

Abstract
The centrosome is a cytoplasmic nonenveloped organelle functioning as one of the microtubule-organizing centers and composing a centriole center surrounded by pericentriolar material (PCM) granules. PCM consists of many centrosomal proteins, including PCM1 and centrosomal protein 131 (CEP131), and helps maintain centrosome stability. Zika virus (ZIKV) is a flavivirus of the family Flaviviridae whose RNA and viral particles are replicated in the cytoplasm. However, how ZIKV interacts with host cell components during its productive infection stage is incompletely understood. Here, using several primate cell lines, we report that ZIKV infection disrupts and disperses the PCM granules. We demonstrate that PCM1- and CEP131-containing granules are dispersed in ZIKV-infected cells, whereas the centrioles remain intact. We found that ZIKV does not significantly alter cellular skeletal proteins, and, hence, these proteins may not be involved in the interaction between ZIKV and centrosomal proteins. Moreover, ZIKV infection decreased PCM1 and CEP131 protein, but not mRNA, levels. We further found that the protease inhibitor MG132 prevents the decrease in PCM1 and CEP131 levels and centriolar satellite dispersion. Therefore, we hypothesized that ZIKV infection induces proteasomal PCM1 and CEP131 degradation and thereby disrupts the PCM granules. Supporting this hypothesis, we show that ZIKV infection increases levels of mind bomb 1 (MIB1), previously demonstrated to be an E3 ubiquitin ligase for PCM1 and CEP131 and that ZIKV fails to degrade or disperse PCM in MIB1-ko cells. Our results imply that ZIKV infection activates MIB1-mediated ubiquitination that degrades PCM1 and CEP131, leading to PCM granule dispersion.
Keywords: centrosome, flavivirus, deubiquitylation (deubiquitination), E3 ubiquitin-protein ligase RNF8 (RNF8), infection, centrosomal protein 131 (CEP131), mind bomb 1 (Mib1), pericentral material 1 (PCM1), Zika virus (ZIKV), pericentrin (PCNT), ubiquitination, proteasomal degradation, single-stranded RNA virus, microtubule-organizing center (MTOC)
Introduction
Zika virus (ZIKV),3 an emerging mosquito-transmitted pathogen, is causatively associated with neonatal microcephaly when the infection occurs in pregnant women (1–3). ZIKV, together with West Nile virus, yellow fever virus, Japanese encephalitis virus, and Dengue fever virus, belongs to the family Flaviviridae (4, 5). It is likely that interactions of viral proteins with host factors determine the fate and/or efficiency of infection, pathogenicity, transmission, and epidemic potential. This family of viruses has an enveloped icosahedral capsid that contains a single-stranded RNA genome (about 11,000 nucleotides) with positive sense (6). Therefore, the infected viral RNA can be directly translated to a large polyprotein precursor, which is co- and post-translationally processed by viral and cellular proteases into three structural and seven nonstructural (NS) proteins.
The centrosome is a cytoplasmic unenveloped organelle functioning as one of the microtubule-organizing centers and hence is an important regulator of cell-cycle progression (7–9). It has been reported that the centrosome is important in different biological processes, including cell proliferation, differentiation, and development (10). The centrosome is essential for cell-cycle regulation in eukaryotic cells because the perturbation of core centrosomal or centrosome-associated proteins results in cell-cycle dysregulation and certain cancers (11). A centrosome contains a core of two centrioles arranged orthogonally. These two centrioles remain attached until mitosis. Each centriole is composed of a nine-triplet microtubule assembled in a tubular structure, with other components such as centrin, cenexin, and tektin (10). Pericentriolar materials (PCM) are the proteins surrounding the centriole, especially the γ-tubulin ring complexes (γ-TuRCs). EM revealed that PCM proteins form electron-dense granules of around 70–100 nm in diameter localizing around the centrosome in various cell types (12). The granules were initially detected next to newly appearing daughter centrioles during centriole duplication in replicating cells (12). The PCM proteins include γ-tubulin, pericentrin, and ninein and are responsible for microtubule nucleation (13). Pericentriolar material 1 (PCM1, also known as PTC4 or RET/PCM-1) and CEP131 (also called AZI1) are components of centriolar satellites (14), which presents as cytoplasmic granules around the centrosome (15–19).
PCM1 is a 228-kDa coiled-coil protein with multiple isoforms that interacts with the cellular centrosome through the G1, S, and part of G2 phases of the cell cycle, but disassociates with the centriole at the late G2 and M phases (20). Studies by depletion, mutation, or dysfunction of PCM1 demonstrated that PCM1 comprises a structural platform for centriolar satellites, is important for satellite particle assembly, and is essential for the correct localization of several centrosomal proteins and for anchoring microtubules to the centrosome (10). PCM-1 interacts with more than 50 different proteins, one of which is CEP131. CEP131, a 131-kDa protein, is recruited to centriolar satellites by PCM1 (21). The biological function of CEP131 is largely unknown. Because ZIKV has been demonstrated to affect stem cell division and proliferation, it is important to know whether ZIKV infection could affect these proteins that are related to centrosome.
Recently, we and other groups have investigated the ZIKV-triggered centrosome abnormality. It was reported that ZIKV infection caused disruption of the centrosome (22) and that infection of ZIKV resulted in a significant increase of foci harboring centriolar markers and impaired spindle positioning (23, 24). Furthermore, an ultrastructural study revealed that ZIKV infection caused cytoskeletal changes that may have unidentified effects on centrosomal function (25). Our present studies show that ZIKV infection disrupts the association of PCM1 and CEP131 with the centrosome and causes the Mib1-mediated ubiquitination and proteasome-dependent degradation of PCM1 and CEP131.
Results
ZIKV infection disperses PCM1 from centrosome in SK-N-SH, U-251MG, Vero, ARPE-19, neural stem cells (NSCs), and MRC-5 cells
The centriole is a barrel-like structure, composed of multiple proteins, that resides at the core of the centrosome (26). Its functions include recruiting PCM proteins and nucleating centrosomes. Antibodies against pericentrin (PCNT), centrosomal protein 135 (CEP135), or γ-tubulin are commonly used to show the loci of centrioles, and antibodies against CEP131 or PCM1 are used to show the pericentriolar materials by immunocytochemistry (ICC). As shown in Fig. 1, ICC with anti-PCNT antibody shows only one or two condensed dots in each SK-N-SH cell (Fig. 1A2). Other centrosome-related proteins are needed for centrosome structure and maturation and stability, such as PCM1 (Fig. 1A3). PCM1 is distributed in the cytoplasm with a center domain where it associates with PCNT (Fig. 1, A1–A3). The association between PCNT and PCM1 at the centriole can be observed in many other cells, such as MRC-5, U-251MG, ARPE-19, NSC, and Vero cells (Fig. 1D). We set out to know whether this association could be affected by ZIKV infection. Both MR766 (African strain) and PRVABC59 (Asian strain) strains of ZIKV were used in this study, and the results from the two strains are the same. We present the results from MR766 in Fig. 1. At 24 h after MR766 infection of SK-N-SH cells, ICC assays were performed to show the viral E protein and PCM1 (as shown in Fig. 1B) or E protein and PCNT (Fig. 1C). The center condensed domain of PCM1 disappears with a dispersed distribution around the cytosol of the ZIKV-infected SK-N-SH cell, whereas the uninfected cell (leftmost cell in Fig. 1B3) retains an intense PCM1 center. Although the PCM1 structure is disrupted, the PCNT core distribution remains intact after ZIKV infection (Fig. 1C3). A similar effect of ZIKV MR766 on PCM1 has been observed in NSC, as shown in Fig. 2B. ZIKV infection clearly dispersed the PCM1 granules in the NSC but had no effects on PCNT (Fig. 2A) that colocalizes with PCM1 in uninfected cells as shown in Fig. 2C.
