

Abstract
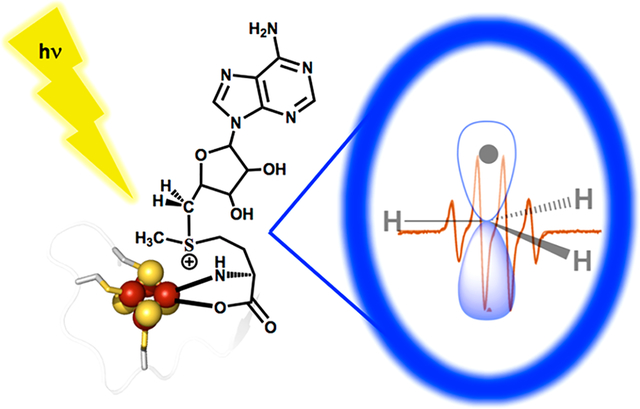
Radical SAM (RS) enzymes use S-adenosyl-l-methionine (SAM) and a [4Fe–4S] cluster to initiate a broad spectrum of radical transformations throughout all kingdoms of life. We report here that low-temperature photoinduced electron transfer from the [4Fe–4S]1+ cluster to bound SAM in the active site of the hydrogenase maturase RS enzyme, HydG, results in specific homolytic cleavage of the S–CH3 bond of SAM, rather than the S–C5′ bond as in the enzyme-catalyzed (thermal) HydG reaction. This result is in stark contrast to a recent report in which photoinduced ET in the RS enzyme pyruvate formate-lyase activating enzyme cleaved the S–C5′ bond to generate a 5′-deoxyadenosyl radical, and provides the first direct evidence for homolytic S–CH3 bond cleavage in a RS enzyme. Photoinduced ET in HydG generates a trapped •CH3 radical, as well as a small population of an organometallic species with an Fe–CH3 bond, denoted ΩM. The •CH3 radical is surprisingly found to exhibit rotational diffusion in the HydG active site at temperatures as low as 40 K, and is rapidly quenched: whereas 5′-dAdo• is stable indefinitely at 77 K, •CH3 quenches with a half-time of ~2 min at this temperature. The rapid quenching and rotational/translational freedom of •CH3 shows that enzymes would be unable to harness this radical as a regio- and stereospecific H atom abstractor during catalysis, in contrast to the exquisite control achieved with the enzymatically generated 5′-dAdo•.
Graphical Abstract
INTRODUCTION
Adenosylcobalamin (coenzyme B12) enzymes1–3 and radical S-adenosyl-l-methionine (SAM) (RS) enzymes4–6 both initiate radical reactions in biology through generation of the reactive 5′-deoxyadenosyl (5′-dAdo•) radical, as created either by homolysis of the Co–C5′ bond of B12 or by reductive cleavage of the sulfonium S–C5′ bond of SAM. This radical then abstracts an H atom from substrate. The deep parallelism of the two processes has been established by the recent discovery of a primary and mechanistically central organometallic RS intermediate Ω that contains an Fe–C5′ bond, and liberates 5′-dAdo• through homolysis of this bond in precise analogy to the liberation of 5′-dAdo• by homolysis of the Co–C5′ bond of B12.7–9
We recently sought to generate 5′-dAdo• through photolysis and homolytic Fe–C5′ cleavage of Ω, in analogy to the Co–C5′ photohomolysis of coenzyme B12.10–13 However, while doing so, we instead discovered that photoinduced electron transfer (ET) from the [4Fe–4S]1+ cluster to bound SAM in pyruvate formate-lyase activating enzyme (PFL-AE) at 12 K directly cleaved the S–C5′ bond of SAM to generate 5′-dAdo• (Figure 1).14 In short, the energy of light allowed the system to overcome the intrinsic thermodynamic barrier associated with electron transfer from the [4Fe–4S]+ cluster (midpoint potential ~ −450 to −550 mV)15–20 to SAM (cathodic peak potential ~ −1.4 V).21 The key result of this process was that the 5′-dAdo• radical thus generated was trapped next to the diamagnetic [4Fe–4S]2+ cluster in the PFL-AE active site and could be completely characterized by electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) spectroscopies, thus realizing a goal sought for over half a century.14 An underlying lesson from this experiment was that the photoinduced ET resulted in reductive cleavage of the same S–C5′ bond of SAM as is cleaved thermally during RS catalysis in PFL-AE.22,23
Figure 1.

Reductive cleavage of SAM and the structure of HydG. (A) The SAM-cluster interaction in RS enzymes (left), and the three different radical products that can result from reductive S–C bond cleavage in SAM (right). Canonical RS enzymes cleave the S–C5′ bond (red) to generate the 5′-dAdo• radical intermediate, while the noncanonical RS enzyme Dph2 cleaves the S–C(γ) bond (green) to yield an ACP radical fragment that adds to substrate. (B) Overview of the structure of HydG (PDB 4WCX), with the radical SAM cluster (top) and the auxiliary cluster (bottom) connected by a long active site tunnel (purple mesh).
