

Abstract
Hydrogen-atom transfer (HAT) from a substrate carbon to an iron(IV)-oxo (ferryl) intermediate initiates a diverse array of enzymatic transformations. For outcomes other than hydroxylation, coupling of the resultant carbon radical and hydroxo ligand (oxygen rebound) must generally be averted. A recent study of FtmOx1, a fungal iron(II)- and 2-(oxo)glutarate-dependent oxygenase that installs the endoperoxide of verruculogen by adding O2 between carbons 21 and 27 of fumitremorgin B, posited that tyrosine (Tyr or Y) 224 serves as HAT intermediary to separate the C21 radical (C21•) and Fe(III)–OH HAT products and prevent rebound. Our re-investigation of the FtmOx1 mechanism revealed, instead, direct HAT from C21 to the ferryl complex and surprisingly competitive rebound. The C21-hydroxylated (rebound) product, which undergoes deprenylation, predominates when low [O2] slows C21•–O2 coupling in the next step of the endoperoxidation pathway. This pathway culminates with addition of the C21-O–O• peroxyl adduct to olefinic C27 followed by HAT to the C26• from a Tyr. The last step results in sequential accumulation of Tyr radicals, which are suppressed without detriment to turnover by inclusion of the reductant, ascorbate. Replacement of each of four candidates for the proximal C26 H• donor (including Y224) with phenylalanine (F) revealed that only the Y68F variant (i) fails to accumulate the first Tyr• and (ii) makes an altered major product, identifying Y68 as the donor. The implied proximities of C21 to the iron cofactor and C26 to Y68 support a new structural model of the enzyme-substrate complex that is consistent with all available data.
Graphical Abstract
INTRODUCTION
Compounds possessing the cyclic dialkylperoxide (endoperoxide) functionality have important bioactivities.1 Endoperoxide lipids are precursors to prostaglandin hormones linked to the inflammatory response, and inhibition of the endoperoxide-installing enzymes (hereafter, termed endoperoxide synthases), cyclooxygenase (COX) 1 and COX 2,2,3 is the mechanism of action of important, over-the-counter, non-steroidal (NSAID) painkillers such as aspirin.4 Artemisinin, an endoperoxide-bearing sesquiterpene lactone produced by Artemisia annua, is used (as are its synthetic derivatives) to treat malaria,5–7 and other endoperoxide natural products show promise for treatment of cancer.1,8 Biocatalytic endoperoxidation could be exploited in large-scale production of drugs, as in recently reported semisynthetic routes to artemisinin.9 The most well-studied endoperoxide synthases are the aforementioned COX isozymes,3,10,11 but recent studies have identified examples of fungal, non-heme iron(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenases that install this rare bioactive functional group.12,13 Endoperoxide synthases and their mechanisms clearly warrant further study: neither is known for the case of artemisinin biosynthesis, for example.
The COX isoforms convert arachidonic acid (AA) to prostaglandin H2 (PGH2) via prostaglandin G2 (PGG2). These enzymes catalyze two distinct reactions that constitute the first steps in the prostaglandin biosynthetic pathway.3,10,11 The first reaction, a typical peroxidase-type transformation at the heme cofactor, reduces the hydroperoxide of PGG2 to the alcohol of PGH2 (Scheme S1A) and activates the enzyme for the second (endoperoxidation) reaction, which catalytically converts AA to PGG2 (Scheme S1B). Activation occurs by formation of a quasi-stable tyrosyl radical (Tyr•), which initiates endoperoxidation by abstracting hydrogen (H•) from C13 of arachidonic acid to produce a resonance-stabilized C11-C15 dienyl radical. Sequential couplings of two O2 molecules to carbon radicals on C11 and C15 are interrupted by C–O– and C–C–coupling steps involving addition of radicals (C11–O–O• and C8•) to olefins (at C9 and C12, respectively). The resultant C15 peroxyl radical (C15–O–O•) takes H• back from the tyrosine to regenerate the Tyr• for another turnover. In a fraction of events, a different H• donor quenches the peroxyl radical, leaving the enzyme in the Tyr•-free form, which must be re-activated for endoperoxidation by another round of the PGG2-peroxidase reaction.3,10 Features that distinguish this mechanism from those of most other C–H-activating heme enzymes include the initial hydrogen-atom transfer (HAT) from the substrate to the Tyr• intermediate and the regeneration of this oxidant at the end of the reaction, without intervention of the heme cofactor. These features make endoperoxidation self-sustaining (catalytic) except when the adventitious “quenching” of the Tyr• periodically intervenes.
The more recent identification of Fe/2OG endoperoxide synthases12 expanded the already impressive repertoire of reactions attributed to members of this versatile enzyme family.14 These enzymes share a largely conserved HX(D/E)XnH ligand motif located in close proximity to a substrate binding site, which typically presents to the iron cofactor an unactivated aliphatic C–H bond to be cleaved by HAT.15,16 Although the best-studied Fe/2OG oxygenases mediate hydroxylation, a range of other outcomes17–30 are known. In all Fe/2OG enzymes studied to date, addition of O2 to the 2OG-coordinated Fe(II) cofactor yields CO2 and a succinate-coordinated Fe(IV)–oxo (ferryl) intermediate.14,31 In most cases, HAT from a substrate carbon to the ferryl then produces a Fe(III)–OH/carbon-radical (C•) state that is a key branch point to the different outcomes.14 In the hydroxylation pathway, coupling of the C• with the Fe(III)-coordinated hydroxo, a step commonly referred to as oxygen rebound,32 yields the alcohol product and regenerates the Fe(II) state of the cofactor for the next turnover.33 In more recently elucidated non-hydroxylation outcomes, the rebound step, which is believed to have a low activation barrier,34 must be averted. In a few cases, specific adaptations to disfavor rebound have been identified and disabled to unleash the default hydroxylation outcome.18,22,26,35
The fungal Fe/2OG oxygenase, FtmOx1, installs an endoperoxide moiety between C21 and C27 of its substrate, fumitremorgin B (2), to produce verruculogen (3) on the biosynthetic pathway to fumitremorgin A (Scheme 1).12,36,37 FtmOx1 and the more recently discovered NvfI from the novofumigatonin biosynthetic pathway13 are the first two examples of Fe/2OG endoperoxide synthases. A recent study of FtmOx1 suggested that it avoids the generally facile rebound step by implementing a COX-like route to the endoperoxide (Scheme 2A).38 The study posited an active-site Tyr (Y224) serving as intermediary between the ferryl complex and C21 of 2 to form the C21 radical (C21•) in two sequential HAT steps. With rebound disfavored (at least in part) by the spatial separation of the radical and hydroxo ligand, the C21• could instead capture O2, a well-known reactivity of alkyl radicals.3,39 The resultant C21–O–O• peroxyl radical would then add to olefinic C27, and the C26• would be resolved by HAT from Y224. Although the proposed role of Y224 in HAT, first to the cofactor and subsequently from, and back to, the substrate, has direct precedent in the COX mechanism, it would embody a new strategy for control of outcome among members of the versatile Fe/2OG oxygenase family. Moreover, the COX-like mechanism for FtmOx1 would potentially allow for catalytic turnover following decarboxylation of a single equivalent of 2OG, a phenomenon also not previously described for a member of the Fe/2OG oxygenase family.
Scheme 1.

Reactions Catalyzed by FtmOx1 in the Biosynthesis of Fumitremorgin A.
Scheme 2.

Possible Mechanisms of Installation of the Endoperoxide in Verruculogen by FtmOx1.
Yan et al. also considered, but apparently disfavored, a second mechanism for endoperoxide installation by FtmOx1, in which the ferryl complex would serve its more usual function of HAT acceptor directly from the substrate (from C21). In this alternative mechanism (Scheme 2B), O2 coupling to the C21•, addition of the resultant peroxyl radical to C27, and H• donation to C26 by a Tyr residue would produce verruculogen but leave the enzyme in a Fe(III)/Tyr• state incompetent for subsequent turnover. This mechanism has obvious analogy to that previously elucidated for the stereoinversion reaction catalyzed by carbapenem synthase (CarC). In this reaction – the first of two sequential steps catalyzed by CarC (the second being a C–C desaturation) in the biosynthetic pathway to carbapenem antibiotics – the ferryl complex abstracts H• from the bridgehead carbon (C5) of the bicyclic substrate, (3S,5S)-carbapenam-3-carboxylate, and Y165 then quenches the radical by HAT to the opposite face, producing the 3S,5R diastereomer.26 This outcome leaves the enzyme in an Fe(III)/Tyr165•-containing state, making the reaction stoichiometric rather than catalytic in the absence of an exogenous reductant (e.g., ascorbate) to regenerate the O2-reactive [Fe(II)/Tyr165] form. The mechanism for FtmOx1 disfavored by Yan, et al. would differ from the CarC mechanism only by the intervention of a pair of C–O-coupling steps between the two HAT steps. Because we judged certain observations from their study to be potentially more consistent with this CarC-like mechanism than with the favored COX-like mechanism, we re-examined the structure and mechanism of FtmOx1.
Six key observations from this re-examination strongly favor the CarC-like mechanism. First, chromatographic and mass-spectrometric (GC/LC-MS) product analyses of reactions with varying [16O2] or in the presence of 18O2 showed that oxygen rebound does, in fact, compete with bimolecular trapping of O2 by the C21•. The surprising facility of the rebound step implies close proximity of C21 to the cofactor and direct HAT to the ferryl complex. Second, transient absorption spectra (similar to those reported in the earlier study) suggested accumulation of two tyrosyl radicals in sequence, and the results of freeze-quench EPR experiments with FtmOx1 containing β-deuterium-labeled Tyr (3,3-d2-Tyr) established that the absorbing species are indeed radicals derived from Tyr. Third, inclusion of ascorbate enhanced conversion of 2 to 3 (just as CarC requires a reductant for multiple turnovers) but also suppressed the transient Tyr radicals, indicating that they form concomitantly with or after (but not before, as in the COX-like mechanism) the product, 3. Fourth, when we substituted (with Phe) any of the four Tyr residues that could potentially serve as H• donor or acceptor in the reaction, we found that none of the Tyr → Phe variants was completely incompetent to consume the substrate; all could make either 3 or an unidentified alternative product. By contrast, variants of COX and fatty acid α-oxygenase lacking the Tyr residues that abstract H• directly from their substrates were reportedly devoid of activity.40–42 Fifth, for the Y68F variant of FtmOx1, but not for any of the other Tyr → Phe variants (including Y224F), the rapidly developing, lower-energy absorption features attributed to the first of the sequentially forming Tyr radicals was eliminated, production of 3 was drastically diminished, and a new, unidentified product became predominant, implicating Y68 as the C26 H• donor. Analogously to the H•-donating Y165 in CarC, Y68 of FtmOx1 is positioned on a mobile, peripheral loop element. Finally, although we concluded from re-examination of the crystallographic data deposited by Yan, et al. that their structural model for the FtmOx1•Fe(II)•2OG•2 reactant complex is not reliable and were unable to solve an experimental structure ourselves, we generated from our structure of the FtmOx1•Fe(II)•2OG ternary complex a docking model of the complete reactant state, in which the mechanistically implied proximities of C21 to the cofactor and C26 to Y68 were readily accommodated. In sum, the new data and analysis weigh heavily against a COX-like mechanism and in favor of a CarC-like pathway for endoperoxide installation by FtmOx1.