Figure 1.

ICC to show the interaction of ZIKV with centrosome proteins. A1–A4, relationship of PCNT and PCM1 in SK-N-SH cells. A1, merged image; A2, PCNT to show the centriole; A3, PCM1 to show the PCM1 granules; A4, DAPI to show the nuclei. B, effects of ZIKV MR766 on PCM1 granules in SK-N-SH cells. B1, merged image; B2, E protein to show the viral infection; B3, PCM1 to show the PCM1 granules; B4, DAPI to show the nuclei. C, effects of ZIKV MR766 on centriole in SK-N-SH cells. C1, merged image; C2, E protein to show the ZIKV infection; C3, PCNT to show centriole; C4, DAPI. D, distribution of PCM1 and PCNT in different cells. ICC was performed to show the relationship of PCNT (green) and PCM1 (red) in the cell line as indicated. PCNT and PCM1 colocalize in all of the cells: U251 MG, MRC-5, Vero, ARPE-19, and NSC. E, percentile of the cells in which the PCM1 was dispersed in different cells: MRC-5, U-251 MG, SK-N-SH, Vero, ARPE-19, and NSC. The degree of significance was as follows: *, p < 0.001. F, same as B, but photographs were taken with a ×40 lens so that more cells could be visualized. Scale bar, 10 μm. Error bars, S.E.
Figure 2.

ICC to show the interaction of ZIKV with centrosome proteins in NSC. A, PCNT was not affected by ZIKV infection. PCNT is shown in red, and ZIKV infection is shown by E protein in green. DAPI was used to show the nuclei. B, effects of ZIKV MR766 on PCM1 granules in NSC cells. C, colocalization of PCNT and PCM1. Scale bar, 10 μm.
We performed similar experiments in other ZIKV-permissive cell lines (MRC-5, U-251MG, ARPE-19, and Vero) but extended the infection time to 48 h, and the results were not different among the different cells. We then counted the number of cells in which the PCM1 center domain was dispersed in ZIKV-infected cells versus noninfected cells. The rate of PCM1 dispersion in noninfected cells is ∼5%, but the rate of PCM1 dispersion in ZIKV-infected cells ranges from 70 to 90% (Fig. 1E). Statistical analysis was carried out using a pairwise two-tailed t test to compare the two groups (ZIKV-infected cells versus noninfected cells), and the differences were significant (Fig. 1E; *, p < 0.001). Therefore, our experimental results demonstrate that ZIKV infection causes dispersion of PCM1 from the centriole. SK-N-SH, Vero, MRC-5, ARPE-19, and U-251MG are all permissive for ZIKV replication (27). SK-N-SH is a neuroblastoma cell line that displays epithelial morphology, grows very well in adherent culture (28), and is frequently used to study neural stem cell differentiation, so we chose SK-N-SH cells for subsequent experiments.
As mentioned above, there were still a small portion of ZIKV-infected cells that had intact PCM1. To show this, we took a photograph under the confocal immunofluorescence microscope with a ×40 lens so that more cells could be visualized. As shown in Fig. 1F, SK-N-SH cells were infected with ZIKV at an MOI of 1 for 24 h. IFA was performed to detect the viral protein (NS3) and cellular protein (PCM1). Among the 14 ZIKV-infected cells (shown by anti-NS3 antibody in green as indicated by the stars), only five of them contain intact PCM1 granules (indicated by red stars). PCM1 granules in all of the other ZIKV-infected cells are dispersed as indicated by white stars. If the viral infection is extended to 48 h, the dispersion of PCM1 can reach higher rates. We also demonstrated that ZIKV PRVABC59 had PCM1-dispersing effects as shown in Fig. 3.
Figure 3.

ICC to examine the interaction of ZIKV PRVABC59 and PCM1. SK-N-SH cells were grown on coverslips and infected with ZIKV PRVABC59 at an MOI of 0.5 for 24 h. The cells were then fixed for IFA to check viral protein (E protein in green) and cellular proteins (PCM1 in red). Scale bar, 10 μm.
Like PCNT, CEP135 is another centriolar protein because it forms a core structure within nascent centrioles (29, 30), so we further examined the centriole using ICC to test whether ZIKV infection modifies the morphology of CEP135. After ZIKV MR766 infection for 48 h at an MOI of 0.5, the SK-N-SH cells were stained for CEP135 in green and NS3 in red (Fig. 4A). One or two centrioles can be seen in the infected cells (top two cells) and are intact as in uninfected cells (bottom one cell). No significant modifications in numbers and volume of the CEP135 were observed upon ZIKV infection. These results are consistent with the observation for PCNT as shown in Fig. 1C. Cytoskeleton proteins are reported to be related to biogenesis of centrosome (31). To know whether ZIKV has any effects on cellular morphology, we further examined the ZIKV infection in SK-N-SH cells for cytoskeleton proteins, including α-tubulin, β-actin, and acetyl-α-tubulin. As shown in Fig. 4 (B–D), the pictures were taken to include both infected and uninfected cells. We did not observe a significant modification of the cytoskeleton proteins by ZIKV infection. The results are reproducible in PRVABC59 infected cells. First, PCM1 was dispersed by ZIKV PRVABC59 at 24 hpi in SKN-SH cells as shown in Fig. 3. In addition, ZIKV PRVABC59 infection caused no significant modification to cellular skeletal proteins, such as CEP135, α-tubulin, acetyl-α-tubulin, and β-actin (Fig. 5). Therefore, the results from the ICC assays suggest that ZIKV infection causes the dispersion of PCM1, which is not due to ZIKV infection–caused cytoskeleton alteration.
Figure 4.

ICC to examine the interaction of ZIKV and cellular proteins. SK-N-SH cells were grown on coverslips and infected with ZIKV at an MOI of 0.5 for 24 h. The cells were then fixed for IFA to check viral protein (NS3) and cellular proteins. A, CEP135 to show centriole; B, α-tubulin; C, acetyl-α-tubulin; D, β-actin. Scale bar, 10 μm.
Figure 5.

ICC to examine the interaction of ZIKV PRVABC59 and cellular proteins. SK-N-SH cells were grown on coverslips and infected with ZIKV PRVABC59 at an MOI of 0.5 for 24 h. The cells were then fixed for IFA to check viral protein (NS3) and cellular proteins. A, CEP135, to show centriole; B, α-tubulin; C, acetyl-α-tubulin; D, β-actin. Top, ZIKV-infected cells; bottom, mock-infected cells.
More interestingly, we were curious about whether other flaviviruses disperse PCM1 granules. For that purpose, we infected SK-N-SH cells with DENV-2 for 24 h. The cells were fixed and stained for DENV E protein in red and PCM1 in green as shown in Fig. 6. As can be seen, the PCM1 dotlike structure remains intact in both DENV- and mock-infected cells. Therefore, it might be specific for ZIKV to cause dispersion of PCM1.
