

Abstract
Loss-of-function mutations in the gene encoding human protein DJ-1 cause early onset of Parkinson's disease, suggesting that DJ-1 protects dopaminergic neurons. The molecular mechanisms underlying this neuroprotection are unclear; however, DJ-1 has been suggested to be a GSH-independent glyoxalase that detoxifies methylglyoxal (MGO) by converting it into lactate. It has also been suggested that DJ-1 serves as a deglycase that catalyzes hydrolysis of hemithioacetals and hemiaminals formed by reactions of MGO with the thiol and amino groups of proteins. In this report, we demonstrate that the equilibrium constant of reaction of MGO with thiols is ∼500 m−1 at 37 °C and that the half-life of the resulting hemithioacetal is only 12 s. These thermodynamic parameters would dictate that a significant fraction of free MGO will be present in a fast equilibrium with hemithioacetals in solution. We found that removal of free MGO by DJ-1's glyoxalase activity forces immediate spontaneous decomposition of hemithioacetals due to the shift in equilibrium position. This spontaneous decomposition of hemithioacetals could be mistaken for deglycase activity of DJ-1. Furthermore, we demonstrate that higher initial concentrations of hemithioacetals are associated with lower rates of DJ-1–mediated conversion of MGO, ruling out the possibility that hemithioacetals are DJ-1 substrates. Experiments with CRISPR/Cas-generated DJ-1–knockout HEK293 cells revealed that DJ-1 does not protect against acute MGO toxicity or carboxymethylation of lysine residues in cells. Combined, our results suggest that DJ-1 does not possess protein deglycase activity.
Keywords: Parkinson disease; Parkinson disease (autosomal recessive, early onset) 7 (PARK7); thiol; glycation; neurodegeneration; deglycase; DJ-1; glyoxalase; hemiaminal; hemithioacetal
Introduction
Several loss-of-function mutants in the human gene PARK7 (1) have been linked to hereditary recessive forms of Parkinson's disease. PARK7 encodes a small (∼20-kDa) protein called DJ-1 that has been shown to protect cells during oxidative stress in several experimental systems (2). The molecular mechanisms underlying the protective effects of DJ-1 are unclear; however, a homolog of DJ-1 in Escherichia coli, the Hsp31 protein, possesses GSH-independent glyoxalase activity (3), converting toxic metabolites methylglyoxal (MGO)2 and glyoxal into lactic and glycolic acids respectively (Fig. 1, Equation 1). Several reports suggest that human DJ-1 is also capable of catalyzing the conversion of MGO and glyoxal, providing a potential explanation for the neuroprotective effects of DJ-1 (4, 5). It has been noted, however, that the glyoxalase activity of DJ-1 is much lower than that of other members of the DJ-1 superfamily (5, 6) and may be insufficient to allow for any meaningful physiological function in the human brain. The low glyoxalase activity of DJ-1 is supported by structural studies: DJ-1 lacks a histidine residue that is a part of the catalytic triad in Hsp31, making it a poor catalyst for efficient detoxification of MGO and glyoxal (7).
Figure 1.
Reactions of methylglyoxal in water solutions. MGO can be transformed into lactic acid by glyoxalase activity of DJ-1 (Equation (Eq.) 1), or it can react with cellular thiols to form hemithioacetals (Equation 2). It has also been suggested that DJ-1 can catalyze the hydrolysis of hemithioacetals according to Equation 3, although this was not confirmed in the present study.
Both MGO and glyoxal are toxic to cells due to the presence of two reactive carbonyl groups that react with thiols to form hemithioacetals (Fig. 1, Equation 2) and amines to form hemiaminals. These reactions are quick and reversible, but further transformation into advanced glycation end products such as carboxymethyl lysine, although slow, may cause the gradual accumulation of damaged and aggregated proteins associated with various health problems. The formation of insoluble aggregates of the cytosolic protein synuclein is a hallmark of progressing Parkinson's disease. Thus, increased production of MGO may be linked to the synuclein aggregation that leads to neuronal death and could explain why the glyoxalase activity of DJ-1 is important for neuroprotection.
More recently, it has been suggested that DJ-1, along with several homologous proteins, serves as a protein deglycase (8, 9) that catalyzes hydrolysis of hemithioacetals and hemiaminals into lactic acid and a corresponding amino acid (Fig. 1, Equation 3). Although these findings have been challenged (10), a number of other recent reports support the idea that DJ-1 is a protein deglycase and suggest that its deglycase activity plays an important protective (11) and regulatory (12, 13) role. Human DJ-1 is now classified as a deglycase in several major databases such as UniProt and NCBI and has been assigned an Enzyme Commission (EC) number of 3.5.1.124 based on its presumed protein deglycase activity.
In the present study, we re-examined the formation and stability of MGO-derived hemithioacetals in solution as well as the catalytic activity of human DJ-1 toward MGO and related hemithioacetals and hemiaminals. We demonstrate that DJ-1 possesses a weak glyoxalase activity. However, its apparent deglycase activity is a result of the removal of MGO by glyoxalase activity that leads to a subsequent decomposition of hemithioacetals and hemiaminals due to the shift in equilibrium position. Cultured human cells lacking the PARK7 gene have the same tolerance toward acute MGO toxicity and identical levels of carboxymethylated lysine residues in proteins. Therefore, we conclude that the neuroprotective function of DJ-1 is not due to its protein deglycase activity and that recent studies suggesting DJ-1 deglycase activity should be reconsidered in light of these results.