EXPERIMENTAL PROCEDURES
Materials.
All reagents were obtained from commercial sources (see the Supporting Information) and used without additional purification. Verruculogen was purchased from Enzo Life Sciences (Farmingdale, NY). Preparation of expression vectors for the Tyr → Phe variant proteins is described in the Supporting Information. Procedures for the overexpression and purification of wild-type (wt) and variant FtmOx1 proteins are also provided in the Supporting Information. Products of the FtmOx1 reactions were analyzed by either liquid or gas chromatography with detection by mass spectrometry (LC/GC-MS). Details of the analytical methods are provided in the Supporting Information.
18O-tracer experiments to determine the mechanism of deprenylation.
All samples were prepared in an anoxic chamber (as described above) to final concentrations of 1.0 mM FtmOx1, 1.0 mM Fe(NH4)2(SO4)2, 5.0 mM 2OG, 0.50 mM 2, and 0.50 mM sodium ascorbate. The reaction was initiated by mixing a 0.20 mL aliquot of this solution with 0.20 mL buffer saturated with either 16O2 or 18O2. The reaction was allowed to proceed for 1 min prior to addition of excess NaBH4 to reduce the solvent-exchangeable aldehyde (prenal) product and trap the oxygen in the exchange-inert alcohol (prenol). The remaining substrate and products were extracted twice into 0.20 mL CHCl3, and the combined organic layers were dried under N2. The compounds were dissolved in ~ 15 μL of CH3CN for GC-MS detection of the prenol product. An aliquot of the same mixture was diluted further for parallel analysis of the fumitremorgin products by LC-MS. Control samples were prepared by adding or omitting NaBH4 to a standard of 3 purchased from Enzo Life Sciences (Farmingdale, NY). The control samples were treated identically to the 16O2 and 18O2 reactions. Details of the GC-MS and LC-MS methods are provided in the Supporting Information.
LC-MS analysis of partition between deprenylation and endoperoxidation with varying [O2].
Unless otherwise indicated, all samples were prepared in an MBraun (Peabody, MA) anoxic chamber with solutions that had been rendered anoxic prior to use. For the samples prepared under low [O2], the final concentrations were 2.0 mM FtmOx1, 2.0 mM Fe(NH4)2(SO4)2, 10 mM 2OG, 0.50 mM 2, and 1.0 mM sodium ascorbate. The mixture was prepared to a final volume of 0.20 mL and transferred to a 0.30 mL glass vial to minimize the headspace volume. The reaction solution was then removed from the anoxic chamber and opened to air for ~ 30 s before the vial was re-sealed. The reaction was allowed to proceed overnight. The remaining substrate and products were extracted twice into 0.20 mL CHCl3, and the combined organic layers were dried under N2. The compounds were dissolved in 0.10 mL of CH3CN prior to LC-MS analysis (see Supporting Information).
For samples prepared under high [O2], chlorite dismutase (Cld) was used to generate O2 in situ, as previously described.43 Each reaction was prepared to a total volume of 0.10 mL with final concentrations of 2.0 μM FtmOx1, 2.0 μM Fe(NH4)2(SO4)2, 5.0 mM 2OG, 0.50 mM 2, 1.0 mM ascorbate, 5.0 mM NaClO2, and 5.0 μM Cld. The reaction solution was constituted stepwise, by initial mixing of FtmOx1 and Fe(NH4)2(SO4)2. A solution of 2OG, 2, ascorbate, and Cld was prepared in a separate tube. The separate enzyme and substrate solutions were removed from the anoxic chamber. The Cld reaction was initiated by addition of NaClO2 to the substrate solution, and the preformed FtmOx1•Fe(II) complex was then immediately added. The reaction was allowed to proceed overnight, and the workup was performed as described above. Both low- and high-[O2] samples were prepared in triplicate.
Analysis of the previously published FtmOx1 structure with 2 modeled in the active site.
The coordinates of the structural model for FtmOx1 with Fe(II) and 2 bound (PDB accession code 4ZON) were downloaded directly from the RCSB PDB and used to generate Figure S1 in the PyMOL molecular graphics software package (Schrödinger, LLC). A 2Fo-Fc electron density map was generated using FFT44 from the deposited structure factors without further refinement. An Fo-Fc omit map was calculated by deletion of 2 from the 4ZON PDB file and subsequent refinement in Refmac545 against the deposited structure factors. Relevant statistics concerning the geometry of 2 modeled in the 4ZON structure and the fit of 2 into the observed electron density from x-ray diffraction experiments were taken directly from the PDB validation report on RCSB PDB (https://www.rcsb.org/structure/4ZON) and are reported in Table S1.
X-ray crystallography of FtmOx1.
We solved more than 30 structures of wt FtmOx1 and its Y140F variant in attempts to obtain a structure of the enzyme with a fully occupied active site. Our efforts included incubating 2 with FtmOx1 before crystallization (co-crystallization), soaking pre-formed crystals of the FtmOx1•Fe(II)•2OG complex with varying concentrations of 2 over different time intervals (30 min to 24 h), and soaking 2 into crystals obtained with the substrate also present during crystallization. The two structures of highest resolution are described here. The electron-density maps calculated from these datasets imply that 2 was still not bound at high occupancy, but we present them here for the purposes of comparison to the published structure (4ZON). In all cases, the Y140F variant diffracted to higher resolution. Both structures reported here are of this variant protein, but we obtained similar results when we applied the same treatment to the wt protein. One dataset originates from a sample that retained the N-terminal His6-tag, and the other was collected on crystals of protein that was expressed as a His6-SUMO fusion and subsequently processed with TEV protease to remove the SUMO domain and tag. Samples of the His6-tagged Y140F variant used for x-ray crystallography were purified by Ni-NTA affinity chromatography and stripped of adventitiously bound metal ions by dialysis against 100 mM Tris-HCl, pH 7.5, containing 5 mM EDTA. The protein was further purified by size-exclusion chromatography (GE HiLoad 16/600 Superdex 200 PG in 20 mM sodium HEPES, pH 7.5, 200 mM NaCl) at a flow rate of 1 mL/min. Its elution volume was consistent with a dimer. The purified protein samples were exchanged into 20 mM sodium HEPES, pH 7.5, rendered anoxic by ~ 30 min of argon gas purge, and transferred into a Coy Laboratory Products anoxic chamber for crystallization trials.
To generate tagless FtmOx1-Y140F samples, the His6-SUMO Ni-NTA affinity-purified protein was diluted to 2 mg/mL in 20 mM sodium HEPES, 200 mM NaCl, 5% glycerol, pH 7.5, and cleavage of the His6 tag was initiated by addition of TEV protease to a final concentration of 0.03 mg/mL. The cleavage reaction was allowed to proceed at 4°C for 16–18 h before the sample was applied to a second Ni-NTA affinity column. The flow-through was collected and concentrated prior to purification by size-exclusion chromatography, as described above. The purified protein was buffer-exchanged and rendered anoxic as described above.
All protein samples were diluted to 12 mg/mL in 20 mM sodium HEPES, pH 7.5, and mixed with 1 molar equivalent (equiv) Fe(NH4)2(SO4)2 and 5 equiv 2OG. In co-crystallization trials with 2 (Quality Phytochemicals, East Brunswick, NJ), 1–2 equiv of 2 was added to the solution. The resulting solution was mixed with an equal volume of a well solution of 25% (w/v) PEG 4000, 0.3 M MgCl2, and 100 mM Tris-HCl, pH 8.5. This solution was allowed to come to equilibrium with 0.50 mL of the well solution by the sitting-drop vapor-diffusion method. Protein crystals appeared within a few days of initial setup and were allowed to grow for another 1–2 weeks. In substrate soaking trials, 2–3 μL of a solution containing 30% (v/v) DMSO, 25% (w/v) PEG 4000, 0.2 M MgCl2, 2 mM 2, and 100 mM Tris-HCl, pH 8.5 was added to the 2 μL protein drop and incubated for 0.5–8 h. Prior to data collection, crystals were mounted on rayon loops and soaked in well solution supplemented with 20–25% (v/v) ethylene glycol. Samples were flash frozen by direct plunge into liquid N2 and stored at 77 K.
X-ray diffraction datasets were collected at the Advanced Photon Source (LS-CAT beam line 21-ID-G and GM/CA-CAT beam line 23-ID-B) and processed using the software package HKL2000.46 All structures were solved by molecular replacement using the published crystal structure of FtmOx1 as a search model (PDB accession code 4ZON) in Phaser MR.47 Subsequent refinement and model building were performed in Refmac5 and COOT,48 respectively. Ramachandran statistics were compiled with Molprobity.49 A summary of the processing and refinement statistics is presented in Table S2.
Both proteins crystallized in the P212121 space group with two molecules in the asymmetric unit. In the His6-FtmOx1-Y140F structure, residues 6–290 and 4–292 could be modeled in chains A and B, respectively. The final model additionally contains 2 iron ions, 2 molecules of 2OG, and 48 water molecules. In the structure of untagged FtmOx1-Y140F, residues 6–290 could be modeled in both chains A and B, and the final model also contains 2 iron ions and 258 water molecules. Inspection of Fo-Fc difference maps after initial refinement did not yield any features above 3.0 σ that could be attributed to bound 2 or 3 (or any of its byproducts).
Stopped-flow absorption experiments.
Stopped-flow absorption (SF-abs) experiments were performed on an Applied Photophysics (Surrey, UK) SX1.8MV instrument at 5°C with a 1 cm path length, as previously described.50 The SF-abs unit was housed in an MBraun anoxic chamber, and spectral datasets were obtained using a photodiode array (PDA) detector. In experiments to detect the previously observed transient species absorbing at 416–420 nm and 410 nm,38 the stopped-flow instrument was used in a single-mix configuration. A solution of 0.65 mM FtmOx1 (wt or Y → F variant), 0.58 mM Fe(NH4)2(SO4)2, 0.58 mM 2, and 5.0 mM 2OG was mixed at 5°C with an equal volume of O2-saturated buffer. For SF-abs experiments with sodium ascorbate, 1.0 mM of the reductant was included in the enzyme syringe.