Figure 6.

ICC to examine the interaction of DENV and PCM1. SK-N-SH cells were grown on coverslips and infected with ZIKV PRVABC59 at an MOI of 0.5 for 24 h. The cells were then fixed for IFA to check viral protein (E protein in red) and cellular proteins (PCM1 in green). Scale bar, 10 μm.
ZIKV infection caused dispersion of CEP131
Another PCM protein, CEP131, is associated with PCM1 (32) and responds to regulation of cell stress. We wanted to determine whether ZIKV infection could cause the similar alteration of CEP131. First, we performed IFA for SK-N-SH cells to visualize the CEP131 and PCM1 at a normal state (uninfected cells). As shown in Fig. 7A, PCM1 (red) and CEP131 (green) presented as dotlike structures in the cytoplasm and co-localized in almost all of the cells. Then we examined the effects of ZIKV infection on CEP131 distribution. The SK-N-SH cells were infected with ZIKV MR766 at an MOI of 0.5 for 24 h, fixed, and immunostained with anti-CEP131 (red) and -NS3 (green). As can be seen in Fig. 7B, the dotlike structure of CEP131 is dispersed in most ZIKV-infected cells. Therefore, CEP131 is another PCM component that is dispersed following ZIKV infection.
Figure 7.

ICC to examine the interaction of CEP131 with PCM1 and ZIKV protein. A, PCM1 and CEP131 colocalize in SK-N-SH cells. The photograph was taken with a ×40 lens. B, CEP131 was dispersed by ZIKV infection (MOI of 0.5) in SK-N-SH cells for 24 h. The photograph was taken with a ×40 lens. Scale bar, 10 μm.
ZIKV infection induced ubiquitination and degradation of PCM1, which can be prevented by a protease inhibitor, MG132
We noticed in the ICC experiments shown in Fig. 1 that PCM1 density is much lower in the ZIKV-infected cells than in mock-infected cells. This reduction may result from protein instability and/or translational defects. To determine whether proteasome-dependent protein degradation is involved in PCM1 dispersion after ZIKV infection, we selected MG132, a well-known inhibitor of proteasome-dependent protein degradation. The SK-N-SH cells were infected with ZIKV MR766 at an MOI of 1, and the medium was changed to contain 10 μm MG132 or no MG132 12 h before collecting samples. Mock-infected cells were used as a control. Whole-cell lysate samples were then collected at 24 or 48 hpi. Western blotting assays were performed to examine the levels of PCM1. As shown in Fig. 8A, the ZIKV-infected cells have much less PCM1. However, when the cells were treated with 10 μm MG132, the PCM1 degradation was prevented. Next, we tested for dose effects of MG132 on protection of PCM1. As shown in Fig. 8B, we infected SK-N-SH cells with ZIKV (MR766 on the left and PRVABC59 on the right) at an MOI of 1 for 12 h, and the medium was then changed to contain different concentrations of MG132 as indicated and incubated for another 12 h. A mock infection was used as control. Clearly, the cells that are treated with more MG132 contain more undegraded PCM1, so the protective effects of MG132 on PCM1 are dose-dependent.
Figure 8.

Degradation of PCM1 by ZIKV in SK-N-SH cells. A, Western blotting assay to examine the levels of PCM1 and Mib1 in SK-N-SH cells that are infected or mock-infected with ZIKV at an MOI of 1 in the presence or absence of MG132 (10 μm). The MG132 was added 12 h before sample collection. NS3 shows viral infection, and tubulin is for control of sample loading. B, Western blotting assay to determine the dose dependence of MG132 in inhibiting the degradation of PCM1 by ZIKV. SK-N-SH cells were infected with ZIKV at an MOI of 1 for 24 h. The MG132 was added 12 h before collecting the samples. C, immunoprecipitation and Western blotting assays to determine ubiquitination of PCM1. The whole-cell lysate from mock-infected SK-N-SH cells, ZIKV-infected SK-N-SH cells, or ZIKV-infected SK-N-SH cells treated with TAK-243 (MLN7243) (purchased from Chemietek, catalog no. AOB87172) was incubated with anti-PCM1 and protein G beads; the beads were washed; and the eluted samples were probed with anti-PCM1 or anti-ubiquitin antibody.
To determine whether ubiquitination is involved in the degradation of PCM1, we performed an immunoprecipitation (IP) assay using anti-PCM1 antibody to pull down the total PCM1 and then blotted with anti-ubiquitin or anti-PCM1 antibody. As shown in Fig. 8C, the immunoprecipitated samples include those from mock-infected cells (first lane), ZIKV-infected cells (second lane), and ZIKV-infected cells treated with TAK-243, which is a ubiquitin-activating enzyme (UAE) inhibitor (third lane). TAK-243 (also called MLN7243) prevents the conjugation of ubiquitin to the protein through inhibition of UAE1 (33). As shown in the input, ZIKV infection decreased the protein level of PCM1, but treatment of TAK-243 rescued the degradation of PCM1. The co-IP results showed that ZIKV infection enhanced the interaction of ubiquitin with PCM1, and the interaction was inhibited by TAK-243. Therefore, ubiquitination is utilized in viral PCM1 degradation.
MG132 prevents the dispersion of PCM1 by ZIKV infection
Next, we asked whether the dispersion of PCM1 granules depends on the proteasomal degradation of PCM1 protein. In doing so, we infected the SK-N-SH cells with ZIKV MR766 at an MOI of 1 for 24 h and then added MG132 at a final concentration of 10 μm, and 12 h later, the cells were fixed for an immunofluorescent assay. A no-MG132 group was used as control as shown on the left side of Fig. 9A. Consistently, as shown in Fig. 9 (A and B), we found that the presence of MG132 prevents the dispersion of PCM1. Therefore, the dispersion of PCM1 induced by ZIKV-infection involves a process of ubiquitination and proteasomal degradation.
Figure 9.

Effect of MG132 on the ZIKV-induced dispersion of PCM1 in SK-N-SH cells. SK-N-SH cells were infected with ZIKV at an MOI of 1 for 24 or 48 h. A, the cells were assayed by ICC to examine PCM1 in red and E protein in green. Left, no treatment of MG132; right, cells were treated with MG132. Scale bar, 10 μm. B, the positively infected cells (24 or 48 hpi) were counted to determine the percentiles of cells in which the PCM1 was dispersed or not dispersed (right). *, p < 0.01. Error bars, S.E.
ZIKV infection induced protein levels of Mib1, but not PCM1 or CEP131
To determine whether a PCM1 gene suppression occurs upon ZIKV infection at the transcriptional level, we performed real-time RT-PCR as shown in Fig. 10 to determine the mRNA level. The total RNA was isolated from either mock-infected or ZIKV-infected (at an MOI of 1) SK-N-SH cells. Real-time RT-PCR was performed for PCM1, PCNT, CEP131, and PCM1-related proteins such as Mib1, USP9X, CEP290, and GAPDH. The mRNA levels of PCM1, PCNT, CEP131, CEP290, USP9X, and Mib1 were normalized to GAPDH and averaged from two independent experiments as shown in Fig. 10. The mRNA levels of PCM1, PCNT, Mib1, CEP290, USP9X, and CEP131 are slightly reduced upon ZIKV infection; only the reductions of CEP290 and PCNT mRNA levels are statistically significant (p < 0.001) after t test. Therefore, ZIKV infection negatively regulated the protein levels of the centrosomal genes moderately at transcriptional level.