Results
Preparation of intact DJ-1 and its glyoxalase activity
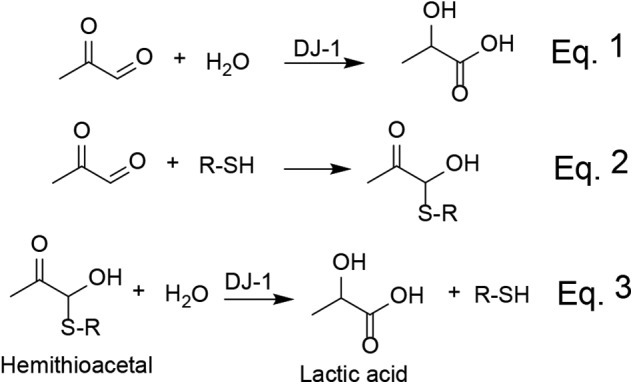
To exclude artifacts from the presence of tags and post-translational modifications of its active-site cysteine, we designed a protocol to prepare a highly pure, untagged DJ-1 with intact Cys106. Untagged, full-length DJ-1 was expressed in E. coli and purified by ammonium sulfate fractionation, hydrophobic interaction, and anion-exchange chromatography. Purified DJ-1 resolves as a single band on an overloaded SDS-PAGE gel (Fig. 2A), and the ESI-MS spectrum of purified DJ-1 shows a single peak with an average molecular mass of 19,759.82 Da (Fig. 2B). This mass corresponds within 0.1 Da to the protein with intact Cys106 but without the initiating methionine residue. The cotranslational removal of its N-terminal methionine is expected because Met1 of DJ-1 is followed by a small amino acid (alanine) and is likely to occur in eukaryotes as well (14). Purified DJ-1 resolves as a dimer on an analytical size-exclusion column (Fig. 2C) and has a high melting point of 60 °C, determined in a Thermofluor assay (Fig. 2D). Together, the above data indicate that purified DJ-1 is properly folded and therefore functional. Enzymatic conversion of MGO into lactate was monitored by 1H NMR spectrometry. We used a robust GSH-independent glyoxalase (Glx3) from Candida albicans (6) to demonstrate that we could detect conversion of MGO into lactate. When 5 mm MGO was incubated with 5 μm Glx3 for 1 h, lactate was easily detected due to the appearance of a first-order quartet at 3.95 ppm (Fig. 3A). When DJ-1 was used instead of Glx3, no lactate was detected under the same conditions; however, when incubation time was increased to 10 h, lactate was readily detectable (Fig. 3A), demonstrating that DJ-1 exhibits a weak glyoxalase activity. To further characterize the glyoxalase activity of DJ-1, we monitored the consumption of MGO using a colorimetric reaction of MGO with 2,3-dinitrophenylhydrazine (DNPH). Consumption of MGO showed linear time dependence even when MGO was depleted by more than 60% (Fig. 3B), allowing kcat at room temperature to be estimated at ∼0.02 s−1. Next, we tested whether the addition of bovine serum albumin (BSA) or N-acetylcysteine (NAC) resulted in an increase in the rate of MGO consumption. Such an increase has been reported by Richarme et al. (8) and was attributed to the protein deglycase activity of DJ-1. We did not detect any increased conversion of MGO by DJ-1 in the presence of either 15 mg/ml BSA or 2 mm N-acetylcysteine (Fig. 3B). The absence of increased rates of MGO conversion by DJ-1 in the presence of NAC or BSA suggested that, contrary to several reports, DJ-1 lacks protein deglycase activity. Therefore, we reinvestigated the kinetic and thermodynamic features of MGO-based hemithioacetal formation and possible deglycase activity of DJ-1 toward hemithioacetals.
Figure 2.

Preparation and characterization of DJ-1. A, Coomassie-stained gel showing purified DJ-1 and Glx3 (5 μg each). Lane M, molecular mass markers. B, electrospray mass spectrum of intact DJ-1 reveals multicharged species. Deconvolution of the spectrum returned an average mass of 19,759.82 Da, which corresponds to full-length DJ-1 without Met1. C, size-exclusion profile of DJ-1 indicates it is a dimer. D, thermal stability of DJ-1 was assessed using SYPRO Orange dye. The maximum (Tm) of the derivative function of fluorescence intensity corresponds to the midpoint of the unfolding (denaturation) curve of the protein. A.U., arbitrary units; mAU, milli-absorbance units; RFU, relative fluorescent units.
Figure 3.
Glyoxalase activity of DJ-1. A, 1H NMR spectra of 5 mm MGO (black line) and the effect of the addition of 5 μm DJ-1 (10 h; red line) or 5 μm Glx3 (1 h; blue line). B, MGO (2 mm) and DJ-1 (25 μm) were incubated in PBS with or without 15 mg/ml BSA or 2 mm NAC at 22 °C. The concentration of MGO was assayed at the indicated time points with DNPH. Kinetic curves are offset on the y axis for clarity. A.U., arbitrary units.
Characterization of the thermodynamic equilibrium of MGO-based hemithioacetals
Concentrations of hemithioacetals in aqueous solutions can be measured at 288 nm; however, the reported extinction coefficients (ϵ) vary between 98 (15) and 249 m−1 cm−1 (16) for N-acetylcysteine, making quantitative analysis impossible. Therefore, we measured an extinction coefficient of MGO-based hemithioacetals at 288 nm using a large concentration of thiols (50 mm) to drive the yield of the reaction (Fig. 4A). The extinction coefficient was found to be 250 m−1 cm−1 for GSH and 300 m−1 cm−1 for NAC, much higher than the values reported previously (15). This discrepancy is explained by the fact that the concentrations of thiols and/or MGO used in previous studies were not high enough to quantitatively shift the equilibrium toward the formation of hemithioacetal (see below). Next, we determined the equilibrium constant, Kc, for formation of hemithioacetals in this setup. Various concentrations of GSH were added to a fixed concentration of MGO (4 mm), and the concentration of GSH-MGO was measured at 37 °C by absorbance at 288 nm (Fig. 4B). The data we obtained could be fit very well (see supporting methods), assuming fast equilibrium between all forms of MGO, GSH, and GSH-MGO, and returned a value for the equilibrium constant Kc of 510 m−1 and ϵ of 278 m−1 cm−1. We note that ϵ = 278 m−1 cm−1 for GSH-MGO is in agreement with the value ϵ = 250 m−1 cm−1 obtained from our independent saturation experiments (Fig. 4A), indicating that our calculations are correct. Similar experiments returned a value of 336 m−1 for the Kc of the formation of NAC-MGO hemithioacetal (NAC-MGO) (Fig. S1). Using the Kc value of 510 m−1, we calculated the concentration of free MGO for equimolar mixtures of MGO and thiols in the range of 1–10 mm because this is a typical concentration range used to produce and study hemithioacetals. Our calculations show that a large portion of free MGO will be present in solution both in terms of molar concentration and percentage of unreacted MGO throughout the entire concentration range (Fig. 4C). For example, a mixture of 5 mm GSH and 5 mm MGO is expected to produce ∼60% hemithioacetal with remaining ∼40% or ∼2 mm MGO present in free form (as a mixture of hydrates).
Figure 4.

Thermodynamic parameters of hemithioacetal formation. A, extinction coefficients for GSH-MGO and NAC-MGO at 288 nm in PBS were determined in the presence of 50 mm thiols to shift equilibrium toward product formation. B, 4 mm MGO in PBS was incubated with the indicated amounts of GSH at 37 °C to determine the concentration of the resulting hemithioacetal. Nonlinear curve fitting was performed as described in supporting methods and produced an excellent fit for the experimental data (red line), yielding an equilibrium constant of 510 m−1. C, calculated values of free MGO expressed in mm (left axis, black line) or as percentage of total MGO (right axis, blue line) are shown as a function of the concentration of GSH and MGO present in equal amounts. Values on the x axis show the concentrations per reactant (e.g. 5 mm reactants corresponds to 5 mm GSH + 5 mm MGO).