Freeze-quench electron paramagnetic resonance (EPR) experiments.
General procedures for freeze-quench (FQ) EPR experiments have been published previously.50 Experiments were carried out at 5°C by mixing a solution of 0.65 mM FtmOx1 (wt or Y → F variant), 0.58 mM Fe(NH4)2(SO4)2, 0.58 mM 2, and 5.0 mM 2OG with an equal volume of O2-saturated buffer and allowing the reaction to proceed for varying time before quenching by injection into 2-methylbutane at − 150°C. Reaction times were chosen so that samples would be highly enriched in one of the two sequentially forming radical species. For the fast-forming species with lower energy absorption (416–420 nm maximum absorbance), a time of 0.43 s was selected for most of the proteins; for the second species (410 nm maximum absorbance, see Supporting Information), a reaction time of 30 s was selected, unless otherwise indicated.
For the preparation of 3,3-d2-L-Tyr-labeled FtmOx1, the pET28a FtmOx1 over-expression plasmid was used to transform an Escherichia coli strain auxotrophic for tyrosine (CGSC Strain # 5974, WU-36, Genetic Stock Center at Yale University). Colonies were grown in M9 minimal medium supplemented with 3,3-d2-L-Tyr (Cambridge Isotopes Laboratories, Tewksbury, MA). The purified protein was analyzed by mass spectrometry to ensure incorporation of the labeled tyrosine (see Supporting Information for details). FQ EPR samples were prepared with the purified 3,3-d2-L-Tyr-containing FtmOx1 according to the procedure described above, with reaction times of 0.44 s and 30 s (with the Y140F variant) for the first and second EPR signal.
X-band (~ 9.5 GHz) EPR spectra were acquired on a Bruker ESP 300 spectrometer with an ER 041 MR microwave bridge and an ER 4116DM resonator. The temperature of the sample was maintained by a continuous flow of liquid helium through an ER 4112-HV Oxford Instruments cryostat.
Reactions of wt FtmOx1 and Y → F variants for product analysis.
All reaction samples were prepared in an anoxic chamber (as described above). The final volume of each sample was 0.10 mL. The reaction was initiated by dilution to the final volume with O2-saturated buffer, giving final concentrations of 10 μM FtmOx1 (wt or Y → F variant), 10 μM Fe(NH4)2(SO4)2, 1 mM 2OG, 0.50 mM 2, and 1 mM ascorbate. The reaction was allowed to proceed for ~ 1 h at ambient temperature, open to the air with gentle stirring. The reaction solution was extracted twice with CHCl3. The combined organic layer was dried under N2 and dissolved in 50 μL of CH3CN for analysis by LC-MS (see the Supporting Information for details).
Ligand docking and enzyme-substrate model construction.
To generate a starting conformation of 2 that is compatible with the FtmOx1 reaction, we began with a structure produced by AceDRG51 from the isomeric SMILES string of 3 (PubChem CID: 13887805). This structure was optimized by steepest-descent minimization with the UFF forcefield and standard parameters in Avogadro.52,53 The minimized product structure was manually edited in Avogadro to convert it to 2 by deleting the peroxide oxygens and adding and deleting hydrogens and bonds as required; the resulting model was minimized again. All atoms were renamed to match the convention in the published FtmOx1 structure (PDB accession code 4ZON).38
To prepare our FtmOx1•Fe(II)•2OG ternary-complex structure for docking, we deleted chain B, its associated cofactors, and all water molecules. We initially placed the model for 2 into the active site by aligning the prepared structure with the published structure (4ZON) using Chimera.54 We then aligned the substrate model to the 4ZON substrate coordinates using atoms N18, C20, and C5. The resulting coordinates containing our experimental FtmOx1•Fe(II)•2OG ternary-complex structure and the starting coordinates for 2 were written to a new PDB file that was used as input to Rosetta.
Because the default pdb components dictionary in Rosetta specifies a neutral protonation state for 2OG that is inappropriate for this system, we manually edited the dictionary file to delete atoms and bonds associated with the two carboxylic-acid hydrogens for this molecule. Although a canned Rosetta protocol for ligand docking has been released,55 this protocol could not accommodate our requirement that the metal coordination environment and 2OG position be maintained during structure refinement. Thus, we used RosettaScripts to write a simpler protocol for ligand docking and refinement with fixed degrees of freedom compatible with the chemistry of the FtmOx1 system.56 Each docking trajectory included the following steps:
Random uniform translation of the substrate from its starting position
Random uniform rotation of the substrate
Rigid-body minimization of the substrate with a soft-repulsive scoring function
Repacking of a fixed set of sidechains in the substrate cavity (excluding metal-coordinating residues) with a hard-repulsive scoring function
Torsion-space minimization of nearby sidechains with rigid-body minimization of the substrate
Cartesian-space of the same set of residues, including backbone atoms
This series of steps was executed in 40,000 independent trajectories. The resulting docked configurations were written to PDB only if they satisfied two distance constraints: (1) < 6 Å between the iron and substrate C21; (2) < 6 Å between the phenolic oxygen of Tyr68 and substrate atom C26. Of the 40,000 docked configurations, 2748 satisfied both distance constraints and were saved. The resulting PDB files were partitioned into 10 clusters based on the heavy-atom Cartesian coordinates of the substrate using the K-means clustering functionality of scikit-learn in Python3.57 The top-scoring three configurations from each cluster were visually inspected in Chimera, and the best was chosen on the basis of chemical intuition regarding molecular interactions and cavities as well as the mechanistic model for the FtmOx1 reaction developed in this work.
RESULTS AND DISCUSSION
Oxygen rebound in competition with C21•–O2 coupling leads to deprenylation of 2.
The first experimentally addressable distinction between the COX-like and CarC-like mechanisms in Scheme 2 is whether the H• donor to the ferryl complex is the phenolic hydroxyl group of Y224 or C21 of 2. The latter case would conform to the paradigm established by studies on a number of other Fe/2OG oxygenases,22,23,58–60 whereas the former case would represent a deviation. Evidence for a direct-HAT mechanism has been obtained in studies of other Fe/2OG enzymes by observation of markedly slower decay of the ferryl complex (by as much as 60-fold) when the donor carbon bears deuterium, a reflection of a very large primary deuterium kinetic isotope effect (D-KIE) arising from quantum-mechanical tunneling in the HAT step. Stabilization of the ferryl complex in the FtmOx1 reaction with 2 bearing deuterium at C21 would thus be anticipated for the CarC-like mechanism, but not for the COX-like mechanism. Unfortunately, this substrate isotopologue was neither commercially available nor synthetically accessible, rendering this key test unfeasible.
A second experimentally addressable distinction between the mechanisms arises from the spatial separation of the C21• and Fe(III)–OH in the COX-like mechanism and their necessary proximity in the CarC-like mechanism. Prior studies of other Fe/2OG oxygenases that mediate non-hydroxylation outcomes by direct-HAT mechanisms have shown that hydroxylation via oxygen rebound generally competes to some extent.22,35,61 The COX-like mechanism, on the other hand, could be immune to competing rebound. The anticipated hemiaminal product of oxygen rebound would be expected to decompose to 12,13-dihydroxyfumitremorgin C (1) by loss of the prenyl substitutent from N1 as prenal (Scheme 3, red arrows). Indeed, both Yan, et al. and the authors of an earlier study detected a considerable quantity of 1 in their FtmOx1 reactions, allowing for the possibility that oxygen rebound does, in fact, occur.36,38 However, more recent studies elucidated an alternative mechanism for C–N-bond cleavage that could potentially be operant in deprenylation of 2.22,66 Electron transfer (ET) from C21• to the Fe(III)–OH complex could produce an N1-iminium species, which would be expected to break down by hydrolysis (Scheme 3, blue arrows). Because ET can be facile even at donor-acceptor distances much greater than would be needed to form bonds (e.g., by rebound), deprenylation by this route could occur in either the COX-like or the CarC-like mechanism and would not differentiate the two. Fortunately, the two possible deprenylation pathways (Scheme 3, blue and red) differ in their predicted origins of the prenal oxygen: O2 for rebound (red) versus solvent for ET/hydrolysis (blue). Therefore, to elucidate the deprenylation mechanism, we determined the origin of the installed oxygen by conducting the reaction under 18O2 in natural-abundance H2O.22,28,35,62,63 A complication was presented by the well-known propensity of aldehyde carbonyl oxygens to exchange with water,64 which might have erroneously suggested a solvent origin of the prenal oxygen, even for the case of its installation by the rebound mechanism. Addition of NaBH4 immediately after initiation of reactions allowed the prenal side product to be reduced to prenol (alcohol), trapping any 18O label incorporated in the deprenylation. Indeed, a mixture of 18O and 16O was detected in the prenol product by GC-MS (Figure 1A–B). The incorporation of any 18O suggests that deprenylation of 2 results from oxygen rebound and not from ET/hydrolysis, because the fraction of 16O incorporated can be explained by solvent exchange of the ferryl oxo ligand, the aldehyde oxygen, or both, prior to the fixing reduction by NaBH4. To ensure that reductive cleavage of the endoperoxide ring of 3 was not the source of the detected 18O-labeled prenol (Figure S2A), we treated commercially obtained 3 with NaBH4 under the same conditions used in the 18O2-tracer experiments. LC-MS analysis of the control samples showed no detectable breakdown of 3 (Figure S2B), and GC-MS analysis revealed no detectable production of prenol (Figure S2C). The results thus confirm that the 18O-labeled prenol detected in analysis of the FtmOx1 reaction under 18O2 did indeed arise from hydroxylation of C21 by competing oxygen rebound (Scheme 3, red arrows).
Scheme 3.

Kinetic and Mechanistic Rationale for Competition between Deprenylation and Endoperoxidation of Fumitremorgin B by FtmOx1.
Figure 1.

Origin of the incorporated oxygen atom in the deprenylation of fumitremorgin B by FtmOx1 and dependence of the partitioning between endoperoxidation and deprenylation on [O2]. (A) GC-MS total-ion chromatograms of the prenol standard (black) and prenol produced from reactions under an atmosphere of 16O2 (blue) or 18O2 (red). (B) Corresponding mass spectra for the 16O2 and 18 O2 reactions, respectively, showing the +2 features at m/z = 73 and 88 resulting from incorporation of 18O in the deprenylation reaction. The paired peaks at m/z = 71 and 86 in the 18O2 reaction reflect solvent exchange either in the ferryl complex or in the initial aldehyde product before it underwent reduction by NaBH4. (C) Ratio of LC-MS peaks for verruculogen (3, black) and 12,13-dihydroxyfumitremorgin C (1, gray) at lowest (left) and highest (right) achievable O2 concentrations (as described in Experimental Procedures). Each bar is the average of three trials, and the error bars are the standard deviation.