Figure 10.
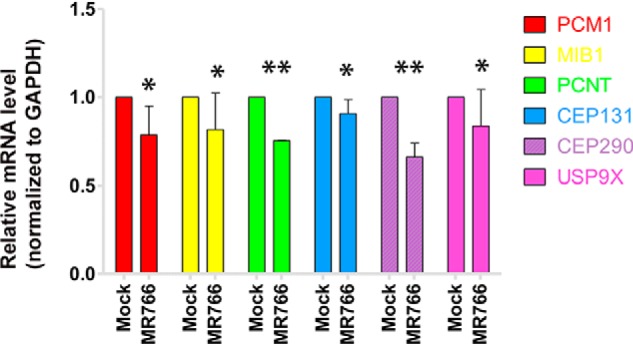
Real-time RT-PCR to determine the levels of PCM1 and Mib1. 1.25 μg of total RNA was used for each RT-PCR to examine the levels of PCM1, Mib1, PCNT, CEP131, CEP290, USP9X, and GAPDH using the primers shown in Table 1. The experiments were performed independently two times, and the average mRNA levels were normalized with that of GAPDH. *, p > 0.05; **, p < 0.001. Error bars, S.E.
Next, we asked whether ZIKV infection could result in alteration of production of PCM1, CEP131, and other centrosomal proteins. For that purpose, we infected SK-N-SH cells with ZIKV MR766 at an MOI of 1. The whole-cell lysates were prepared at different time points as indicated: 24, 48, and 72 hpi. Western blotting assays were performed to examine the levels of cellular and viral proteins. As can be seen in Fig. 11, the protein level (the density of the band) was first normalized to α-tubulin. Then the ratios were obtained by comparing the infected group with the mock groups. A ratio of 1 means no change. We found that PCM1, CEP131, USP9x, and CEP290 are reduced significantly by ZIKV infection. PCNT protein level was moderately decreased. Surprisingly, Mib1 was significantly increased, whereas other protein levels were reduced. Because Mib1 has been identified as the E3 of PCM1 or CEP131 during the ubiquitination procedure, we presume that Mib1 is an important factor for ubiquitin-mediated degradation of centrosomal proteins. Consistently, MG132 treatment abolished the ZIKV-caused decrease of PCM1, CEP131, and CEP290, but not USP9x.
Figure 11.

ZIKV infection resulted in reduction of pericentrosomal proteins: PCM1, CEP290, USP9X, and CEP131. Western blotting assay to examine the levels of PCM1 and Mib1 in SK-N-SH cells that were infected or mock-infected with ZIKV at an MOI of 1 for 24, 48, or 72 h. NS3 was to show viral infection, and tubulin was used to control for sample loading. Left, without MG132 treatment; right, with MG132 treatment 12 h before collecting samples. We have performed two independent experiments; the densitometry was obtained by averaging the two Western blot analyses, and the numbers represent the mean protein expression. The densities of protein bands are first normalized with tubulin, and then the proteins from ZIKV-infected groups were compared with these from mock-infected groups. If the ratio is <1, it means that the protein is reduced by ZIKV.
Mib1 is required for ZIKV to disperse and degrade PCM1
To further demonstrate the importance of Mib1 in degradation of PCM1, we knocked out the Mib1 from SK-N-SH cells (Mib1-ko) using the CRISPR/Cas9 system. As shown in Fig. 12A by Western blotting and ICC, Mib1 cannot be detected in the Mib1-ko cells. Interestingly, the PCM1 in the Mib1-ko cells appears more condensed, as shown by ICC (bottom of Fig. 12A). Then we infected ZIKV (MR766 or PRVABC59) to Mib1-ko or normal SK-N-SH cells for 24 h at an MOI of 1. A Western blotting assay was performed to examine the level of PCM1. As can be seen on the left side of Fig. 12B, PCM1 level is not changed by ZIKV infection in the Mib1-ko cells, but it is clearly reduced in normal SK-N-SH cells. In addition, we performed the co-transfection assays in the Mib1-ko cells as shown on the right side of Fig. 12B. PCM1 (shown by anti-GFP) is not degraded when transfected alone (lane 2) or co-transfected with ubiquitin (shown by anti-Myc, lane 3). PCM1 is clearly degraded when the Mib1 (anti-HA) is co-transfected together with PCM1 and ubiquitin (lane 4). Consistently, the infection of ZIKV fails to disperse PCM1 in Mib1-ko cells, as shown in Fig. 12C, lending further support to the idea that Mib1 is not only essential for ZIKV-mediated degradation of PCM1 but also important for ZIKV dispersal of PCM1.
Figure 12.

Effects of ZIKV on PCM1 in Mib1 knockout cells. SK-N-SH cells were transfected with a mixture of CRISPR/Cas9 plasmids that target the Mib1 gene, and the GFP-positive cells were selected and diluted to generate Mib1-ko cells. A, left, Western blot analysis to test the protein level of Mib1 in SK-N-SH and its Mib1-ko cells; right, ICC to test Mib1 and PCM1 production in SK-N-SH and Mib1-ko cells. B, left, ZIKV (MR766 or PRVABC59) was infected into Mib1-ko or normal SK-N-SH cells for 24 h at an MOI of 1. The whole-cell lysates were collected to run a Western blotting assay to detect PCM1, Mib1, NS3, and tubulin. Right, co-transfections of plasmids expressing protein as indicated into Mib1-ko cells for 24 h. Western blots were performed to examine each protein as indicated on the right. C, ICC to show whether ZIKV infection disperses PCM1 in either SK-N-SH or Mib1-ko cells.
Discussion
Due to the medical significance of ZIKV outbreaks, massive in vitro and in vivo studies have been undertaken recently to elucidate how the ZIKV infection causes congenital disorders (34, 35). It has become clear that ZIKV infection disturbs the proliferation of stem cells, but how ZIKV infection interferes with cellular growth is not fully understood. In addition to the apoptosis induced by ZIKV infection, which has been widely accepted as the major mechanism by which ZIKV causes neurogenic defects, other mechanisms have not been explored thoroughly. The interaction of virus with the infected cells is intricate; ZIKV might have more biological effects on the infected cells than we have yet discovered or imagined. For example, ZIKV might interfere with cell division to prohibit cell proliferation in addition to inducing cell suicide. One of the most important steps in cell proliferation is cell division; centrosomes and centrosomal components are central to cell division. Considering these facts, it is possible that ZIKV infection affects the centrosome.
We studied the interaction of ZIKV with the centrosome. The centrosome is a cytoplasmic organelle that is not enveloped; hence, its components are more dynamic and freer to move inside the cytoplasm. Besides the center of centrosome that constitutes centriole-forming proteins, several proteins have been identified that surround the centriole, dubbed pericentriolar materials (PCM). The protein PCM1 was identified as a granule cytoplasmic protein, and its encoding gene was mapped to chromosome 8p21.3-p22 (36). The distribution of PCM1 in cells is dynamic with the cell cycle (37). Although it was demonstrated that PCM1 is not required for nucleation of microtubules (38) that initiates from γ-TuRC, it may function in maintaining the radial organization of microtubules (37). One of our major discoveries in this study is that centrosomal structures were modified by ZIKV infection. PCM1-related granule structure was dispersed from centrioles in almost all ZIKV-infected cells, whereas the centriole remains intact (Figs. 1–3). Other centrosome-associated proteins tested are not affected by ZIKV, including PCNT, CEP135, and γ-tubulin. Impairment of cellular skeletal proteins, such as β-actin and α-tubulin, was not observed (Figs. 4 and 5). Two strains of ZIKV, MR766 and PRVABC59, both dispersed the PCM1 granules (Figs. 1 and 3). However, our results showed that DENV-2 has no dispersive effects on PCM1 (Fig. 6). More importantly, ZIKV dispersed PCM1 in many kinds of cells, including neural stem cells (Figs. 1 and 2); whether this is related to ZIKV pathogenesis will be further investigated.
