Therefore, if all templates are associated with q, the nominator and denominator in eq 2 are the same, and Cscore(q) is one, that is, 100% confident, even when none of the templates shares high sequence similarity to the query. A more extensive description of these methods is provided in our 2018 report.1
The approximate running times of I-TASSER structure prediction and COFACTOR structure-based function prediction for a protein with 375 residues, the median length of human proteins,27 are 118 and 11 CPU hours, respectively, as determined empirically previously.23
Protein Function Databases Used in COFACTOR Pipeline
The COFACTOR pipeline uses three protein function databases: UniProt, STRING, and BioLiP. We initially started this project using UniProt version 2018_11 for sequence-homology identification in our sequence-based method. However, we later learned that updates to functional annotation, such as GO terms, are always made public in UniProtKB a few months before being incorporated into the neXtProt release. As a result, taking UniProt entries from November 2018 as templates for prediction was equivalent to taking the neXtProt 2019–01 entries from the test set. Therefore, we revised our prediction process by using a safely older UniProt database version 2018_02 as the template database for two component methods of the COFACTOR pipeline. The sequence-based component retrieves UniProt annotated GO terms for (PSI-)BLAST hits of query sequence, while the PPI-based component obtains UniProt GO terms annotated to UniProt sequences homologous to the PPI partner of query. In both cases, only the sequence and GO term annotations from UniProt are used. We do not use UniProt PPI annotation because it is not as comprehensive as STRING database (see below), and we do not use UniProt/neXtProt free-text function annotations for prediction because they are not amenable for programmatic parsing.
The BioLiP structure function database uses GO terms annotated to PDB chains exclusively collected from the SIFTS and PDBsum projects. SIFTS28 and PDBsum29 in turn derive GO terms either by mapping PDB chains to respective UniProt entries or running the same InterPro2GO annotation pipeline as used by UniProt. Therefore, GO term annotation in BioLiP will not be more recent than the corresponding UniProt released at the same date. By rolling back the use of BioLiP to that in Feb 2018, we avoided similar bias.
For the PPI-based pipeline, we used STRING database version 10.0, instead of the most recent STRING version 11 released in Jan 2019, for the same reason. A large portion of the PPI entries in STRING are computational predictions, such as those by text mining, genome neighbor, or expression pattern, many of which could be false PPIs. Therefore, for a pair of proteins in a species, STRING does not simply give a binary yes or no answer. Instead, it reports a confidence score, a “STRING score”, to estimate the probability of interaction, usually ranging between 0.15 and 1.00, which is considered in the weighting of GO term prediction in our PPI-based pipeline, as mentioned earlier. To ensure coverage of the PPI network, we do not filter out computationally predicted PPI pairs; instead, we combine all PPI information and weight them according to the STRING score (eq 2) so that reliable PPI pairs are up-weighted in GO term prediction.
Manual Free-Text Function Interpretation
We follow three steps to assign free-text annotation for automated GO term prediction:
Examine MF and BP GO terms from I-TASSER/COFACTOR, excluding general terms, either those defined in neXtProt SPARQL NXQ_00022 or terms like “cellular process”.
Select the most specific GO term in MF or BP with C-score >0.5. If there is no GO term with C-score >0.5, consider terms with C-score >0.4.
If the aspect (MF/BP) with the term selected in step (b) also has other high confidence unrelated GO terms, proceed to the complementary aspect (BP/MF) and repeat step b.
For example, even though the BP prediction for C1QTNF8 (P60827–1) includes multiple terms with C-score ≥0.5 (GO:0009987 “cellular process”, C-score = 0.93; GO:0048518 “positive regulation of biological process”, C-score = 0.67; GO:0032502 “developmental process”, C-score = 0.65; GO:0044238 “primary metabolic process”, C-score = 0.54; GO:0048584 “positive regulation of response to stimulus”, C-score = 0.53), these GO terms are not informative for the purpose of free-text function interpretation because they only vaguely suggest the protein’s involvement in biological regulation of unspecified pathways. Meanwhile, this protein does not have any MF GO term predicted with C-score ≥0.5 (after excluding the GO terms considered by neXtProt as too general). Therefore, for this protein, we alternatively use MF GO terms predicted with C-score ≥0.4 (GO:0005102 “signaling receptor binding”, C-score = 0. 41) and assign the free-text interpretation “signal receptor binding”.
In the event that predicted GO terms are too diverse in one of the three GO aspects to conclusively interpret the function, other aspects of GO are used for interpretation. For example, BP prediction of RFPL1 (O75677–1) is too diverse (GO:0016567 “protein ubiquitination”, C-score = 0.55; GO:0010468 “regulation of gene expression”, C-score = 0.56; and GO:0002376 “immune system process”, C-score = 0.63); we instead used its high confidence MF GO term predictions, which are exclusively related to ubiquitin-protein transferase activity (GO:0004842, C-score = 0.78).
All free-text interpretations strictly use phrases in the definitions of predicted GO terms selected by the above criteria.
This exercise of our free-text annotation was performed to emulate how biologists would interpret a list of computationally predicted GO terms for a protein. It is only performed for the small neXtProt data set of 44 proteins because manual inspection of the full CAFA3 data set with 20 197 human proteins was impractical. As exemplified by REPL1 (O75677–1) above, to simplify our interpretation, the free-text annotation derived from predicted GO terms only attempted to cover the most likely function of a protein. Therefore, such a free-text annotation may not be as comprehensive as the respective UniProt/neXtProt free-text annotation, which aims to cover as many different functions of a protein as possible so long as there is conclusive literature evidence. This difference in how our free-text annotations and those of UniProt/neXtProt are derived also affects how we evaluate the performance of our free-text annotations, as discussed later.
Assessment Metrics for Function Prediction
Biologically meaningful metrics for assessing protein function prediction should not focus only on the precision of predicted GO terms. For example, a protein function predictor that only predicts shallow and generic GO terms such as “protein binding” or “cellular process” could have a very good precision but is rarely useful in practice. In fact, neXtProt does not consider 11 MF and 2 BP GO terms for being too general and does not use cellular component (CC) at all when retrieving uPE1 proteins (https://www.nextprot.org/proteins/search?mode=advanced&queryId=NXQ_00022). The 11 general MF terms are GO:0005524 “ATP binding”, GO:0000287 “magnesium ion binding”, GO:0005515 “protein binding”, GO:0042802 “identical protein binding”, GO:0008270 “zinc ion binding”, GO:0051260 “protein homooligomerization”, GO:0005509 “calcium ion binding”, GO:0003676 “nucleic acid binding”, GO:0003824 “catalytic activity”, GO:0046914 “transition metal ion binding”, and GO:0046872 “metal ion binding”; the two general BP terms are GO:0007165 “signal transduction”, and GO:0035556 “intracellular signal transduction”. We have accepted those exclusions in this analysis of neXtProt data. To simultaneously assess the precision and recall of our prediction, we follow the standard practice of CAFA and evaluate the accuracy of automatic GO term prediction by maximum F1-score, that is, the Fmax:
| (3) |
| (4) |
In the above equations, pr(t), or “precision”, is the number of correctly predicted GO terms, true positive tp(t), over the number of all GO terms predicted with confidence score ≥t, that is, tp(t) + fp(t). re(t), or “recall”, is tp(t) divided by all true positive plus false negative GO terms annotated to query by UniProt/neXtProt ground truth, that is, tp(t) + fn(t). In Supplementary Figure S1, we display a graphic illustration of this metric.