According to the CarC-like mechanism (Scheme 2B), the step of the endoperoxidation pathway with which unimolecular oxygen rebound competes is C21• ↔ O2 coupling, which is potentially bimolecular and kinetically first-order in [O2]. We tested the implication that the endoperoxidation/deprenylation partition ratio might, accordingly, depend on the concentration of O2, with deprenylation being favored by low [O2] and endoperoxidation by high [O2] (equations at bottom of Scheme 3). Indeed, in reactions carried out by slow infusion of air into an anoxic solution of the reactant complex to give the minimum possible [O2], deprenylated product 1 was favored by a factor of ~ 7 (3:1 ~ 13:87, Figure 1C). Conversely, at the highest [O2] that could be generated (by use of the previously described ClO2−/chlorite-dismutase system43) the endoperoxide product 3 was favored. However, even at this non-physiologically high [O2], 3 predominated by only a factor of ~ 3 (76:24) over the deprenylated product, 1. The results of these two experiments establish that oxygen rebound to C21•, leading to deprenylation of N1, does in fact compete with C21• ↔ O2 coupling and is actually surprisingly efficient.
Re-examination of the published x-ray crystal structure of the FtmOx1•fumitremorgin B complex.
The demonstration of facile oxygen rebound, a strong contraindication to the COX-like mechanism, led us to re-examine the evidence cited in the prior study for the non-canonical pathway. The reported x-ray crystal structure of the FtmOx1•Fe(II)•2 complex (PDB accession code 4ZON38) seemed to provide a sound basis for the hypothesis of a COX-like mechanism: the phenolic oxygen of Y224 appeared well-disposed relative to the iron cofactor and C21 of 2 to act as HAT intermediary. However, our re-analysis of the deposited coordinates and structure factors (Figure S1A) revealed little 2Fo-Fc electron density for 2, even at the lowest acceptable levels of map contouring (1.0 σ). Inspection of the PDB validation report for the 4ZON structure (Table S1) additionally revealed outliers for the real-space correlation coefficient (RSCC) and real-space R (RSR) ligand-validation metrics, which both assess the quality of the fit of a modeled ligand into the experimental electron density.65 The modeled substrate, 2, exhibits a RSCC value of 0.66 and a RSR value of 0.52. Values less than 0.8 and greater than 0.4, respectively, indicate a poor fit. Furthermore, metrics used to analyze ligand geometry produced outlier values for > 10% of bond lengths/angles in 2.66 We also generated our own simple omit electron density map for 2, calculated by deletion of the molecule from the coordinate file and refinement against the published structure factors (Figure S1A–C). This omit map is nearly identical to the 2Fo-Fc electron density map calculated directly from the deposited structure factors and is inconsistent with the presence and orientation of the substrate as modeled. Importantly, none of the maps had electron density to support modeling of the target carbon for the initiating HAT (C21). We concluded from this analysis that (i) the published 4ZON structure cannot be taken as an experimentally validated model of the FtmOx1 reactant complex and (ii) the primarily structural foundation for favoring the COX-like mechanism in that study was not sound.
Four Tyr residues in the active site of FtmOx1 with potential roles in HAT.
In light of these conclusions, we attempted to solve a structure of the complete FtmOx1•Fe(II)•2OG•2 reactant complex with full occupancy of the substrates. We identified new conditions to crystallize the protein either in the presence of all components (including 2) or in the absence of the primary substrate and, for the latter case, to soak 2 into pre-formed crystals to allow it to bind. Neither approach yielded interpretable electron density in the active site consistent with full occupancy by 2. Importantly, addition of water molecules to the model could account for most of the extra electron density observed in the active site (Figure S1C, D). Given the similarity of our observations to those in the previous study,38 we suspect that features in the electron density map attributed to substrate in the 4ZON structure could be explained by ordered solvent. Interestingly, the dimeric asymmetric unit that we observe overlays almost precisely with the previously solved structures,38 even though the crystals were grown under different conditions, resulting in a different space group (Figure 2, top and Figures S3–S4). We considered the possibility that crystal packing might preclude binding of 2, but the observed interaction between molecules likely constitutes a functional dimer. We also generated a variant of FtmOx1, in which Y140, a residue at the dimer interface (Figure 2, bottom left), was replaced with phenylalanine. Although the crystals of FtmOx1-Y140F diffracted x-rays to higher resolution (1.92 Å) than those of the wt enzyme (2.0 – 2.5 Å), we were still unable to observe electron density for 2 that would have allowed it to be modeled in the active site. We also considered the possibility that the N-terminal His6 metal-ion-affinity tag and 21-residue linker, present in both the wt and Y140F proteins, might interfere with substrate binding in the crystal lattice. Trials with the untagged protein yielded crystals of improved diffraction limit (~ 1.5 Å resolution), but we still could not detect 2 after co-crystallization or soaking.
Figure 2.

Structural features of the complex of FtmOx1-Y140F with Fe(II) and 2OG. (Top) A cartoon diagram of the Fe(II)•2OG•FtmOx1 Y140F shows the dimeric asymmetric unit. (Bottom left) The F140 side chain in the variant protein resides far from the active site, near the dimer interface. (Bottom center) The coordination environment around the Fe(II) cofactor reveals an off-line 2OG binding mode. (Bottom right) In addition to Y224, several tyrosine residues surround the active site. Selected side chains and the co-substrate 2OG are shown in stick format. The Fe(II) ion and coordinated water molecule are shown as orange and red spheres, respectively.
Our efforts yielded > 30 high-resolution structures of wt FtmOx1 and its Y140F variant. Although we could not achieve substrate binding in crystallo, other features of the active site proved worthy of closer examination. The C1 carboxylate of 2OG is coordinated trans to the distal histidine ligand (H205) — a carboxylate-offline configuration seen previously in a number of crystal structures of Fe/2OG oxygenases (Figure 2, bottom middle).26,67–69 The same observation by Yan, et al. led them to suggest that FtmOx1 utilizes a geometrically distinct ferryl complex, with the oxo ligand also located offline, trans to H129.62,70 However, it is known that the coordination geometry can change in response to substrate binding or addition of O2.18,67 Consequently, mechanistic hypotheses formulated from analysis of incomplete reactant states must be viewed with greater caution.
The side chains of Y224, Y68, and Y74 all reside in the active site cavity (Figure 2, bottom right). This array of aromatic side chains resembles that seen in the substrate binding pocket of CarC, which is encircled by five Tyr residues (Figure S5).26 In CarC, Y165, the sole Tyr shown to engage directly in chemistry with the substrate, resides on a lid loop that becomes ordered only upon binding of the substrate. In contrast to these observations from CarC, the entire region surrounding the active site of FtmOx1 can be confidently modeled in the substrate-free state. The FtmOx1 structures show that Y224 is fully buried and its position largely fixed. By contrast, the side chains of Y68 and Y74 are on the periphery of the protein, in a lid loop. This loop exhibits elevated B-factors in our structures, suggesting that it could be flexible (Figure S6), as in CarC.26 In addition to potentially participating in catalysis, the residues in this loop could help sequester the otherwise-exposed active site from solvent during the reaction.
Evidence for H• donation by Y68 in the FtmOx1 endoperoxidation reaction.
Both of the mechanisms considered here and by Yan, et al. would involve a Tyr• intermediate, and the previous study presented SF-Abs and FQ-EPR data consistent with this hypothesis. A sharp, transient absorption feature at ~ 420 nm and a broad, transient ĝ 2.0 EPR signal developed within 0.20 s and decayed within ~ 3 s following mixing (at 8°C) of the FtmOx1•Fe(II)•2OG•2 complex with O2. We reproduced (at 5°C) the SF-Abs observations (Figure 3), while also establishing that the presence of 2 is required for these transient features. Mixing of the FtmOx1•Fe(II)•2OG ternary complex (lacking 2) with O2-saturated buffer resulted in only very sluggish development of stable absorption in the ultraviolet regime (Figure S7). Similar behavior in the reactions of other Fe/2OG enzymes in the absence of their primary substrates has been attributed to slow, unproductive oxidation of the Fe(II) cofactors.33 FtmOx1 thus shares with most (if not all) Fe/2OG oxygenases the property of “substrate triggering,” in which binding of the primary substrate markedly accelerates reaction of the cofactor with O2 (by as much as 5,000-fold).59 We also showed that the rate of accumulation of the sharp, transient, ~ 417-nm feature is dependent on the concentration of O2 (Figure S8), suggesting that no intermediate state between the O2-addition step and the presumptive Tyr• (e.g., the canonical ferryl complex) accumulates to a high level under these conditions. The strength of the C21–H bond cleaved in the FtmOx1 reaction is diminished by both the α heteroatom (N1) and the C22–C23 olefin, and so it is perhaps not surprising that the HAT step would be rapid and the ferryl complex would fail to accumulate.
Figure 3.

Time-dependent absorption spectra from the reactions at 5°C of (A) wild-type FtmOx1 and (B) the Y140F, (C) Y74F, (D) Y224F and (E) Y68F variant proteins. The spectra shown were acquired 0.003 s (dashed black), 0.030 s (dotted black), 0.35 s (solid black), 2 s (dotted gray) and 10 s (solid gray) after rapid mixing of an anoxic solution of 0.65 mM enzyme, 0.58 mM Fe(NH4)2(SO4)2, 0.58 mM 2, and 5 mM 2OG with an equal volume of O2-saturated buffer. The dotted red and blue lines indicate the positions of the signature features of the first and second Tyr radicals, respectively.
