

Abstract
Fosmanogepix (APX001) is a first-in-class prodrug molecule that is currently in Phase 2 clinical trials for invasive fungal infections. The active moiety manogepix (APX001A) inhibits the novel fungal protein Gwt1. Gwt1 catalyzes an early step in the GPI anchor biosynthesis pathway. Here we describe the synthesis and evaluation of 292 new and 24 previously described analogs that were synthesized using a series of advanced intermediates to allow for rapid analoging. Several compounds demonstrated significantly (8- to 32-fold) improved antifungal activity against both Cryptococcus neoformans and C. gattii as compared to manogepix. Further in vitro characterization identified three analogs with a similar preliminary safety and in vitro profile to manogepix and superior activity against Cryptococcus spp.
Keywords: APX001, APX001A, Gwt1, GPI anchor biosynthesis, Cryptococcus, antifungal, fosmanogepix, manogepix, MGX
Cryptococcus neoformans is a major cause of illness in people living with HIV/AIDS and it is uniformly fatal unless treated. Widespread availability of Antiretroviral Therapy (ART) reduced the estimated cases of cryptococcal meningitis (CM) to 200,000–300,000 cases per year, but mortality is still >50% when inferior therapeutic regimens are used.1 A recent report from Botswana illustrated that the incidence of CM has continued to be high and stable for the last 5 years despite ART use.2 Even in resource-available regions such as North America and Europe, CM still occurs with a mortality rate in some risk groups of up to 30%.3, 4 The morbidity and long-term health care costs (5 years post-infection) remain substantial.5 Thus there is an urgent need for new treatment options for CM.
In August 2019, the FDA added CM to its priority review voucher program to encourage the development of new treatment options in this area of unmet medical need, and a few companies have entered this development space. Viamet Pharmaceuticals received Fast Track designation for VT-1129 for the treatment of CM. This orally available inhibitor of fungal CYP51 additionally received orphan drug designation for the treatment of CM and has been designated a Qualified Infectious Disease Product (QIDP) by the FDA. Similarly, Amplyx Pharmaceuticals has received Orphan drug and QIDP designations for fosmanogepix (APX001), a first-in class broad spectrum antifungal agent. The active moiety of fosmanogepix inhibits the fungal enzyme Gwt1.
The Gwt1 enzyme catalyzes an early step in the glycosylphosphatidylinositol (GPI)-anchor biosynthesis pathway and was explored by Eisai as an antifungal target6. It has been shown that inhibition of Gwt1 prevents the appropriate localization of fungal cell wall mannoproteins, thus compromising cell wall integrity, germ tube formation, biofilm formation and fungal growth.7, 8 Multiple series of Gwt1 inhibitors have been optimized through extensive medicinal chemistry efforts at Eisai and other groups.9–12 E1210 [now manogepix (MGX), APX001A] is one of the most potent inhibitors and the phosphonooxymethyl prodrug (fosmanogepix, APX001, previously E1211) is currently in clinical development for the treatment of invasive fungal infections. While optimization efforts at Eisai focused on improving the activity against Candida albicans and Aspergillus fumigatus, little was known about the SAR of these compounds against Cryptococcus spp.13
In this study we synthesized a number of analogs of manogepix and evaluated their activity against two species of Cryptococcus. Compounds with minimum inhibitory concentration (MIC) values less than or equal to 0.016 μg/mL against both C. neoformans and C. gattii were assessed for stability in human liver microsomes (HLM) and cytotoxicity using a HepG2 assay. The best performing compounds were then evaluated in a Gwt1 overexpression assay to confirm the mechanism of action (MoA). The final selection criterion was lack of activity against PigW7, 14 (the closest human homolog of Gwt1) to decrease the risk of mechanism-based toxicity in humans.
Data on Gwt1 inhibitors published by Eisai suggest that compounds with potent antifungal activity against Aspergillus and Candida are composed of four structural elements (A to D), which are interconnected as shown in Figure 1.11
Figure 1.

Structural elements of Gwt1 inhibitors and rationale for designing novel analogs.