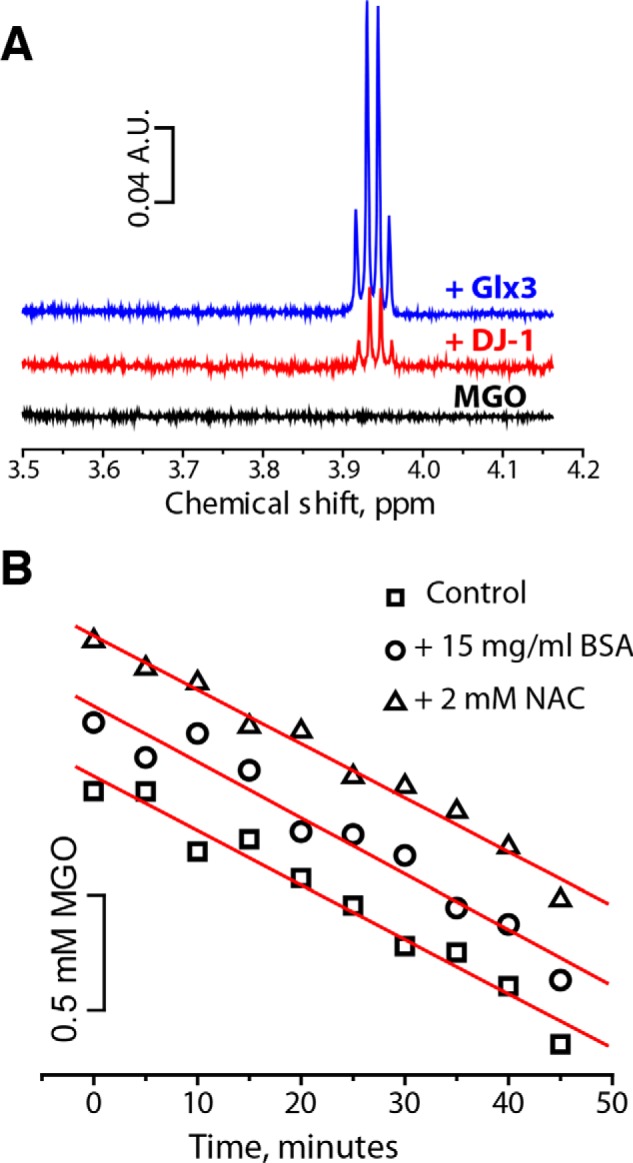
To quantitatively assess the stability of GSH-MGO in aqueous solutions, we incubated a mixture of 3 mm MGO and 3 mm GSH to drive GSH-MGO formation and then rapidly diluted the mixture 2 times to induce the decomposition of GSH-MGO. Upon dilution, the half-life of GSH-MGO is relatively short (∼12 s), whereas the new equilibrium is established within ∼1 min (Fig. 5A). The relatively low value of Kc combined with the rapid decomposition of hemithioacetals (Fig. 5A) indicates that (a) proteins glycated with MGO for 1–2 h and thoroughly purified from MGO are unlikely to have cysteine residues in the form of hemithioacetals and (b) even in the presence of MGO, the amount of a protein's thiol groups reversibly modified by MGO cannot be measured by Ellman's reagent or similar thiol-reactive compounds because they will outcompete MGO and eventually modify all available thiol groups. We confirmed these predictions experimentally: glycated BSA (100 mm MGO; 120 min; 37 °C) purified from MGO by two rounds of desalting on size-exclusion columns had the same number of free sulfhydryl groups as untreated BSA (Fig. S2). Moreover, as expected, MGO slows down the reaction with Ellman's reagent in a dose-dependent manner due to competition but does not affect the amount of available thiol groups when compared with the untreated BSA (Fig. 5B). We conclude that similar to GSH and NAC, the cysteine residue in BSA reacts quickly and reversibly with MGO. The resulting hemithioacetal is unstable; therefore, the significant decreases in the amount of free sulfhydryl groups in BSA measured by Ellman's reagent after short (<2-h) treatment with MGO reported previously (8, 17) are likely due to technical errors.
Figure 5.
MGO-based hemithioacetals are short-lived. A, kinetics of hemithioacetal decomposition after quick dilution. A mixture of 3 mm MGO and 3 mm GSH in PBS was incubated for 10 min before being quickly diluted 2 times. Experimental data were fitted with an exponential decay function (red line), which yielded a half-life value (t½) of 12 s. B, BSA (45 μm) was incubated with the indicated concentrations of MGO for 10 min before the addition of Ellman's reagent (0.5 mm final concentration). Optical absorption at 412 nm was recorded for 10 min and converted into concentration of titratable thiol groups using an extinction coefficient of 14.2 mm−1 cm−1. MGO dose-dependently slows down the reaction of BSA with Ellman's reagent due to the formation of short-lived hemithioacetals with BSA, but Ellman's reagent eventually modifies all thiol groups.
The apparent deglycase activity of DJ-1 is a result of the conversion of free MGO present in fast equilibrium with hemithioacetals and hemiaminals
To study the deglycase activity of DJ-1, NAC and MGO (5 mm each) were incubated for 10 min to form NAC-MGO. When DJ-1 was added to the mixture, the absorbance at 288 nm gradually decreased (Fig. 6A), suggesting the degradation of hemithioacetal by DJ-1, consistent with earlier reports (8, 18). This effect can be mimicked by Tris buffer (Fig. 6B) as shown by Pfaff et al. (10), most likely due to the competing equilibrium of hemiaminal formation by the amino group of Tris. However, in the case of DJ-1, the decrease in hemithioacetal represents true enzymatic activity of DJ-1 because mutation of its active-site cysteine to serine (C106S) abrogates the decrease of absorbance at 288 nm (Fig. 6B). Nevertheless, because formation of hemithioacetals is a highly dynamic and reversible process, their apparent degradation by DJ-1 can be explained by the removal of free MGO present in equilibrium with hemithioacetal by the glyoxalase activity of DJ-1. Removal of free MGO will cause a quick decomposition of NAC-MGO with the release of MGO so that the system reaches a new equilibrium position. To test this hypothesis, we replaced DJ-1 with Glx3 from C. albicans and observed a similar loss of absorbance at 288 nm, indicating that conversion of free MGO into lactate results in the expected loss of NAC-MGO (Fig. 6C). Note that Glx3 is more efficient than DJ-1 in causing an apparent decomposition of NAC-MGO (Fig. 6C), which is consistent with its higher glyoxalase activity (Fig. 3A and Ref. 6).
Figure 6.

Hemithioacetals can be decomposed by DJ-1, Glx3, and Tris through the removal of free MGO. 5 mm MGO was mixed with 5 mm NAC and incubated for 10 min before the addition of DJ-1 (A), the C106S mutant of DJ-1 or Tris (B), or Glx3 (C) at the indicated final concentrations. Protein absorbance at 288 nm was subtracted from all samples.
To determine whether hemithioacetal or free MGO is a substrate for DJ-1, we studied DJ-1–induced GSH-MGO decomposition under two different conditions. When 2.5 mm MGO was mixed with a large excess (25 mm) of GSH, ∼94% of MGO was converted into GSH-MGO, leaving ∼0.16 mm free MGO. In contrast, at 2.5 mm GSH, only ∼40% of MGO formed hemithioacetal. Kinetic curves for DJ-1–induced hemithioacetal decomposition for both conditions are shown in Fig. 7A. At a higher initial concentration of GSH-MGO (Fig. 7A, red curve), the rate of [GSH-MGO] decomposition induced by DJ-1 is slightly faster than in conditions with lower initial concentration of GSH-MGO (Fig. 7A, black curve). These results could lead to the erroneous conclusion that GSH-MGO is a substrate for DJ-1. However, when kinetic curves for free MGO are calculated for the same data sets using the assumption of a fast equilibrium between MGO, GSH, and GSH-MGO, the concentration of free MGO decreases much faster at the lower initial GSH-MGO concentration (Fig. 7B, black curve). A kinetic profile of total MGO in the system (free MGO + GSH-MGO) can be obtained by summing up both curves (Fig. 7C). The resulting kinetic profile demonstrates that the total rate of MGO conversion by DJ-1 calculated using the sum of MGO and GSH-MGO concentrations is much higher at the lower initial concentration of GSH-MGO, providing strong evidence that GSH-MGO is not a true substrate for DJ-1.