Two further clarifications should be made for the Fmax, as a measure of consistency between our prediction and the UniProt/neXtProt GO annotation. First, although Fmax = 0.5 means half of the predicted GO terms exactly match GO terms annotated by UniProt/neXtProt, and half of the UniProt/neXtProt GO terms are among the predicted GO terms, a predictor achieving Fmax = 0.5 should not be interpreted as being no better than guessing the two faces of a flipped coin. Unlike a flipped coin whose probability for the landing of the two faces are half-half, the average probability for a GO term to get annotated (or not annotated) to a protein is far from half–half in the database: for 92.1%, 98.5%, and 99.8% of the 47 340 GO terms defined by the Gene Ontology Consortium, each of them is annotated to less than 0.1%, 1%, and 10% of any UniProt proteins, respectively. Therefore, predicting GO terms with 50% precision is indeed a challenging task and a significant success.
Second, Fmax should not be confused with C-score. The C-score is for each predicted GO term of a query protein, while Fmax is an overall statistic for a protein or a set of proteins. C-score is estimated by COFACTOR without knowing the ground-truth, while Fmax can only be calculated if we know both the predicted GO terms and the ground-truth GO terms.
Compared to GO term evaluation, assessment of free-text annotation is more challenging as there is no agreed-upon metric to quantify the similarity between two free-text biological function descriptions. Moreover, free-text function annotations for a protein, especially one that performs multiple functions or is involved in complicated pathways, are affected by subjective judgment by the function curators for both our predictions and by UniProt/neXtProt curators. In this blinded analysis, we compared both GO terms and free-text interpretation from I-TASSER/COFACTOR prediction and from the UniProt/neXtProt literature-based free-text curation. Another complication for head-to-head comparison between the two kinds of free-text annotation is that, as mentioned above, our free-text interpretation from I-TASSER/COFACTOR only attempts to cover the most likely function of the target protein, while UniProt/neXtProt free-text annotation attempts to more comprehensively cover different functions of a protein. Therefore, if free-text interpretation from I-TASSER/COFACTOR matches at least part of the neXtProt free-text annotation for a target protein by manual inspection, we consider that the pair of free-text annotations is consistent (see Supplementary Table S1).
While both free-text and MF/BP GO terms are considered “function annotations”, free-text annotations curated by UniProt/neXtProt may not be fully reflected by the GO terms annotated for the same protein, partly due to the complexity of data source and curation process. neXtProt function annotations have the following major sources. First, all manual annotations performed by Swiss-Prot curators from experimental papers are generally captured as free-text, MF/BP GO terms (using the closest possible terms), keywords, and, in the case of enzymes, enzyme commission (EC) numbers. Sometimes there is no existing GO term to describe a particular function, resulting in only a free text description without GO terms, which happens to 17 and 3 for MF and BP, respectively, for the 25 blindly tested neXtProt targets. In most cases, GO terms assigned in this way are more generic than the respective free-text. Second, MF and BP GO terms are also manually annotated by other members of the Gene Ontology Consortium such as HGNC and MGI. Finally, MF and BP annotations computationally assigned by UniProt or the Gene Ontology Consortium are considered. Apart from free-text and GO terms, neXtProt includes other function annotations such as pathway annotations from KEGG and Reactome, and transporter classification from TCDB. For this paper, we mainly focus on GO terms and free-text.
RESULTS AND DISCUSSION
Data Sets
This study used three data sets: one benchmark neXtProt data set for recalibrating the C-score of COFACTOR, and two time-elapsed blindly tested data sets from CAFA3 human targets and newly annotated PE1 entries from neXtProt 2019–01. The “recalibration” set is used to establish the relation between C-score and function prediction precision in COFACTOR, while the performance of I-TASSER/COFACTOR is evaluated on the two blindly tested data sets.
The recalibration set consists of 1995 well-annotated human PE1 proteins with up to 750 residues in neXtProt release 2019–01. Similar to the benchmark set of 100 Chromosome 17 PE1 proteins in our 2018 report,1 each protein in this recalibration set has ≥3 Gold MF terms, ≥ 3 Gold BP terms, and ≥3 Gold CC terms.
The blindly tested CAFA3 human data set included 20 197 human protein targets, among which 267, 912, and 347 targets acquired new GO terms in UniProt between 2017 and 02–02 and 2017–11–15 for MF, BP, and CC, respectively (see supplementary_data/cafa3/benchmark20171115.tar at https://figshare.com/articles/Supplementary_data/8135393). These targets are further divided into two types: 147, 240, and 214 “No Knowledge” targets do not have any experimental GO annotation before CAFA3 for MF, BP, and CC, respectively; 120, 672, and 133 “Limited Knowledge” targets have at least one experimental GO annotation before CAFA3 for MF, BP, and CC. Statistical analysis of function predictions on this data set, released by the CAFA Consortium in May 2019 on bioRxiv,30 is evaluated based on the GO term predictions our group submitted during the CAFA3 challenge before 2017–02–02.
As of the neXtProt release 2019–01, 25 of the 44 proteins submitted for curation in 2018 acquired new function annotations. While all 25 targets receive free-text function annotation in neXtProt, only 8 and 22 acquired GO terms for MF and BP, respectively, excluding GO annotations deemed by neXtProt as being too general in the neXtProt SPARQL query NXQ_00022 as explained above in the Materials and Methods. We make available our predictions for all 44 (https://zhanglab.ccmb.med.umich.edu/COFACTOR/nx2019addition/GOterm.html#3) so that comparison with future neXtProt releases will be facilitated. Among these 25 recently curated neXtProt targets, the function annotation for one target (P0C870–1, https://www.uniprot.org/uniprot/P0C870?version=85&version=87&diff=true) was updated by UniProt on 2019–02–13 and was not in time to be included in neXtProt release 2019–01. For this particular target, we use the more recent UniProt annotation on 2019–02–13 instead of that from neXtProt 2019–01. We do not separately evaluate our result on Gold GO terms and on Gold plus Silver GO terms as in previous study1 because all newly annotated MF and BP GO terms for this set of 8 and 22 targets have Gold status.
Recalibration of COFACTOR C-Score for Human Proteins
While COFACTOR assigns a C-score for each predicted GO term for a target protein, the C-score is strongly correlated with, but does not strictly equal, the probability of the GO term being associated with the target. When we originally reported the GO term prediction method of COFACTOR, we calibrated this C-score to the corresponding probability of GO term association, that is, the precision of GO term prediction, given the C-score, on a prokaryotic set of 1244 E. coli proteins.3 Because of the later improvement of our function prediction method23 and the change of species of interest (E. coli to human), it became necessary to recalibrate the current COFACTOR algorithm with the recalibration set of 1995 human proteins. To calculate GO term prediction precision given C-score, all GO term predictions for each of the three aspects (MF, BP, and CC) were grouped into 10 bins by C-score with bin width = 0.1. To examine whether the calibration depends on the availability of close homology templates, we performed two separate calibration runs by excluding function templates sharing ≥0.3 and ≥0.9 sequence identity (ID) to the query. The calibration curve and the precision-recall curve are shown in Figure 1. For the PPI-based pipeline in COFACTOR, there are two rounds of sequence homologue searches: the first round maps query sequence to its STRING entry, which is used to identify PPI partners interacting with the query; the second round of sequence search maps PPI partners to UniProt proteins to obtain the GO annotations. Because the function annotations in the PPI-based pipeline are eventually derived only from the PPI partner homologues in UniProt, the sequence identity cutoffs in Figure 1 are applied only between query and the PPI partner homologues in UniProt.