Interestingly, the dispersion can be prevented by adding MG132 into the cell culture as shown in Fig. 9. This study also clarified that the dispersion of PCM1 from centriole is mediated by degradation of PCM1 and CEP131; MG132 blocked the degradation of PCM1, and the blocking effect was in an MG132 dose–dependent manner. Therefore, we conclude that ZIKV disperses PCM1 from the centriole by degrading PCM1. Mib1 has been found to have activity of E3 ubiquitin ligase of several proteins and be involved in degradation of PCM1 (39–41). We found that Mib1 is required for ZIKV to degrade or disperse PCM1 because Mib1 knockout cells can still be infected by ZIKV, but the PCM1 remained intact (Fig. 12). We demonstrated that ZIKV induces expression of Mib1 and decreases the protein levels of PCM1 and CEP131 (Figs. 8, 11, and 12). The reduction of PCM1 or CEP131 does not occur at the transcriptional level because real-time RT-PCR results show that the mRNA levels of PCM1 or CEP131 are only moderately attenuated by ZIKV infection.
More than 100 centrosomal proteins congregate around the centriole and largely relate to cell division. They form granular structures and localize around the centriole (12). Several centrosomal proteins have been reported to be associated with ZIKV infection. For example, it was reported that ZIKV infection of spinal cord neuroepithelial stem cells and radial glial cells resulted in centrosomal depletion and mitochondrial sequestration of phospho-TBK1 during mitosis (42). Our present study demonstrates that ZIKV disrupts PCM1 granular structure. PCM1 is related to cell cycle and cell division because its distribution was dispersed during early G2 phase and concentrated in the centrosome at the G1 phase (17). Therefore, our experimental results suggest that ZIKV infection might have functional effects on the centrosome to cause microcephaly. However, we did not find that ZIKV infection affected any other centrosomal proteins or related proteins, such as tubulin and actin (Figs. 4 and 5).
It has been determined that ZIKV is the causative pathogen of microcephaly in infants born to ZIKV-infected mothers, although the occurrence rate is only 5% among ZIKV-infected pregnancies (43). In vitro and in vivo experiments have demonstrated that ZIKV infection causes defects of NSC proliferation (44–52). The widely accepted pathogenic mechanism causing the defect of the NSCs is that ZIKV infection induces apoptosis of NSCs (34). However, the interaction between ZIKV and host has not been fully understood. A dozen genes have been identified to be related to primary microcephaly (MCPH). More than half of the MCPH proteins are directly related to the centrosome (26, 52, 53). Our results from the present studies suggest a connection between centrosomal proteins and ZIKV infection; centrosomal proteins are affected by ZIKV through Mib1-mediated proteasomal degradation.
In the future, we will further investigate the consequences of the interaction of ZIV with centrosome at in vivo level. It is also important to identify which ZIKV protein is responsible for the effects of ZIKV on centrosomal proteins.
Materials and methods
Cell lines, tissue culture, and viruses
Vero cells (ATCC®, CCL-81TM), SK-N-SH (ATCC®, HTB-11TM), U-251MG (Sigma, 09063001), MRC-5 (ATCC®, CCL-171TM), and HEK 293T (ATCC®, CRL-1573TM) were purchased from ATCC or Sigma. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and penicillin (100 IU/ml)-streptomycin (100 μg/ml) and amphotericin B (2.5 μg/ml) (54). ZIKV strains MR766 (55) and PRVABC59 (56) were obtained from ATCC.
NSC culture
Human fetal primary NSCs/NPCs (neural progenitor cells) were purchased from the Comprehensive NeuroAids Center (CNAC) at Temple University. Before culture of the cells, plates or dishes were coated with Matrigel (BD Biosciences, catalog no. 356230; 1:400 dilution with DMEM/F-12 before use) at 37 °C for 2 h. The primary NSCs/NPCs were expanded by monolayer in DMEM/F-12 NSC/NPC proliferation medium (Cellgro, 10-092-CV) containing 20 ng/ml epidermal growth factor, 10 ng/ml basic fibroblast growth factor, and B27 supplement (50×; Gibco, 12587-010). To dissociate the cells for propagation, the monolayer cells were treated with Accutase (Sigma, A6964-100) at room temperature. The dissociated single cells from the third to fifth passaged NSC/NPC were plated on Matrigel-coated 24-well chamber slides (for immunocytochemistry) and 6-well plates (for Western blotting). The cells were treated as follows: mock or ZIKV infection. Each group contained 2–4 replicates. Experiments were repeated at least three times for statistical analyses.
Knockout of Mib1 from SK-N-SH cells using CRISPR/Cas9 technique
SK-N-SH cells were seeded in a well of a 24-well plate. When the cells reached 50% confluence, we transfected a plasmid mixture of human Mib1 CRISPR/Cas9 ko plasmid (Santa Cruz Biotechnology, Inc., sc-404769). The plasmid also expresses GFP as a marker. If GFP expression was detected, the cells were trypsinized, diluted, and distributed into a 96-well plate. When the cell clones grew to 30% of the well, the cells were then transferred into a 48-well plate, and each clone was distributed into two wells of the 48-well plate. When the cells grew to 60%, one well was lysed for a Western blotting assay to check the production of Mib1. The Mib1-negative cell clones were selected and named as Mib1-ko for further experiments.
Antibodies
Anti-PCNT (ab4448), anti-ubiquitin (ab7780), and anti-cyclin D3 (ab63535), anti-CEP135 (ab75005), and anti-γ-tubulin (ab84355) were purchased from Abcam (Cambridge, MA). Rabbit anti-PCM1 (H-262, sc-67204), mouse anti-Mib1 (D6), and mouse anti-PCM1 (G-6, sc-398365) for showing pericentrosomal materials; anti-β-tubulin (sc-5274); anti-β-actin (sc-47778); anti-Myc (sc-40); anti-acetyl α-tubulin (sc-23950); and anti-α-tubulin (4G1, sc-58666) were purchased from Santa Cruz Biotechnology, Inc. The anti-ZIKV antibody for ICC was generated from the hybridoma cell line D1-4G2-4-15 (ATCC® HB-112TM) in our laboratory. The anti-ZIKV NS3 antibody for Western blotting is a mAb generated and purified in our laboratory. Rabbit anti-NS3 (GTX133309) was purchased from GeneTex (Irvine, CA).
ICC assay
Immunostaining was performed on cells grown on coverslips after fixation with 1% paraformaldehyde (10 min at room temperature) and permeabilization in 0.2% Triton (20 min on ice) by sequential incubation with primary and Texas Red–labeled secondary antibodies (Vector Laboratories, Burlingame, CA) for 30 min each (all solutions in PBS). Finally, cells were equilibrated in PBS, stained for DNA with Hoechst 33258 (0.5 μg/ml), and mounted in Fluoromount G (Fisher).