Within the described compounds, most potent analogs have an aminopyridine or diaminopyridine head group A directly connected to a 5-membered aromatic heterocycle B, preferentially an isoxazole. This two-ring system is connected via a -CH2-group to a 6-membered (hetero-)aryl ring C, preferably a phenyl ring, which is substituted at the meta- or para-position with group D. Group D consists of a one or two atom linker attached to a 5- or 6-membered (hetero-)aryl ring. A large variety of different linkers and aryl groups are tolerated at position D while maintaining potent antifungal activity against Aspergillus and Candida.
In order to establish SAR for manogepix based inhibitors of Gwt1 against Cryptococcus, we synthesized a collection of analogs of manogepix and evaluated their activity against C. neoformans and C. gattii. Based on the analysis above, we opted to hold constant the aminopyridine as head group A and the isoxazole as heterocycle B and focused on modifying groups C and D. Eisai has published several patents outlining the syntheses of manogepix related compounds.11, 15 While the synthetic routes were highly convergent, they did not allow for rapid exploration of group D analogs for various A-B-C-ring scaffolds. Therefore, we adopted Eisai’s procedures for the syntheses of the advanced intermediates I to V. From these intermediates, final compounds were accessible in only one synthetic step. The synthesis of the intermediates is outlined in Scheme 1. Eisai introduced the Boc-protected chloromethylisoxazole 115, which was used in a Suzuki coupling reaction in the synthesis of manogepix (previously known as E1210), compound 3. We observed that the hydroxypyridyl group in compound 3 (manogepix) would substitute quantitatively with chloride when refluxed in dioxane in presence of concentrated HCl. Thus, we used this method for the synthesis of the advanced intermediate I after following Eisai’s procedure for the synthesis of compound 3. The Boc-protected chloromethylisoxazole 1 also reacted smoothly with a variety of other boronic acids and boronic esters. Direct Boc-deprotection of the coupling products 4, 6 and 7 in formic acid gave access to the intermediates II, IV and V. The global deprotection of coupling product 5 was achieved by using 4 M aq. HCl in dioxane to form advanced intermediate III.
Scheme 1.

Synthesis of advanced intermediates I to V. Reagents and conditions: (i) 2 M sodium carbonate in water, palladium(0)tetrakis(triphenylphosphine), dimethoxyethane, 90 °C; (ii) formic acid, 21–25 °C; (iii) 12 M aq. HCl, dioxane, reflux; (iv) 4 M aq. HCl, dioxane, 50 °C.
With the advanced intermediates I to V in hand, we synthesized 24 previously described Gwt1 inhibitors and 292 novel compounds. Intermediate I was most versatile and underwent Suzuki coupling reactions to form either 1- or 2-carbon atom linked analogs (8a and 8b), as well as substitution reactions with a variety of nucleophiles, such as pyrazoles, phenols, anilines or amines. In that manner we were able to synthesize -CH2-linked pyrazole 9a, -CH2O-linked aryloxy analog 9b and the -CH2NH-linked derivatives 10a and 10b (Scheme 2). The free amino group in intermediate II allowed for nucleophilic aromatic substitution reactions with a variety of activated 5- and 6-membered aromatic heterocycles forming the -CH2NH-linked analogs 11a and 11b. In a similar fashion we reacted the free phenol in intermediate III to furnish the aryl ethers 12a and 12b. Phenol III also reacted smoothly with several benzyl or alkyl halides delivering -OCH2-linked compounds 12c and 12d. The fluoropyridine intermediates IV and V were equally reactive and underwent nucleophilic aromatic substitutions with a large variety of alcohols, phenols, amines and anilines to give analogs 13a-n and 14a-b, respectively (Scheme 3). Schemes 2 and 3 exemplify representative reactions for each of the intermediates, while additional analogs were synthesized in the same manner. Table 1 summarizes the activities for selected compounds against C. neoformans and C. gattii. MIC values for manogepix were of 0.25 μg/mL and 0.125 μg/mL, respectively. More than half of the synthesized compounds had significantly improved MIC values against both strains, demonstrating the great