

Summary
Mechanisms of sex chromosome dosage compensation (SCDC) differ strikingly among animals. In Drosophila flies, chromosome-wide transcription is doubled from the single X chromosome in hemizygous (XY) males whereas in Caenorhabditis nematodes, expression is halved for both X copies in homozygous (XX) females [1, 2]. Unlike other female-heterogametic (WZ female / ZZ male) animals, moths and butterflies exhibit sex chromosome dosage compensation patterns typically seen only in male-heterogametic species [3]. The monarch butterfly carries a newly-derived Z chromosome segment that arose from an autosomal fusion with the ancestral Z [4]. Using a highly contiguous genome assembly, we show that gene expression is balanced between sexes along the entire Z chromosome, but with distinct modes of compensation on the two segments. On the ancestral Z segment, depletion of H4K16ac corresponds to nearly halving of biallelic transcription in males, a pattern convergent to nematodes. Conversely, the newly-derived Z segment shows a Drosophila-like mode of compensation, with enriched H4K16ac levels corresponding to doubled monoallelic transcription in females. Our work reveals that, contrary to the expectation of co-opting regulatory mechanisms readily in place, the evolution of plural modes of dosage compensation is also possible along a single sex chromosome within a species.
Keywords: monarch butterfly, sex chromosome evolution, dosage compensation, H4K16 acetylation, neo Z chromosome
In Brief
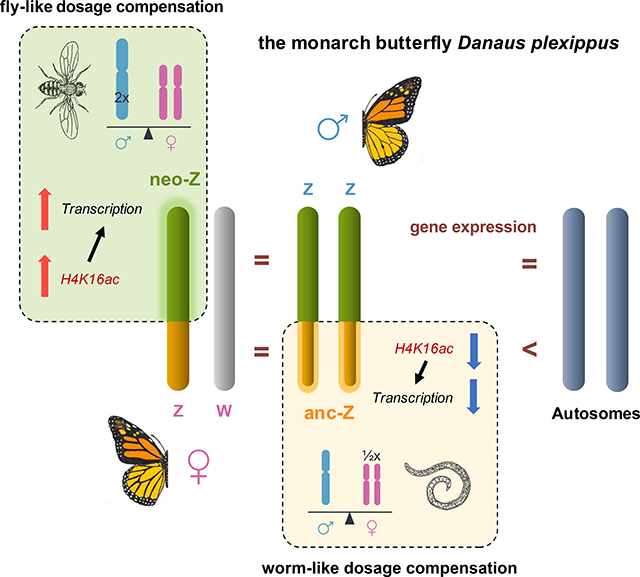
Gu et al. use genomic, transcriptomic and epigenomic tools to analyze sex chromosome dosage compensation in the monarch butterfly which carries a neo Z chromosome. They find that the newly-derived neo-Z and ancestral-Z segments have evolved distinct modes of dosage compensation that mirrors Drosophila flies and nematodes, respectively.
Graphical Abstract
Results and Discussion
Sex chromosome dosage compensation (SCDC) in Lepidoptera presents the only known exception among female-heterogametic taxa
The evolution of heterogametic sex chromosomes results in unbalanced gene dosage between sexes that can potentially disrupt gene expression networks between sex-linked and autosomal genes. This “peril of hemizygosity” [5] is often mitigated by sex-specific regulatory processes on the X (or Z) chromosome, a mechanism broadly referred to as sex chromosome dosage compensation. It has been hypothesized that the initial X (Z) up-regulation to recapitulate ancestral expression levels in the heterogametic sex would also cause over-expression in the homogametic sex, and as a response, some form of X (Z) repression would evolve to balance the expression between sexes [5–7]. These two steps correspond to two distinct aspects of SCDC: dosage compensation (in its strictest sense), which is the compensatory up-regulation of X (Z) expression to achieve X:Autosome parity; and dosage balance, which is the equalization of X (Z) expression between sexes.