Closer inspection of the time-dependent absorption spectra for the complete wt FtmOx1 reaction and comparison to the analogous data from the reactions of a set of four Y → F variants revealed that, in fact, two sharp, partially-resolved features in the ~ 380–450-nm region developed and decayed on different timescales (Figure 3, red and blue dotted lines). Both features resembled signatures of known Tyr radicals, including association of the sharp peak with shoulders at slightly higher energy. The more rapidly developing signal was broader and slightly more red-shifted than for most known Tyr radicals,26,71,72 having an absorption peak at ~ 417 nm and a shoulder at ~ 398 nm (red lines). The second signal was more typical for Tyr radicals, with a sharp peak at 410 nm and shoulder at ~ 390 nm (blue lines). Both features were readily seen in the time-dependent spectra from the reaction of the wt protein (Figure 3A), but the red-shifted first signature was most prominent early (at ~ 0.4 s) in the reaction of the Y140F variant (Figure 3B, solid black spectrum), whereas the unshifted second signature was particularly evident later (~ 2–10 s) in the reaction of the Y74F variant (Figure 3C, gray spectra). In fact, each of the four Y → F substitutions was seen to perturb the kinetics of the reaction and resultant transient absorption spectra in a unique way (Figure S9), as we also saw in our previous work on CarC.26 Both signatures were markedly enhanced in the reaction of the Y140F variant (Figure 3B, solid black and gray spectra). The first, red-shifted chromophore was readily seen early (at ~ 0.03 s) in the reaction of the Y74F variant (Figure 3C, black dotted spectrum) but developed less intensity because it gave way more rapidly (within ~ 0.3 s) to the second signal (solid black spectrum) than in the reaction of the wt protein (compare to Figure 3A, solid black spectrum). Importantly, the Y224F variant, which lacks the Tyr purported by Yan, et al. to mediate HAT from the substrate to the ferryl complex, gave early spectra (Figure 3D, black spectra) and kinetics (Figure S9) that most closely matched those of the wt protein, before subsequently developing significantly less (if any) of the second, 410-nm signature (Figure 3D, gray spectra). By contrast, the Y68F variant was most severely perturbed at early reaction times (Figure 3E, black spectra), failing to develop any hint of the early, red-shifted signature before developing the 410-nm feature somewhat more rapidly than in the wt enzyme (Figure 3E, gray spectra; Figure S9). The fact that only the Y68F substitution abolished the early chromophore implies a primary role for this Tyr, whereas the absence of an early effect of the Y224F substitution suggests that this Tyr may have only a secondary role (as elaborated below).
Consistent with these conclusions, we found that only the Y68F variant among the four Y → F variant proteins that we examined is compromised in its endoperoxide synthase activity. Whereas the Y224F, Y74F, and Y140F variants generated quantities of endoperoxide 3 similar to that produced by the wt protein (Figure 4), the Y68F variant made much less (~ 5%) of this product under the same conditions (red arrow). Importantly, replacement of Y68 did not result in complete inactivity. Rather, the Y68F variant converted the substrate primarily to a different product with m/z = 510 (Figures 4 and 5, marked by asterisks). Although this value matches the m/z of 4, the previously reported ketone product resulting from further oxidation of diol 3, the FtmOx1-Y68F product was resolved from 4 (as well as from 1-3) in the chromatographic analysis (Figure S10), implying that it is a different compound (of unknown structure). Moreover, the Y68F variant could completely consume 2.5 equiv of 2 under favorable conditions (Figure 5). The effect of the Y68F substitution in FtmOx1 thus almost precisely mirrors that of the Y165F substitution in CarC, which we found in prior work to block accumulation of Tyr• and cause formation of a different primary product without inactivating the enzyme outright.
Figure 4.

LC-MS chromatograms from the reactions of wild-type FtmOx1 (black) and its Y140F (purple), Y74F (blue), Y224F (green) and Y68F (red) variants. Reactions contained 0.010 mM protein, 0.010 mM Fe(NH4)2(SO4)2, 1.0 mM 2OG, 0.50 mM 2, and 1.0 mM ascorbate. The red arrow highlights the drastically diminished yield of verruculogen (3) uniquely in the reaction of the Y68F variant, and the asterisk marks the new major (as-yet-uncharacterized) product generated by this variant. The apparatus and conditions for the chromatographic analysis are provided in Experimental Procedures.
Figure 5.

LC-MS chromatograms illustrating the effect of ascorbate on the products of (A) the wild-type and (B) the Y68F variant FtmOx1 proteins. The complete reactions (blue) contained 0.25 mM protein, 0.20 mM Fe(II) [from Fe(NH4)2(SO4)2], 0.50 mM 2, 1.0 mM 2OG, and 1.0 mM ascorbate and were carried out as described in Experimental Procedures. The effect of omission of ascorbate (red) or 2OG (black) can be seen by comparison to the middle and lower trace, respectively.
We verified by FQ experiments on the wt and four Y → F variant proteins that both transient absorption features in the FtmOx1 reaction are associated with g ~ 2 EPR signals (Figure 6). Our X-band spectra of samples quenched early (~ 0.4 s) in the reaction of the wt protein (Figure 6A, black spectrum) essentially reproduced those reported by Yan, et al.38 As we also saw in the SF-Abs experiments, the Y224F variant and wt enzyme behaved almost identically in the early part of the reaction when monitored by EPR. Samples frozen at 0.43 s, near the time of maximum intensity of the early, red-shifted absorption signature, exhibited nearly identical g = 2 EPR line-shapes and intensities (Figure 6A, compare green and black spectra). The Y140F variant, shown in the SF-Abs experiments to develop more of the first chromophore, also supported development of this EPR signal, and with enhanced intensity (Figure 6A, purple spectrum). This correlation associates the first absorption signature with a radical. Much later in the reactions of the wt and Y140F proteins (at 30 s), correlating with when the red-shifted chromophore fully decayed and the 410-nm signature developed, a different EPR spectrum was observed (Figure 6B, black and purple spectra). Again, the greater intensity in the spectrum from the reaction of the Y140F variant (purple spectrum) correlated with its enhanced 410-nm absorption feature, supporting the assignment of the second species as a radical. By contrast, in the reaction of the Y224F variant, which failed to develop the 410-nm signature, this EPR signal was not observed, suggesting that Y224 could be involved in the formation of (and perhaps even be the site of) the second radical. Instead, an intense, narrow EPR singlet developed in its place (Figure 6B, green spectrum). As already implied by the activity assays, the fact that its replacement exerted an effect only late in the reaction is more consistent with a secondary role for Y224 than with the primary role in HAT proposed by Yan, et al.38 Hints of the sharp, narrow singlet could also be seen in the later spectra of reactions of other variant proteins (e.g., Y140F, purple spectrum).
Figure 6.

X-band EPR spectra (at 40 K) of freeze-quenched samples from the reactions of wt FtmOx1 and its Y → F variants. Samples were prepared by rapid mixing of a solution containing 0.65 mM protein, 0.58 mM Fe(NH4)2(SO4)2, 0.58 mM 2, and 5.0 mM 2OG with an equal volume of O2-saturated buffer and rapidly freezing after reaction times of (A) 0.43 s, (B) 30 s (B), and (C) 0.090 s. The dotted purple trace in panel B is the spectrum of the 0.43-s species from panel A, reproduced for ease of comparison. All scaling factors are relative to the wild-type trace in panel A. The spectrometer and conditions are provided in the Experimental Procedures.
The Y74F and Y68F substitutions both affected development of the first chromophore: it was visible only early (e.g., at 0.03 s) in the reaction of the Y74F variant, whereas it appeared not to develop at all in the reaction of the Y68F variant. EPR spectra of samples freeze-quenched early (at 0.09 s) in the two reactions (to avoid interference from the second radical) confirmed that the Y74F protein formed the first radical (Figure 6C, blue spectrum). By contrast, the Y68F variant developed a much less intense signal with a different lineshape (red spectrum). These EPR data are again consistent with an important role for Y68 in early events.
The EPR spectra of known Tyr radicals in the dynamically restrictive environment of proteins are well understood.73 At X-band frequencies, their signals are approximately 7–8 mT wide and exhibit fine structure due to hyperfine couplings with the 1H nuclei (I = 1/2) of the Tyr. The shape of the signal strongly depends on the orientation of the two hydrogens on C3 relative to the phenoxyl ring, because the magnitude of the hyperfine coupling to these hydrogens depends on the dihedral angles between the ring, the C4–C3 bond, and the C3–H bonds, which change with rotation around the C3–C4 bond. In the presence of a nearby paramagnetic center, the EPR features of the Tyr• can also be broadened by dipolar coupling between the two electron spins, with the extent of the broadening depending on the spatial separation of the two spin centers.26,74 This broadening can qualitatively change the line shape of the spectra relative to those seen for magnetically-isolated Tyr radicals.
The breadth of the two EPR signals developed in the FtmOx1 reaction is greater than 1.2 mT and fully consistent with the presence of dipolar coupling between the Tyr• and the high-spin Fe(III) cofactor expected also to be present at that stage of the reaction; the spectra do not exhibit the typical EPR-spectroscopic features of magnetically isolated Tyr•. To provide additional evidence that the radicals are derived from Tyr, we incorporated 3,3-[2H2]-L-tyrosine (β-d2-Tyr) into wt FtmOx1 and its Y140F variant by producing the proteins in the Tyr-auxotrophic E. coli strain, WU-36 (see Experimental Procedures and the Supporting Information for details). The X-band spectrum of the rapidly forming radical in the β-d2-Tyr-containing wt enzyme (Figure 7A, red spectrum) was subtly perturbed by the replacement of the two I = 1/2 1H nuclei with I = 1 2H nuclei (compare to black spectrum). This observation confirms that the radical spin density resided at least partially on one or more Tyr residue (or a species derived therefrom). Similarly, the spectrum of the later-forming radical (produced in the Y140F variant for increased yield) was also subtly affected by the isotopic substitution (Figure 7B, compare black and red spectra). The combined results of the SF-Abs, FQ EPR, and deuterium-substitution experiments suggest that both radical chromophores involve a Tyr (Y68 for the first species and Y224 for the second).
Figure 7.

X-band EPR spectra (at 40 K) of freeze-quenched samples from the reactions of (A) wt FtmOx1 and (B) its Y140F variant illustrating the effect of the presence of 3,3-d2-Tyr in the protein on the lineshape of the early (A, reaction time of 0.43 s) and late (B, reaction time of 30 s) radical species. The samples were prepared and spectra acquired as described in the Figure 6 caption and the Experimental Procedures.
The observations that the hyperfine couplings to the C3 1H nuclei are less obvious, and the effects of their replacement by 2H less pronounced, than has typically been seen for other Tyr• systems can be rationalized by the posited magnetic interaction of the Tyr radical(s) with the high-spin (S = 5/2) Fe(III) form of the cofactor, which, for the case of a Y68 radical, would be separated by ~ 9 Å (Fe to Y68-Oη distance in our structures). In addition, it is also possible, that the radical could be more heterogeneous than those interrogated in other systems. Heterogeneity could arise in one of two ways. First, the radical could be distributed among multiple Tyr residues, possibly in equilibrium at the temperature of the reaction. Upon rapid freezing, trapping at multiple sites would lead to a spectrum representing the superposition of the individual Tyr• spectra, each with its characteristic coupling. Distribution of the radical might also explain the marked effect on the reaction kinetics of substituting any of the Tyr residues, observed here and also in our previous work on CarC. Alternatively, the radical could be localized on a single Tyr (Y68) that does not have a single fixed conformation in solution. Rapid freezing of a distribution of conformations could result in a population-weighted, averaged spectrum, potentially obscuring the coupling in any given conformation. It would appear that a radical harbored on Y68 could, barring a major conformational change associated with binding of 2, have unusually facile phenoxyl ring dynamics. If so, it would be expected also to remain solvent exposed and reactive toward reductants, as probed below.