Immunoblot analysis
Proteins were separated by SDS-7.5% PAGE (57) (10–20 μg loaded in each lane), transferred to nitrocellulose membranes (Amersham Biosciences), and blocked with 5% nonfat milk for 60 min at room temperature. Membranes were incubated overnight at 4 °C with primary antibody followed by incubation with a horseradish peroxidase–coupled secondary antibody (Amersham Biosciences) and detection with enhanced chemiluminescence (Pierce), according to standard methods. Membranes were stripped with stripping buffer (100 mm β-mercaptoethanol, 2% SDS, 62.5 mm Tris-HCl, pH 6.8), washed with PBS plus 0.1% Tween 20, and used to detect additional proteins.
Co-IP assay
The SK-N-SH cells were lysed with a lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 5% glycerol) supplemented with protease inhibitor mixture (catalog no. p8340, Sigma) on ice for 10 min. The lysates were then centrifuged at 3,000 × g for 5 min, and the supernatants were transferred to new tubes. The supernatants were incubated with antibodies overnight in a cold room with rolling. The incubations were coupled to protein G–Sepharose beads (Amersham Biosciences) according to the manufacturer's instructions for 3 h. The beads were washed three times in PBS plus 0.1% BSA and resuspended in a mixture of PBS and 2× Laemmli buffer (20 μl of each). After heating at 95 °C for 5 min, beads were removed by centrifugation, and supernatants were analyzed by SDS-PAGE and immunoblotting.
RNA isolation and real-time RT-PCR
Total RNA was isolated using TRI Reagent (Ambion) from SK-N-SH cells that were mock- or ZIKV-infected for 24 h. The RNA was treated with DNase I (RNase-free, Invitrogen, catalog no. 18047-019) according to the manufacturer's instructions.
The total RNA concentration was measured with the Thermo Fisher Qubit 4 Fluorometer, and a quality check was conducted using electrophoresis to ensure that the 28S, 18S, and 5S rRNA bands are clearly seen. One μg of the total RNA was used for reverse transcription, which was carried out using an iScriptTM cDNA synthesis kit (Bio-Rad, catalog no. 170-8891) according to the manufacturer's protocol, and 25 ng of cDNA was used for real-time PCR. To quantitatively examine the mRNA levels of PCM1, MIB1, CEP131, PCNT, CEP290, and USP9X, a real-time PCR was undertaken using the SsoAdvancedTM Universal SYBR Green Supermix kit (Bio-Rad, catalog no. 172-5271). The reactions of quantitative PCR were as follows: 94 °C for 30 s, followed 40 cycles of 94 °C for 15 s; 60 °C for 30 s. Following the amplification cycles, a melting temperature curve analysis was obtained by measuring the fluorescence during a period of warming from 65 to 95 °C. All samples were run in duplicate. Data were collected, and relative gene expression level was analyzed by the 2−ΔΔCT method using the CFX software (Bio-Rad), with GADPH as the reference gene. The primers used to determine transcriptional levels of PCM1, MIB1, USP9X, CEP131, PCNT, CEP290, and GAPDH are listed in Table 1.
Table 1.
Primers for RT-PCR
| Primer | Sequence |
|---|---|
| PCM1_Fw | 5′-AGTGAAGTTTCTACCATCC-3′ |
| PCM1_Rv | 5′-GCACAAGATTGTATCACTCC-3′ |
| CEP131_Fw | 5′-TGATGCTCTTCGAGGGCAG-3′ |
| CEP131_Rv | 5′-GGAACTCCGGGCATTGGAT-3′ |
| USP9X_Fw | 5′-AAGTGAAGCATGTCAGCGATT-3′ |
| USP9X_Rv | 5′-GCCACACATAGCTCCACCA-3′ |
| CEP290_Fw | 5′-AGATGCTCACCGAACAAGTAGA-3′ |
| CEP290_Rv | 5′-ATGAGTCTGTTGAGAAAGGGTTG-3′ |
| PCNT_Fw | 5′-TTGTGGTCCCAGCTTGATTCT-3′ |
| PCNT_Rv | 5′-CTCCAGTTCGGACTCATGCT-3′ |
| GAPDH_Fw | 5′-GAAGGTGAAGGTCGGAGTC-3′ |
| GPDH_Rv | 5′-GAAGATGGTGATGGGATTTC-3′ |
| Mib1_Fw | 5′-AAGTGAGAGGGTTCTGTTAGAG-3′ |
| Mib1_Rv | 5′-AGTCCTGGATTTCTGTTACCT-3′ |
Confocal microscopy
Cells were examined with a Leica TCS SPII confocal laser-scanning system. Two or three channels were recorded simultaneously and/or sequentially and controlled for possible breakthrough between the FITC and Texas Red signals and between the blue and red channels.
Statistical studies
Statistical details of experiments are found in the corresponding figure legends. Viral titers in treated and untreated cells were analyzed by Student's t test. All statistical data were analyzed with GraphPad Prism version 5.01 (GraphPad Software, Inc., La Jolla, CA) using the pairwise two-tailed t test. Data are given as mean ± S.E. as indicated, and the significant difference is indicated in each legend.
Author contributions
F. W. formal analysis; F. W., N. A., W. H., L. A. O., K. I., H. U., and Q. T. investigation; F. W., N. A., W. H., R. C.-C., L. A. O., K. I., H. U., M.-H. L., and A. S. methodology; F. W., N. A., W. H., and Q. T. writing-original draft; W. H. and H. U. validation; R. C.-C., K. I., and M.-H. L. resources; M.-H. L., A. S., and Q. T. writing-review and editing; A. S. and Q. T. supervision; Q. T. conceptualization; Q. T. data curation; Q. T. project administration.
Acknowledgments
We acknowledge the instrumental support of the PSM Molecular Biology Core Laboratory and thank Dr. Andrew Boileau for critical reading. We thank Dr. Azad Bonni (Washington University School of Medicine) and Dr. Simon Holst Bekker-Jensen (University of Copenhagen) for generous gifts of plasmids.
This study was supported by NIAID, National Institutes of Health, Grant SC1AI112785 (to Q. T.) and NIMHD, National Institutes of Health, Grant 2U54MD007597. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ZIKV
- Zika virus
- PCM
- pericentriolar material(s)
- γ-TuRC
- γ-tubulin ring complex
- NSC
- neural stem cell
- PCNT
- pericentrin
- CEP135
- centrosomal protein 131
- ICC
- immunocytochemistry
- hpi
- hours postinfection
- IP
- immunoprecipitation
- UAE
- ubiquitin-activating enzyme
- MOI
- multiplicity of infection
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ko
- knockout
- MCPH
- microcephaly
- DMEM
- Dulbecco's modified Eagle's medium
- DAPI
- 4′,6-diamidino-2-phenylindole
- ko
- knockout.