Considering this distinction between compensation and balance, SCDC patterns can be categorized into three basic types [3]. In the first type, or “Drosophila-like” SCDC, both dosage balance and complete compensation are achieved via regulation only in the heterogametic sex, i.e, two-fold hyper-transcription along the entire single X copy in males. This type of SCDC has so far only been reported among XX/XY species [8–12]. The second type is “nematode-like” SCDC, as best-known in nematodes [13] and therian mammals [14, 15]. It presents as dosage balance with partial compensation, and involves mechanisms operating in both sexes. Animals with “nematode-like” SCDC balance X expression between sexes by halving biallelic X transcription in the homogametic sex to equalize expression with the single X copy in the heterogametic male, either by repressing both X copies (nematodes) or silencing one of them (mammals). Concomitantly, limited X up-regulation in both sexes leaves reduced expression on the X relative to autosomes, reflecting partial compensation. The third type, or “avian-like” SCDC, lacks both balance and complete compensation. In contrast to chromosome-wide mechanisms seen in Drosophila-like and nematode-like SCDC, a minority of dosage-sensitive sex-linked loci in species with avian-like SCDC are locally up-regulated in the heterogametic sex while the homogametic expression remains unaffected. Notably, while all three patterns of SCDC can be found among XX/XY species, almost all WZ/ZZ taxa examined so far show avian-like SCDC [3]. The singular known exception is the insect order of Lepidoptera (moths and butterflies) [16–22], which exhibits nematode-like SCDC.
The distinction between lepidopteran insects and other WZ/ZZ taxa, and their convergence of nematode-like SCDC pattern with XX/XY taxa, raises the important question of whether there is also convergence in underlying molecular mechanisms among these systems. Investigations of SCDC mechanism, which have primarily focused on only a few model species, have revealed a common theme among chromosome-wide mechanisms (i.e., Drosophila-like and nematode-like SCDC): both sex- and sex chromosome-specific chromatin remodeling that leads to global hyper- or hypo-transcription/silencing. While a variety of histone marks are involved across species, one commonality among these systems is the modulation of two H4 histone modifications: acetylation of H4 lysine 16 (H4K16ac) and mono-methylation of H4 lysine 20 (H4K20me1) [23]. In particular, H4K16ac and H4K20me1 are both directly modulated by the dosage compensation machineries in male Drosophila melanogaster [24] and hermaphrodite Caenorhabditis elegans [25], respectively.
For Lepidoptera, the only information regarding SCDC mechanisms comes from a study on sex determination in the silkworm (Bombyx mori), which shows that RNAi knockdown of Masc, the primary masculinizing gene, results in broadly increased gene expression on the Z chromosome in males (ZZ), without changes in autosomal expression [22]. This result suggests that dosage balance observed in B. mori and other lepidopteran species [17, 19–21] is achieved by transcriptional repression of the Z in males. Furthermore, it seems likely the mechanism involves partial suppression of both Z chromosomes, like in nematodes, rather than silencing one Z chromosome, like the mammalian X-inactivation. The absence of Z-inactivation in Lepidoptera is also supported by a large body of cytogenetic studies on moths and butterflies revealing no evidence for a heterochromatinized Z chromosome in males resembling the iconic Barr body of the silenced X in mammals [26].
To shed further light on molecular mechanisms of SCDC in Lepidoptera, we used the monarch butterfly Danaus plexippus as a model, and generated a highly contiguous genome assembly to evaluate SCDC using spatial patterns of both gene expression and histone modifications. D. plexippus is of particular interest for its large neo Z chromosome, which arose from a fusion between the ancestral Z and an autosome [4] (Figure 1A). The monarch neo Z system provides a unique opportunity to contrast two groups of sex-linked genes with distinct evolutionary histories on a single sex chromosome. Throughout analyses, we partitioned the monarch Z chromosome into two segments corresponding to the ancestral Z (anc-Z, which is also the Z in other lepidopteran genera), and the portion of recent autosomal origin (neo-Z, which is autosomal in other lepidopteran genera).
Figure 1. Chromosome-level assembly of D. plexippus genome confirms the neo Z chromosome.

(A) Neo sex chromosome in D. plexippus. Anc-Z and neo-Z segments of the monarch Z chromosome are homologous to the conserved Z and an autosome respectively in non-danaid species. Lepidopterans chromosomes are holocentric (without centromeres). (B) Female to male coverage ratio of 30 chromosomal-length scaffolds. For each scaffold, mean M:F read count ratio is calculated across non-overlapping 500 bp windows. The longest chromosome is the Z, with M:F coverage ratio of 2.15. (C) Synteny between D. plexippus and S. litura. Each vertical line represents a homology block with > 80% nucleotide sequence identity. The D. plexippus Z chromosome shows bipartite mapping to S. litura chromosomes 30 and 31 (Z), reflecting its history of autosomal fusion. (D) Allelic heterozygosity in transcriptome. Female Z transcripts from both anc-Z and neo-Z are overwhelmingly homozygous compared to male or autosomal transcripts, indicating the female Z is mono-allelically expressed. For sites that were identified as polymorphic among all samples, the proportion of single nucleotide polymorphism (SNP) sites that were heterozygous in an individual was approximately 1/3 for all autosomes in both sexes as well as the Z in males. See also Table S1.