Essentially all characterized canonical RS enzymes initiate radical catalysis via reductive cleavage of the S–C5′ bond of SAM. This regioselectivity has been proposed to result from the geometry of the SAM-[4Fe–4S] cluster interaction, in which the S–C5′ bond of SAM is trans to the S–Fe interaction, making the unique iron, sulfonium sulfur, and C5′ approximately in the colinear arrangement required for a radical displacement mechanism.24 Configurational interaction between the [4Fe–4S]1+ donor orbital and the S–C5′ σ* acceptor orbital is thought to facilitate specifically S–C5′ bond cleavage.25 The mechanistic formation of Ω during the reductive cleavage of SAM by RS enzymes may help to define this regioselectivity.8,9 In contrast, a different regioselectivity is observed in the noncanonical RS enzyme Dph2, where the S–C(γ) bond to the methionine side chain is cleaved during catalysis (Figure 1).26 This S–C(γ) regioselectivity, like that in canonical RS enzymes, appears to have a mechanistic origin reflecting the specific geometry of the SAM-cluster interaction, and also involves an organometallic intermediate.27
The observation of S–C5′ cleavage in canonical radical SAM enzymes, and S–C(γ) bond cleavage in Dph2, raises the question of whether enzymatic systems exist that might generate a SAM-derived methyl radical, •CH3, via reductive cleavage of the third S–C bond of SAM (Figure 1).28 Such a possibility was considered by Kampmeier, who proposed that an alternate SAM-cluster interaction geometry would lead to such homolytic S–CH3 bond cleavage;24 further, a SAM-derived •CH3 radical has been proposed as a possible intermediate during NifB-catalyzed carbon insertion into the iron–molybdenum cofactor of nitrogenase.29 However, in possible opposition to these ideas, while an entire subclass of radical SAM enzymes are known to carry out methylation chemistry using the methyl of SAM, these methylation reactions go via SN2 rather than radical mechanisms, with subsequent radical chemistry initiated via 5′-dAdo•.30
HydG is a radical SAM enzyme that catalyzes a key reaction required for maturation of the H-cluster of the [FeFe]-hydrogenase: the synthesis of the CO and CN− ligands of the H-cluster. HydG uses SAM as a cosubstrate, converting it to 5′-deoxyadenosine and methionine while initiating the radical decomposition of tyrosine to produce CO, CN−, and p-cresol; the CO and CN− are ultimately incorporated into the active-site H-cluster of the [FeFe]-hydrogenase.31–35 Two HydG [4Fe–4S] clusters are involved in this process. As is typical in RS enzymes, the [4Fe–4S]RS cluster binds SAM through the methionyl amino acid and initiates radical chemistry via the reductive cleavage of SAM to provide a 5′-dAdo• that abstracts H• from tyrosine to initiate its decomposition.33 An auxiliary [4Fe–4S]AUX cluster is involved in subsequent chemistry,33,36 and can bind a fifth, cysteine-coordinated iron to produce an S = 5/2 [4Fe–4S]AUX[FeCys] cluster.37 The fifth “dangler” iron of the [4Fe–4S]AUX[FeCys] cluster has been proposed to serve as a site for formation of an [Fe(CO)2(CN)] synthon that is transferred to the scaffold protein HydF during hydrogenase maturation.38–40 The [4Fe–4S]RS and [4Fe–4S]AUX clusters are at opposite ends of an active site cavity/tunnel spanning the central TIM barrel of HydG, with the [4Fe–4S]RS at the N-terminal end and [4Fe–4S]AUX at the C-terminal end; the distance between the two clusters is approximately 24 Å (Figure 1).37,41
We previously showed that during catalytic turnover of HydG with SAM and tyrosine, the [4Fe–4S]1+RS cluster reductively cleaves the S–C5′ bond of SAM homolytically to generate the Ω intermediate, which subsequently liberates 5′-dAdo• through Fe–C5′ bond homolysis.8 The [4Fe–4S]1+RS cluster of HydG does not reductively cleave SAM in the absence of tyrosine.42 We now report that the low-temperature photoinduced electron transfer from the [4Fe–4S]1+RS cluster to bound SAM in the substrate-free HydG active site results in the specific cleavage of the S–CH3 bond of SAM, rather than the S–C5′ bond cleaved during catalysis,31–33,43 to generate primarily a trapped •CH3 radical, with a small concentration of an Ω- like organometallic species with an Fe–CH3 bond, denoted ΩM, also observed. The •CH3 radical and ΩM species and their isotopologues have been characterized by EPR and ENDOR spectroscopies. The •CH3 radical is rapidly quenched even at 77 K and is surprisingly free to tumble in the HydG active site at temperatures as low as 40 K. We discuss the implications of these observations for the evolution of regioselective SAM S–C5′ bond cleavage for the regio- and stereospecific H atom abstraction from substrates in RS enzymes by 5′-dAdo•.
MATERIALS AND METHODS
All chemicals and other materials used herein were from commercial sources and of the highest purity available. Triton-X100 and imidazole were obtained from Alfa Aesar (Ward Hill, MA). NaCl was obtained from Sigma-Aldrich (St. Louis, MO). HEPES, potassium phosphate, IPTG, PMSF, tryptone, yeast extract, MOPS, DTT, glucose, and streptomycin were obtained from RPI (Mt. Prospect, IL). Ferrous ammonium sulfate was obtained from J.T. Baker Chemical Company (Phillipsburg, New Jersey). MgCl2, KCl, and glycerol were obtained from EMD (Gibbstown, NJ). Sodium dithionite and sodium sulfide were obtained from Acros Organics (Fair Lawn, NJ). DNase I, RNase A, and lysozyme (hen egg) were obtained from Roche (Indianapolis, IN). Iron(III) chloride and l-cysteine were obtained from Fisher Scientific (Fair Lawn, NJ).
Wild-Type HydGΔEF Expression and Purification.
HydG was overexpressed and purified with some modifications to previously published procedures.31,33,34,44 The Clostridium acetobutylicum hydG gene (CAC1356) was cloned into a pCDFDuet vector to allow for the expression of the N-terminally, 6×-histidine tagged form of the protein;44 the vector was transformed into E. coli BL21(DE3) (Stratagene) cells. This expression system of the HydG enzyme in the absence of HydE and HydF is denoted HydGΔEF. Fresh agar plates were streaked and single colonies were utilized for 50 mL overnight cultures in media supplemented with 50 μg/mL streptomycin. The following morning, the overnight cultures were used to inoculate 9 L LB that was buffered with 50 mM potassium phosphate, pH 7.5, and additionally contained 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl, 5 g/L glucose, and 50 μg/mL streptomycin. These cultures were then grown aerobically at 37°C with 230 rpm shaking. When OD600 values were between 0.5–0.6, 0.09 g/L of ferrous ammonium sulfate (FAS) and IPTG (1 mM final concentration) were added. The cultures continued to grow under aerobic conditions for an additional 2.5 h at 37°C with 230 rpm shaking. At this time, the cultures were cooled to room temperature and supplemented with an additional aliquot of 0.09 g/L FAS. The cultures were then transferred to a 4°C refrigerator and actively sparged with N2(g) for 15 h. The following morning, the cells were harvested via centrifugation and resulting cell pellets were flash frozen in liquid N2 and stored at −80°C until lysis and purification.