References
- 1. Mlakar J., Korva M., Tul N., Popović M., Poljšak-Prijatelj M., Mraz J., Kolenc M., Resman Rus K., Vesnaver Vipotnik T., Fabjan Vodušek V., Vizjak A., Pižem J., Petrovec M., and Avšič Županc T. (2016) Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 10.1056/NEJMoa1600651 [DOI] [PubMed] [Google Scholar]
- 2. Petersen E., Wilson M. E., Touch S., McCloskey B., Mwaba P., Bates M., Dar O., Mattes F., Kidd M., Ippolito G., Azhar E. I., and Zumla A. (2016) Rapid spread of Zika virus in the Americas: implications for public health preparedness for mass gatherings at the 2016 Brazil Olympic Games. Int. J. Infect. Dis. 44, 11–15 10.1016/j.ijid.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 3. Weaver S. C., Costa F., Garcia-Blanco M. A., Ko A. I., Ribeiro G. S., Saade G., Shi P. Y., and Vasilakis N. (2016) Zika virus: history, emergence, biology, and prospects for control. Antiviral Res. 130, 69–80 10.1016/j.antiviral.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wikan N., and Smith D. R. (2016) Zika virus: history of a newly emerging arbovirus. Lancet Infect. Dis. 16, e119–e126 10.1016/S1473-3099(16)30010-X [DOI] [PubMed] [Google Scholar]
- 5. Plourde A. R., and Bloch E. M. (2016) A literature review of Zika virus. Emerg. Infect Dis. 22, 1185–1192 10.3201/eid2207.151990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faye O., Freire C. C., Iamarino A., Faye O., de Oliveira J. V., Diallo M., Zanotto P. M., and Sall A. A. (2014) Molecular evolution of Zika virus during its emergence in the 20th century. PLoS Negl. Trop. Dis. 8, e2636 10.1371/journal.pntd.0002636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stearns T. (2001) Centrosome duplication: a centriolar pas de deux. Cell 105, 417–420 10.1016/S0092-8674(01)00366-X [DOI] [PubMed] [Google Scholar]
- 8. Rodrigues-Martins A., Riparbelli M., Callaini G., Glover D. M., and Bettencourt-Dias M. (2007) Revisiting the role of the mother centriole in centriole biogenesis. Science 316, 1046–1050 10.1126/science.1142950 [DOI] [PubMed] [Google Scholar]
- 9. Naghavi M. H., and Walsh D. (2017) Microtubule regulation and function during virus infection. J. Virol. 91, e00538–00517 10.1128/JVI.00538-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hori A., and Toda T. (2017) Regulation of centriolar satellite integrity and its physiology. Cell Mol. Life Sci. 74, 213–229 10.1007/s00018-016-2315-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badano J. L., Teslovich T. M., and Katsanis N. (2005) The centrosome in human genetic disease. Nat. Rev. Genet 6, 194–205 10.1038/nrg1557 [DOI] [PubMed] [Google Scholar]
- 12. Theg D. E. (1964) Cytoplasmic microtubules in different animal cells. J. Cell Biol. 23, 265–275 10.1083/jcb.23.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rieder C. L., Faruki S., and Khodjakov A. (2001) The centrosome in vertebrates: more than a microtubule-organizing center. Trends Cell Biol. 11, 413–419 10.1016/S0962-8924(01)02085-2 [DOI] [PubMed] [Google Scholar]
- 14. Kubo A., Sasaki H., Yuba-Kubo A., Tsukita S., and Shiina N. (1999) Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 147, 969–980 10.1083/jcb.147.5.969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Romei C., and Elisei R. (2012) RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front. Endocrinol. (Lausanne) 3, 54 10.3389/fendo.2012.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corvi R., Berger N., Balczon R., and Romeo G. (2000) RET/PCM-1: a novel fusion gene in papillary thyroid carcinoma. Oncogene 19, 4236–4242 10.1038/sj.onc.1203772 [DOI] [PubMed] [Google Scholar]
- 17. Bärenz F., Mayilo D., and Gruss O. J. (2011) Centriolar satellites: busy orbits around the centrosome. Eur. J. Cell Biol. 90, 983–989 10.1016/j.ejcb.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 18. Kodani A., Yu T. W., Johnson J. R., Jayaraman D., Johnson T. L., Al-Gazali L., Sztriha L., Partlow J. N., Kim H., Krup A. L., Dammermann A., Krogan N. J., Walsh C. A., and Reiter J. F. (2015) Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife 4, e07519 10.7554/eLife.07519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mennella V., Agard D. A., Huang B., and Pelletier L. (2014) Amorphous no more: subdiffraction view of the pericentriolar material architecture. Trends Cell Biol. 24, 188–197 10.1016/j.tcb.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balczon R., Bao L., and Zimmer W. E. (1994) PCM-1, a 228-kD centrosome autoantigen with a distinct cell cycle distribution. J. Cell Biol. 124, 783–793 10.1083/jcb.124.5.783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Staples C. J., Myers K. N., Beveridge R. D., Patil A. A., Lee A. J., Swanton C., Howell M., Boulton S. J., and Collis S. J. (2012) The centriolar satellite protein Cep131 is important for genome stability. J. Cell Sci. 125, 4770–4779 10.1242/jcs.104059 [DOI] [PubMed] [Google Scholar]
- 22. Gabriel E., Ramani A., Karow U., Gottardo M., Natarajan K., Gooi L. M., Goranci-Buzhala G., Krut O., Peters F., Nikolic M., Kuivanen S., Korhonen E., Smura T., Vapalahti O., Papantonis A., et al. (2017) Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20, 397–406 e395 10.1016/j.stem.2016.12.005 [DOI] [PubMed] [Google Scholar]
- 23. Wolf B., Diop F., Ferraris P., Wichit S., Busso C., Missé D., and Gönczy P. (2017) Zika virus causes supernumerary foci with centriolar proteins and impaired spindle positioning. Open Biol. 7, 160231 10.1098/rsob.160231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou W., Cruz-Cosme R., Armstrong N., Obwolo L. A., Wen F., Hu W., Luo M. H., and Tang Q. (2017) Molecular cloning and characterization of the genes encoding the proteins of Zika virus. Gene 628, 117–128 10.1016/j.gene.2017.07.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortese M., Goellner S., Acosta E. G., Neufeldt C. J., Oleksiuk O., Lampe M., Haselmann U., Funaya C., Schieber N., Ronchi P., Schorb M., Pruunsild P., Schwab Y., Chatel-Chaix L., Ruggieri A., and Bartenschlager R. (2017) Ultrastructural characterization of Zika virus replication factories. Cell Rep. 18, 2113–2123 10.1016/j.celrep.2017.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thornton G. K., and Woods C. G. (2009) Primary microcephaly: do all roads lead to Rome? Trends Genet. 25, 501–510 10.1016/j.tig.2009.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hou W., Armstrong N., Obwolo L. A., Thomas M., Pang X., Jones K. S., and Tang Q. (2017) Determination of the cell permissiveness spectrum, mode of RNA replication, and RNA-protein interaction of Zika virus. BMC Infect. Dis. 17, 239 10.1186/s12879-017-2338-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biedler J. L., Helson L., and Spengler B. A. (1973) Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 33, 2643–2652 [PubMed] [Google Scholar]