Chromosome-level assembly of the monarch genome
In order to facilitate chromosome-level analyses, we employed Chicago and Hi-C data to scaffold contigs from the D. plexippus v3 assembly [27] into a new assembly (v4) with greatly improved contiguity, including 30 chromosome-length scaffolds that range from 3.4 Mb to 15.6 Mb (Figure 1B and C, and Table S1). Previous evidence from both cytogenetic and resequencing data have indicated little, if any, sequence similarity between the entire Z and the W in D. plexippus [4]. By analyzing patterns of transcript heterozygosity using RNA-seq data, we further confirmed the mono-allelic expression of Z-linked genes in females (Figure 1D).
Pattern of dosage compensation on the anc-Z, but not the neo-Z, is consistent with other Lepidoptera
Comparing levels of gene expression between the Z and autosomes in D. plexippus reveals surprisingly distinct SCDC patterns on the two Z segments (Figure 2A). On the anc-Z, expressed genes (defined here as FPKM > 0.01), exhibit on average nearly half the autosomal levels in both sexes, as expected given previous investigations of Lepidoptera [17, 19–21]. In contrast, there is no such reduction for neo-Z genes in either sex, suggesting complete dosage compensation has evolved in heterogametic females on this recently-derived segment. This pattern is pronounced even in comparisons of the anc-Z and neo-Z with individual autosomes (Figure S1), and is robust across a range of minimum FPKM cut-offs for gene inclusion (Figure S2). The distributions of Female:Male (F:M) expression ratios, which provides a direct assessment of dosage balance, are similar between the autosomes (median AA:AA = 0.99) and either of the Z segments (median Z:ZZ = 0.96 for anc-Z and 0.94 for neo-Z, Figure 2B). Although these small absolute differences were both statistically significant, some comparisons of individual autosomes to the rest also yielded significant differences in F:M ratio (Table S2). Therefore, while a subtle dosage effect on the Z chromosome may exist, it did not appear to be qualitatively distinct from interchromosomal variations among autosomes.
Figure 2. Gene expression patterns reveal contrasting patterns of dosage compensation between anc-Z versus neo-Z segments in the D. plexippus.

(A) Gene expression levels by linkage class. Violin plots show the median, interquartile range, and distribution of log2(FPKM) for each linkage class. Dotted lines denote the median values of autosomal expression. Number of genes (n) are noted above the boxplots. Median Z:A ratios are noted under the plots. Significance of differences were contrasted between autosomes and Z segments using a Mann-Whitney U test (***P < 0.001). (B) Gene-wise correlation between female and male expression levels. Lines represent the linear regression between female and male expression. See also Figures S1, S2 and Table S2.
Given the unexpected pattern of complete compensation on the neo-Z, we further sought validation using interspecific comparative analyses, which contrast the expression levels of present-day sex-linked genes with their orthologs that have remained autosomal in another lineage [19]. Comparing monarch to Manduca sexta [17], we found no significant differences in orthologous expression ratios between the neo-Z and autosomes in both sexes (Figure 3A), thereby corroborating the assessment of complete compensation on the neo-Z based on intraspecific patterns. This comparative analysis with D. plexippus draws intriguing contrasts and parallels with the codling moth Cydia pomonella, which carries an independently evolved neo-Z segment [28]. Unlike D. plexippus, neo-Z expression in C. pomonella is reduced in both sexes when compared to M. sexta, indicating only partial compensation [19]. However, as observed in D. plexippus, anc-Z expression in C. pomonella is approximately 30% lower relative to M. sexta. No such difference in anc-Z expression was observed when making a comparison between M. sexta and Heliconius melpomene, two species both bearing the conserved ancestral Z chromosome (Figure 3B). This pattern is also reflected in intraspecific SCDC patterns across Lepidoptera. Specifically, anc-Z:A ratios are close to 0.5 in both D. plexippus and C. pomonella (indicating near absence of compensation), but are much higher in M. sexta (~0.8) and other species with the ancestral Z karyotype, consistent with partial compensation [16–18, 20, 21]. Therefore, it appears that both Z-autosome fusions in C. pomonella and D. plexippus have caused further reduction of anc-Z expression.