Cell lysis and protein purification were carried out in a Coy chamber (Grass Lake, MI) that was maintained with a ~97% N2(g), ~3% H2(g) atmosphere and housed inside a 4°C walk-in cold room. Frozen cell pellets were transferred to the Coy chamber and then thawed and resuspended in a lysis buffer containing 50 mM Tris pH 8.0, 0.25 M KCl, 5% glycerol, and 10 mM imidazole (Buffer A). Lysis mixtures also contained 20 mM MgCl2, 1 mM PMSF, 0.8% Triton X-100, 0.4 mg lysozyme per gram of cells, and 0.07 mg DNase and RNase per gram of cells. This mixture was then stirred for 60 min, transferred into gastight centrifuge bottles, and centrifuged for 30 min at 18 000 rpm. Clarified supernatants were then passed over a 5 mL HisTrap Ni2+-affinity column (GE Healthcare, Piscataway, NJ) using an ÄKTA Basic 100 FPLC (GE Healthcare). Purified HydG was eluted from the column using an imidazole gradient with increasing amounts of Buffer B (50 mM Tris, pH 8.0, 0.25 M KCl, 5% glycerol, 500 mM imidazole). Purification fractions were immediately pooled together and buffer exchanged into a 50 mM Tris, pH 8.0, 0.25 M KCl, 5% glycerol buffer using a Sephadex G25 resin column. Eluted fractions were concentrated using Amicon Ultra 10 kDa molecular weight cutoff filter devices (Millipore). Concentrated protein aliquots were then flash frozen in liquid N2 and stored at −80°C until further use. Protein was quantified via Bradford assays using a bovine serum albumin standard solution (Thermo Scientific). Iron in purified protein samples was quantified via a Varian SpectrAA 220 FS flame atomic absorption spectrometer in comparison to a 0.4–2.0 ppm standard curve created from a 1000 ppm iron AA standard solution (Ricca Chemical Company).
Chemical Reconstitution of HydG.
HydG typically purifies with substoichiometric amounts of Fe loading. For example, the WT HydGΔEF enzyme utilized herein contained 3.2 ± 0.1 Fe/protein in its as-purified state. It was therefore necessary to chemically reconstitute protein samples in order to improve [Fe–S] cluster loading. Reconstitutions were carried out with minor modifications to our previously published procedures.31,33,34 Briefly, HydG enzyme (≤150 μM) was incubated with a 6-fold excess of FeCl3 and Na2S in the presence of 5 mM DTT in 50 mM Tris, pH 8.0, 0.25 M KCl, 5% glycerol buffer. The reconstitutions were carried out in scintillation vials for 2.5–3.5 h with gentle stirring in an anaerobic Coy chamber. Following the reconstitution period, mixtures were centrifuged to pellet bulk FeS precipitates and resulting supernatants were passed over a Sephadex G-25 column to remove adventitiously bound iron, sulfide, and DTT. Gel filtered proteins were then concentrated using Amicon Ultra 10 kDa molecular weight cutoff filter devices (Millipore). Aliquots of protein were then flash frozen in liquid N2 and stored at −80°C until spectroscopic characterization. Bradford analysis and iron quantitation were performed as described above. Following reconstitution, WT HydGΔEF harbored 7.7 ± 0.2 Fe atoms/protein.
Preparation of Dangler Loaded HydG.
Reconstituted HydGΔEF was loaded with the dangler iron via modification of previously published protocols.39,45 Dangler loading was accomplished by treating as-reconstituted HydG (150 μM) with 2 mM NaDT, 1.25 equiv of FeCl3, 10 equiv of SAM, and 7 equiv of l-cysteine. The mixture was incubated at 4°C with stirring in a Coy anaerobic chamber for 45 min. Following the incubation period, the sample was immediately passed over a Sephadex G-25 column, and key fractions were pooled together, and then concentrated using Amicon Ultra 10 kDa molecular weight cutoff filters (Millipore). The resulting protein stocks were then aliquoted, flash frozen in liquid N2, and stored at −80°C until spectroscopic characterization. Final iron quantitation numbers were 9.4 ± 0.3 Fe/protein.
Sample Preparation.
In an anaerobic Coy chamber, HydG samples for intracavity photolysis and EPR spectroscopic analysis were prepared by mixing enzyme with Na-dithionite to reduce the clusters to the 1+ state, SAM, and buffer (50 mM Tris (pH 8.0), 250 mM KCl, 10% glycerol), to final concentrations of 100 μM protein, 3 mM DT, 5.5 mM SAM, respectively. These samples were then briefly centrifuged, transferred to EPR tubes, flash frozen, and stored in liquid N2.
EPR and ENDOR Measurements.
These measurements were carried out as described recently.14 X-band CW EPR spectroscopy was conducted on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910, while Q-band CW EPR spectroscopy was conducted on a Bruker EMX spectrometer equipped with an Oxford Instruments Mercury iTC continuous helium flow cryostat. Typical experimental parameters were at 12 and 40 K, 9.38 or 34.0 GHz, and 10 G modulation amplitude. EPR simulations were performed with the EasySpin5,2,23 program operating in Matlab.46 35 GHz CW and pulse ENDOR spectroscopic data were collected on spectrometers, described previously,47–49 that are equipped with liquid helium immersion dewars for measurements at 2 K. The CW measurements employed 100 kHz field modulation and dispersion-mode detection under rapid passage conditions. 1H CW ENDOR spectra employed bandwidth broadening of the RF to 100 kHz to improve signal-to-noise.50
RESULTS
Photoinduced ET in the HydG/[4Fe–4S]1+RS/SAM Complex Generates •CH3.
Irradiation of the SAM-bound [4Fe–4S]+ state of substrate-free HydG with 450 nm light at 12 K in the EPR cavity results in rapid loss of the signal from the [4Fe–4S]1+RS cluster (Figure 2A), while leaving undiminished the signal from the second [4Fe–4S]AUX cluster. This is accompanied by the appearance of a new free-radical signal (Figure 2B). Figure 2C shows that the time course for the loss of [4Fe–4S]1+RS signal directly correlates with that of the appearance of the new radical signal. Measurements of the radical signal intensity generated by comparable total flux of light of 400, 450, and 520 nm showed a ratio of intensities of I(400)/I(450)/I(520) ~ 1/0.6/0.1 (Figure S1), in rough correspondence with the wavelength dependence of the absorbance spectrum.
Figure 2.

X-band EPR spectra of (A) ([4Fe–4S]+ + SAM) HydG complex before and (B) after photolysis at 12 K with 450 nm LED for 1 h. (C) Time course for decay of ([4Fe–4S]+ + SAM) (■) upon photolysis monitored at 3640 G, and increase of •CH3 (●) monitored at 3306 G. The photo reaction appears to be complete at ~90 m, as the EPR spectrum does not change upon further photolysis. The two progress curves are then normalized and fit to stretched-exponential decay, I = exp(−[t/τ]n), and rise, I = 1 − exp(−[t/τ]n), functions with the same parameters, τ = 7 ± 1 min and n = 0.52 ± 0.02. In (B), the unchanged signal from [4Fe–4S]AUX[(Cys)Fe] cluster and that from •CH3 are both identified. EPR conditions: microwave frequency, 9.38 GHz; modulation 10 G; T = 12 K.