Figure 7.

GSH-MGO hemithioacetal is not a substrate for DJ-1. GSH and MGO at the indicated concentrations were mixed in PBS to form GSH-MGO and incubated with 20 μm DJ-1 at 30 °C. A, kinetic curves of GSH-MGO decomposition by DJ-1 were obtained using optical absorbance at 288 nm as described in supporting methods. B, concentrations of free MGO were calculated for the data sets shown in A, assuming a fast equilibrium between MGO and GSH-MGO. In the case where MGO and GSH-MGO are initially present in comparable amounts (black curves), the decrease in GSH-MGO is accompanied by a significant decrease in free MGO due to fast equilibrium. C is a sum of the curves in A and B, which corresponds to the kinetic profile of total MGO (free + hemithioacetal) in both samples. Note that the concentration of total MGO decreases faster in the case of a lower initial concentration of GSH-MGO (black curves), a result inconsistent with GSH-MGO being a substrate of DJ-1.
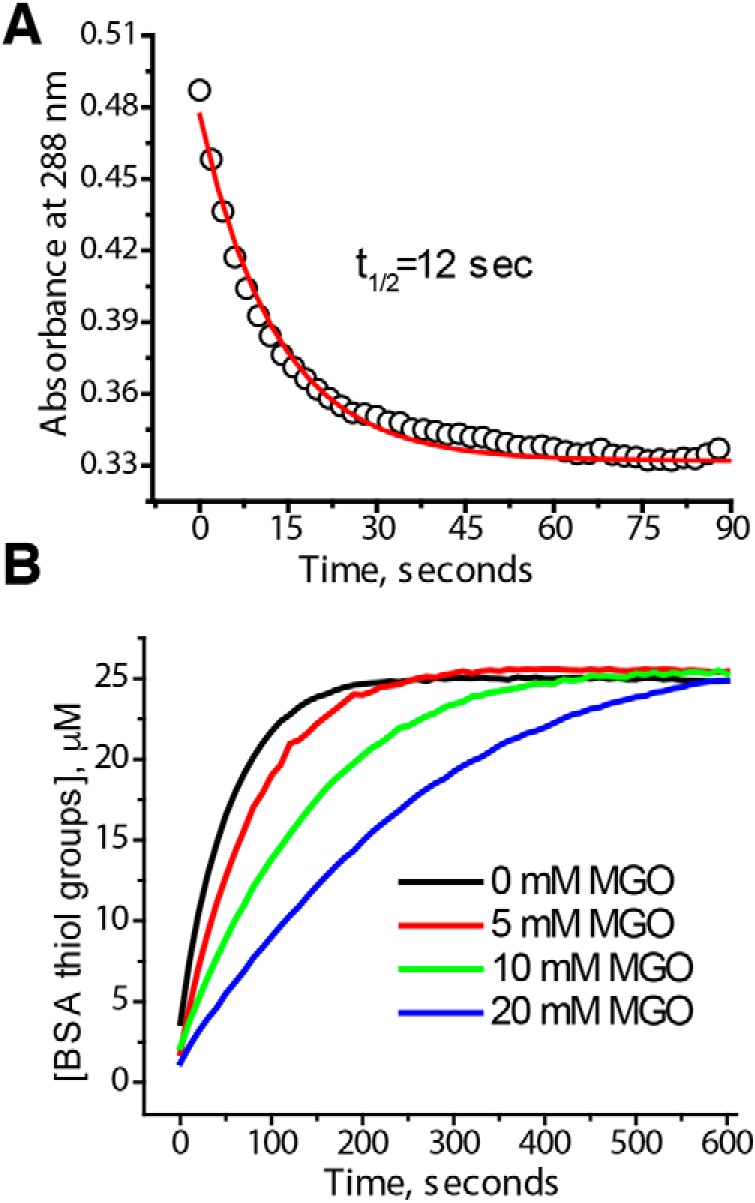
Hemiaminals are structurally similar to hemithioacetals, so it is likely that apparent protein deglycase activity of DJ-1 toward hemiaminals is also due to the equilibrium involving free MGO. To study deglycation of BSA by DJ-1, we glycated 100 μm BSA with 100 mm MGO and removed excess MGO by two rounds of quick buffer exchange on size-exclusion spin columns. Next, this glycated BSA solution was placed into a protein concentrator and subjected to short centrifugation runs (∼1 min) to obtain BSA-free samples for analysis of free MGO at different times after purification. We found that glycated and purified BSA initially contained ∼0.6 molar eq of free MGO, most likely due to the release of MGO from hemiaminals after purification (Fig. 8A). The amount of free MGO gradually decreased and stabilized at ∼0.3 molar eq of BSA, possibly due to a newly established equilibrium in the reaction with a single cysteine residue of BSA present in thiol form. Next, the same sample of glycated and purified BSA was incubated with DJ-1 or Glx3, and the concentration of l-lactate was determined by a lactate oxidase/peroxidase-based assay. We have found that amounts of l-lactate that are produced in these reactions (0.4–0.5 molar eq of BSA) are similar to the amounts of free MGO in this sample for both DJ-1 and Glx3 (Fig. 8B). DJ-1–catalyzed conversion of MGO was reported to produce only l-lactate regardless of the presence of thiols (5, 18), whereas catalysis by Glx3 produces both d- and l-lactate in comparable amounts (6). This means that more MGO has been converted by Glx3 compared with DJ-1, which is consistent with higher activity of Glx3. Together, our results indicate that small amounts of MGO are present in dynamic equilibrium with amino and thiol groups of BSA even after purification by desalting. This free MGO can be converted to lactate by the glyoxalase activity of Glx3; however, no additional lactate could be released from glycated BSA using DJ-1. These results again argue against DJ-1 protein deglycase activity.
Figure 8.