Figure 1.

(A) Calibration curve for GO term prediction precision versus C-score for MF (blue up-pointing triangles), BP (red circles), and CC (green down-pointing triangles). Curve for template sequence identity (ID) <0.3 and <0.9 to the query are shown in hollow and solid markers, respectively. The lower legend shows the Pearson’s correlation coefficient (PCC) between precision and C-score. (B) Precision-recall curve for GO term prediction. The lower legend shows the Fmax for each curve (see Materials and Methods).
As shown in Figure 1B, GO term prediction accuracy of COFACTOR, as measured by Fmax, is higher by 24%, 28%, and 11% when high sequence identity (ID < 0.9) templates are available, compared to low sequence identity cutoff (ID < 0.3) cases. Nevertheless, the values are still quite high for the lower cutoff. On the other hand, the correlation between precision and C-score does not have as strong a dependency on sequence identity cutoff, even though the low sequence identity cases still have slightly lower precision given the same C-score. For example, for 0.4 < C-score ≤ 0.5, the precision is 0.69, 0.64, and 0.57 for MF, BP, and CC for ID < 0.9, compared with 0.60, 0.57, and 0.57 for ID < 0.3 (Figure 1A). Considering the fact that most poorly characterized or uncharacterized proteins have sequence identity around 0.3 to the closest functionally characterized homologue (see our earlier methodology paper23), we recommend the use of the recalibration curve obtained at ID < 0.3 for interpretation of COFACTOR function prediction for human targets.
Performance of GO Term Prediction by COFACTOR in CAFA3
A preliminary version of COFACTOR3 was tested in the 2017 CAFA3 experiment, the latest CAFA community-wide challenge for protein function prediction, by our team “Zhang-Freddolino lab”. In the official CAFA3 result30 released in May 2019, our team was ranked as one of the top performing groups (ranked second, third, fourth, and fifth for prediction of motility, biofilm formation, CC, and BP, respectively) among 68 teams (see Figures 3 and 4 of the CAFA3 report30). We note that such performance was obtained by a partial implementation of COFACTOR with just the sequence- and PPI-based pipeline for 82 903 (63.4%) of the 130 827 prediction targets from 23 species, as the structure-based pipeline of COFACTOR was not ready for high-throughput prediction when we participated in CAFA3 in 2017.31 To further save time, among the reduced CAFA3 set of 47 924 structure-based function prediction targets, the full length structure models of query proteins were generated by LOMETS threading followed by MODELLER32 homology modeling for 43 953 targets (91.7%), while the full I-TASSER pipeline was only used for the remaining 3971 targets (8.3%). The lack or lower quality of structure information is part of the reasons for our limited CAFA3 performance in MF because the specificity of molecular function such as biomolecule binding and catalytic activity is determined by structure.
Figure 3.

Fmax of MF (blue triangle) and BP (red circle) GO term prediction versus (A) sequence length, (B) global sequence identity of closest PSI-BLAST hit, (C) highest PPI interaction score (STRING score), and (D) TM-score between query structure and the closest BioLiP template. A pair of error bars marks the standard deviation of Fmax at each bin. Inside each figure legend, the two numbers are the PCC and its p-value, respectively.33 (D) Among the set of 267 and 912 CAFA3 human targets for MF and BP, all were subjected to function prediction based on sequence and PPI by COFACTOR; only 88 and 227 targets, respectively, were predicted by the structure-based pipeline of COFACTOR.
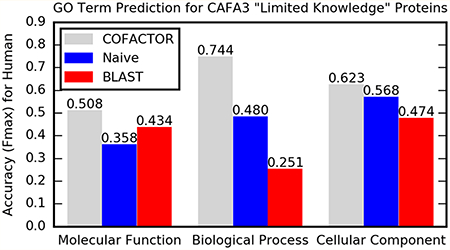
Our performance on the human subset of CAFA3 is shown in Figure 2. Since prediction models from other CAFA3 predictor teams are not publicly available, we compare our predictions obtained during CAFA3 challenge in 2017–02–02 with two baseline methods implemented by CAFA3 assessors: (l) the “BLAST” method searching a query against UniProt, where the prediction C-score of GO term q equals the sequence identity at the BLAST-aligned region between query and the top BLAST hit annotated with q; and (2) the “Naive” method, equivalent to the background distribution of GO terms: for any target, “Naive” predicts every GO term, where the C-score of GO term q equals the number of UniProt proteins experimentally annotated with q divided by the total number of experimentally annotated UniProt proteins.
Figure 2.

Fmax for (A, B) MF, (C, D) BP, and (E, F) CC GO term prediction by COFACTOR (Zhang-Freddolino lab) and two baseline methods, “Naive” and “BLAST” for (A, C, E) “No Knowledge” and (B, D, F) “Limited Knowledge” targets. Fmax calculations exclude GO terms annotated before 2017–02–02. (See Supplementary Text S1 for how to retrieve these data.)
As shown in Figure 2, COFACTOR prediction consistently outperformed the baseline methods in CAFA3 for all assessment categories. The advantage is particularly evident for BP, where our Fmax was 54% and 55% higher than the best performing baseline methods for “No Knowledge” and “Limited Knowledge” types. Moreover, COFACTOR outperforms “Naïve” on CC by 15% and 10% for “No Knowledge” and “Limited Knowledge” targets. No computational method outperformed “Naive” in CAFA2.9 Even though our predictions ultimately derive function annotation from UniProt annotated GO terms similar to the “Naïve” and “BLAST” baseline methods, COFACTOR more effectively identifies functional templates and combines their GO annotations, instead of relying on simple sequence similarity search (“BLAST”) or accepting background distribution of GO terms (“Naïve”).
To understand better why the functions of some targets are easier to predict than other targets, we computed the Pearson’s correlation coefficient (PCC) between various features of the query protein in the target set and the Fmax of GO term prediction accuracy for MF and BP (Figure 3) based on the supplementary data accompanying the CAFA330 report (https://figshare.com/articles/Supplementary_data/8135393, see Supplementary Text S1 for how to retrieve the data). For a meta-server (such as COFACTOR) that combines multiple features (identities of multiple sequence homologues, STRING scores of PPI partners, and similarities of structure templates) to derive a consensus prediction, it is often impossible for the consensus prediction to be dependent only on one feature. Nevertheless, it is still possible to identify whether the quality of a feature affects the accuracy of final prediction in a statistically significant manner. For example, while we did not observe significant dependence of Fmax on query sequence length (Figure 3A), Fmax of the sequence-, PPI-, and structure-based component methods of COFACTOR is significantly dependent upon the availability of templates or interaction partners, as quantified by their sequence identity (Figure 3B), STRING score (Figure 3C), and TM-score (Figure 3D), especially when the template score is modest (sequence identity <0.5, STRING score <0.7, or TM-score <0.6). However, for all three methods, the correlation coefficient between Fmax and the score of first template is not high (|PCC| ≤ 0.3), partly because each of the three component methods is a consensus approach to simultaneously consider all template hits (eq 2) so that the prediction result for a GO term will not be completely biased by a single high scoring template.