- 29. Ohta T., Essner R., Ryu J. H., Palazzo R. E., Uetake Y., and Kuriyama R. (2002) Characterization of Cep135, a novel coiled-coil centrosomal protein involved in microtubule organization in mammalian cells. J. Cell Biol. 156, 87–99 10.1083/jcb.200108088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y. D., and Nigg E. A. (2007) Plk4-induced centriole biogenesis in human cells. Dev. Cell 13, 190–202 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 31. Gudi R., Zou C., Li J., and Gao Q. (2011) Centrobin-tubulin interaction is required for centriole elongation and stability. J. Cell Biol. 193, 711–725 10.1083/jcb.201006135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villumsen B. H., Danielsen J. R., Povlsen L., Sylvestersen K. B., Merdes A., Beli P., Yang Y. G., Choudhary C., Nielsen M. L., Mailand N., and Bekker-Jensen S. (2013) A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 32, 3029–3040 10.1038/emboj.2013.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barghout S. H., Patel P. S., Wang X., Xu G. W., Kavanagh S., Halgas O., Zarabi S. F., Gronda M., Hurren R., Jeyaraju D. V., MacLean N., Brennan S., Hyer M. L., Berger A., Traore T., et al. (2019) Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia 33, 37–51 10.1038/s41375-018-0167-0 [DOI] [PubMed] [Google Scholar]
- 34. Ming G. L., Tang H., and Song H. (2016) Advances in Zika virus research: stem cell models, challenges, and opportunities. Cell Stem Cell 19, 690–702 10.1016/j.stem.2016.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrison T. E., and Diamond M. S. (2017) Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol. 91, e00009–17 10.1128/JVI.00009-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohata H., Fujiwara Y., Koyama K., and Nakamura Y. (1994) Mapping of the human autoantigen pericentriolar material 1 (PCM1) gene to chromosome 8p21.3-p22. Genomics 24, 404–406 10.1006/geno.1994.1640 [DOI] [PubMed] [Google Scholar]
- 37. Dammermann A., and Merdes A. (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159, 255–266 10.1083/jcb.200204023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Q., Hansen D., Killilea A., Joshi H. C., Palazzo R. E., and Balczon R. (2001) Kendrin/pericentrin-B, a centrosome protein with homology to pericentrin that complexes with PCM-1. J. Cell Sci. 114, 797–809 [DOI] [PubMed] [Google Scholar]
- 39. Wang L., Lee K., Malonis R., Sanchez I., and Dynlacht B. D. (2016) Tethering of an E3 ligase by PCM1 regulates the abundance of centrosomal KIAA0586/Talpid3 and promotes ciliogenesis. Elife 5, e12950 10.7554/eLife.12950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luxán G., Casanova J. C., Martínez-Poveda B., Prados B., D'Amato G., MacGrogan D., Gonzalez-Rajal A., Dobarro D., Torroja C., Martinez F., Izquierdo-García J. L., Fernández-Friera L., Sabater-Molina M., Kong Y. Y., Pizarro G., et al. (2013) Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 19, 193–201 10.1038/nm.3046 [DOI] [PubMed] [Google Scholar]
- 41. Itoh M., Kim C. H., Palardy G., Oda T., Jiang Y. J., Maust D., Yeo S. Y., Lorick K., Wright G. J., Ariza-McNaughton L., Weissman A. M., Lewis J., Chandrasekharappa S. C., and Chitnis A. B. (2003) Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67–82 10.1016/S1534-5807(02)00409-4 [DOI] [PubMed] [Google Scholar]
- 42. Onorati M., Li Z., Liu F., Sousa A. M. M., Nakagawa N., Li M., Dell'Anno M. T., Gulden F. O., Pochareddy S., Tebbenkamp A. T. N., Han W., Pletikos M., Gao T., Zhu Y., Bichsel C., et al. (2016) Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 16, 2576–2592 10.1016/j.celrep.2016.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Oliveira W. K., de França G. V. A., Carmo E. H., Duncan B. B., de Souza Kuchenbecker R., and Schmidt M. I. (2017) Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: a surveillance-based analysis. Lancet 390, 861–870 10.1016/S0140-6736(17)31368-5 [DOI] [PubMed] [Google Scholar]
- 44. Qian X., Nguyen H. N., Song M. M., Hadiono C., Ogden S. C., Hammack C., Yao B., Hamersky G. R., Jacob F., Zhong C., Yoon K. J., Jeang W., Lin L., Li Y., Thakor J., et al. (2016) Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 10.1016/j.cell.2016.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olagnier D., Muscolini M., Coyne C. B., Diamond M. S., and Hiscott J. (2016) Mechanisms of Zika virus infection and neuropathogenesis. DNA Cell Biol. 35, 367–372 10.1089/dna.2016.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garcez P. P., Loiola E. C., Madeiro da Costa R., Higa L. M., Trindade P., Delvecchio R., Nascimento J. M., Brindeiro R., Tanuri A., and Rehen S. K. (2016) Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 10.1126/science.aaf6116 [DOI] [PubMed] [Google Scholar]
- 47. Aliota M. T., Caine E. A., Walker E. C., Larkin K. E., Camacho E., and Osorio J. E. (2016) Characterization of lethal Zika virus infection in AG129 mice. PLoS Negl. Trop. Dis. 10, e0004682 10.1371/journal.pntd.0004682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hickman H. D., and Pierson T. C. (2016) Zika in the brain: new models shed light on viral infection. Trends Mol. Med. 22, 639–641 10.1016/j.molmed.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lazear H. M., Govero J., Smith A. M., Platt D. J., Fernandez E., Miner J. J., and Diamond M. S. (2016) A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 10.1016/j.chom.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li C., Xu D., Ye Q., Hong S., Jiang Y., Liu X., Zhang N., Shi L., Qin C. F., and Xu Z. (2016) Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 672 10.1016/j.stem.2016.10.017 [DOI] [PubMed] [Google Scholar]
- 51. Miner J. J., Sene A., Richner J. M., Smith A. M., Santeford A., Ban N., Weger-Lucarelli J., Manzella F., Rückert C., Govero J., Noguchi K. K., Ebel G. D., Diamond M. S., and Apte R. S. (2016) Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep. 16, 3208–3218 10.1016/j.celrep.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Faheem M., Naseer M. I., Rasool M., Chaudhary A. G., Kumosani T. A., Ilyas A. M., Pushparaj P., Ahmed F., Algahtani H. A., Al-Qahtani M. H., and Saleh Jamal H. (2015) Molecular genetics of human primary microcephaly: an overview. BMC Med. Genomics 8, Suppl. 1, S4 10.1186/1755-8794-8-S1-S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mochida G. H. (2009) Genetics and biology of microcephaly and lissencephaly. Semin. Pediatr. Neurol. 16, 120–126 10.1016/j.spen.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Bruyn Kops A., and Knipe D. M. (1988) Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55, 857–868 10.1016/0092-8674(88)90141-9 [DOI] [PubMed] [Google Scholar]
- 55. Kuno G., and Chang G. J. (2007) Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 152, 687–696 10.1007/s00705-006-0903-z [DOI] [PubMed] [Google Scholar]
- 56. Lanciotti R. S., Lambert A. J., Holodniy M., Saavedra S., and Signor Ldel C. (2016) Phylogeny of Zika Virus in Western Hemisphere, 2015. Emerg. Infect. Dis. 22, 933–935 10.3201/eid2205.160065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Münch K., Messerle M., Plachter B., and Koszinowski U. H. (1992) An acidic region of the 89K murine cytomegalovirus immediate early protein interacts with DNA. J. Gen. Virol. 73, 499–506 10.1099/0022-1317-73-3-499 [DOI] [PubMed] [Google Scholar]