Effects of ascorbate consistent with CarC-like mechanism.
The last issue we addressed to distinguish between possible CarC-like and COX-like mechanisms for FtmOx1 was the effect of the reductant, ascorbate, on the reaction stoichiometry and transient-kinetic behavior. As noted above, the COX-like mechanism would have a Tyr• forming early in the reaction sequence, in advance of the product 3, and could potentially allow each enzyme molecule to mediate multiple turnovers following production of the Tyr• by the ferryl intermediate, as occurs in COX. By contrast, we observed in our prior work on CarC that the Tyr• formed concomitantly with the product and that the reaction was stoichiometric with enzyme in the absence of a reductant to return the Fe(III)/Y165• enzyme form back to the Fe(II)/Y165 state, ready for the next turnover. The multiple outcomes of the FtmOx1 reaction (Scheme 1) complicate the question of stoichiometry relative to the case of CarC. Whereas endoperoxide formation is redox neutral with respect to the protein and its iron cofactor, the competing conversion of 2 to 1 (deprenylation) and further oxidation of the verruculogen diol, 3, to its ketone analogue, 4 (seen previously and also in this study), both effectively transfer two electrons from the substrate to the enzyme. Another turnover could ensue following either of these two outcomes, thereby affecting the overall stoichiometry. Employing conditions of high [O2] to minimize deprenylation, we challenged the wt enzyme and Y68F variant with 2.5 equiv [relative to Fe(II)] of 2 in the presence of excess 2OG and O2 and allowed the reactions to proceed to completion (Figure 5). Both reactions had ~ half of the added 2 remaining at completion (red traces), suggesting that neither protein is capable of consuming substantially more than ~ 1 equiv of the substrate in the absence of a reductant. By contrast, in the presence of excess ascorbate (at twice the concentration of 2), both the wt and Y68F proteins fully consumed all 2.5 equiv of 2 (blue traces). These results imply that, if the COX-like cycle deploying the H•-abstracting Tyr• is, in fact, operant, the Tyr• must not support multiple turnovers once installed, unlike in COX.
A second impact of ascorbate on the outcome of the wt FtmOx1 reaction is suppression of the further oxidation of 3 to 4 (Figure 5A, compare red and blue traces). As discussed below, the reductant effectively quenches the otherwise persistent Tyr• formed when H• donation completes the verruculogen endoperoxide. The suppression of 4 production implies that the persistent Tyr• is involved in this subsequent oxidation. Importantly, the altered product of the FtmOx1-Y68F variant is not suppressed by ascorbate (Figure 5B, compare red and blue traces), providing additional confirmation that it is the initial product in this reaction and not a byproduct of further oxidation.
Our final assessment of the effect of ascorbate was on the kinetics of the transient Tyr radicals. Despite its capacity to enhance turnover, as little as 1 mM ascorbate was found to completely prevent accumulation of both Tyr• species (Figure 8). The implication is that the reductant quenches the initial Tyr• rapidly with respect to its formation. For that event not to impede the endoperoxidation outcome, the Tyr• must accumulate (in the absence of ascorbate) as a result of either the same step in which the endoperoxide product is completed or a later step. This conclusion is more consistent with the CarC-like mechanism (Scheme 2B) than with the previously proposed COX-like mechanism (Scheme 2A), in which Y224 oxidation by the ferryl complex was suggested to precede the first substrate-processing step. In addition, because reducing conditions normally prevail in the cell, the conclusion that the downstream radical species observed in the absence of reductant are unlikely to be important in vivo. Thus, a CarC-like mechanism, in which the ferryl complex abstracts H• directly from C21, Y68 quenches the C26 radical after addition of O2 between C21 and C27 (Scheme 2B), and the resultant Y68• is then quenched by a reductant, is consistent with all the available experimental data.
Figure 8.

Kinetic traces illustrating the complete suppression of both Tyr• absorption signatures by ascorbate. The panels show “dropline” treatments applied to the raw SF-abs data to extract the heights of the peaks (A) at 416 nm (reference wavelengths of 406 and 426 nm), arising from the first Tyr• and (B) at 410 nm (reference wavelengths of 404 and 416 nm), arising from the second Tyr•. The reactions were carried out by mixing an anoxic solution of 0.65 mM protein, 0.58 mM Fe(NH4)2(SO4)2, 0.58 mM 2, 5.0 mM 2OG, and (solely for the blue traces) 1.0 mM ascorbate at 5°C with an equal volume of O2-saturated buffer.
A computational model of the FtmOx1 reactant complex is consistent with a CarC-like mechanism.
Our favored mechanistic proposal would demand proximity of C21 to the iron cofactor and C26 to Y68. In the absence of an experimental structure of the complete reactant complex, we sought to evaluate whether the active site of FtmOx1 can bind 2 in a mode that satisfies these constraints. The structures of FtmOx1•Fe(II)•2OG (previously reported and solved in this work) reveal a sizeable cavity above the iron cofactor that would appear capable of accommodating a range of substrate binding modes (Figure 9A). We generated a series of computational models for the full FtmOx1•Fe(II)•2OG•2 reactant complex by docking a model of the substrate into the structure of the FtmOx1•Fe(II)•2OG ternary complex, using the coordinates of our highest-resolution structure of the Y140F variant as the protein input. We initially considered two approaches to obtaining the starting model for 2. Use of the JLigand extension of the COOT software package48,75 yielded a structure that was severely bent about the central 6-membered C ring and had a number of bond angles and lengths deviating from their ideal values, particularly in the C ring. The three largest outliers are shown in Figure S11A. The structure that we generated in JLigand overlays almost precisely with the substrate in the 4ZON structure from the Yan, et al. study (Figure S12A), suggesting that the authors might have applied a similar approach. In principle, a more favorable substrate conformation might have evolved during refinement of the 4ZON structure, had there been sufficiently clear electron density in the active site against which to compare its molecular geometry. It appears that such substrate refinement did not occur.
Figure 9.

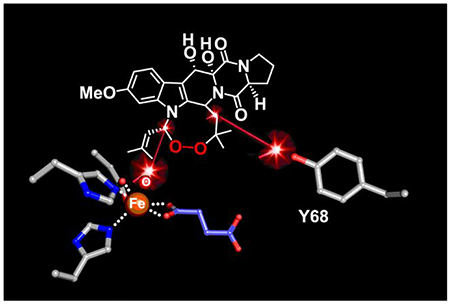
Structural analysis of potential locations and modes of binding of the substrate, fumitremorgin B (2), in the active site of FtmOx1 as it is configured in its ternary complex with Fe(II) and 2OG. (A) Cavity that could accommodate 2 (green contour) seen in the crystal structure of the Y140F variant, created using the program HOLLOW.78 (B) Preferred computationally-derived model of the reactant complex from docking of the substrate (green sticks) into the observed cavity. (C) Mechanistically relevant distances and potential enzyme-substrate interactions (dotted lines) seen in the preferred docking model. (D) Zoomed-in view showing the disposition of the substrate and cofactor in the preferred model.
Given the problems with the computationally-generated starting model for 2, we elected instead to use a model of the substrate generated in AceDRG from the SMILES description of a small-molecule crystal structure of 3, with editing first to remove the endoperoxide bridge and then to obtain the minimum-energy conformation (Figure S12B–D). The latter steps were performed in Avogadro. The resulting coordinates retain the extended conformation of the fused ring system found in the product structure with reasonable geometry for all atoms, including those in the central 6-membered ring (Figure S11B). With this model for 2, we then used Rosetta to evaluate 40,000 ligand poses, from which we chose ~ 2,700 candidate complexes that exhibited Fe–C21 and Y68-O–C26 distances of < 6 Å. We clustered the docked substrate models into 10 groups and visually inspected the top three lowest-energy complexes in each cluster for optimal geometry relative to active-site components. A single model that fits the criteria is shown in Figure 9B–D. We note, however, that this model is not the sole possibility and was judged only by visual inspection to be most optimal on the basis of chemical intuition and the mechanistic constraints outlined above.
Intriguingly, the hypothetical binding mode of 2 exposes the proline and 2,5-diketopiperazine rings to solvent outside the protein architecture (Figure 9B). A similar situation was observed in the x-ray crystal structures of the Fe/2OG halogenase, WelO5,18 and the Fe/2OG epimerase, SnoN.27 These enzymes also target bulky natural-product substrates with polyheterocyclic cores, and their substrates protrude from the active site while they simultaneously appear to shield it from solvent. By contrast, in Fe/2OG oxygenases that process smaller substrates, such as amino acids or derivatives thereof, shielding of the active site is typically achieved solely by the protein and most often by the closure or ordering of a dynamic lid-loop in response to substrate binding.76,77 The docking analysis also highlights the hydrophobic nature of the active site, showing the substrate in a binding pocket lined by the side chains of L64, Y68, F72, F115, L222, Y224, and F233 (Figure 9C). This docked position of 2 places C21 5.4 Å from the iron cofactor and the phenolic oxygen of Y68 4.5 Å from C26. Both distances are reasonable for the initial and final HAT steps (Figure 9D). Although we reiterate that this model is not a definitive rendering of the FtmOx1 reactant complex, the analysis supports the notion that the active site can simultaneously accommodate the geometrically favorable, extended conformation of 2 and the requirements of the proposed CarC-like mechanism (i) that C21 be close enough to the iron cofactor for direct HAT (and competing rebound) and (ii) that C26 be positioned to accept H• from Y68 to quench the radical produced by endoperoxide ring closure.
CONCLUSIONS
The structural and mechanistic analysis of FtmOx1 by Yan, et al.38 posited an evolutionary adaptation that would have been quite remarkable for an Fe/2OG oxygenase. Although precedented by the heme-dependent COX1 and COX2 isoenzymes, the proposed use of a tyrosine residue as substrate-to-cofactor HAT intermediary would have represented a strategy to enhance both chemoselectivity (by disfavoring rebound leading to oxidative deprenylation) and efficiency (by enabling catalytic use of 2OG and independence of reductant) not previously seen in a member of this versatile enzyme family. By contrast, our demonstration that the competing deprenylation observed in all three in vitro studies of FtmOx1 results from oxygen rebound is strong evidence for close approximation of C21 to the cofactor and implies that endoperoxidation is initiated by the usual direct HAT between them. In fact, our data show that FtmOx1 is both inefficient, in the sense that it is dependent on a general reductant for multiple turnovers, and remarkably promiscuous, in its partition between endoperoxidation and deprenylation, even at high [O2]. It is interesting to consider that, as an enzyme in a pathway to a toxic secondary metabolite, FtmOx1 might not have been subjected to strong evolutionary pressure toward catalytic efficiency, much less toward elimination of a competing side reaction (deprenylation) that is, in effect, merely a one-step setback in the overall pathway (Scheme 1).