In short, our analysis indicates that, while COFACTOR is a good function predictor that goes far beyond simple sequence homology searching, it still has the intrinsic limitation of a template-based predictor, where target proteins with less reliable templates are more difficult to model.
Evaluation of Free-Text and GO Term Prediction Using Newly Annotated uPE1 Proteins in neXtProt 2019–01–11
While CAFA3 provides a large blindly tested set for relatively robust statistical analysis of our GO term prediction performance, we could not evaluate the performance of free-text function as it was neither required nor feasible given the very large set of targets in the CAFA3 challenge. To complement the CAFA3 evaluation and contribute to the C-HPP uPE1 CP50 Challenge,34 we assessed the I-TASSER/COFACTOR pipeline on a narrowly focused blindly tested set of 25 previously unannotated proteins with new function annotation in the neXtProt 2019–01–11 release. The detailed findings are presented in Supplementary Table S1, while a simplified table is presented in Table 1, with the proteins listed in descending order of Fmax values.
Table 1.
Comparison of I-TASSER/COFACTOR Function Annotation and UniProt/neXtProt Curation for 25 uPE1 with Newly Provided Function Annotation in neXtProt release 2019–01–11a
| # | accession, gene | * | our annotation | Fmax MF/BP | excerpt of UniProt/neXtProt annotation |
|---|---|---|---|---|---|
| 1 | Q96M27–1, PRRC1 | * | Protein kinase A regulation | 1.00, 0.88 | Activation of protein kinase A activity. Protein kinase A regulatory subunit binding. |
| 2 | P0C870–1, JMJD7 | * | Histone demethylation | 0.55, 0.90 | Endopeptidase cleaving histones N-terminal tails at the carboxyl side of methylated arginine or lysine residues. Fe2+ and 2-oxoglutarate-dependent monoxygenase. |
| 3 | Q7Z5A7–1, FAM19A5 | * | Regulation of microglial cell activation | 0.67, 0.50 | Stimulates chemotactic migration of macrophages. Blocks osteoclast formation from macrophages. Negatively regulating vascular smooth muscle cell (VSMC) proliferation and migration. Inhibits injury-induced cell proliferation and neointima formation in the femoral arteries |
| 4 | Q5T0D9–1, TPRG1L | + | Phosphatidylinositol-4-phosphate phosphatase | NA, 0.55 | Regulates synaptic release probability by decreasing the calcium sensitivity of release. |
| 5 | Q96D15–1, RCN3 | Catalytic activity, acting on a protein | NA, 0.47 | Molecular chaperone assisting protein biosynthesis and transport in endoplasmic reticulum. Pulmonary surfactant homeostasis. Anti-fibrotic activity by negatively regulating the secretion of collagens. | |
| 6 | Q8WTR8–1, NTN5 | * | Anatomical structure morphogenesis | NA, 0.44 | Neurogenesis. Prevents motor neuron cell body migration out of the neural tube. |
| 7 | Q9C0D6–1, FHDC1 | * | Binding of cytoskeleton | 0.44, 0.33 | Microtubule-associated formin. Regulates actin and microtubule dynamics. Induces microtubule acetylation and stabilization and actin stress fiber formation. Regulates Golgi ribbon formation. Required for normal cilia assembly. |
| 8 | O75363–1, BCAS1 | (for CC: neuron part) | NA, 0.40 | Myelination. | |
| 9 | P60827–1, C1QTNF8 | * | Signaling receptor binding | NA, 0.40 | Relaxin receptor RXFP1 binding. |
| 10 | Q8IUY3–1, GRAMD2A | Binding of GTPase from Ras superfamily | 0.11, 0.38 | Organization of endoplasmic reticulum-plasma membrane contact sites. STIM1 recruitment and calcium homeostasis. | |
| 11 | Q9BZH6–1, WDR11 | NA, 0.35 | Involved in Hedgehog (Hh) signaling pathway. Essential for normal ciliogenesis. | ||
| 12 | Q6ZNE9–2, RUFY4 | Regulation of protein folding | 0.26, 0.32 | Positively regulates macroautophagy in primary dendritic cells. Increases autophagic flux by stimulating autophagosome formation and facilitating tethering with lysosomes. Binds to phosphatidylinositol 3-phosphate (PtdIns3P). | |
| 13 | Q9GZU8–1, FAM192A | Hydrolase of protein | NA, 0.32 | Promotes the association of the proteasome activator complex subunit PSME3 with the 20S proteasome and regulates its activity. Inhibits PSME3-mediated degradation of proteasome substrates. | |
| 14 | Q494U1–1, PLEKHN1 | Transmembrane transport of nucleotide | 0.05, 0.29 | Controls the stability of the leptin mRNA harboring an AU-rich element (ARE) in its 3′ UTR. | |
| 15 | Q8IUW5–1, RELL1 | * | Regulation of apoptosis through TNF | NA, 0.29 | Induces activation of MAPK14/p38 cascade. |
| 16 | Q8NDM7–1, CFAP43 | NA, 0.29 | Flagellar protein involved in sperm flagellum axoneme organization and function. | ||
| 17 | Q8TDG2–1, ACTRT1 | Regulation of chromosome organization either though histone acetylation or binding of cytoskelton used in chromsome segregation | 0.03, 0.29 | Negatively regulates the Hedgehog (SHH) signaling. Binds to the promoter of the SHH signaling mediator, GLI1, and inhibits its expression. | |
| 18 | O75677–1, RFPL1 | * | Ubiquitin-protein transferase activity | NA, 0.27 | Negatively regulates the G2-M phase transition, by promoting cyclin B1/CCNB1 and CDK1 proteasomal ubiquitin-dependent degradation. |
| 19 | Q5VTQ0–1, TTC39B | * | Protein ubiquitination regulation | NA, 0.26 | Regulates high density lipoprotein (HDL) cholesterol metabolism by promoting the ubiquitination and degradation of the oxysterols receptors. |
| 20 | Q96S16–1, JMJD8 | Histone demethylation | NA, 0.21 | Positive regulator of TNF-induced NF-κB signaling. Regulates angiogenesis and cellular metabolism. | |
| 21 | Q9H9L7–1, AKIRIN1 | By binding to RNA polymerase, regulate expression of genes such as cytokines | NA, 0.18 | Signal transducer for MSTN during skeletal muscle regeneration and myogenesis. Regulates chemotaxis of macrophages and myoblasts by reorganising actin cytoskeleton, leading to more efficient lamellipodia formation via a PI3 kinase dependent pathway. | |
| 22 | Q96KV7–1, WDR90 | Regulation of transcription by nucleic acid binding | NA, 0.17 | Required for efficient primary cilium formation. | |
| 23 | Q6AI39–1, BICRAL | Sodium:potassium ion transporter | NA, NA | Enzyme component of SWI/SNF chromatin remodeling subcomplex GBAF, changing chromatin structure by altering DNA-histone contacts in an ATP-dependent manner. | |
| 24 | Q96J88–1, EPSTI1 | Cytoskeleton binding | NA, NA | M1 macrophage polarization. Regulation of gene expression during macrophage differentiation. RELA/p65 and STAT1 phosphorylation and nuclear localization upon activation of macrophages. | |
| 25 | Q9BZD6–1, PRRG4 | Serine-type endopeptidase | NA, NA | Axon guidance across the CNS. Prevents the delivery of ROBO1 at the cell surface and downregulates its expression. |
Full detail of this table is available as Supplementary Table S1.
