

Abstract
Posttranslational modifications of proteins, such as phosphorylation and dephosphorylation, play critical roles in cellular functions through diverse cell signaling pathways. Protein kinases and phosphatases have been described early on as key regulatory elements of the phosphorylated state of proteins. Tight spatial and temporal regulation of protein kinase and phosphatase activities has to be achieved in the cell to ensure accurate signal transduction. We demonstrated previously that phosphorylation of a membrane protein can lead to its topological rearrangement. Additionally, we found that both the rate and extent of topological rearrangement upon phosphorylation are lipid charge– and lipid environment–dependent. Here, using a model membrane protein (the bacterial lactose permease LacY reconstituted in proteoliposomes) and a combination of real-time measurements and steady-state assessments of protein topology, we established a set of experimental conditions to dissect the effects of phosphorylation and dephosphorylation of a membrane protein on its topological orientation. We also demonstrate that the phosphorylation-induced topological switch of a membrane protein can be reversed upon protein dephosphorylation, revealing a new regulatory role for phosphorylation/dephosphorylation cycles. Furthermore, we determined that the rate of topological rearrangement reversal is correlated with phosphatase activity and is influenced by the membrane's lipid composition, presenting new insights into the spatiotemporal control of the protein phosphorylation state. Together, our results highlight the importance of the compartmentalization of phosphorylation/dephosphorylation cycles in controlling membrane protein topology and, therefore, function, which are influenced by the local lipid environment of the membrane protein.
Keywords: membrane protein, phospholipid, posttranslational modification (PTM), protein folding, phospholipid vesicle, charge–balance rule, topogenesis
Introduction
Signal transduction pathways constitute a complex network of information integration, processing, and transmission, which is essential for homeostasis, allowing a cell to respond to external stimuli. Protein phosphorylation is one of the main mechanisms used by cells to properly and rapidly transduce these signals (1). Protein phosphorylation is a dynamic, reversible posttranslational modification that plays key roles in several physiological processes, including cell growth, proliferation, differentiation, and apoptosis (2–6). Protein phosphorylation is one of the most studied posttranslational modifications, partially because kinases are often overexpressed and/or mutated in disease. Aberrant phosphorylation profiles have been observed in the context of cancer, diabetes, and inflammatory disorders (7–9).
Mass spectrometry–based phosphoproteomics studies have shown that a large percentage of cellular proteins can be phosphorylated (more than 6,000 proteins), including many membrane proteins (10, 11). Despite the discovery of more than 20,000 unique protein phosphorylation sites, the abundance of individual phosphorylated forms (the fraction of a given protein phosphorylated at a given site) is frequently low (10, 12), underlining the functional significance of phosphorylation/dephosphorylation dynamics.
Although equally important in establishing and controlling the subtle balance between phosphorylated and dephosphorylated states of proteins in the cell, protein phosphatases have received less attention than protein kinases. Recent findings demonstrated that protein phosphatases are equally critical in setting the levels of protein phosphorylation in cells, thus participating in the regulation of many physiological processes (4, 13, 14). In particular, functional and imaging studies have demonstrated that protein phosphatases show high levels of functional regulation, with specific subcellular localization and tissue expression determined for specific protein phosphatase isoforms (15) and modulation of activity and specificity via formation of multiprotein complexes (16, 17).
Signals transduced by kinases depend on the extent and duration of substrate phosphorylation. Therefore, tight regulation of protein kinase and phosphatase activity in space and time has to be achieved in the cell to ensure accurate signal transduction. This is achieved through a diversity of mechanisms, such as compartmentalization of both the protein kinase and phosphatase (18). There are many kinase- and phosphatase-binding proteins, and their role is to tether their enzyme-binding partners to cellular sublocalization, thus streamlining signal transduction by placing these enzymes in close and targeted proximity to their substrates. Two archetypical examples of this process are type 1 protein phosphatase–targeting subunits (19) and protein kinase A–anchoring proteins (20, 21). Together, these findings led to the conclusion that signal transduction through the cell does not simply occur via protein diffusion (22) but that multiple cellular proteins are phosphorylated and then dephosphorylated at different cellular locations (23), and signal integration occurs via several mechanisms, such as multisite phosphorylation of the target protein, temporal and spatial encoding of information, and communication between kinases (24, 25).
Protein phosphorylation regulates the activity, location, and properties of a protein in multiple and complex ways. The effects of phosphorylation on a protein include changes in structural properties through allosteric effects, modification of protein–protein binding properties, and disorder–order– and order–disorder–coupled transitions (26). We recently demonstrated that phosphorylation of a membrane protein can trigger its topological switch, with rates of topological change occurring on a scale of seconds (27). Our results demonstrated how posttranslational modifications may influence membrane protein topology in a lipid-dependent manner, both along the organelle trafficking pathway and at their final destination.