Whether a COX-like mechanism could ever be viable in an Fe/2OG oxygenase remains an open question. Whereas COX is reactivated following adventitious reduction of its catalytic Tyr• by reaction of the mid-valent [Fe(III)] state of its cofactor with its own hydroperoxide product, Tyr• generation in FtmOx1 (or any Fe/2OG enzyme) would demand the reduced [Fe(II)] metal (thus requiring reductant), 2OG, and O2. In other words, the “self-healing” trait of COX, made possible by its operation in the Fe(III/V) catalytic manifold and generation of a reactive oxidant as its product, seems less obviously feasible in the Fe(II/IV) manifold of an Fe/2OG enzyme.
The alternative, CarC-like mechanism implicated by our analysis is, to the best of our knowledge, just the second example of an Fe/2OG-oxygenase reactivity in which the initiating H• removal from the substrate is redox balanced by H• donation by the enzyme (from a Tyr residue) in a subsequent step, creating a need for an additional two electrons from a general reductant, such as ascorbate, for catalytic function. It seems likely that more examples in this manifold await discovery. For example, a sequence of ferryl-mediated C–H cleavage, attack of the carbon radical on an olefin, and donation by a Tyr to the other carbon of the olefin would permit C–C coupling to produce an aliphatic carbocycle, a reaction that has, to the best of our knowledge, not yet been demonstrated. Inspection for the presence of two elements shared by CarC and FtmOx1 – an Fe/2OG oxygenase scaffold with one or more Tyr residue on an apparently flexible, peripheral lid-loop element – might be a means to prospect for unknown enzymes that conform to this mechanistic logic.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Searle Scholars Program (to A.K.B.), and the NIH (GM113106 to J.M.B./C.K., GM127079 to C.K., and GM119707 to A.K.B.). Portions of this work were conducted at the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. GM/CA at APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by the NIH-Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289-01A1). Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant (085P1000817). We thank Alexey Silakov for advice and technical assistance in the EPR experiments.
Footnotes
Supporting Information
Materials and Methods, Scheme S1, Tables S1–S2, and Figures S1–S12 (PDF). This information is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing interests.
REFERENCES
- (1).Casteel DA Peroxy natural products. Nat. Prod. Rep 1999, 16 (1), 55. [DOI] [PubMed] [Google Scholar]
- (2).Czechowski T; Larson TR; Catania TM; Harvey D; Brown GD; Graham IA Artemisia annua mutant impaired in artemisinin synthesis demonstrates importance of nonenzymatic conversion in terpenoid metabolism. Proc Natl Acad Sci U S A 2016, 113 (52), 15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rouzer CA; Marnett LJ Cyclooxygenases: structural and functional insights. J. Lipid Res 2009, 50, S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vane JR Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biology 1971, 231 (25), 232. [DOI] [PubMed] [Google Scholar]
- (5).Covello PS; Teoh KH; Polichuk DR; Reed DW; Nowak G Functional genomics and the biosynthesis of artemisinin. Phytochemistry 2007, 68 (14), 1864. [DOI] [PubMed] [Google Scholar]
- (6).White NJ Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother 1997, 41 (7), 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tu Y The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nature Medicine 2011, 17, 1217. [DOI] [PubMed] [Google Scholar]
- (8).Bu M; Yang BB; Hu LM Natural endoperoxides as drug lead compounds. Curr. Med. Chem 2016, 23 (4), 383. [DOI] [PubMed] [Google Scholar]
- (9).Paddon CJ; Westfall PJ; Pitera DJ; Benjamin K; Fisher K; McPhee D; Leavell MD; Tai A; Main A; Eng D; Polichuk DR; Teoh KH; Reed DW; Treynor T; Lenihan J; Fleck M; Bajad S; Dang G; Dengrove D; Diola D; Dorin G; Ellens KW; Fickes S; Galazzo J; Gaucher SP; Geistlinger T; Henry R; Hepp M; Horning T; Iqbal T; Jiang H; Kizer L; Lieu B; Melis D; Moss N; Regentin R; Secrest S; Tsuruta H; Vazquez R; Westblade LF; Xu L; Yu M; Zhang Y; Zhao L; Lievense J; Covello PS; Keasling JD; Reiling KK; Renninger NS; Newman JD High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496 (7446), 528. [DOI] [PubMed] [Google Scholar]
- (10).van der Donk WA; Tsai AL; Kulmacz RJ The cyclooxygenase reaction mechanism. Biochemistry 2002, 41 (52), 15451. [DOI] [PubMed] [Google Scholar]
- (11).Dubois RN; Abramson SB; Crofford L; Gupta RA; Simon LS; Van De Putte LBA; Lipsky PE Cyclooxygenase in biology and disease. Faseb J 1998, 12 (12), 1063. [PubMed] [Google Scholar]
- (12).Steffan N; Grundmann A; Afiyatullov S; Ruan H; Li SM FtmOx1, a non-heme Fe(II) and alpha-ketoglutarate-dependent dioxygenase, catalyses the endoperoxide formation of verruculogen in Aspergillus fumigatus. Org Biomol Chem 2009, 7 (19), 4082. [DOI] [PubMed] [Google Scholar]
- (13).Matsuda Y; Bai TX; Phippen CBW; Nodvig CS; Kjaerbolling I; Vesth TC; Andersen MR; Mortensen UH; Gotfredsen CH; Abe I; Larsen TO Novofumigatonin biosynthesis involves a non-heme iron-dependent endoperoxide isomerase for orthoester formation. Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bollinger JM Jr.; Chang W.-c.; Matthews ML; Martinie RJ; Boal AK; Krebs C In 2-oxoglutarate-dependent oxygenases; Hausinger RP, Schofield CJ, Eds.; The Royal Society of Chemistry: London, 2015, p 95. [Google Scholar]
- (15).Que L Jr. One motif--many different reactions. Nat Struct Biol 2000, 7 (3), 182. [DOI] [PubMed] [Google Scholar]
- (16).Koehntop KD; Emerson JP; Que L Jr. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem 2005, 10 (2), 87. [DOI] [PubMed] [Google Scholar]
- (17).Vaillancourt FH; Yin J; Walsh CT SyrB2 in syringomycin E biosynthesis is a nonheme FeII a-ketoglutarate- and O2-dependent halogenase. Proc Natl Acad Sci U S A 2005, 102 (29), 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mitchell AJ; Zhu Q; Maggiolo AO; Ananth NR; Hillwig ML; Liu X; Boal AK Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat Chem Biol 2016, 12, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ishikawa N; Tanaka H; Koyama F; Noguchi H; Wang CC; Hotta K; Watanabe K Non-heme dioxygenase catalyzes atypical oxidations of 6,7-bicyclic systems to form the 6,6-quinolone core of viridicatin-type fungal alkaloids. Angew Chem Int Ed Engl 2014, 53 (47), 12880. [DOI] [PubMed] [Google Scholar]
- (20).Busby RW; Townsend CA A single monomeric iron center in clavaminate synthase catalyzes three nonsuccessive oxidative transformations. Bioorg. Med. Chem 1996, 4 (7), 1059. [DOI] [PubMed] [Google Scholar]
- (21).Liao HJ; Li JK; Huang JL; Davidson M; Kurnikov I; Lin TS; Lee JL; Kurnikova M; Guo YS; Chan NL; Chang W -c. Insights into the desaturation of cyclopeptin and its C3 epimer catalyzed by a non-heme iron enzyme: structural characterization and mechanism elucidation. Angew. Chem.-Int. Edit 2018, 57 (7), 1831. [DOI] [PubMed] [Google Scholar]
- (22).Dunham NP; Chang W.-c.; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Jr. Two distinct mechanisms for C-C desaturation by iron(II)- and 2-(oxo)glutarate-dependent oxygenases: importance of alpha-heteroatom assistance. J Am Chem Soc 2018, 140 (23), 7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pan J; Bhardwaj M; Zhang B; Chang W. c.; Schardl CL; Krebs C; Grossman RB; Bollinger JM Jr. Installation of the ether bridge of lolines by the iron- and 2-oxoglutarate-dependent oxygenase, LolO: regio- and stereochemistry of sequential hydroxylation and oxacyclization reactions. Biochemistry 2018, 57 (14), 2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hashimoto T; Matsuda J; Yamada Y Two-step epoxidation of hyoscyamine to scopolamine is catalyzed by bifunctional hyoscyamine 6-beta-hydroxylase FEBS Lett 1993, 329 (1–2), 35. [DOI] [PubMed] [Google Scholar]
- (25).McCulloch KM; McCranie EK; Smith JA; Sarwar M; Mathieu JL; Gitschlag BL; Du Y; Bachmann BO; Iverson TM Oxidative cyclizations in orthosomycin biosynthesis expand the known chemistry of an oxygenase superfamily. Proc Natl Acad Sci U S A 2015, 112 (37), 11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chang W.-c.; Guo Y; Wang C; Butch SE; Rosenzweig AC; Boal AK; Krebs C; Bollinger JM Jr. Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science 2014, 343 (6175), 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Siitonen V; Selvaraj B; Niiranen L; Lindqvist Y; Schneider G; Metsa-Ketela M Divergent non-heme iron enzymes in the nogalamycin biosynthetic pathway. Proc Natl Acad Sci U S A 2016, 113 (19), 5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hollenhorst MA; Bumpus SB; Matthews ML; Bollinger JM Jr.; Kelleher NL; Walsh CT The nonribosomal peptide synthetase enzyme DdaD tethers N-beta-fumaramoyl-L-2,3-diaminopropionate for Fe(II)/alpha-ketoglutarate-dependent epoxidation by DdaC during dapdiamide antibiotic biosynthesis J Am Chem Soc 2011, 133 (5), 1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chang W.-c.; Yang Z-J; Tu Y-H; Chien T-C Reaction mechanism of a nonheme iron enzyme catalyzed oxidative cyclization via C–C bond formation. Org Lett 2019, 21 (1), 228. [DOI] [PubMed] [Google Scholar]
- (30).Chen L; Yue Q; Li Y; Niu XM; Xiang MC; Wang WZ; Bills GF; Liu XZ; An ZQ Engineering of Glarea lozoyensis for exclusive production of the pneumocandin B-0 precursor of the antifungal drug caspofungin acetate. Appl. Environ. Microbiol 2015, 81 (5), 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Jr. Non-heme Fe(IV)-oxo intermediates. Acc Chem Res 2007, 40 (7), 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Groves JT Key elements of the chemistry of cytochrome P-450: The oxygen rebound mechanism. J Chem Ed 1985, 62 (11), 928. [Google Scholar]