Figure 3. Interspecific comparisons of orthologous gene expression confirm complete compensation on the D. plexippus neo-Z.

(A) D. plexippus ~ M. sexta comparison. (B) H. melpomene ~ M. sexta comparison. Boxplots show the median (black bar), interquartile range (box), and whiskers extending one times the interquartile ranges; outliers are not plotted. Horizontal dashed lines denote the ratio of one. Number of orthologous pairs (n) are noted above the boxplots. Significance of differences (MWU test) were contrasted between autosomes and Z (segments). ***P < 0.001; F: female; M: male; Aut: autosomes.
H4K16ac, but not H4K20me1, is associated with chromosome-wide SCDC in the monarchs
Motivated by the prominent role of two H4 histone modifications (H4K16ac and H4K20me1) in mediating dosage compensation in other taxa, we next performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) in monarch heads, the same tissue used for gene expression analyses. ChIP signal intensity (using only autosomal loci) correlated positively with gene expression levels, and there were clear enrichment profiles along the gene body for substantially expressed gene (defined here as FPKM>1) that were absent for weakly- and non-expressed genes (FPKM<1) (Figure S3). Genome-wide distribution profiles of ChIP signal also mirrored the distribution profile of gene density (Figure 4A). These patterns are consistent with roles for both H4K16ac and H4K20me1 in mediating gene activation in the monarch butterfly.
Figure 4. ChIP-seq reveals contrasting levels of H4K16ac, but not H4K20me1, between the two Z segments in D. plexippus.

(A) Gene density (counts in 100 Kbp windows) and ChIP profiles (average signal per 10 Kbp window) across the genome. (B) Metagene profiles. Solid lines represent normalized ChIP score in 50 bp bins averaged across loci for each linkage group. TSS: transcription start site; TES: transcriptional end site. (C) H4K16ac enrichment levels 500 bp 5’ of TSS. In each sex, each expressed gene (FPKM > 0.01) is represented by averaging ChIP signal across ten 50-bp bins. Boxplots show median (black bar), interquartile range (box), and whiskers extending one times interquartile ranges; outliers are not plotted. Horizontal dotted lines denote median values for autosomal genes. Significance of differences (MWU test) were contrasted between autosomes and Z segments (***P < 0.001). See also Figures S3 and S4.
Next, we compared enrichment levels of these two H4 marks in relation to both linkage and sex. H4K20me1 enrichment level did not differ between males and females on either Z segment (Figure 4A and B, and Figure S4A for all chromosomes plotted individually), implying that it is unlikely to have a role in mediating SCDC in D. plexippus. In contrast, we observe striking differences in H4K16ac levels between the two Z segments, with distinct patterns between sexes (Figure 4A and B, and Figure S4A), all of which coincide with the expression patterns reported above. We further quantified these patterns by focusing on the profile peak regions (a 500 bp window 5’ of the transcription start site (TSS)) to statistically compare linkage classes in each sex (Figure 4C). In females, the neo-Z exhibited a prominent and significant enrichment of H4K16ac while the anc-Z was comparable to autosomes. This contrasting levels of H4K16ac enrichment is consistent with a neo-Z specific epigenetic compensation mechanism causing increased female transcription. Conversely, the absence of such a mechanism on the anc-Z results in little or no specific modulation of transcription, and thus an effectively monoallelic dose of expression. An opposing pattern was observed in males, where the anc-Z was significantly depleted for H4K16ac relative to autosomes, while the neo-Z was comparable to autosomes. This reduction of H4K16ac levels on the anc-Z is in accord with the comparative expression analyses described above and evidence from elsewhere [19, 22], and lends further support to the hypothesis that gene expression from the (ancestral) lepidopteran Z chromosome is epigenetically suppressed in males to match that of the single Z copy in females. The lack of such a reduction on the male neo-Z also coincides the full biallelic expression, matching both the autosomal expression and the enhanced female neo-Z expression.