As previously,14 these results indicate that photoinduced electron transfer (ET) from the [4Fe–4S]1+RS to SAM causes reductive cleavage of SAM to generate the diamagnetic [4Fe–4S]2+RS cluster and a cryotrapped radical. However, this radical is not 5′-dAdo•, as previously observed upon photolysis of the SAM-bound [4Fe–4S]1+ state of PFL-AE.14 Rather, the species generated in HydG exhibits a quartet EPR signal, Figure 2B, better resolved at 40 K, Figure 3A, that is quite different from that of 5′-dAdo•. This signal is precisely simulated by a four-line pattern with 1/3/3/1 intensity ratio as predicted for isotropic hyperfine coupling to three equivalent 1H with aiso = 63.6 MHz. This pattern and splitting value are characteristic of the •CH3 radical,51 thereby identifying the signal as arising from this species. This identification is confirmed by the collapse of the 1H splittings when CD3-SAM is used, Figure 3B, and the appearance of an overlapping doublet of quartets, generated by a 13C hyperfine coupling, a3(13C) = 80 MHz, when 13CH3–SAM is used, Figure 3C. Photoreduction has cleaved the S–CH3 bond of SAM, not the S–C5′ bond.
Figure 3.

EPR spectra (black) and simulations (red). (A) Spectrum of rapidly tumbling •CH3 generated with natural abundance SAM, obtained by subtraction of the properly scaled signal from the persistent signal (see text). (B) •CD3 generated with methyl-d3-SAM. (C) •13CH3 generated with 13C-methyl-SAM. Conditions: T = 40 K, modulation amplitude, 5 G; see Figure 4 for spectra with lower modulation. EasySpin simulation parameters: function, chili; g = [2.0015, 2.0015, 2.0055,], (giso = 2.003). For spectra (A) and (C), A(1Ha,b,c) = −[86, 44, 62] MHz (aiso(1Ha,b,c) = −63.3 MHz), (α,β,γ) = (120, 0, 0) for 1Ha, (α,β,γ) = (−120, 0, 0) for 1Hb, (α,β,γ) = (0, 0, 0) for 1Hc; for spectrum (C), A(13C) = [−10, −10, 260] MHz. τC = 3.2 ns; uniform Gaussian line widths 21 MHz.
The quartet signal from the •CH3 radical rapidly decays at 77 K, with a half-time of a few minutes, and progressively more rapidly at higher temperatures (Figure S2). The decay of this signal on annealing reveals a weak underlying minority signal, which persists up to 150 K and has been subtracted from the spectra shown in Figure 3. The signal that persists after annealing, Figure 4, is not that of a “free” methyl radical. It is narrower (Figure S3), does not show resolved 1H hyperfine splittings (Figures 4, S3), and can be simulated with an envelope that arises from an anisotropic g-tensor, g = [2.021, 2.0047, 1.987] (Figure 4A). This anisotropy is distinctly less than that of the organometallic intermediate Ω.7 However, when this species is generated with 13CH3–SAM the signal broadens significantly; the broadening can be simulated by the addition of a 13C hyperfine coupling, aiso(13C) ~ 23 MHz (Figure 4B). This observation is confirmed by 13C ENDOR (CW, 35 GHz, 2 K): despite the low population of this species, we were able to obtain a 13C ENDOR signal that corresponds to a weakly anisotropic coupling with aiso(13C) ≈ 19 MHz, Figure 4C. Although this aiso(13C) is about twice the coupling to the 13C5-dAdo• bound to an Fe of Ω,7 both couplings are much smaller than the coupling expected for an isolated, immobilized •13CH3 radical, which also would be highly anisotropic, rather than nearly isotropic, and with much larger couplings, A(13C) ≈ [−10, −10, 260] MHz.7 The most plausible interpretation of these observations is that during cleavage of the S–CH3 bond of SAM a small fraction of the •CH3 reacts with the RS cluster, forming an Fe–CH3 bond to generate an organometallic species, ΩM, analogous to Ω. However, for completeness we note an alternative formulation, in which the •CH3 reacts with the RS cluster to form an S–CH3 bond.29
Figure 4.

X-band EPR spectra. (A) ΩM (12CH3); (B) ΩM (13CH3); (C) 35 GHz 13C CW ENDOR Spectra of ΩM (13CH3). Inverted triangle, 13C Larmor frequency; “goalpost”, half the hyperfine coupling, aiso/2. EPR conditions: T = 40 K; modulation amplitude, 5 G; microwave frequency, 9.38 GHz. ENDOR conditions: T = 2 K; modulation amplitude, 4 G; scan speed, 3 MHz/s; scan direction, forward; microwave frequency, 34.8 GHz. EasySpin EPR simulations: function, pepper; g = [2.021, 2.0047,1.987]; Gaussian line widths 30 MHz; for spectrum (B), A(13C) = [20, 30, 20] MHz; Hstrain = [60, 30, 60] MHz.
Mobility of •CH3 in HydG.
As discussed below, the extent of the motional freedom of •CH3 in the active-site during catalysis at near-ambient temperatures is important in understanding the properties of RS enzymes. As this freedom will be far greater under those conditions than at cryogenic temperature, we have examined the low-temperature results as giving insights into and a lower limit to the rotational and translational mobility during catalysis.
The statistical 1/3/3/1 intensity pattern of the •CH3 radical at 40 K and above (Figures 3A and 5) implies that the radical is undergoing rapid rotational diffusion within the active site even down to these low temperatures. Although •CH3 is roughly disk-shaped and presumably the rotational diffusion is to some extent anisotropic, the spectra of •12CH3 and •13CH3 at 40 K can actually be jointly described by the simplification of treating reorientation as a fully three-dimensional (classical; isotropic) rotational diffusion. As seen in Figure 3, both these 40 K spectra are satisfactorily reproduced with a rotational correlation time of τc ≈ 3 ns, at least 10-fold longer (slower rotation) than would be expected at ambient temperature, although nonetheless in the “fast-motion” regime. The lines in the ·13CH3 spectrum are broader because of less complete averaging of the 13C hyperfine anisotropy, whose magnitude is ~6.4 times greater ((A∥ − A⊥) ~ 270 MHz) than that of an α-proton (|Amax − Amin| ~ 42 MHz). Some additional remarks about 3D vs 1D •CH3 rotation are presented in the SI (SI text, Figure S4).
Figure 5.

Temperature dependence of the •CH3 signal. Conditions: modulation amplitude, 2G to avoid modulation broadening in the higher-T spectra; power adjusted at each temperature to avoid saturation; because of this adjustment, the intensities are simply normalized to equal height.