- (33).Price JC; Barr EW; Hoffart LM; Krebs C; Bollinger JM Jr. Kinetic dissection of the catalytic mechanism of taurine:alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 2005, 44 (22), 8138. [DOI] [PubMed] [Google Scholar]
- (34).Groves JT High-valent iron in chemical and biological oxidations. J. Inorg. Biochem 2006, 100 (4), 434. [DOI] [PubMed] [Google Scholar]
- (35).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr. Substrate positioning controls the partition between halogenation and hydroyxlation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci U S A 2009, 106 (42), 17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kato N; Suzuki H; Takagi H; Uramoto M; Takahashi S; Osada H Gene disruption and biochemical characterization of verruculogen synthase of Aspergillus fumigatus. Chembiochem 2011, 12 (5), 711. [DOI] [PubMed] [Google Scholar]
- (37).Mundt K; Wollinsky B; Ruan HL; Zhu TJ; Li SM Identification of the Verruculogen Prenyltransferase FtmPT3 by a Combination of Chemical, Bioinformatic and Biochemical Approaches. Chembiochem 2012, 13 (17), 2583. [DOI] [PubMed] [Google Scholar]
- (38).Yan W; Song H; Song F; Guo Y; Wu CH; Sae Her A; Pu Y; Wang S; Naowarojna N; Weitz A; Hendrich MP; Costello CE; Zhang L; Liu P; Zhang YJ Endoperoxide formation by an alpha-ketoglutarate-dependent mononuclear non-haem iron enzyme. Nature 2015, 527 (7579), 539. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (39).Rajakovich LJ; Nørgaard H; Warui DM; Chang W.-c.; Li N; Booker SJ; Krebs C; Bollinger JM Jr.; Pandelia M-E Rapid reduction of the diferric-peroxyhemiacetal intermediate in aldehyde-deformylating oxygenase by a cyanobacterial ferredoxin: evidence for a free-radical mechanism. J Am Chem Soc 2015, 137 (36), 11695. [DOI] [PubMed] [Google Scholar]
- (40).Shimokawa T; Kulmacz RJ; DeWitt DL; Smith WL Tyrosine 385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J Biol Chem 1990, 265 (33), 20073. [PubMed] [Google Scholar]
- (41).Gupta A; Mukherjee A; Matsui K; Roth JP Evidence for protein radical-mediated nuclear tunneling in fatty acid α-oxygenase. J Am Chem Soc 2008, 130 (34), 11274. [DOI] [PubMed] [Google Scholar]
- (42).Koeduka T; Matsui K; Akakabe Y; Kajiwara T Catalytic properties of rice α-oxygenase: a comparison with mammalian prostaglandin H synthases J Biol Chem 2002, 277 (25), 22648. [DOI] [PubMed] [Google Scholar]
- (43).Dassama LMK; Yosca TH; Conner DA; Lee MH; Blanc B; Streit BR; Green MT; DuBois JL; Krebs C; Bollinger JM Jr. O(2)-evolving chlorite dismutase as a tool for studying O(2)-utilizing enzymes. Biochemistry 2012, 51 (8), 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ten Eyck LF Fast Fourier transform calculation of electron density maps. Methods Enzymol 1985, 115, 324. [DOI] [PubMed] [Google Scholar]
- (45).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 1997, 53, 240. [DOI] [PubMed] [Google Scholar]
- (46).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307. [DOI] [PubMed] [Google Scholar]
- (47).McCoy AJ; Grosse-Kunstleve RW; Storoni LC; Read RJ Likelihood-enhanced fast translation functions. Acta Crystallogr D 2005, 61 (4), 458. [DOI] [PubMed] [Google Scholar]
- (48).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D 2004, 60, 2126. [DOI] [PubMed] [Google Scholar]
- (49).Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 2010, 66 (Pt 1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Price JC; Barr EW; Tirupati B; Bollinger JM Jr.; Krebs C The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42 (24), 7497. [DOI] [PubMed] [Google Scholar]
- (51).Long F; Nicholls RA; Emsley P; Grazulis S; Merkys A; Vaitkus A; Murshudov GN AceDRG: a stereochemical description generator for ligands. Acta Crystallogr D 2017, 73, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rappe AK; Casewit CJ; Colwell KS; Goddard WA; Skiff WM UFF, a full periodic table force-field for molecular mechanics and molecular dynamics simulations J Am Chem Soc 1992, 114 (25), 10024. [Google Scholar]
- (53).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25 (13), 1605. [DOI] [PubMed] [Google Scholar]
- (55).Lemmon G; Meiler J In Computational Drug Discovery and Design; Baron R, Ed.; Humana Press Inc: Totowa, 2012; Vol. 819, p 143. [Google Scholar]
- (56).Fleishman SJ; Leaver-Fay A; Corn JE; Strauch EM; Khare SD; Koga N; Ashworth J; Murphy P; Richter F; Lemmon G; Meiler J; Baker D RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. Plos One 2011, 6 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay E Scikit-learn: machine learning in Python. J. Mach. Learn. Res 2011, 12, 2825. [Google Scholar]
- (58).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JM Jr. Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:alpha-ketoglutarate dioxygenase (TauD). J Am Chem Soc 2003, 125 (43), 13008. [DOI] [PubMed] [Google Scholar]
- (59).Matthews ML; Krest CM; Barr EW; Vaillancourt FH; Walsh CT; Green MT; Krebs C; Bollinger JM Jr. Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry 2009, 48 (20), 4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mitchell AJ; Dunham NP; Martinie RJ; Bergman JA; Pollock CJ; Hu K; Allen BD; Chang WC; Silakov A; Bollinger JM Jr.; Krebs C; Boal AK Visualizing the Reaction Cycle in an Iron(II)- and 2-(Oxo)-glutarate-Dependent Hydroxylase. J Am Chem Soc 2017, 139 (39), 13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhou J; Kelly WL; Bachmann BO; Gunsior M; Townsend CA; Solomon EI Spectroscopic Studies of Substrate Interactions with Clavaminate Synthase 2, a Multifunctional alpha-KG-Dependent Non-Heme Iron Enzyme: Correlation with Mechanisms and Reactivities. J Am Chem Soc 2001, 123, 7388. [DOI] [PubMed] [Google Scholar]
- (62).Hausinger RP Fe(II)/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39 (1), 21. [DOI] [PubMed] [Google Scholar]
- (63).Dunham NP; Mitchell AJ; Del Río Pantoja JM; Krebs C; Bollinger JM Jr.; Boal AK α-Amine desaturation of D-drginine by the iron(II)- and 2-(oxo)glutarate-dependent L-arginine 3-hydroxylase, VioC. Biochemistry 2018, 57 (46), 6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Li N; Chang W.-c.; Warui DM; Booker SJ; Krebs C; Bollinger JM Jr. Evidence for only oxygenative cleavage of aldehydes to alk(a/e)nes and formate by cyanobacterial aldehyde decarbonylases. Biochemistry 2012, 51 (40), 7908. [DOI] [PubMed] [Google Scholar]
- (65).Smart OS; Horsky V; Gore S; Varekova RS; Bendova V; Kleywegt GJ; Velankar S Validation of ligands in macromolecular structures determined by X-ray crystallography. Acta Crystallogr D 2018, 74, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bruno IJ; Cole JC; Kessler M; Luo J; Motherwell WDS; Purkis LH; Smith BR; Taylor R; Cooper RI; Harris SE; Orpen AG Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci 2004, 44 (6), 2133. [DOI] [PubMed] [Google Scholar]
- (67).Zhang Z; Ren J.-s.; Harlos K; McKinnon CH; Clifton IJ; Schofield CJ Crystal structure of a clavaminate synthase-Fe(II)-2-oxoglutarate-substrate-NO complex: evidence for metal centered rearrangements. FEBS Lett 2002, 517, 7. [DOI] [PubMed] [Google Scholar]
- (68).Martinez S; Fellner M; Herr CQ; Ritchie A; Hu J; Hausinger RP Structures and mechanisms of the non-heme Fe(II)- and 2-oxoglutarate-dependent ethylene-forming enzyme: substrate binding creates a twist. J Am Chem Soc 2017, 139 (34), 11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Zhang Z; Smart TJ; Choi H; Hardy F; Lohans CT; Abboud MI; Richardson MSW; Paton RS; McDonough MA; Schofield CJ Structural and stereoelectronic insights into oxygenase-catalyzed formation of ethylene from 2-oxoglutarate. Proc Natl Acad Sci U S A 2017, 114 (18), 4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Boal AK; Bollinger JM Jr.; Chang W -c. Assembly of the unusual oxacycles in the orthosomycin antibiotics. Proc Natl Acad Sci U S A 2015, 112 (39), 11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cotruvo JA Jr.; Stubbe J Escherichia coli class Ib ribonucleotide reductase contains a dimanganese(III)-tyrosyl radical cofactor in vivo. Biochemistry 2011, 50 (10), 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bollinger JM Jr.; Edmondson DE; Huynh BH; Filley J; Norton JR; Stubbe J Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science 1991, 253 (5017), 292. [DOI] [PubMed] [Google Scholar]
- (73).Svistunenko DA; Cooper CE A new method of identifying the site of tyrosyl radicals in proteins (vol 87, pg 582, 2004). Biophys. J 2004, 87 (5), 3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Krebs C; Chen SX; Baldwin J; Ley BA; Patel U; Edmondson DE; Huynh BH; Bollinger JM Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 2. Evidence for and consequences of blocked electron transfer in the W48F variant. Journal of the American Chemical Society 2000, 122 (49), 12207. [Google Scholar]
- (75).Lebedev AA; Young P; Isupov MN; Moroz OV; Vagin AA; Murshudov GN JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D 2012, 68 (Pt 4), 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Strieker M; Kopp F; Mahlert C; Essen LO; Marahiel MA Mechanistic and structural basis of stereospecific Cß-hydroxylation in the calcium dependent antibiotic, a daptomycin-type lipopeptide. ACS Chem Biol 2007, 2 (3), 187. [DOI] [PubMed] [Google Scholar]
- (77).Helmetag V; Samel SA; Thomas MG; Marahiel MA; Essen LO Structural basis for the erythro-stereospecificity of the L-arginine oxygenase VioC in viomycin biosynthesis. FEBS J 2009, 276 (13), 3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Ho BK; Gruswitz F HOLLOW: Generating Accurate Representations of Channel and Interior Surfaces in Molecular Structures. BMC Struct. Biol 2008, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.