Considering the ChIP-profiles outside the peak region reveals additional curious aspects of the system. First, in females, increased H4K16ac levels on the neo-Z appears to be global, in that both genic and intergenic regions exhibit a broadly consistent pattern of elevated H4K16ac levels relative to autosomes and the anc-Z (Figure 4B, and Figure S4B for intergenic regions). On the male neo-Z, however, the prominent differences in H4K16ac levels between neo-Z and anc-Z appear to be localized around TSSs. Elsewhere, H4K16ac levels are similar for the neo-Z and anc-Z, and substantially lower than autosomes. This variable pattern on the male neo-Z could reflect the interaction of two distinct epigenetic mechanisms, where ancestral chromosome-wide repression is counter-acted specifically on the neo-Z by a TSS-localized mechanism, mediated at least in part by hyperacetylation of H4K16. If so, it raises the important question of whether males and females share a mechanism for increased neo-Z transcription (as predicted by theory [5–7]) or, alternatively, the intriguing possibility that sex-specific mechanisms underlie these observations.
Conclusions
Our analyses revealed dichotomous SCDC on the single monarch Z chromosome. Anc-Z expression in the male (ZZ) is down-regulated by nearly two-fold, but with little up-regulation in the female (WZ). This nematode-like SCDC pattern on the ancestral portion of the monarch Z is generally consistent with other lepidopteran species carrying the ancestral Z karyotype, except for the extent of compensation. In contrast, the neo-Z exhibits female-specific two-fold transcriptional upregulation that correlates with a global enrichment of the activating histone mark H4K16ac, while expression and H4K16ac levels in the male remain comparable to autosomes. This Drosophila-like SCDC with complete compensation on the neo-Z, is an unprecedented observation not only among Lepidoptera, but also for all other female heterogametic taxa surveyed to date. For both the monarch butterfly and the codling moth, the presence of the neo-Z appears to constrain the compensation on the anc-Z. Additionally, the male-specific down-regulation of gene expression in monarchs is associated with global depletion of H4K16ac levels only on the ancestral portion of the Z. Unlike in C. elegans, however, monarch SCDC does not seem to involve modulation of H4K20me1, which is associated with gene activation in D. plexippus but mediates gene repression in C. elegans.
What is most surprising is the coexistence of two distinct modes of SCDC on a single sex chromosome. Neo sex chromosomes are expected to co-opt existing mechanisms rather than evolve novel ones, as seen in Drosophila systems [29, 30]. Of particular interest is how H4K16ac level is differentially regulated between both the two Z segments and between sexes in D. plexippus. Addressing this question through further detailed investigations of the molecular processes underpinning SCDC in D. plexippus and other Lepidoptera will greatly inform how SCDC evolved and differs across taxa.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Liuqi Gu (lg356@cornell.edu). This study did not generate new unique reagents.
Method Details
• Hi-C assembly
Danaus plexippus samples were kindly provided by the Monarch Watch (www.monarchwatch.org), where a large outbred colony of captive monarchs is regularly supplemented with individuals from natural populations. Chicago [31] and Hi-C [32] libraries were prepared by Dovetail Genomics (Santa Cruz, California, USA) from flash-frozen female D. plexippus, and sequenced, producing 207 and 223 million read pairs, respectively. The Chicago reads were aligned to the monarch v3 assembly [27] (GenBank assembly accession: GCA_000235995.2), misassemblies were identified and broken, and joins were made using the Dovetail HiRise pipeline. This Chicago-scaffolded assembly was then used as input along with the Hi-C reads to perform another round of misassembly detection, breaking, and scaffolding using the Dovetail HiRise pipeline, yielding the D. plexippus v4 assembly presented here. Only the 30 chromosome-size scaffolds (98.6% of the total assembly length) were included in all subsequent analyses. Further, 99.2% (15006 out of 15130) of the official gene set from the v3 assembly [27] were mapped and transferred to the new V4 assembly using GMAP (2018–07-04 release). By combining partition information from both synteny and gene models at the boundary of the previously identified breakpoint, we localized the fusion point to within a 3,051 bp window centered on position 5,685,560.
• RNA-seq
Total RNA was extracted from fresh adult (within three days of emergence) whole heads using a Qiagen® RNAeasy Kit. Each replicate represents an individual, and a total of three replicates were used for each sex. Sequencing libraries were constructed using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB) and pooled to be sequenced on NextSeq 500 platform at Cornell University Biotechnology Resource Center (Ithaca, New York, USA) with 37 bp paired-end reads. Approximately 22 million high-quality reads were generated for each library.