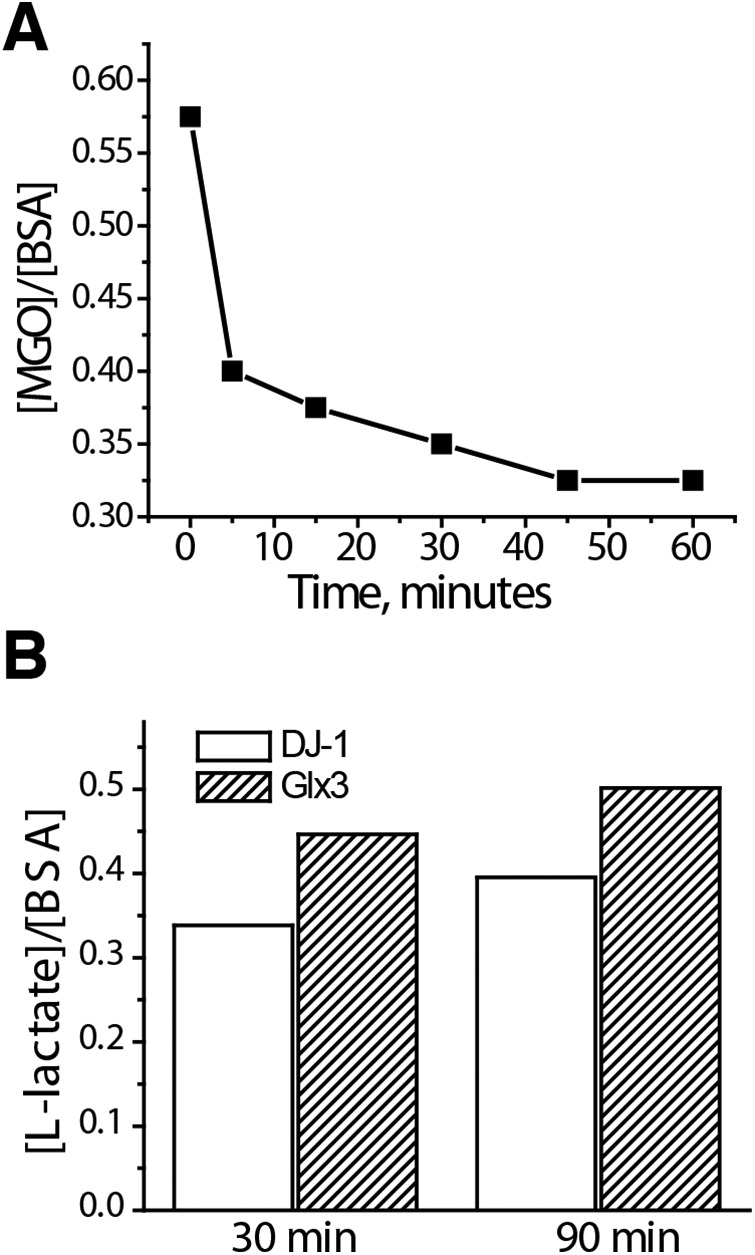
DJ-1 does not deglycate BSA. A, 1.5 ml of 100 μm BSA was incubated with 100 mm DJ-1 in PBS for 2 h at 37 °C. BSA was purified by two rounds of size exclusion on PD-10 columns and placed into a protein concentrator. At the indicated time points, ∼20 μl of flow-through solution was used to measure MGO concentration by DNPH. B, the same sample of glycated and purified BSA as in A was incubated with 30 μm DJ-1 or 10 μm Glx3. Aliquots were taken at 30 and 90 min to measure l-lactate using a lactate peroxidase assay. The experiment was repeated three times; representative data are shown. In all cases, the amount of l-lactate produced by DJ-1 or Glx3 did not significantly exceed the amount of free MGO right after purification of glycated BSA.
DJ-1 does not protect mammalian cells from acute MGO toxicity
To investigate whether DJ-1 is required for protection of mammalian cells against MGO toxicity, we generated stable HEK293 DJ-1–null cell lines using CRISPR/CAS9-mediated genome editing. To exclude the possibility of off-target effects of selected guide RNAs, we targeted two different exons of human DJ-1 independently: exon 2 (KO1) and exon 3 (KO2). Western blot analysis confirmed that the expression of DJ-1 in both KO1 and KO2 stable clones was successfully abolished (Fig. 9B) compared with control clones transfected with empty plasmid (Fig. 9A). To evaluate the relevance of DJ-1 in the detoxification of MGO, we compared cell sensitivity to MGO in HEK293 control and DJ-1–null cells using an MTT-based viability assay. A typical viability curve of HEK293 cells after 24-h exposure to different concentrations of MGO is shown in Fig. 9C. Both control and DJ-1–null cells showed similar sensitivity toward MGO with LD50 of 1.2–1.5 mm MGO, indicating that the presence of DJ-1 does not confer any measurable resistance toward MGO toxicity (Fig. 9D). Quantitative analysis of carboxymethyllysine (CML) content in proteins also argues against protein deglycase activity of DJ-1 because CML content in control and knockout clones was the same (Fig. 9, E and F). Although a significant increase in CML levels was observed in control clones after treatment with MGO, both knockout clones did not show any increase in cellular levels of CML when compared with control clones (Fig. 9F), indicating that physiological levels of DJ-1 are not necessary to prevent CML formation in response to MGO treatment.
Figure 9.

DJ-1 does not confer protection against acute MGO toxicity in live cells. A, sequencing of DNA from CRISPR/Cas9-generated DJ-1–null cells shows the interruption of sequence where nonhomologous end joining introduced indels in both chromosomes. Fragments of sequence from exon 3 in control (C2) and DJ-1–null (KO2) clones are shown. The protospacer adjacent motif (PAM) sequence is underlined, asterisk indicates the site of the expected indel. B, immunoblot analysis of DJ-1 expression in control (C1 and C2) and DJ-1–null (KO1 and KO2) cells. Bands corresponding to the molecular weight of DJ-1 are absent in both the KO1 and KO2 cell clones. GAPDH was used as a loading control. C, typical viability curve of HEK293 cells exposed to varying concentrations of methylglyoxal. D, LD50 values for MGO in control and DJ-1–null cells are not significantly different, arguing against a significant biological role for DJ-1 in detoxification of MGO. E, immunoblot analysis with anti-CML antibodies of total cell extracts from control and DJ-1–null cells grown in the absence (−) or presence (+) of 1 mm MGO. GAPDH was used as a loading control. F, quantification of total CML protein levels normalized to GAPDH signal. Data from three independent experiments are represented as mean ± S.E. (error bars). Asterisks indicate statistically significant differences between control and MGO treated sample groups with p < 0.05 according to two-sample t-test.
Discussion
Given the lack of consensus whether DJ-1 exerts its protective effects through an enzymatic detoxification of MGO and/or adducts of MGO with amino acids, we re-examined the glyoxalase and deglycase activities of DJ-1. Although we could readily detect GSH-independent glyoxalase activity of DJ-1 by NMR as well as by MGO and lactate assays, we have not found any evidence supporting DJ-1 protein deglycase activity. Moreover, we demonstrate that the glyoxalase activity of DJ-1 can account for apparent DJ-1 deglycase activity as discussed below.