potential for optimization of this compound series towards the activity against Cryptococcus. The obtained MIC values result from interactions of our compounds with whole fungal cells and thus are a summary of multiple processes, such as cellular penetration (active/passive transport), efflux and target engagement. Each of these events reacts differently to structural changes within our compounds. This inherent drawback of phenotypical assays made it difficult to extract sharp SAR, however, we identified several trends. We did not observe any strong preference for the type (-CH2-, -O-, -NH-) or the length of the linker (1-, 2- or 3-atoms) between the C- and D-ring of the compounds with respect to their activity against Cryptococcus, though there was some interplay between various heteroaryl rings and linker types that affected activity. Meta fluoro- and chloro-substituents on the D-ring were well tolerated or improved activity, while any ortho-substituents larger than a fluorine atom decreased potency. The ortho-methyl group in 13k, for example, showed about 100-fold weaker antifungal activity compared to the unsubstituted analog 13j. Highly polar D-rings (4-amino-2-methylpyrimidine in 11b) or basic amines (10b) led to inactive compounds. Decreased antifungal activity was also observed when substituents were introduced to the linker between ring C and group D. For example, the addition of a methyl-group to the -OCH2-linker reduced the activity by 8- to 16-fold (13e vs. 13f). Compounds derived from intermediate V (linker to D-ring is meta substituent on the C-ring) were generally less active than the corresponding analogs derived from intermediate IV. A total of 11 compounds demonstrated MIC values less than or equal to 0.016 μg/mL against both strains and were advanced to in vitro metabolic stability studies. The HLM half-life (T1/2) for these compounds was determined according to the methods described under b) HLM half-life. In this experiment, the half-life of manogepix was about 50 min and we decided to advance compounds with a half-life of at least 40 min. Compound 8d with an unsubstituted furan as ring D had an extremely short half-life of about 3 min. To increase metabolic stability, a fluorine atom was introduced. The half-life of fluoro-analog 8e increased to 14 min and the compound maintained its antifungal activity. For five compounds with a half-life of 40 min or longer, we assessed the cytotoxicity in a HepG2 viability assay as described under c) Cytotoxicity assay. Only analog 12b showed significant liability at a concentration of 3 μM and was not further evaluated. The remaining four compounds were, alongside manogepix, subjected to MoA studies. Overexpression/resistance assays are a valuable tool for confirming the target-based activity of inhibitors.16 Overexpression of a target in bacteria or fungi often leads to enhanced resistance to compounds that inhibit that enzyme target. We evaluated the MIC of compounds against S. cerevisiae strains overexpressing high and low levels of Gwt1. MIC values of the compounds were determined for each strain followed by determining the ratio of the MIC values. An overexpression ratio of ≥8 is consistent with on-target activity. 12a, 13a and 13b showed ratios of 8-fold or greater against the overexpression strain, which are comparable to manogepix and are consistent with Gwt1 as the target of these inhibitors.
Scheme 2.

Representative examples for the synthesis of analogs from intermediate I. Reagents and conditions: (i) 2 M sodium carbonate in water, palladium(0)tetrakis(triphenylphosphine), dimethoxyethane, 90 °C, analogous procedures for 8b–8e; (ii) sodium hydride (60% in mineral oil), NMP, 21–25 °C then 50 °C, analogous procedure for 9b; (iii) DMF, 90 °C, analogous procedure for 10b.
Scheme 3.

Representative examples for the synthesis of analogs from intermediates II to V. Reagents and conditions: (i) N-ethyl-N-isopropylpropan-2-amine, DMSO, 120 °C, analogous procedure for 11b; (ii) potassium tert-butoxide (1M in THF), DMF, 21–25 °C then 90 °C, analogous procedures for 12b–12d; (iii) potassium tert-butoxide (1 M in THF), analogous procedures for 13b-13n and 14b.
Table 1.
Antifungal activity of compounds targeting Gwt1 against Cryptococcus spp.