• Chromatin immunoprecipitation followed by sequencing (ChIP-seq)
Fresh monarch butterfly head samples were prepared from live adults of both sexes. Native (without formaldehyde crosslinking) ChIP was performed with antibodies to H4K16ac (Santa Cruz Biotechnology:sc-8662, Dallas, Texas, USA) and H4K20me1 (Abcam:ab9051, Cambridge, United Kingdom), using SimpleChIP® Enzymatic Chromatin IP Kit with magnetic beads from CellSignaling (Danvers, Massachusetts, USA) following both manufacturer’s instruction and [33] with modifications. Aliquots of the same chromatin preparations without IP (male and female) were saved as input controls and sequenced alongside IP-ed samples. DNA sequencing libraries were constructed using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®. Libraries were pooled and sequenced on NextSeq 550 platform at the University of Kansas Genome Sequencing Core (Lawrence, Kansas, USA) with 37bp paired-end reads. High-quality reads were yielded averaging 29 million reads per library.
Quantification and Statistical Analysis
• Coverage and synteny analyses
Illumina resequencing data sets for two each of female and male D. plexippus were downloaded from the NCBI’s SRA database (accession numbers: SRR1549526, SRR1548578, SRR1548504 and SRR1552222). These 100-bp paired-end reads were sequenced on Illumina HiSeq 2000 and have 22~25x genomic depth of coverage. Raw reads were mapped to the monarch v4 reference genome using Bowtie2 (v2.2.9), keeping only concordantly mapped read pairs. Male:Female coverage ratios per scaffold were calculated as median M:F ratio for 500 bp non-overlapping windows across the scaffold. In each window, median-normalized read counts were averaged within sex; windows with fewer than 10 reads in all samples were ignored.
For synteny mapping, the D. plexippus v4 genome was compared with the Spodoptera. litura genome [34] for nucleotide sequence similarity search using Satsuma (v3.1). Regions with at least 80% similarity were then used for plotting in R (v3.5.1). The tobacco cutworm S. litura was chosen for reference here because it currently has the most contiguous (chromosomal-level) genome assembly among lepidopteran species that retain the ancestral karyotype of 31 chromosomes.
• RNA-seq data processing and quantification of gene expression
These raw reads were trimmed with Trimmomatic (v0.36) for adapter sequences and end bases with low-quality and aligned to the reference genome using STAR (v2.5.2b), and quantified using express (v1.5.1). Read counts were normalized using trimmed mean of M values in fragments per kilobase of transcript per million mapped reads (FPKM) [35]. As the correlations between the three replicates for each sex were high (Pearson’s correlation 0.98–0.99 for female replicates and 0.94–0.98 for male replicates), mean values across replicates for each sex were used in all downstream analyses.
• Transcriptomic heterozygosity analysis
Variant calling and filtering on RNA-seq data was carried out following GATK (v3.7) workflow (https://software.broadinstitute.org/gatk/documentation/article.php?id=3891) [36], using BAM files of mapped RNA-seq reads as described above. Specifically, read duplicates were removed and variants were called with HaplotypeCaller. Variants were then filtered per the pipeline’s recommendation to remove clusters of at least three single nucleotide polymorphisms (SNPs) in between a 35-base window, as well as those with Fisher Strand values > 30.0 or Qual-By-Depth values < 2.0. For the purpose of this analysis, we only considered SNP sites in subsequent analyses (no indels), which were carried out in R using the Bioconductor package VariantAnnotation (v3.8). A minimum read depth of 10 was required for a SNP site to be counted in a given sample. For each library that represents an individual, percentages of heterozygous sites and homozygous sites among all SNP sites were calculated by chromosomes. Mean values across three replicates of each sex were then used for plotting.
• Comparative analysis of dosage compensation patterns
Comparative analyses were conducted as previously described in [19]. In brief, reciprocal best hit BLAST (under E-value of 1e-5) was first performed to predict 1:1 orthologs between species. A total of 10,154 1:1 orthologs were identified between the D. plexippus – Manduca sexta pair, which include 76.7% of all monarch neo-Z genes. For the Heliconius melpomene – M. sexta pair, a total of 8,952 1:1 orthologs were identified. Among these ortholog pairs, those that are considered to be expressed (FPKM > 0) in both species of the same sex were used for analysis. For each sex, all FPKM values in each species were scaled by a factor so that the median ratio of autosomal ortholog pairs equals 1. After scaling, median values of FPKM ratios between ortholog pairs (n) were contrasted using Mann-Whitney U test (MWU) for statistical significance and plotted by linkage class.