The value of the equilibrium constant for the formation of hemithioacetals (Kc), ∼500 m−1, is ∼140 times lower than the previously reported value of 7.3 × 104 m−1 due at least in part to an incorrectly determined extinction coefficient by Lo et al. (15). When MGO is mixed with 2–10 mm GSH or NAC, a highly dynamic equilibrium between free MGO and hemithioacetals is quickly established in which free MGO and hemithioacetals are present in comparable amounts. Any intervention that lowers the concentration of free MGO will lead to a rapid decomposition of hemithioacetals, observable as a decrease of absorbance at 288 nm. This is especially clear from the effect of Tris because its reaction with MGO reduces the concentration of hemithioacetals. The pKa value of Tris is roughly the same as that of GSH or NAC, making it a good nucleophile at physiological pH. However, because hemiaminals do not have a significant absorbance at 288 nm, the shift in equilibrium position leads to a concomitant decrease in absorbance. Similarly, when DJ-1 (or any other glyoxalase) reduces the concentration of free MGO by converting it into lactate, spontaneous decomposition of hemithioacetals is unavoidable, and as a result any glyoxalase has a potential to be mistakenly classified as a deglycase (9, 18–20).
The kinetic profile of total MGO content during hemithioacetal decomposition induced by DJ-1 also argues against DJ-1 deglycase activity. We designed our experiments to drive the concentration of free MGO lower by increasing concentration of GSH. The rate of total MGO conversion by DJ-1 is decreased at low (∼0.16 mm) concentrations of free MGO compared with high (∼1.2 mm) concentrations. These data can be explained assuming that DJ-1 is a glyoxalase with a Km for MGO well below 1 mm. In contrast, these data are inconsistent with deglycase activity of DJ-1 because the rate of total MGO conversion by DJ-1 is lower at higher initial concentrations of GSH-MGO (Fig. 7B).
The side chains of lysine and arginine are almost completely protonated at physiological pH, so they are unlikely to be modified by MGO to a significant extent. This is why extremely high concentrations of MGO (100 mm) along with long incubation times are often used to obtain detectable amounts of hemiaminals. We have used a similar approach and could detect ∼0.5 molar eq of free MGO in a sample of BSA that underwent two rounds of purification from MGO by desalting (Fig. 8A). Because the concentration of free MGO continued to gradually decrease in this sample, we assume that immediately following purification, hemithioacetals have largely decomposed due to the efficient removal of MGO by desalting, but some MGO released from slower-decomposing hemiaminals was later recaptured by free cysteine. The addition of DJ-1 to this sample did not release l-lactate in amounts that are significantly larger than the amount of free MGO detectable right after purification (Fig. 8B). These data, combined with the fact that the presence of BSA does not lead to an increased rate of MGO conversion by DJ-1 (Fig. 3B), support the conclusion that DJ-1 cannot catalyze the hydrolysis of protein-based hemiaminals and hemithioacetals.
Although the transient/unstable nature of hemithioacetals and hemiaminals is known, it is often neglected in many experimental designs involving glycation of biomolecules with MGO. For example, given the rapid equilibrium between MGO and hemithioacetals, it is unclear how several studies measured the amount of thiol groups not modified by MGO (8, 17, 19) using thiol-reactive probes such as Ellman's reagent because Ellman's reagent should outcompete MGO in a properly designed experiment. Indeed, we show that even in the presence of 20 mm MGO, Ellman's reagent will modify 100% of all available thiol groups if given enough time (Fig. 5B), underscoring the transient nature of protein-based hemithioacetals.
The rapid equilibrium between hemithioacetals, hemiaminals, and MGO invites questions about the existence of protein deglycases and their necessity. First, it is hard to imagine an enzyme that can efficiently recognize MGO-modified amino acid chains within the context of various protein surfaces and especially inside active centers of enzymes as suggested by Richarme et al. (8). Second, as has been suggested previously (21), even if DJ-1 deglycation exists, it is unlikely to be more kinetically efficient than rapid spontaneous decomposition of MGO adducts followed by removal of MGO by glyoxalases. Conversely, given that the equilibrium constant of hemithioacetal formation is much lower than previously assumed, it is clear that GSH-independent glyoxalase activity of DJ-1 and related members of its superfamily may play an important role inactivating MGO without the need for a direct reaction between MGO and GSH. For example, GSH-independent glyoxalases may constitute an additional line of defense under oxidative stress if cellular levels of GSH drop enough to reduce the efficiency of GSH-dependent glyoxalases.
The question regarding the true molecular function of DJ-1 remains open. We found DJ-1 glyoxalase activity to be low (kcat is ∼0.02 s−1) in accordance with earlier reports (5, 6). It is unclear why an enzyme with such a low activity is crucial for neuroprotection. One possibility is that DJ-1 may detoxify an as yet unidentified neurotoxic molecule structurally similar to MGO. It is also possible that the function of DJ-1 is unrelated to the detoxification of MGO as many of the mutants found in patients with early development of Parkinson's disease have unaltered glyoxalase activity (18). DJ-1 may also serve as a sensor for oxidant stress because its active-site cysteine is prone to oxidation (22, 23). Although it is not clear whether and how post-translational modifications of DJ-1 are sensed by the cell, at least one post-translational modification of active-site cysteine alters the binding properties of DJ-1 and may therefore participate in signal transduction (24).
In conclusion, our data show that MGO is not as reactive toward thiols and amines as has been previously assumed. Moreover, significant quantities of MGO in its free form are likely present in the cytosol in fast equilibrium with hemiaminals and hemithioacetals. These hemithioacetals and hemiaminals are intrinsically unstable and will quickly decompose, releasing MGO until a new equilibrium point is reached. Due to this rapid equilibrium, cellular levels of protein-based hemithioacetals and hemiaminals can be effectively regulated by glyoxalases without the need for a special deglycase. The apparent decomposition of hemithioacetals by DJ-1, Glx3, or any other glyoxalase is a manifestation of this principle because conversion of free MGO into lactate by glyoxalase activity is sufficient to quickly restore thiol groups modified my MGO. Although the role of its glyoxalase activity in neuroprotection is not clear, our results definitively demonstrate that DJ-1 is not a protein deglycase. Therefore, to prevent misdirection in the search for its true molecular function, we propose a functional reclassification of DJ-1.
Experimental procedures
Antibodies
Rabbit polyclonal antibodies against human DJ-1 were produced, purified, and tested at National Biotechnology Center (Nur-Sultan, Kazakhstan). Rabbit anti-carboxymethyllysine antibody (ab27684) and Alexa Fluor 680–labeled mouse mAb were purchased from Abcam. For Western blotting, primary antibodies were used at 1:1,000 dilution in 5% nonfat milk, phosphate-buffered saline (PBS), Tween 20. Anti-mouse IRDye 800CW antibodies were purchased from LI-COR Biosciences and used at 1:5,000 dilution.
Reactions with methylglyoxal and protein assays
Methylglyoxal (Sigma) was freshly diluted from a 40% stock solution and standardized with N-acetylcysteine (16). All reactions were carried out in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4). N-Acetylcysteine and GSH (free acids) were prepared as 100 mm stock solutions in PBS and neutralized with NaOH. To prepare hemithioacetals, thiols were mixed with MGO in PBS and incubated for 10 min. Hemithioacetals were quantified by optical absorbance at 288 nm. Detailed procedures for calculations of equilibrium constants, extinction coefficients, and concentrations of free MGO from spectrophotometric data are described in supporting methods.