| Compda | 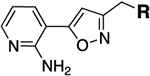 |
MIC (μg/mL)b | Compda | 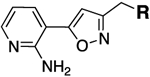 |
MIC (μg/mL)b | ||
|---|---|---|---|---|---|---|---|
| C. neoformans | C. gattii | C. neoformans | C. gattii | ||||
| MGX | 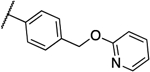 |
0.25 | 0.125 | 13a | 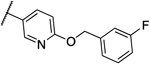 |
0.016 | 0.016 |
| 8a | 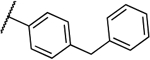 |
0.031 | 0.008 | 13b | 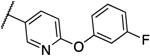 |
0.008 | 0.016 |
| 8b | 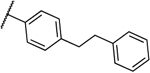 |
0.5 | 0.5 | 13c | 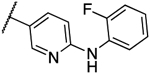 |
0.016 | 0.016 |
| 8c | 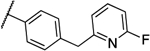 |
0.031 | 0.016 | 13d | 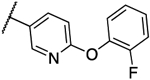 |
0.016 | 0.031 |
| 8d | 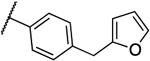 |
0.016 | 0.016 | 13e | 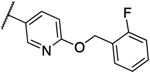 |
0.031 | 0.031 |
| 8e | 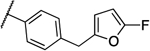 |
0.016 | 0.016 | 13f | 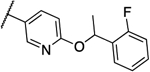 |
0.25 | 0.5 |
| 9a | 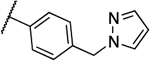 |
0.25 | 0.063 | 13g | 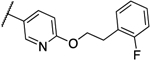 |
0.031 | 0.031 |
| 9b | 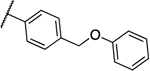 |
0.125 | 0.063 | 13h | 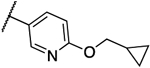 |
0.031 | 0.016 |
| 10a | 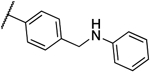 |
0.125 | 0.063 | 13i | 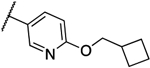 |
0.016 | 0.008 |
| 10b | 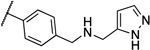 |
> 4 | > 4 | 13j | 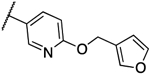 |
0.016 | 0.031 |
| 11a | 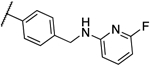 |
0.25 | 0.25 | 13k | 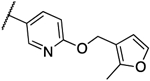 |
2 | 2 |
| 11b | 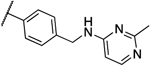 |
> 4 | > 4 | 13l | 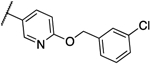 |
0.016 | 0.016 |
| 12a | 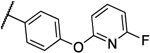 |
0.008 | 0.004 | 13m | 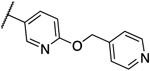 |
0.008 | 0.008 |
| 12b | 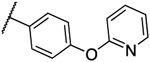 |
0.016 | 0.008 | 13n | 0.063 | 0.063 | |
| 12c | 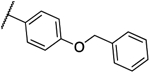 |
0.031 | 0.031 | 14a | 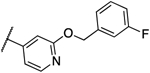 |
0.031 | 0.031 |
| 12d | 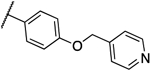 |
0.016 | 0.008 | 14b | 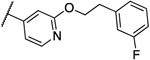 |
0.5 | 1 |
Italicized compound numbers indicate a compound that has been previously synthesized by Eisai.
MIC values were determined according to the methods under a) Antifungal susceptibility testing. Values in bold fulfill the selection criteria.
Abbreviation: MGX, manogepix
Interestingly, compound 13l, which only showed a 4-fold shift in MIC values in the overexpression assay, was also the only compound that showed significant activity against the human homolog PigW7, 14 and was deprioritized. Compounds 12a, 13a and 13b are currently under further investigation.
In summary, we have synthesized over 300 analogs of manogepix and evaluated their activity against of C. neoformans and C. gattii. We were able to improve the antifungal activity up to 32-fold against both strains while maintaining a preliminary PK and toxicity profile similar to manogepix. We have selected three compounds for further in vivo studies.
Supplementary Material
Table 2.
Metabolic stability, Cytotoxicity, and On-target activity of selected compounds.
| Compda | T1/2 HLM (min)b | HepG2 cytotoxicityc | Gwt1 Overexpression ratiod | PigW Inhibitione |
|---|---|---|---|---|
| MGX | 49.9 | 98 | 8 | 0 |
| 8d | 3.15 | - | - | - |
| 8e | 14.4 | - | - | - |
| 12a | 43.0 | 93 | 16 | 0 |
| 12b | 61.5 | 77 | - | - |
| 12d | 17.3 | - | - | - |
| 13a | 62.3 | 97 | 8 | 5 |
| 13b | 93.9 | 102 | 8 | 0 |
| 13c | 37.1 | - | - | - |
| 13i | 4.65 | - | - | - |
| 13l | 68.2 | 98 | 4 | 80 |
| 13m | 20.3 | - | - | - |
Italicized compound numbers indicate a compound that has been previously synthesized by Eisai. MGX, manogepix. Values were determined according to the methods for
under b) HLM half-life, for
under c) Cytotoxicity assay (% viability at 3 μM)), for
under d) Overexpression assay and for
under e) PigW assay (% inhibition at 16 μM). Values in bold fulfill the selection criteria.