The rapid tumbling of the •CH3 radical seen here at ~40 K and above has commonly been observed at liquid nitrogen temperatures and above, as illustrated by a few examples: •CH3 in frozen solutions of solvents ranging from nonpolar to aqueous;52 at 77 K generated by X-irradiation of CH3I;53 at 210 K trapped in zeolite cages;54 and as transiently generated through photolysis of MeCob in aqueous solution at 284 K.55 In this last case, the lines of the quartet at 280 K are extremely narrow, thus demonstrating the expected enhanced rotational mobility near ambient temperatures.
As the sample is cooled to ~30 K and below, the quartet seen above 30 K does not simply broaden, as would be expected if τc for tumbling smoothly increased, nor does the spectrum convert to that characteristic of a low-temperature quantum rotor (for example, refs 56, 57). Instead, the spectrum appears to progressively convert to a broadened quartet that can be assigned to a more slowly tumbling •CH3. Such a transition would appear to reflect progressive conversion of the site into a conformation exhibiting greater hindrance to tumbling, with conversion nearly complete by ~4 K, Figure 5. For completeness, in the SI, we make further comments about •CH3 rotation as exhibited here.
The •CH3 radical signal rapidly decays at 77 K (Figure S2), in clear contrast to the behavior of 5′-dAdo• formed photochemically in PFL-AE; the 5′-dAdo• radical is indefinitely stable at 77 K and only decays at temperatures above ~100 K.14 This decay is not accompanied by the appearance of any new paramagnetic species, other than ΩM as discussed above. We considered the possibility that the rapid •CH3 decay might reflect the existence of a “tunnel” between the RS and AUX [4Fe–4S] clusters of HydG (Figure 1B). This offered the possibility that •CH3 formed at the [4Fe–4S]RS cluster might diffuse through the tunnel to, and react with, the [4Fe–4S]AUX[(Cys)Fe] cluster, and that this reaction might cause the observed decay of the •CH3 signal. We tested this possibility by measuring the intensity of the [4Fe–4S]AUX[(Cys)Fe] cluster signals, both the S = 1/2 signal seen in Figure 2, and the S = 5/2 signals that appear at low fields,37 after 77 K annealing had eliminated the •CH3 signal: the intensity of the [4Fe–4S]AUX[(Cys)Fe] cluster signals had not changed from those seen promptly after photolysis, ruling out this possibility, Figure S5. We thus conclude that •CH3 is intrinsically more reactive than 5′-dAdo•. This is consistent with its greater C–H bond-dissociation free energy, but is more likely associated with its ability to undergo rapid translational diffusion “in search” of a reaction partner. Except when •CH3 is strongly confined by a rigid, covalently bonded environment, for example, when isolated in a zeolite cage54 or adsorbed on a silica surface,58 it diffuses quite freely and in consequence quenches rapidly, as seen here in HydG.
DISCUSSION
We report here that cryogenic photoinduced electron transfer from the [4Fe–4S]1+RS cluster to bound SAM in the active site of HydG results in the cleavage of the S–CH3 bond of SAM, rather than the S–C5′ bond as is cleaved in enzymatic catalysis by nearly all RS enzymes,6 including HydG,34,42 or in photolysis of SAM-bound [4Fe–4S]1+ PFL-AE.14 The S–CH3 bond cleavage reported here is also distinct from the S–C(γ) bond cleaved in the noncanonical RS enzyme Dph2.26 The radical product of HydG/SAM photolysis is, remarkably, a •CH3 free radical trapped in the HydG active site. While a methyl radical has been suggested as a possible intermediate during catalysis by the radical SAM enzyme NifB,29 which inserts the methyl carbon of SAM as the central carbide of the iron–molybdenum cofactor of nitrogenase, no prior evidence for reductive homolytic cleavage of the S–CH3 bond of SAM in a RS enzyme has been reported. Further, while a methyl radical mechanism has been proposed for methyl-coenzyme M reductase,59 no direct evidence for a methyl radical has been reported for this or other enzymatic systems.
The •CH3 cleavage product of HydG/[4Fe–4S]+/SAM photolysis is highly reactive and rotationally and translationally mobile, and clearly could not reliably carry out regioselective H atom abstraction from a substrate. We have been unable to carry out photolysis of HydG/[4Fe–4S]+ in the presence of both SAM and tyrosine due to the rapid reaction of the ternary complex, analogous to the limitations on photolysis of the PFL-AE/[4Fe–4S]+/SAM/PFL complex.14 However, decades of study of •CH3 in the widest range of environments and temperatures have clearly shown that because of its small size and weak interactions with its surroundings,52–55 •CH3 could not be effectively harnessed. As indicated above, the many published studies of •CH3 show that the small •CH3 radical would not only be intrinsically unstable under turnover conditions, but would also undergo rapid rotation within the active site.52–55 Clearly, even in the presence of substrate tyrosine, •CH3 would be highly mobile and not subject to enzymatic control: it could not be constrained to adopt the precise positioning required for selective H atom abstraction.60
The present study thus provides a powerful reason for nature’s “choice” of radical initiator in RS enzymes: despite the fact that •CH3 would be a somewhat more potent H atom abstractor than 5′-dAdo• based on their relative C–H bond energies,61 enzymatic RS reactions require precise control of H atom abstraction from substrate.6 In canonical RS enzymes, the large 5′-dAdo• species fits this requirement, as it can be efficiently directed to its target through nonbonded and H-bonding contacts with active-site residues. Indeed, as we have shown,60 once the 5′-dAdo• free radical is liberated it is “never free”, but is tightly guided through an atom- and regio-/stereospecific reaction with substrate. The enzyme avoids the insurmountable problems associated with formation of •CH3 by using active-site control to ensure the reductive cleavage of S–C5′ to form the relatively easy to control 5′-dAdo• radical, and not cleavage of S–C(Me) to form •CH3.24,60 The 3-amino,3-carboxypropyl (ACP) radical formed in the noncanonical RS enzyme Dph2 by S–C(γ) bond cleavage is anchored by amino and carboxylate coordination to the unique iron of the [4Fe–4S] cluster, which enhances control but also limits its ability to react with the wide range of substrates dealt with in the full RS superfamily.