Thermofluor assays were performed as described (25) except PBS was used instead of Tris buffer. Analytical size-exclusion chromatography on a Superdex 200 10/300 column was performed at 0.4 ml/min. 1H NMR spectra were recorded on a JNM-ECA 500 series FT NMR spectrometer (JEOL) in the presence of 10–80% D2O. To measure MGO concentration by DNPH assay, 10-μl aliquots of reaction mixture were mixed with 10 μl of a 10 mm solution of DNPH in 2 m HCl and incubated for 15 min. Next, 0.98 ml of 2 m NaOH was added, and absorbance was measured at 550 nm.
Determination of l-lactate concentration
l-Lactate concentration was measured using a l-lactate assay kit (Vital Development, Russia) according to the manufacturer's instructions. This end-point assay utilizes bacterial l-lactate oxidase that catalyzes conversion of molecular oxygen and l-lactate into pyruvate and H2O2. Next, H2O2 is quantitatively reduced in peroxidase-catalyzed oxidative coupling of 4-aminoantipyrine with p-chlorophenol, resulting in a strongly absorbing quinoneimine. To measure l-lactate, 20 μl of sample was added to 0.98 ml of solution containing 0.2 unit/ml l-lactate oxidase, 2 units/ml horseradish peroxidase, 1.2 mm p-chlorophenol, 0.4 mm 4-aminoantipyrine, and 10 mm Tris, pH 7.5. The mixture was incubated for 5 min at 37 °C, and the concentration of lactate was determined spectrophotometrically at 505 nm from calibration curves.
Protein expression and purification
The human PARK7 gene was cloned into a pET28(b) vector using the NcoI and HindIII sites to express full-length, untagged DJ-1. BL21(DE3)-RIPL cells were transformed with the DJ-1 expression construct and plated on LB agar plates containing 50 μg/ml kanamycin and 25 μg/ml chloramphenicol. Transformed cells were transferred into terrific broth medium with antibiotics and grown at 37 °C to an OD600 of ∼3.0. Thereafter, expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 0.3 mm, after which the cultures were grown for a further 3 h. Cells were harvested by centrifugation; resuspended in 20 mm Tris, pH 8.0, 7 mm 2-mercaproethanol (buffer A); and frozen at −80 °C. For purification, cell pellets were thawed, lysed with lysozyme, sonicated in the presence of Complete protease inhibitor mixture (Roche Applied Science), and centrifuged for 30 min at 30,000 × g. One volume of saturated solution of ammonium sulfate was added to the clarified lysate, and the precipitate was separated by centrifugation. The supernatant was loaded onto a 4-ml Phenyl-Sepharose column, and unbound proteins were washed off with buffer A saturated to 50% with ammonium sulfate. DJ-1 was eluted with buffer A saturated to 35% with ammonium sulfate, desalted into buffer A plus 20 mm NaCl using PD-10 columns, passed through a 3-ml DEAE gravity column to remove nucleic acids, and dialyzed into PBS. DNA encoding glyoxalase 3 from C. albicans was synthesized and cloned into pET28(+)-TEV vector by GenScript. Glyoxalase 3 was expressed and purified on nickel-nitrilotriacetic acid and Phenyl-Sepharose columns and dialyzed into PBS. The glyoxalase 3 His tag was not removed.
LC-MS ESI of DJ-1
Samples of DJ-1 were desalted using ZipTip-C18 (Millipore) according to the manufacturer's instructions. A trapping column setup (AcclaimPepMap100 C18 precolumn) with a C18 analytical column (150 mm × 75 μm) and Dionex HPLC pump were used for chromatography.
Protein was eluted using a gradient of 0–90% aqueous acetonitrile in the presence of 0.1% formic acid. An unmodified Captive Spray ion source was used to interface the LC system to an Impact II mass spectrometer (Bruker). Data Analysis software was used for charge deconvolution.
CRISPR/Cas-mediated generation of DJ-1–null cell lines
To avoid artifacts due to off-target mutations, we designed target sequences for exons 2 and 3 of human DJ-1 (see supporting methods). The target sequences were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid (Addgene plasmid 42230) under the U6 promoter using BbsI restriction sites. HEK293 cells were transfected with either control pX330 vector or vectors targeting DJ-1 exons using a calcium phosphate transfection method. DJ-1–negative stable clones of transfected cells were isolated by single-cell dilution and screened for efficient DJ-1 knockout by immunoblot analysis. To validate whether DJ-1 knockout in single-cell clones was due to CRISPR/Cas9-generated indels, the genomic region surrounding exon 2 or 3 of DJ-1 was PCR-amplified and sequenced.
Cell culture and viability assay
HEK293 cells were cultured in high-glucose Dulbecco's modified Eagle's medium (Gibco) supplemented with GlutaMAXTM, 1 mm sodium pyruvate (Gibco), 100 units/ml penicillin, 100 μg/ml streptomycin (Gibco), and 10% fetal bovine serum (16000044, Gibco). Cells were maintained at 37 °C in 5% CO2. For viability assays, HEK293 cells were seeded in 96-well plates at a density of 20,000 cells/well and incubated for 2 h. MGO was added to attached cells and incubated for a further 24 h. Next, growth medium was replaced with fresh medium with 0.45 mg/ml MTT, and incubation continued for 4 h. Formazan crystals were solubilized in DMSO, and absorbance was measured at 570 nm. For Western blot analysis, cells grown in 6-well plates were treated with the indicated concentrations of MGO for 24 h and lysed in Laemmli buffer.
Author contributions
A. A. and Z. B. formal analysis; A. A. and D. U. supervision; A. A., Z. B., N. O., T. B., G. Y., M. A., D. T., and D. U. investigation; A. A., Z. B., M. A., and D. U. methodology; A. A., Z. B., D. T., and D. U. writing-original draft; Z. B. and D. U. conceptualization; Z. B. and D. U. validation; D. U. data curation; D. U. funding acquisition; D. U. visualization; D. U. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Aizhan Tkirova for excellent technical assistance. We thank the Proteomics and Mass Spectrometry Laboratory of National Biotechnology Center, Nur-Sultan city, for help with mass spectrometric experiments and the Core facility of Nazarbayev University for help with NMR spectroscopy experiments, oligonucleotide synthesis, and DNA sequencing.
This work was supported by a Nazarbayev University Oak Ridge Associated Universities grant (to D. U.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting methods and Figs. S1 and S2.