Asterisk (*) marks a target if our free-text annotation matches neXtProt free text annotation.
Plus (+) marks a target whose Fmax for either MF or BP is >0.5 but the free-text annotation does not match. Fmax for MF/BP quantitatively measures the consistency between COFACTOR predicted GO terms and neXtProt curated GO terms. NA, or not applicable, means neXtProt did not assign GO term for a target. The table is ranked in descending order of Fmax.
In the last column, phrases at top are free-text annotations, followed by MF and BP GO terms. Red shades indicate free-text phrases consistent between I-TASSER/COFACTOR prediction and neXtProt annotation.
Among the 25 targets in this time-elapsed blindly tested set, 3 have I-TASSER models that are predicted to have correct structure topology (estimated TM-score >0.5: #2, 17, 18 in Supplementary Table S1), while another 10 are predicted to have approximately correct fold (estimated TM-score in the range [0.4,0.5]: #1, 4, 5, 8, 9, 10, 12, 14, 20, 24 in Supplementary Table S1).
Among the 25 targets, we did not assign free-text function annotation for 3 (O75363–1, Q8NDM7–1, and Q9BZH6–1; #8, 16, and 11, respectively, in Table 1) because the GO terms we predicted for these targets are too general to infer the function. For the remaining 22 targets, our manual free-text function interpretations are consistent with neXtProt annotation for 9 of them, as marked by asterisks (*) in Table 1 (#1, 2, 3, 6, 7, 9, 15, 18, 19). Meanwhile, of the 8 and 22 targets with UniProt/neXtProt curated MF or BP GO terms, 3 (#1, 2, 3) for MF and 4 (#1, 2, 3, 4) for BP have Fmax ≥0.5 for our GO term prediction. That makes a total of 4 different targets of the 25 with good matches for GO terms, only one (#4) of which is in addition to the 9 above with good matches for free-text, making a total of 10 that have good matches by either free-text or GO terms or both.
Of course, these newly annotated proteins represent the 7% of PE1 proteins that have resisted functional annotation. Thus, the overall low Fmax of agreement between GO term predictions and literature curation (0.19 and 0.23 for MF and BP, respectively, for the 8 and 22 proteins) is partly attributable to incompleteness in GO term annotation. In fact, our BP prediction accuracy is >15% higher than three state-of-the-art GO term prediction programs—GoFDR,5 GOtcha,6 and DeepGOplus35 (Table 2). In many scenarios, both our method and UniProt/neXtProt curation may only capture some of the many functions a protein performs. For example, target RFPL1 (O75677–1, #18 in Table 1) regulates the cell cycle by promoting ubiquitin-dependent protein degradation according to UniProt/neXtProt. While our MF prediction inferred the ubiquitin-dependent protein degradation function, our BP term prediction did not correctly predict the cell cycle regulation function, resulting in a low Fmax of 0.27 for BP GO terms despite partially consistent function annotation.
Table 2.
Comparison of GO Terms Prediction Accuracy (Fmax) between I-TASSER/COFACTOR, Our Function Annotation by I-TASSER/COFACTOR, and State-of-the-Art Methods for 8 and 22 neXtProt Proteins with Newly Annotated MF and BP GO Termsa
| program | Fmax for MF of 8 proteins | Fmax for BP of 22 proteins |
|---|---|---|
| I-TASSER/COFACTOR | 0.19 | 0.23 |
| GoFDR | 0.28 | 0.20 |
| GOtcha | 0.20 | 0.11 |
| DeepGOplus | 0.17 | 0.16 |
Bold font indicates the most accurate algorithm in each aspect for this data set. While COFACTOR is on average more accurate than both GoFDR5 and GOtcha6 for all three aspects of GO terms as shown in large-scale benchmark studies,1,3 its MF prediction accuracy (Fmax) is lower than GoFDR and GOtcha for this set of targets in MF prediction, probably due to the very small data set size of only 8 proteins. GoFDR infers functions from sequence homologues identified based on similarity of functional discriminating residues. GOtcha predicts GO terms from homologues detected by BLAST search followed by species-specific confidence score normalization. DeepGOplus implements deep neural networks to predict GO terms from sequence itself.35
Such incompleteness of GO term annotation is not uncommon for UniProt/neXtProt literature curation. C1QTNF8 (P60827–1, #9 in Table 1) binds the G proteincoupled receptor RXFP1 (MF) to regulate cell motility (BP). Swiss-Prot curators annotated the protein with the free text “May play a role as ligand of RXFP1” to convey its molecular function without a GO term; the GO consortium annotated GO:2000147 “positive regulation of cell motility” for BP based on the same experimental paper.36 This causes the lack of an appropriate MF GO term for this protein such as GO:0001664 “G protein-coupled receptor binding” or GO:0005102 “signaling receptor binding”. Consequently, even though COFACTOR indeed predicts GO:0005102, we cannot calculate Fmax for MF and have a modest Fmax = 0.40 for BP, despite our consistent free-text interpretation “signaling receptor binding”. While the incompleteness of function curation partly accounts for the low Fmax on this data set, our earlier benchmark performed last year on 100 chromosome 17 PE1 proteins resulted in much higher Fmax of 0.69 and 0.57 for MF and BP, respectively,1 as that benchmark data set included only deeply annotated targets with at least 3 Gold GO terms for each of the three GO terms aspects (MF, BP, and CC). Partly due to incompleteness of GO term annotation in the small data set reported in this study, Fmax of COFACTOR GO term prediction does not have apparent correlation with features of targets such as template availability (Supplementary Figure S2).