The tight regioselectivity of the enzymatic reductive cleavage of SAM observed across the diverse RS superfamily raises the question: why is the S–CH3 bond cleaved, with full regiospecificity, in the current study, whereas photoinduced ET of the PFL-AE/[4Fe–4S]1+/SAM complex results in specific S–C5′ bond cleavage? The results for these two systems indeed suggest that photoexcitation of the [4Fe–4S]1+RS/SAM complex in RS enzymes yields an excited state that is capable of transferring an electron to cleave any of the three S–C bonds of SAM. Although the factors that determine the regioselectivity of S–C bond cleavage during photoinduced ET of [4Fe–4S]1+RS/SAM complexes are not currently understood, it appears that in contrast to the enzymatically catalyzed reactions, this selectivity does not require major differences in the geometry of the SAM-cluster interaction, as this geometry is very similar in PFL-AE and HydG (Figure S6).37,62 In addition, while the S–C bond energies in SAM are not currently known, it appears that differences in these energies are not responsible for the S–C bond cleavage regioselectivity during photoinduced ET, as we observed 100% regioselectivity for S–C(Me) bond cleavage in the current study, yet complete regioselectivity for S–C5′ bond cleavage in our recent study of PFL-AE.14
It is of interest to note that the direct product of catalytic cleavage of both the S–C5′ and S–C(γ) carbons in RS enzymes and the noncanonical RS enzyme Dph2, respectively, is Ω or an Ω-like organometallic intermediate, yet the photoinduced S–C5′ cleavage in PFL-AE generates 5′-dAdo• without forming Ω,14 and the photoinduced cleavage of S–CH3 in HydG yields only a small amount of the ΩM species in addition to •CH3. These variations in cleavage product demonstrate just how subtle are the active-site interactions that determine the radical created by SAM cleavage. Of central importance to the present considerations, the generation of Ω during catalytic cleavage of SAM S–C5′ and at least partial generation of ΩM during photoinitiated cleavage of S–CH3 provides a strong connection between the catalytic and photoinitiated processes.
In summary, active-site control of SAM positioning dictates the cleavage of the S–C5′ bond of SAM to form 5′-dAdo• during catalysis by RS enzymes,24 and control of 5′-dAdo• positioning guides the stereospecific reactions of this agent with the broad range of substrate targets within the RS superfamily.60 Photoinitiated electron transfer to SAM in the substrate-free [4Fe–4S]1+ HydG active site instead induces cleavage of SAM to exclusively generate •CH3, some of which forms the organometallic species ΩM with its Fe–CH3 bond.
The photoinduced ET reported here for the HydG [4Fe–4S]1+/SAM complex, as well as that in our recent study of PFL-AE,14 are among the first examples of photochemistry involving iron–sulfur clusters.63–65 The remarkable specificity and efficiency of the photoinduced ET in both these enzymes leads us to wonder whether nature might have taken advantage of such processes. For example, might there be radical SAM enzymes that are directly activated by light to initiate radical reactions? Could such enzymes in this ancient superfamily have been involved in photochemical reactions during the early evolution of life? It is interesting to note that adenosylcobalamin, which exhibits remarkable parallels to the RS [4Fe–4S]/SAM system,9 has recently been found to take advantage of its inherent photochemistry to function as a light sensor.66 Although to date no evidence for a light-activated RS enzyme has been reported, our ability to use light to quantitatively generate organic radicals in enzyme active sites could very well lead to novel approaches to initiating radical reactions for synthetic or other applications.
The rapid quenching and rotational/translational freedom of •CH3 shows that the HydG active site would be unable to effectively harness this radical as a regio- and stereospecific H atom abstractor during catalysis, thus highlighting the importance of enzymatic control of both the formation and the reactions of 5′-dAdo•.
Supplementary Material
ACKNOWLEDGMENTS
EPR, ENDOR, and photolysis experiments were funded by the NIH (GM 111097 to BMH). All work related to the preparation of HydG was funded by the U.S. Department of Energy, Office of Basic Energy Sciences grant DE-SC0005404 (to JBB and EMS). The synthesis of labeled SAMs was supported by the NIH (GM 54608 and 131889 to JBB). The authors thank Ryan Swimley (MSU) for his assistance with the preparation of HydG.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08541.
Supplementary figures, methods, and text (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Banerjee R; Ragsdale SW The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem 2003, 72, 209–247. [DOI] [PubMed] [Google Scholar]
- (2).Brown KL Chemistry and Enzymology of Vitamin B12. Chem. Rev 2005, 105, 2075–2149. [DOI] [PubMed] [Google Scholar]
- (3).Toraya T Radical catalysis in coenzyme B-12-dependent isomerization (eliminating) reactions. Chem. Rev 2003, 103, 2095–2127. [DOI] [PubMed] [Google Scholar]
- (4).Frey PA Lysine 2,3-aminomutase: is adenosylmethionine a poor man’s adenosylcobalamin? FASEB J. 1993, 7, 662–670. [DOI] [PubMed] [Google Scholar]
- (5).Frey PA; Magnusson OT S-Adenosylmethionine: A wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem. Rev 2003, 103, 2129–2148. [DOI] [PubMed] [Google Scholar]
- (6).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-adenosylmethionine enzymes. Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Horitani M; Shisler KA; Broderick WE; Hutcheson RU; Duschene KS; Marts AR; Hoffman BM; Broderick JB Radical SAM catalysis via an organometallic intermediate with an Fe-[5′-C]-deoxyadenosyl bond. Science 2016, 352, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Byer AS; Yang H; McDaniel EC; Kathiresan V; Impano S; Pagnier A; Watts H; Denler C; Vagstad AL; Piel J; Duschene KS; Shepard EM; Shields TP; Scott LG; Lilla EA; Yokoyama K; Broderick WE; Hoffman BM; Broderick JB Paradigm shift for radical S-adenosyl-L-methionine reactions: The organometallic intermediate Ω is central to catalysis. J. Am. Chem. Soc 2018, 140, 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Broderick WE; Hoffman BM; Broderick JB Mechanism of radical initiation in the radical S-adenosyl-l-methionine superfamily. Acc. Chem. Res 2018, 51, 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rury AS; Wiley TE; Sension R Energy cascades, excited state dynamics, and photochemistry in cob(III)alamins and ferric porphyrins. Acc. Chem. Res 2015, 48, 860–867. [DOI] [PubMed] [Google Scholar]