- MGO
- methylglyoxal
- Glx3
- glyoxalase III from C. albicans
- ESI
- electrospray ionization
- DNPH
- 2,3-dinitrophenylhydrazine
- NAC
- N-acetylcysteine
- KO
- knockout
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CML
- carboxymethyllysine
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C., Squitieri F., Ibanez P., Joosse M., van Dongen J. W., Vanacore N., van Swieten J. C., Brice A., Meco G., et al. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 10.1126/science.1077209 [DOI] [PubMed] [Google Scholar]
- 2. Canet-Avilés R. M., Wilson M. A., Miller D. W., Ahmad R., McLendon C., Bandyopadhyay S., Baptista M. J., Ringe D., Petsko G. A., and Cookson M. R. (2004) The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108 10.1073/pnas.0402959101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Subedi K. P., Choi D., Kim I., Min B., and Park C. (2011) Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 81, 926–936 10.1111/j.1365-2958.2011.07736.x [DOI] [PubMed] [Google Scholar]
- 4. Lee J. Y., Song J., Kwon K., Jang S., Kim C., Baek K., Kim J., and Park C. (2012) Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 21, 3215–3225 10.1093/hmg/dds155 [DOI] [PubMed] [Google Scholar]
- 5. Choi D., Kim J., Ha S., Kwon K., Kim E. H., Lee H. Y., Ryu K. S., and Park C. (2014) Stereospecific mechanism of DJ-1 glyoxalases inferred from their hemithioacetal-containing crystal structures. FEBS J. 281, 5447–5462 10.1111/febs.13085 [DOI] [PubMed] [Google Scholar]
- 6. Hasim S., Hussin N. A., Alomar F., Bidasee K. R., Nickerson K. W., and Wilson M. A. (2014) A glutathione-independent glyoxalase of the DJ-1 superfamily plays an important role in managing metabolically generated methylglyoxal in Candida albicans. J. Biol. Chem. 289, 1662–1674 10.1074/jbc.M113.505784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith N., and Wilson M. A. (2017) Structural biology of the DJ-1 superfamily. Adv. Exp. Med. Biol. 1037, 5–24 10.1007/978-981-10-6583-5_2 [DOI] [PubMed] [Google Scholar]
- 8. Richarme G., Mihoub M., Dairou J., Bui L. C., Leger T., and Lamouri A. (2015) Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 290, 1885–1897 10.1074/jbc.M114.597815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richarme G., and Dairou J. (2017) Parkinsonism-associated protein DJ-1 is a bona fide deglycase. Biochem. Biophys. Res. Commun. 483, 387–391 10.1016/j.bbrc.2016.12.134 [DOI] [PubMed] [Google Scholar]
- 10. Pfaff D. H., Fleming T., Nawroth P., and Teleman A. A. (2017) Evidence against a role for the parkinsonism-associated protein DJ-1 in methylglyoxal detoxification. J. Biol. Chem. 292, 685–690 10.1074/jbc.M116.743823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma N., Rao S. P., and Kalivendi S. V. (2019) The deglycase activity of DJ-1 mitigates α-synuclein glycation and aggregation in dopaminergic cells: role of oxidative stress mediated downregulation of DJ-1 in Parkinson's disease. Free Radic. Biol. Med. 135, 28–37 10.1016/j.freeradbiomed.2019.02.014 [DOI] [PubMed] [Google Scholar]
- 12. Zheng Q., Omans N. D., Leicher R., Osunsade A., Agustinus A. S., Finkin-Groner E., D'Ambrosio H., Liu B., Chandarlapaty S., Liu S., and David Y. (2019) Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 10, 1289 10.1038/s41467-019-09192-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galligan J. J., Wepy J. A., Streeter M. D., Kingsley P. J., Mitchener M. M., Wauchope O. R., Beavers W. N., Rose K. L., Wang T., Spiegel D. A., and Marnett L. J. (2018) Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. U.S.A. 115, 9228–9233 10.1073/pnas.1802901115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varland S., Osberg C., and Arnesen T. (2015) N-terminal modifications of cellular proteins: the enzymes involved, their substrate specificities and biological effects. Proteomics 15, 2385–2401 10.1002/pmic.201400619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lo T. W., Westwood M. E., McLellan A. C., Selwood T., and Thornalley P. J. (1994) Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with Nα-acetylarginine, Nα-acetylcysteine, and Nα-acetyllysine, and bovine serum albumin. J. Biol. Chem. 269, 32299–32305 [PubMed] [Google Scholar]
- 16. Wild R., Ooi L., Srikanth V., and Münch G. (2012) A quick, convenient and economical method for the reliable determination of methylglyoxal in millimolar concentrations: the N-acetyl-l-cysteine assay. Anal. Bioanal. Chem. 403, 2577–2581 10.1007/s00216-012-6086-4 [DOI] [PubMed] [Google Scholar]
- 17. Aćimović J. M., Stanimirović B. D., Todorović N., Jovanović V. B., and Mandić L. M. (2010) Influence of the microenvironment of thiol groups in low molecular mass thiols and serum albumin on the reaction with methylglyoxal. Chem. Biol. Interact. 188, 21–30 10.1016/j.cbi.2010.07.013 [DOI] [PubMed] [Google Scholar]
- 18. Matsuda N., Kimura M., Queliconi B. B., Kojima W., Mishima M., Takagi K., Koyano F., Yamano K., Mizushima T., Ito Y., and Tanaka K. (2017) Parkinson's disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci. Rep. 7, 12816 10.1038/s41598-017-13146-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mihoub M., Abdallah J., Gontero B., Dairou J., and Richarme G. (2015) The DJ-1 superfamily member Hsp31 repairs proteins from glycation by methylglyoxal and glyoxal. Biochem. Biophys. Res. Commun. 463, 1305–1310 10.1016/j.bbrc.2015.06.111 [DOI] [PubMed] [Google Scholar]
- 20. Richarme G., Abdallah J., Mathas N., Gautier V., and Dairou J. (2018) Further characterization of the Maillard deglycase DJ-1 and its prokaryotic homologs, deglycase 1/Hsp31, deglycase 2/YhbO, and deglycase 3/YajL. Biochem. Biophys. Res. Commun. 503, 703–709 10.1016/j.bbrc.2018.06.064 [DOI] [PubMed] [Google Scholar]
- 21. Rabbani N., and Thornalley P. J. (2015) Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 458, 221–226 10.1016/j.bbrc.2015.01.140 [DOI] [PubMed] [Google Scholar]
- 22. Meulener M. C., Xu K., Thomson L., Ischiropoulos H., and Bonini N. M. (2006) Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. U.S.A. 103, 12517–12522 10.1073/pnas.0601891103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blackinton J., Lakshminarasimhan M., Thomas K. J., Ahmad R., Greggio E., Raza A. S., Cookson M. R., and Wilson M. A. (2009) Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J. Biol. Chem. 284, 6476–6485 10.1074/jbc.M806599200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mussakhmetov A., Shumilin I. A., Nugmanova R., Shabalin I. G., Baizhumanov T., Toibazar D., Khassenov B., Minor W., and Utepbergenov D. (2018) A transient post-translational modification of active site cysteine alters binding properties of the parkinsonism protein DJ-1. Biochem. Biophys. Res. Commun. 504, 328–333 10.1016/j.bbrc.2018.08.190 [DOI] [PubMed] [Google Scholar]
- 25. Utepbergenov D., Hennig P. M., Derewenda U., Artamonov M. V., Somlyo A. V., and Derewenda Z. S. (2016) Bacterial expression, purification and in vitro phosphorylation of full-length ribosomal S6 kinase 2 (RSK2). PLoS One 11, e0164343 10.1371/journal.pone.0164343 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.