Among the 25 proteins in this time-elapsed neXtProt blindly tested set, we highlight three representative function predictions. As the first example, we discuss P0C870–1 (JMJD7, #2 in Table 1), a recently characterized endopeptidase and monooxygenase37,38 to illustrate the importance of structure template alignment and local sequence homologue hits in function prediction. As an endopeptidase, JMJD7 cuts histones at methylated arginine residues (GO:0035064 “methylated histone binding”, GO:0004177 “aminopeptidase activity”, GO:0004175 “endopeptidase activity” for MF); as a Fe2+ and 2-oxoglutarate-dependent monooxygenase, JMJD7 catalyzes hydroxylation of DRG1 and DRG2 translation factors (GO:0016706 “2-oxoglutarate-dependent dioxygenase activity”, GO:0004497 “monooxygenase activity” for MF, GO:0018126 “protein hydroxylation” for BP). The I-TASSER structure model of JMJD7 displays a typical “Jelly roll” fold and shares a high TM-score39 of 0.98 for both of its recently solved structures (PDB IDs 5nfn Chain A and 5nfo Chain A, Supplementary Figure S3A), even though neither of the two experimental structures was used in the I-TASSER modeling or function prediction. The structure of JMJD7 is similar to two human oxidoreductases: PDB IDs 3al5 Chain B (TM-score = 0.69, Supplementary Figure S3B), and PDB ID 4b7e Chain A (TM-score = 0.70, Supplementary Figure S3C), which are tRNA hydroxylase and hypoxia-inducible factor-asparagine dioxygenase, respectively. Despite the matching of these two structure analogs and the correct prediction of GO:0016706 “2-oxoglutarate-dependent dioxygenase activity” at C-score = 0.32 by COFACTOR structure-based method, the I-TASSER model also shares high structure similarity to many other proteins that perform other unrelated functions such as GO:0070492 “oligosaccharide binding” and GO:0005215 “transporter activity”, both at C-score = 0.53 by structure-based method, partly because the Jelly roll fold is a common topology in a wide variety of proteins. In the sequence-based method, the closest oxidoreductase hit is Lysine-specific demethylase 8 (UniProt ID B2GUS6), with only 27% sequence identity at the aligned region. Despite the low sequence identity with the top hit, 56% of the BLAST and PSI-BLAST hits are annotated with oxidoreductase activity (GO:0016706), resulting in highly confident prediction of this term at C-score = 0.62 for sequence-based method and C-score = 0.76 for the final consensus COFACTOR prediction. Although our predicted GO terms for both MF and BP overlap very well with neXtProt annotation (Fmax = 0.55 and 0.90, respectively), our blinded manual free-text interpretation process chose the term “histone demethylase activity” (GO:0032452, C-score = 0.63) to derive the free-text function annotation “histone demethylation”, which is not fully consistent with UniProt/neXtProt annotation, even though it correctly indicates the oxidoreductase activity of JMJD7 on methylated histones. This reflects the difficulty of manual interpretation of the function given only partially correct GO term predictions. Nevertheless, in Table 1, we designated this protein (#2) as partially matching the free-text annotations from the curators.
Q5VTQ0–1 (TTC39B, #19 in Table 1) regulates high density lipoprotein (HDL) cholesterol metabolism by promoting the ubiquitination and degradation of the oxysterol receptors LXR (NR1H2 and NR1H3).40 I-TASSER/COFACTOR correctly predicts its protein ubiquitination regulation (but unfortunately not the cholesterol metabolism regulation function, resulting in low Fmax of 0.26 for MF). For this target, the prediction of protein ubiquitination regulation (GO:0006508 “proteolysis”, C-score = 0.52; GO:0016567 “protein ubiquitination”, C-score = 0.50) is mainly due to its structure similarity to anaphase-promoting complex subunits (Apc/C, Supplementary Figure S4). This protein also has an asterisk for free-text annotation matching curators.
Q8IUW5–1 (RELL1, #15 in Table 1) is a receptor of tumor necrosis factor (TNF) and induces activation ofMAPK14/p38 cascade and apoptosis.41,42 Our prediction correctly describes regulation of apoptosis through tumor necrosis factor (TNF) but does not include the MAPK14/p38 cascade regulation. On the other hand, neXtProt BP GO term annotation only includes “positive regulation of p38MAPK cascade” (GO:1900745) but does not include the TNF-mediated apoptosis, resulting in low Fmax = 0.29 for BP prediction. The prediction of TNF-mediated apoptosis regulation (GO:0097190 “apoptotic signaling pathway”, C-score = 0.51, for BP and GO:0005031 “tumor necrosis factor-activated receptor activity”, C-score = 0.40, for MF) is not due to one single highly significant hit but due to multiple consensus (PSI-)BLAST hits with consistent GO term annotations. The closest sequence homologue is TNF receptor superfamily member 19L (UniProt ID Q969Z4), which shares 30% sequence identity with the query. This protein ends up with a match for free-text but not for GO terms.
CONCLUSIONS
For this report, we performed two sets of rigorous time-elapsed blind tests of our previously reported I-TASSER/COFACTOR pipeline, using results from the CAFA3 Challenge and from 25 neXProt newly annotated PE1 proteins. On the very recently published results of blind testing hundreds of CAFA3 human targets,30 a partial implementation of COFACTOR consistently achieved higher Fmax than the best baseline methods. This result shows that COFACTOR goes far beyond simple sequence homology search.
Meanwhile, being template- and PPI-based, the performance of COFACTOR is dependent on the availability of high scoring template and PPI partners. Without such templates and PPI partners appropriately identified, the accuracy of the function prediction will be low. To address this issue, we are working on the development of new approaches based on machine learning such as deep neural networks (NNs).35,43,44 NNs are shown to be able to extract features solely from query protein sequences without homologous template information when trained with known sequences in the functional databases and exploit the implicit relation between sequence and function through NN training. These approaches will be integrated with the current I-TASSER/COFACTOR pipeline to increase its ability and accuracy for modeling the non- and distant-homologous protein targets.
For the much smaller set of 25 neXtProt targets that acquired new function annotation in neXtProt release 2019–01 and could be used for the assessment, our free-text and specific GO term function annotation derived solely from automated I-TASSER/COFACTOR pipeline predicted good matches with the curators for 10 of them. This result is remarkable, given the variety of GO terms and limited depth of GO terms in the UniProt/neXtProt cases and the lack of structural information for most of these proteins.
Because free-text annotations are not readily amenable for computational parsing, they cannot be utilized directly by template-based function prediction algorithms such as COFACTOR. Thus, text-mining tools need to be developed to automate the conversion of free-text annotations into GO term annotations and vice versa, as we have initiated with GPCR proteins.45
The C-HPP CP50 initiative on functional annotation of 1254 remaining uPE1 proteins is a very large task. The utilization of such an automated pipeline, even for clues and insights to organize experimental studies, seems quite promising, as was demonstrated in CAFA3 for model organisms.30,46 In one such study,46 protein function predictions helped biologists to narrow down candidate genes potentially associated with long-term memory in Drosophila from approximately 14 thousand genes in the whole genome to 29 genes for experimental validation, of which 12 (41%) were eventually confirmed to be involved in long-term memory. Although more than half of the function predictions are incorrect, these predicted models managed to convert an experimental project from infeasible to fruitful. To assist investigators, we make available the algorithms and pipeline at the Web servers https://zhanglab.ccmb.med.umich.edu/I-TASSER/ and https://zhanglab.ccmb.med.umich.edu/COFACTOR/. neXtProt has already introduced links to precomputed and annually updated I-TASSER/COFACTOR predictions for proteins lacking function annotation as illustrated for JMJD7 (NX_P0C870) at https://www.nextprot.org/entry/NX_P0C870/gh/zhanglabs/COFACTOR; “NX_P0C870” can be replaced by the neXtProt ID to obtain function prediction for any uPE1 target of interest to the neXtProt user.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Xiaoqiong Wei for involvement in our earlier benchmark study1 and Dr. Peter L. Freddolino for collaboration during the CAFA3 challenge.31 This work used the Extreme Science and Engineering Discovery Environment (XSEDE),47 which is supported by the National Science Foundation (ACI-1548562). This work was supported in part by the National Institutes of Health (AI134678, GM083107, and GM116960 to Y.Z.; P30ES017885 and U24CA210967 to G.S.O.), the National Science Foundation (DBI1564756 to Y.Z.), and SIB Swiss Institute of Bioinformatics and University of Geneva (to L.L.).
ABBREVIATIONS
- uPE1
unknown function with PE1 evidence
- GO
Gene Ontology
- MF
molecular function
- BP
biological process
- CC
cellular component
- CAFA
critical assessment of function annotation
- CASP
critical assessment of protein structure prediction
- PDB
Protein Data Bank
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.9b00537.