- (11).Robertson WD; Warncke K Photolysis of adenosylcobalamin and radical pair recombination in ethanolamine ammonia-lyase probed on the micro- to millisecond time scale by using time-resolved optical absorption spectroscopy. Biochemistry 2009, 48, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Robertson WD; Wang M; Warncke K Characterization of protein contributions to cobalt-carbon bond cleavage catalysis in adenosylcobalamin-dependent ethanolamine ammonia-lyase by using photolysis in the ternary complex. J. Am. Chem. Soc 2011, 133, 6968–6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jones AR The photochemistry and photobiology of vitamin B12. Photochem. Photobiol. Sci 2017, 16, 820–834. [DOI] [PubMed] [Google Scholar]
- (14).Yang H; McDaniel EC; Impano S; Byer AS; Jodts RJ; Yokoyama K; Broderick WE; Broderick JB; Hoffman BM The elusive 5′-deoxyadenosyl radical: captured and characterized by electron paramagnetic resonance and electron nuclear double resonance spectroscopies. J. Am. Chem. Soc 2019, 141, 12139–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ugulava NB; Gibney BR; Jarrett JT Biotin synthase contains two distinct iron-sulfur cluster binding sites: Chemical and spectroelectrochemical analysis of iron-sulfur cluster interconversions. Biochemistry 2001, 40, 8343–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang SC; Frey PA Binding energy in the one-electron reductive cleavage of S-adenosylmethionine in lysine 2,3-aminomutase, a radical SAM enzyme. Biochemistry 2007, 46, 12889–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Maiocco SJ; Grove TL; Booker SJ; Elliott SJ Electrochemical resolution of the [4Fe–4S] centers of the AdoMet radical enzyme BtrN: Evidence of proton coupling and an unusual, low-potential auxiliary cluster. J. Am. Chem. Soc 2015, 137, 8664–8667. [DOI] [PubMed] [Google Scholar]
- (18).Blaszczyk AJ; Silakov A; Zhang B; Maiocco SJ; Lanz ND; Kelly WL; Elliott SJ; Krebs C; Booker SJ Spectroscopic and electrochemical characterization of the iron-sulfur and cobalamin cofactors of TsrM, an unusual radical S-adenosylmethionine methylase. J. Am. Chem. Soc 2016, 138, 3416–3426. [DOI] [PubMed] [Google Scholar]
- (19).Maiocco SJ; Arcinas AJ; Landgraf BJ; Lee K-H; Booker SJ; Elliott SJ Transformations of the FeS clusters of the methylthiotransferases MiaB and RimO, detected by direct electrochemistry. Biochemistry 2016, 55, 5531–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Walker LM; Kincannon WM; Bandarian V; Elliott SJ Deconvoluting the reduction potentials for the three [4Fe–4S] clusters in an AdoMet radical SCIFF maturase. Biochemistry 2018, 57, 6050–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Miller SA; Bandarian V Analysis of electrochemical properties of S-adenosylmethionine and implications for its role in radical SAM enzymes. J. Am. Chem. Soc 2019, 141, 11019–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Knappe J; Schmitt T A novel reaction of S-adenosyl-L-methionine correlated with the activation of pyruvate formate-lyase. Biochem. Biophys. Res. Commun 1976, 71, 1110–1117. [DOI] [PubMed] [Google Scholar]
- (23).Frey M; Rothe M; Wagner AFV; Knappe J Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. J. Biol. Chem 1994, 269, 12432–12437. [PubMed] [Google Scholar]
- (24).Kampmeier JA Regioselectivity in the homolytic cleavage of S-adenosylmethionine. Biochemistry 2010, 49, 10770–2. [DOI] [PubMed] [Google Scholar]
- (25).Dey A; Peng Y; Broderick WE; Hedman B; Hodgson KO; Broderick JB; Solomon EI S K-edge XAS and DFT calculations on SAM dependent pyruvate formate-lyase activating enzyme: nature of interaction between the Fe4S4 cluster and SAM and its role in reactivity. J. Am. Chem. Soc 2011, 133, 18656–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Y; Zhu X; Torelli AT; Lee M; Dzikovski B; Koralewski RM; Wang E; Freed J; Krebs C; Ealick SE; Lin H Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 2010, 465, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dong M; Kathiresan V; Fenwick MK; Torelli AT; Zhang Y; Caranto J; Dzikovski B; Sharma A; Lancaster KM; Freed JH; Ealick SE; Hoffman BM; Lin H Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis. Science 2018, 359, 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Broderick JB A radically different enzyme. Nature 2010, 465, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wiig JA; Hu Y; Lee CC; Ribbe MW Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 2012, 337, 1672–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bauerle MR; Schwalm EL; Booker SJ Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem 2015, 290, 3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shepard EM; Duffus BR; George SJ; McGlynn SE; Challand MR; Swanson KD; Roach PL; Cramer SP; Peters JW; Broderick JB [FeFe]-Hydrogenase maturation: HydG-catalyzed synthesis of carbon monoxide. J. Am. Chem. Soc 2010, 132, 9247–9249. [DOI] [PubMed] [Google Scholar]
- (32).Driesener RC; Challand MR; McGlynn SE; Shepard EM; Boyd ES; Broderick JB; Peters JW; Roach PL [FeFe]-hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew. Chem., Int. Ed 2010, 49, 1687–90. [DOI] [PubMed] [Google Scholar]
- (33).Driesener RC; Duffus BR; Shepard EM; Bruzas IR; Duschene KS; Coleman NJ-R; Marrison APG; Salvadori E; Kay CWM; Peters JW; Broderick JB; Roach PL Biochemical and kinetic characterization of radical S-adenosylmethionine enzyme HydG. Biochemistry 2013, 52, 8696–8707. [DOI] [PubMed] [Google Scholar]