Acknowledgements.
We thank Dr. Katsura Hata and Eisai Co. Ltd. for providing strains for the overexpression assays.
Research Support. This project was supported by Amplyx Pharmaceuticals and in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under SBIR grant award 5R44AI131864-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement. The authors are current employees of Amplyx Pharmaceuticals which has Gwt1 antifungal programs.
Methods and Experimental details for the synthesis of key intermediates and compounds are available in the Supporting Information document.
References.
- 1.Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A and Boulware DR, Lancet Infect Dis, 2017, 17, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tenforde MW, Mokomane M, Leeme T, Patel RKK, Lekwape N, Ramodimoosi C, Dube B, Williams EA, Mokobela KO, Tawanana E, Pilatwe T, Hurt WJ, Mitchell H, Banda DL, Stone H, Molefi M, Mokgacha K, Phillips H, Mullan PC, Steenhoff AP, Mashalla Y, Mine M and Jarvis JN, Clin Infect Dis, 2017, 65, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bratton EW, El Husseini N, Chastain CA, Lee MS, Poole C, Sturmer T, Weber DJ, Juliano JJ and Perfect JR, Antimicrob Agents Chemother, 2013, 57, 2485–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bratton EW, El Husseini N, Chastain CA, Lee MS, Poole C, Sturmer T, Juliano JJ, Weber DJ and Perfect JR, PLoS One, 2012, 7, e43582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charalambous LT, Premji A, Tybout C, Hunt A, Cutshaw D, Elsamadicy AA, Yang S, Xie J, Giamberardino C, Pagadala P, Perfect JR and Lad SP, J Med Microbiol, 2018, 67, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsukahara K, Hata K, Nakamoto K, Sagane K, Watanabe NA, Kuromitsu J, Kai J, Tsuchiya M, Ohba F, Jigami Y, Yoshimatsu K and Nagasu T, Mol Microbiol, 2003, 48, 1029–1042. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe NA, Miyazaki M, Horii T, Sagane K, Tsukahara K and Hata K, Antimicrob Agents Chemother, 2012, 56, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLellan CA, Whitesell L, King OD, Lancaster AK, Mazitschek R and Lindquist S, ACS Chem Biol, 2012, 7, 1520–1528. [DOI] [PubMed] [Google Scholar]
- 9.Mann PA, McLellan CA, Koseoglu S, Si Q, Kuzmin E, Flattery A, Harris G, Sher X, Murgolo N, Wang H, Devito K, de Pedro N, Genilloud O, Kahn JN, Jiang B, Costanzo M, Boone C, Garlisi CG, Lindquist S and Roemer T, ACS Infect Dis, 2015, 1, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamoto K, Tsukada I, Tanaka K, Matsukura M, Haneda T, Inoue S, Murai N, Abe S, Ueda N, Miyazaki M, Watanabe N, Asada M, Yoshimatsu K and Hata K, Bioorg Med Chem Lett, 2010, 20, 4624–4626. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, Inoue S, Murai N, Matsukura M, Nakamoto K, Shirotori S, Abe S “Heterocycles substituted pyridine derivatives and antifungal agent containing thereof”. US Patent 7,691,882 B2. April6, 2010. [Google Scholar]
- 12.McLellan L, Mazitschek R, Whitesell L, Lindquist SL “Compounds for treating infectious diseases”. PCT Int.Appl (2013), WO 2013192517 A2. [Google Scholar]
- 13.Miyazaki M, Horii T, Hata K, Watanabe NA, Nakamoto K, Tanaka K, Shirotori S, Murai N, Inoue S, Matsukura M, Abe S, Yoshimatsu K and Asada M, Antimicrob Agents Chemother, 2011, 55, 4652–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sagane K, Umemura M, Ogawa-Mitsuhashi K, Tsukahara K, Yoko-o T and Jigami Y, J Biol Chem, 2011, 286, 14649–14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsukura M “Heterocyclic ring and phosphonoxymethyl group substituted pyridine derivatives and antifungal agent containing same”. US Patent 8,513,287 B2. August20, 2013. [Google Scholar]
- 16.Prelich G, Genetics, 2012, 190, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.