Retrieval of Fmax data; graphic explanation of Fmax; Fmax versus features of target protein; I-TASSER models; ; comparison of our function annotation by I-TASSER/COFACTOR and by UniProt/neXtProt(PDF)
REFERENCES
- (1).Zhang C; Wei X; Omenn GS; Zhang Y Structure and Protein Interaction-based Gene Ontology Annotations Reveal Likely Functions of Uncharacterized Proteins on Human Chromosome 17. J. Proteome Res 2018, 17 (12), 4186–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zheng W; Zhang C; Bell EW; Zhang Y I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener Comput. Syst 2019, 99, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang C; Freddolino PL; Zhang Y COFACTOR: improved protein function prediction by combining structure, sequence and protein—protein interaction information. Nucleic Acids Res. 2017, 45 (W1), W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; et al. Gene Ontology: tool for the unification of biology. Nat. Genet 2000, 25 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gong QT; Ning W; Tian WD GoFDR: A sequence alignment based method for predicting protein functions. Methods 2016, 93, 3–14. [DOI] [PubMed] [Google Scholar]
- (6).Martin DMA; Berriman M; Barton GJ GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinf. 2004, 5 (1), 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang C; Omenn GS; Zhang Y Structure and Protein Interaction-based Gene Ontology Annotations Reveal Likely Functions of Uncharacterized Proteins on Human Chromosome 17; HUPO World Congress: Orlando, FL, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Moult J; Fidelis K; Kryshtafovych A; Schwede T; Tramontano A Critical assessment of methods of protein structure prediction (CASP)—Round XII. Proteins: Struct., Funct., Genet 2018, 86 (S1), 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jiang Y; Oron TR; Clark WT; Bankapur AR; D’Andrea D; Lepore R; Funk CS; Kahanda I; Verspoor KM; Ben-Hur A; et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016, 17 (1), 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kahanda I; Funk CS; Ullah F; Verspoor KM; Ben-Hur A A close look at protein function prediction evaluation protocols. GigaScience 2015, 4 (1), 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schnoes AM; Ream DC; Thorman AW; Babbitt PC; Friedberg I Biases in the Experimental Annotations of Protein Function and Their Effect on Our Understanding of Protein Function Space. PLoS Comput. Biol 2013, 9 (5), e1003063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Beasley L; Manda P Comparison of Natural Language Processing Tools for Automatic Gene Ontology Annotation of Scientific Literature; International Conference on Biomedical Ontology (ICBO): Corvallis, OR, 2018. [Google Scholar]
- (13).Gaudet P; Michel P-A; Zahn-Zabal M; Britan A; Cusin I; Domagalski M; Duek PD; Gateau A; Gleizes A; Hinard V; et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017, 45 (D1), D177–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Duek P; Gateau A; Bairoch A; Lane L Exploring the Uncharacterized Human Proteome Using neXtProt. J. Proteome Res 2018, 17 (12), 4211–4226. [DOI] [PubMed] [Google Scholar]
- (15).Paik YK; Overall CM; Corrales F; Deutsch EW; Lane L; Omenn GS Toward Completion of the Human Proteome Parts List: Progress Uncovering Proteins That Are Missing or Have Unknown Function and Developing Analytical Methods. J. Proteome Res 2018, 17 (12), 4023–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).neXtProt. New release with variant frequency data, new protein digestion tool and more; neXtProt, 2019. https://www.nextprot.org/news/new-release-with-variant-frequency-data-new-protein-digestion-tool-and-more (retrieved September 4, 2019).
- (17).Wu S; Zhang Y LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35 (10), 3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang Y; Skolnick J SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem 2004, 25 (6), 865–871. [DOI] [PubMed] [Google Scholar]
- (19).Zhang J; Liang Y; Zhang Y Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 2011, 19 (12), 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008, 9 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang Y; Skolnick J TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research 2005, 33 (7), 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang JY; Roy A; Zhang Y BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2012, 41 (D1), D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang C; Zheng W; Freddolino PL; Zhang Y MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein-protein network mapping. J. Mol. Biol 2018, 430 (15), 2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Altschul SF; Madden TL; Schaffer AA; Zhang JH; Zhang Z; Miller W; Lipman DJ Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17), 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Szklarczyk D; Franceschini A; Wyder S; Forslund K; Heller D; Huerta-Cepas J; Simonovic M; Roth A; Santos A; Tsafou KP; Kuhn M; Bork P; Jensen LJ; von Mering C STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43 (D1), D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bateman A; Martin MJ; O’Donovan C; Magrane M; Alpi E; Antunes R; Bely B; Bingley M; Bonilla C; Britto R; Bursteinas B; Bye-A-Jee H; Cowley A; Da Silva A; De Giorgi M; Dogan T; Fazzini F; Castro LG; Figueira L; Garmiri P; Georghiou G; Gonzalez D; Hatton-Ellis E; Li WZ; Liu WD; Lopez R; Luo J; Lussi Y; MacDougall A; Nightingale A; Palka B; Pichler K; Poggioli D; Pundir S; Pureza L; Qi GY; Rosanoff S; Saidi R; Sawford T; Shypitsyna A; Speretta E; Turner E; Tyagi N; Volynkin V; Wardell T; Warner K; Watkins X; Zaru R; Zellner H; Xenarios I; Bougueleret L; Bridge A; Poux S; Redaschi N; Aimo L; Argoud-Puy G; Auchincloss A; Axelsen K; Bansal P; Baratin D; Blatter MC; Boeckmann B; Bolleman J; Boutet E; Breuza L; Casal-Casas C; de Castro E; Coudert E; Cuche B; Doche M; Dornevil D; Duvaud S; Estreicher A; Famiglietti L; Feuermann M; Gasteiger E; Gehant S; Gerritsen V; Gos A; Gruaz-Gumowski N; Hinz U; Hulo C; Jungo F; Keller G; Lara V; Lemercier P; Lieberherr D; Lombardot T; Martin X; Masson P; Morgat A; Neto T; Nouspikel N; Paesano S; Pedruzzi I; Pilbout S; Pozzato M; Pruess M; Rivoire C; Roechert B; Schneider M; Sigrist C; Sonesson K; Staehli S; Stutz A; Sundaram S; Tognolli M; Verbregue L; Veuthey AL; Wu CH; Arighi CN; Arminski L; Chen CM; Chen YX; Garavelli JS; Huang HZ; Laiho K; McGarvey P; Natale DA; Ross K; Vinayaka CR; Wang QH; Wang YQ; Yeh LS; Zhang J; Consortium, U. UniProt: the universal protein knowledge-base. Nucleic Acids Res. 