- (34).Duffus BR; Ghose S; Peters JW; Broderick JB Reversible H atom abstraction catalyzed by the radical S-adenosylmethionine enzyme HydG. J. Am. Chem. Soc 2014, 136, 13086–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kuchenreuther JM; George SJ; Grady-Smith CS; Cramer SP; Swartz JR Cell-free H-cluster synthesis and [FeFe] hydrogenase activation: All five CO and CN- ligands derive from tyrosine. PLoS One 2011, 6, No. e20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pagnier A; Martin L; Zeppieri L; Nicolet Y; Fontecilla-Camps JC CO and CN- syntheses by [FeFe]-hydrogenase maturase HydG are catalytically differentiated events. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dinis P; Suess DLM; Fox SJ; Harmer JE; Driesener RC; De La Paz L; Swartz JR; Essex JW; Britt RD; Roach PL X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kuchenreuther JM; Myers WK; Suess DLM; Stich TA; Pelmenschikov V; Shiigi SA; Cramer SP; Swartz JR; Britt RD; George SJ The HydG enzyme generates an Fe(CO)2(CN) synthon in assembly of the FeFe hydrogenase H-cluster. Science 2014, 343, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Suess DLM; Pham CC; Bürstel I; Swartz JR; Cramer SP; Britt RD The radical SAM enzyme HydG requires cysteine and a dangler iron for generating an organometallic precursor to the [FeFe]-hydrogenase H-cluster. J. Am. Chem. Soc 2016, 138, 1146–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rao GD; Tao LZ; Suess DLM; Britt RDA [4Fe–4S]-Fe(CO)(CN)-L-cysteine intermediate is the first organometallic precursor in [FeFe] hydrogenase H-cluster bioassembly. Nat. Chem 2018, 10, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nicolet Y; Pagnier A; Zeppieri L; Martin L; Amara P; Fontecilla-Camps JC Crystal structure of HydG from Carboxydothermus hydrogenoformans: A trifunctional [FeFe]-hydrogenase maturase. ChemBioChem 2015, 16, 397–402. [DOI] [PubMed] [Google Scholar]
- (42).Pilet E; Nicolet Y; Mathevon C; Douki T; Fontecilla-Camps JC; Fontecave M The role of the maturase HydG in [FeFe]-hydrogenase active site synthesis and assembly. FEBS Lett. 2009, 583, 506–511. [DOI] [PubMed] [Google Scholar]
- (43).Rubach JK; Brazzolotto X; Gaillard J; Fontecave M Biochemical characterization of the HydE and HydG iron-only hydrogenase maturation enzymes from Thermatoga maritima. FEBS Lett. 2005, 579, 5055–5060. [DOI] [PubMed] [Google Scholar]
- (44).McGlynn SE; Shepard EM; Winslow MA; Naumov AV; Duschene KS; Posewitz MC; Broderick WE; Broderick JB; Peters JW HydF as a scaffold protein in [FeFe] hydrogenase H-cluster biosynthesis. FEBS Lett. 2008, 582, 2183–2187. [DOI] [PubMed] [Google Scholar]
- (45).Suess DLM; Bürstel I; De La Paz L; Kuchenreuther JM; Pham CC; Cramer SP; Swartz JR; Britt RD Cysteine as a ligand platform in the biosynthesis of the FeFe hydrogenase H cluster. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11455–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Stoll S; Schweiger A EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (47).Werst MM; Davoust CE; Hoffman BM Ligand Spin Densities in Blue Copper Proteins by Q-band 1H and 14N ENDOR Spectroscopy. J. Am. Chem. Soc 1991, 113, 1533–1538. [Google Scholar]
- (48).Zipse H; Artin E; Wnuk S; Lohman GJS; Martino D; Griffin RG; Kacprzak S; Kaupp M; Hoffman B; Bennati M; Stubbe J; Lees N Structure of the nucleotide radical formed during reaction of CDP/TTP with the E441Q-a2b2 of E. coli ribonucleotide reductase. J. Am. Chem. Soc 2009, 131, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Davoust CE; Doan PE; Hoffman BM Q-Band Pulsed Electron Spin-Echo Spectrometer and Its Application to ENDOR and ESEEM. J. Magn. Reson., Ser. A 1996, 119, 38–44. [Google Scholar]
- (50).Hoffman BM; DeRose VJ; Ong JL; Davoust CE Sensitivity enhancement in field-modulated CW ENDOR via RF bandwidth broadening. J. Magn. Reson., Ser. A 1994, 110, 52–57. [Google Scholar]
- (51).McKenzie I; Brodovitch J-C; Ghandi K; McCollum BM; Percival PW Hyperfine coupling in methyl radical isotopomers. J. Phys. Chem. A 2007, 111, 10625–10634. [DOI] [PubMed] [Google Scholar]
- (52).Greatorex D; Kemp TJ Electron spin resonance studies of photo-oxidation by metal ions in rigid media at low temperatures. Part 3. - Ce(IV) photo-oxidations of aldehydes, ketones, esters and amides. J. Chem. Soc., Faraday Trans 1 1972, 68, 121–129. [Google Scholar]
- (53).Cole T; Pritchard HO; Davidson NR; McConnell HM Structure of the methyl radical. Mol. Phys 1958, 1, 406–409. [Google Scholar]
- (54).Danilczuk M; Pogocki D; Lund A; Michalik J EPR and DFT study on the stabilization of radiation-generated methyl radicals in dehydrated Na-A zeolite. J. Phys. Chem. B 2006, 110, 24492–24497. [DOI] [PubMed] [Google Scholar]
- (55).Bussandri AP; Kiarie CW; Van Willigen H Photoinduced bond homolysis of B12 coenzymes. An FT-EPR study. Res. Chem. Intermed 2002, 28, 697–710. [Google Scholar]
- (56).Kubota S; Iwaizumi M; Ikegami Y; Shimokoshi K Anisotropic hyperfine interaction in the electron spin resonance spectrum of the methyl radical trapped in CH3COONa•3D2O crystal at low temperatures. J. Chem. Phys 1979, 71, 4771–4776. [Google Scholar]
- (57).Yamada Y; Komaguchi K; Shiotani M; Benetis NP; Sørnes AR High-resolution EPR and quantum effects on CH3, CH2D, CHD2, and CD3 radicals under argon matrix isolation conditions. J. Phys. Chem. A 1999, 103, 4823–4829. [Google Scholar]
- (58).Turkevich J; Fujita Y Methyl radicals: Preparation and stabilization. Science 1966, 152, 1619–1621. [DOI] [PubMed] [Google Scholar]
- (59).Wongnate T; Sliwa D; Ginovska B; Smith D; Wolf MW; Lehnert N; Raugei S; Ragsdale SW The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science 2016, 352, 953–958. [DOI] [PubMed] [Google Scholar]
- (60).Horitani M; Byer AS; Shisler KA; Chandra T; Broderick JB; Hoffman BM Why nature uses radical SAM enzymes so widely: electron nuclear double resonance studies of lysine 2,3-aminomutase show the 5′-dAdo• “free radical” is never free. J. Am. Chem. Soc 2015, 137, 7111–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Blanksby SJ; Ellison GB Bond dissociation energies of organic molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (62).Vey JL; Yang J; Li M; Broderick WE; Broderick JB; Drennan CL Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16137–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mao Z; Liou S-H; Khadka N; Jenney FEJ; Goodin DB; Seefeldt LC; Adams MWW; Cramer SP; Larsen D Cluster-dependent charge-transfer dynamics in iron-sulfur proteins. Biochemistry 2018, 57, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lukoyanov D; Khadka N; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Reversible photoinduced reductive elimination of H2 from the nitrogenase dihydride state, the E4(4H) Janus intermediate. J. Am. Chem. Soc 2016, 138, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Lukoyanov D; Khadka N; Dean DR; Raugei S; Seefeldt LC; Hoffman BM Photoinduced reductive elimination of H2 from the nitrogenase dihydride (Janus) state involves a FeMo-cofactor-H2 intermediate. Inorg. Chem 2017, 56, 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Jost M; Simpson JH; Drennan CL The transcription factor CarH safeguards use of adenosylcobalamin as a light sensor by altering the photolysis products. Biochemistry 2015, 54, 3231–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.