• ChIP-seq analysis
Sequencing reads were trimmed for adapter sequences and end bases with low-quality with Trimmomatic (v0.36) and aligned to the reference genome using STAR (v2.5.2b). Downstream analyses and plotting were carried out using custom bash scripts, CIRCOS (v0.69–6), deepTools suite (v2.0) and R (v3.5.1). In brief, after filtering out non-uniquely mapped reads and duplicate reads, coverage of ChIP samples was normalized using input samples following the signal extraction scaling method as described in [37]. For each histone mark, ChIP signal was then calculated using log-transformed epitope to input coverage ratio in 50 bp bins across the genome per sex. For H4K16ac, the promoter proximal region within the signal peak was further quantified for genes expressed in each sex (FPKM > 0). Each gene was represented by the mean value of ten 50 bp bins from 500 bp 5’ of transcription start site, and genes (n) were contrasted using MWU for statistical significance and plotted by linkage class.
Data and Code Availability
The D. plexippus v4 assembly is available in GenBank under BioProject PRJNA564985.
Sequencing reads for RNA-seq and ChIP-seq generated from this study are available at NCBI Short Read Archive under BioProjects PRJNA522622 and PRJNA565786.
Supplementary Material
Highlights.
A new chromosome-level assembly is generated for the monarch butterfly.
The neo-Z expression is up-regulated by two-fold in females (WZ).
The ancestral-Z expression is down-regulated by nearly two-fold in males (ZZ).
H4K16ac is enriched on neo-Z in females but depleted on ancestral-Z in males.
Acknowledgements
We thank Dr. David Soderlund and Dr. Douglas Knipple for accommodating the ChIP-seq experiments in their laboratories. Monarch Watch (www.monarchwatch.org) kindly supplied monarch butterfly samples. Florian Termin contributed to the illustration art. Two anonymous reviewers provided valuable comments on the previous manuscript draft. This work was funded by National Science Foundation (NSF-DEB 1457758 and NSF-DBI 1661454 to J.R.W.) and National Institutes of Health (R01 GM115523 to P.A.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lau AC, and Csankovszki G (2015). Balancing up and downregulation of the C. elegans X chromosomes. Curr. Opin. Genet. Dev 31, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucchesi JC, and Kuroda MI (2015). Dosage compensation in Drosophila. Cold Spring Harb. Perspect. Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu L, and Walters JR (2017). Evolution of sex chromosome dosage compensation in animals: a beautiful theory, undermined by facts and bedeviled by details. Genome Biol. Evol 9, 2461–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mongue AJ, Nguyen P, Volenfkova A, and Walters JR (2017). Neo-sex chromosomes in the monarch butterfly, Danaus plexippus. G3 (Bethesda) 7, 3281–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohno S (1967). Sex chromosomes and sex-linked genes (New York: Springer-Verlag; ). [Google Scholar]
- 6.Charlesworth B (1996). The evolution of chromosomal sex determination and dosage compensation. Curr. Biol 6, 149–162. [DOI] [PubMed] [Google Scholar]
- 7.Mank JE, Hosken DJ, and Wedell N (2011). Some inconvenient truths about sex chromosome dosage compensation and the potential role of sexual conflict. Evolution 65, 2133–2144. [DOI] [PubMed] [Google Scholar]
- 8.Vicoso B, and Bachtrog D (2015). Numerous transitions of sex chromosomes in Diptera. PLoS. Biol 13, e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahajan S, and Bachtrog D (2015). Partial dosage compensation in Strepsiptera, a sister group of beetles. Genome Biol. Evol 7, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pal A, and Vicoso B (2015). The X chromosome of hemipteran insects: conservation, dosage compensation and sex-biased expression. Genome Biol. Evol 7, 3259–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richard G, Legeai F, Prunier-Leterme N, Bretaudeau A, Tagu D, Jaquiery J, and Le Trionnaire G (2017). Dosage compensation and sex-specific epigenetic landscape of the X chromosome in the pea aphid. Epigenetics Chromatin 10, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marin R, Cortez D, Lamanna F, Pradeepa MM, Leushkin E, Julien P, Liechti A, Halbert J, Bruning T, Mossinger K, et al. (2017). Convergent origination of a Drosophila-like dosage compensation mechanism in a reptile lineage. Genome Res. 27, 1974–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albritton SE, Kranz AL, Rao P, Kramer M, Dieterich C, and Ercan S (2014). Sex-biased gene expression and evolution of the X chromosome in nematodes. Genetics 197, 865–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Julien P, Brawand D, Soumillon M, Necsulea A, Liechti A, Schutz F, Daish T, Grutzner F, and Kaessmann H (2012). Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS Biol. 10, e1001328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin F, Xing K, Zhang J, and He X (2012). Expression reduction in mammalian X chromosome evolution refutes Ohno’s hypothesis of dosage compensation. Proc. Natl. Acad. Sci. USA 109, 11752–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walters JR, and Hardcastle TJ (2011). Getting a full dose? Reconsidering sex chromosome dosage compensation in the silkworm, Bombyx mori. Genome Biol. Evol 3, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith G, Chen YR, Blissard GW, and Briscoe AD (2014). Complete dosage compensation and sex-biased gene expression in the moth Manduca sexta. Genome Biol. Evol 6, 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopinath G, Srikeerthana K, Tomar A, Sekhar SMC, and Arunkumar KP (2017). RNA sequencing reveals a complete but an unconventional type of dosage compensation in the domestic silkworm Bombyx mori. R. Soc. Open Sci 4, 170261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu L, Walters JR, and Knipple DC (2017). Conserved patterns of sex chromosome dosage compensation in the Lepidoptera (WZ/ZZ): insights from a moth neo-Z chromosome. Genome Biol. Evol 9, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walters JR, Hardcastle TJ, and Jiggins CD (2015). Sex chromosome dosage compensation in Heliconius butterflies: global yet still incomplete? Genome Biol. Evol 7, 2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huylmans AK, Macon A, and Vicoso B (2017). Global dosage compensation is ubiquitous in Lepidoptera, but counteracted by the masculinization of the Z chromosome. Mol. Biol. Evol 34, 2637–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiuchi T, Koga H, Kawamoto M, Shoji K, Sakai H, Arai Y, Ishihara G, Kawaoka S, Sugano S, Shimada T, et al. (2014). A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509, 633–636. [DOI] [PubMed] [Google Scholar]
- 23.Wells MB, Csankovszki G, and Custer LM (2012). Finding a balance: how diverse dosage compensation strategies modify histone H4 to regulate transcription. Genet. Res. Int 2012, 795069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelbart ME, Larschan E, Peng S, Park PJ, and Kuroda MI (2009). Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat. Struct. Mol. Biol 16, 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vielle A, Lang J, Dong Y, Ercan S, Kotwaliwale C, Rechtsteiner A, Appert A, Chen QB, Dose A, Egelhofer T, et al. (2012). H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS Genet. 8, e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyon MF (1961). Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190, 372–373. [DOI] [PubMed] [Google Scholar]
- 27.Zhan S, and Reppert SM (2013). MonarchBase: the monarch butterfly genome database. Nucleic Acids Res. 41, D758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen P, Sýkorová M, Šíchová J, Kůta V, Dalíková M, Čapková Frydrychova R, Neven LG, Sahara K, and Marec F (2013). Neo-sex chromosomes and adaptive potential in tortricid pests. Proc. Natl. Acad. Sci. USA 110, 6931–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bone JR, and Kuroda MI (1996). Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 144, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marin I, Franke A, Bashaw GJ, and Baker BS (1996). The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 383, 160–163. [DOI] [PubMed] [Google Scholar]
- 31.Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. (2016). Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis JJ, van der Burg KRL, Mazo-Vargas A, and Reed RD (2016). ChIP-Seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in Lepidoptera. Cell Rep. 16, 2855–2863. [DOI] [PubMed] [Google Scholar]
- 34.Cheng T, Wu J, Wu Y, Chilukuri RV, Huang L, Yamamoto K, Feng L, Li W, Chen Z, Guo H, et al. (2017). Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat. Ecol. Evol 1, 1747–1756. [DOI] [PubMed] [Google Scholar]
- 35.Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brouard JS, Schenkel F, Marete A, and Bissonnette N (2019). The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J. Anim. Sci. Biotechnol 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz A, Park K, Lim DA, and Song JS (2012). Normalization, bias correction, and peak calling for ChIP-seq. Stat. Appl. Genet. Mol. Biol 11, Article 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu TD, and Watanabe CK (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. [DOI] [PubMed] [Google Scholar]
- 39.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grabherr MG, Russell P, Meyer M, Mauceli E, Alfoldi J, Di Palma F, and Lindblad-Toh K (2010). Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 26, 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts A, and Pachter L (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 10, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, and Morgan M (2014). VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The D. plexippus v4 assembly is available in GenBank under BioProject PRJNA564985.
Sequencing reads for RNA-seq and ChIP-seq generated from this study are available at NCBI Short Read Archive under BioProjects PRJNA522622 and PRJNA565786.