2017, 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Brocchieri L; Karlin S Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33 (10), 3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dana JM; Gutmanas A; Tyagi N; Qi GY; O’Donovan C; Martin M; Velankar S SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019, 47 (D1), D482–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Laskowski RA; Jablonska J; Pravda L; Varekova RS; Thornton JM PDBsum: Structural summaries of PDB entries. Protein Sci 2018, 27 (1), 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou N; Jiang Y; Bergquist TR; Lee AJ; Kacsoh BZ; Crocker AW; Lewis KA; Georghiou G; Nguyen HN; Hamid MN; Davis L; Dogan T; Atalay V; Rifaioglu AS; Dalkiran A; Cetin-Atalay R; Zhang C; Hurto RL; Freddolino PL; Zhang Y; Bhat P; Supek F; Fernandez JM; Gemovic B; Perovic VR; Davidovic RS; Sumonja N; Veljkovic N; Asgari E; Mofrad MR; Profiti G; Savojardo C; Martelli PL; Casadio R; Boecker F; Kahanda I; Thurlby N; McHardy AC; Renaux A; Saidi R; Gough J; Freitas AA; Antczak M; Fabris F; Wass MN; Hou J; Cheng J; Hou J; Wang Z; Romero AE; Paccanaro A; Yang H; Goldberg T; Zhao C; Holm L; Toronen P; Medlar AJ; Zosa E; Borukhov I; Novikov I; Wilkins A; Lichtarge O; Chi P-H; Tseng W-C; Linial M; Rose PW; Dessimoz C; Vidulin V; Dzeroski S; Sillitoe I; Das S; Lees JG; Jones DT; Wan C; Cozzetto D; Fa R; Torres M; Vesztrocy AW; Rodriguez JM; Tress ML; Frasca M; Notaro M; Grossi G; Petrini A; Re M; Valentini G; Mesiti M; Roche DB; Reeb J; Ritchie DW; Aridhi S; Alborzi SZ; Devignes M-D; Emily Koo DC; Bonneau R; Gligorijevic V; Barot M; Fang H; Toppo S; Lavezzo E; Falda M; Berselli M; Tosatto SC; Carraro M; Piovesan D; Rehman HU; Mao Q; Zhang S; Vucetic S; Black GS; Jo D; Larsen DJ; Omdahl AR; Sagers LW; Suh E; Dayton JB; McGuffin LJ; Brackenridge DA; Babbitt PC; Yunes JM; Fontana P; Zhang F; Zhu S; You R; Zhang Z; Dai S; Yao S; Tian W; Cao R; Chandler C; Amezola M; Johnson D; Chang J-M; Liao W-H; Liu Y-W; Pascarelli S; Frank Y; Hoehndorf R; Kulmanov M; Boudellioua I; Politano G; Di Carlo S; Benso A; Hakala K; Ginter F; Mehryary F; Kaewphan S; Björne J; Moen H; Tolvanen MEE; Salakoski T; Kihara D; Jain A; Smuc T; Altenhoff A; Ben-Hur A; Rost B; Brenner SE; Orengo CA; Jeffery CJ; Bosco G; Hogan DA; Martin MJ; O’Donovan C; Mooney SD; Greene CS; Radivojac P; Friedberg I The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019, DOI: 10.1101/653105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang C; Freddolino PL; Zhang Y Protein Function Prediction by COFACTOR in CAFA3; Intelligent Systems for Molecular Biology (ISMB): Prague, Czech Republic, 2017. [Google Scholar]
- (32).Sali A; Blundell TL Comparative Protein Modeling by Satisfaction of Spatial Restraints. J. Mol. Biol 1993, 234 (3), 779–815. [DOI] [PubMed] [Google Scholar]
- (33).SciPy developers. scipy.stats.pearsonr; SciPy, 2019. https://docs.scipy.org/doc/scipy/reference/generated/scipy.stats.pearsonr.html (retrieved Aug 5, 2019).
- (34).Paik YK; Lane L; Kawamura T; Chen YJ; Cho JY; LaBaer J; Yoo JS; Domont G; Corrales F; Omenn GS; Archakov A; Encarnacion-Guevara S; Lui SQ; Salekdeh GH; Cho JY; Kim CY; Overall CM Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res. 2018, 17 (12), 4042–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kulmanov M; Hoehndorf R DeepGOPlus: improved protein function prediction from sequence. Bioinformatics 2019, btz595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Glogowska A; Kunanuvat U; Stetefeld J; Patel TR; Thanasupawat T; Krcek J; Weber E; Wong GW; Del Bigio MR; Hoang-Vu C; Hombach-Klonisch S; Klonisch T C1q-tumour necrosis factor-related protein 8 (CTRP8) is a novel interaction partner of relaxin receptor RXFP1 in human brain cancer cells. J. Pathol 2013, 231 (4), 466–479. [DOI] [PubMed] [Google Scholar]
- (37).Markolovic S; Zhuang QQ; Wilkins SE; Eaton CD; Abboud MI; Katz MJ; McNeil HE; Lesniak RK; Hall C; Struwe WB; Konietzny R; Davis S; Yang M; Ge W; Benesch JLP; Kessler BM; Ratcliffe PJ; Cockman ME; Fischer R; Wappner P; Chowdhury R; Coleman ML; Schofield CJ The Jumonji-C oxygenase JMJD7 catalyzes (3S)-lysyl hydroxylation of TRAFAC GTPases. Nat. Chem. Biol 2018, 14 (7), 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liu HL; Wang C; Lee S; Deng Y; Wither M; Oh S; Ning FK; Dege C; Zhang QQ; Liu XJ; Johnson AM; Zang JY; Chen ZZ; Janknecht R; Hansen K; Marrack P; Li CY; Kappler JW; Hagman J; Zhang GY Clipping of arginine-methylated histone tails by JMJD5 and JMJD7. Proc. Natl. Acad. Sci U. S. A 2017, 114 (37), E7717–E7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang Y; Skolnick J Scoring function for automated assessment of protein structure template quality. Proteins: Struct., Funct, Genet 2004, 57 (4), 702–710. [DOI] [PubMed] [Google Scholar]
- (40).Hsieh J; Koseki M; Molusky MM; Yakushiji E; Ichi I; Westerterp M; Iqbal J; Chan RB; Abramowicz S; Tascau L; Takiguchi S; Yamashita S; Welch CL; Di Paolo G; Hussain MM; Lefkowitch JH; Rader DJ; Tall AR TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature 2016, 535 (7611), 303–U282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Moua P; Checketts M; Xu LG; Shu HB; Reyland ME; Cusick JK RELT family members activate p38 and induce apoptosis by a mechanism distinct from TNFR1. Biochem. Biophys. Res. Commun 2017, 491 (1), 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cusick JK; Mustian A; Goldberg K; Reyland ME RELT induces cellular death in HEK 293 epithelial cells. Cell. Immunol 2010, 261 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cao RZ; Freitas C; Chan L; Sun M; Jiang HQ; Chen ZX ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22 (10), 1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fa R; Cozzetto D; Wan C; Jones DT Predicting human protein function with multi-task deep neural networks. PLoS One 2018, 13 (6), e0198216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wu JS; Yin Q; Zhang CX; Geng JJ; Wu HJ; Hu HF; Ke XY; Zhang Y Function Prediction for G Protein-Coupled Receptors through Text Mining and Induction Matrix Completion. Acs Omega 2019, 4 (2), 3045–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kacsoh BZ; Barton S; Jiang YX; Zhou NH; Mooney SD; Friedberg I; Radivojac P; Greene CS; Bosco G New Drosophila Long-Term Memory Genes Revealed by Assessing Computational Function Prediction Methods. G3: Genes, Genomes, Genet 2019, 9 (1), 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD; Roskies R; Scott JR; Wilkins-Diehr N XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng 2014, 16 (5), 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.