The goal of this study was to test the effects of phosphorylation/dephosphorylation cycles on the topological (re)orientation of a membrane protein. We therefore determined the effects of dephosphorylation on the reversibility of phosphorylation-induced topological switching of a membrane protein, the impact of kinase/phosphatase compartmentalization, the consequences of variable dephosphorylation rates, and the influence of membrane lipid composition on membrane protein topological inversion rates and yields. To tackle these questions, we used a model membrane protein (lactose permease; LacY reconstituted in proteoliposomes, where we can control the lipid composition, the occurrence of phosphorylation (kP, Fig. 1) and the location and rate of dephosphorylation (kUP1 when the phosphatase is located outside of the proteoliposomes and kUP2 when the phosphatase is located in the proteoliposomes). Our strategy enabled us to determine rates of membrane protein topological switches from correct to incorrect (kF1) and from incorrect to correct (kF2).
Figure 1.
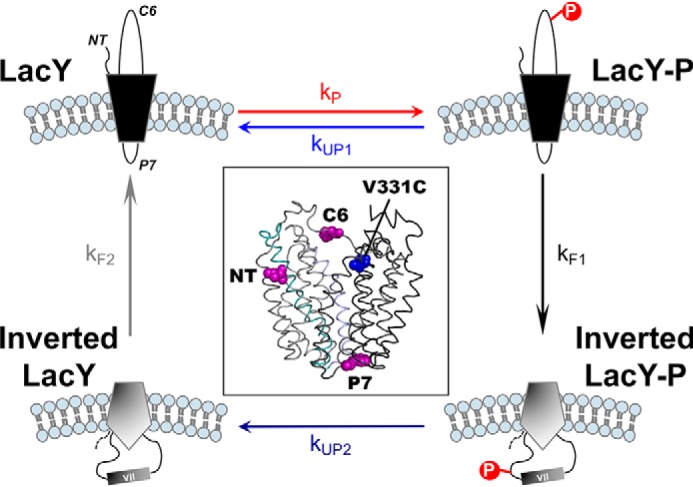
Schematic of the events occurring during phosphorylation/dephosphorylation-induced topological switching of a membrane protein in proteoliposomes. LacY topology is shown when assembled in proteoliposomes containing E. coli native lipids (correct LacY, top left) before and during phosphorylation by a kinase located outside of the proteoliposomes. The NT, C6, and P7 domains are indicated. Membrane phospholipids are depicted in light blue. LacY-P, phosphorylated LacY. The boxed area depicts a side view of LacY (PDB code 2CFQ) with the diagnostic Trp replacements introduced in EMD NT, C6, and P7 (magenta spheres) and the IAEDANS label at Cys-331 (blue spheres).
We uncovered a new dynamic and regulatory role of posttranslational phosphorylation and dephosphorylation in the structure and function of membrane proteins that is linked to the local lipid environment of the protein. Our findings further our understanding of the roles played by membrane lipids in membrane protein topology and present new insights into the mechanisms of cellular signaling through posttranslational modifications of membrane proteins.
Results
We first developed a FRET-based assay to monitor extramembrane domain (EMD)3 flipping of LacY in proteoliposomes induced by a post-reconstitution change in lipid composition (28). Trp residues were introduced in EMD NT, C6, or P7 (Fig. 1). V331C was labeled by 1,5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (IAEDANS) in the C-terminal transmembrane domain (TMD) bundle, whose topology is insensitive to the lipid environment. When assessing NT and C6 EMDs, high FRET indicates correct orientation, whereas low FRET reflects the inverted orientation. Controls demonstrated that the Trp/IAEDANS LacY derivatives behave identically to near-WT LacY with respect to topology inversion, as determined previously by the substituted cysteine accessibility method for determining TMD orientation (SCAMTMD).
We previously established that changing the net charge of EMDs C2/C4/C6 from +2/+2/+2 to −2/−2/−2 of LacY leads to topological reorientation of the N-terminal six-TMD bundle of LacY accompanied by solvent exposure of TMD VII in PE-containing cells, whereas a change to −2/0/−2 did not result in inversion (29). This observation led us to use two different LacY charge templates to test the effects of posttranslational phosphorylation on topology. Using LacY charge templates −2/0/−2 and −2/+2/+2, where we engineered either one or two phosphorylation sites in EMD C6, we established that the sensitivity of a given membrane protein to posttranslational phosphorylation topological reorientation correlates with its native EMD charge state and the level of phosphorylation to which it may be subjected (27). To minimize complexity, we opted to focus on LacY templates containing a single engineered phosphorylation site and investigate the impact of a single phosphorylation event regarding the potential for a given template to reorient across the lipid bilayer. Because LacY does not contain any native phosphorylation sites, we engineered a phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylation site in EMD C6 of LacY derivatives containing the above FRET pairs. The C-terminal His6-tagged LacY derivative can be purified to homogeneity and reconstituted into liposomes of Escherichia coli native lipid composition as well as liposomes mimicking that of eukaryotic membranes. Addition of excess kinase resulted in flipping of the C6 EMD followed by slower flipping of the NT EMD on a time scale of seconds (27), as observed previously for lipid-induced flipping (28).
Here we determined whether this phosphorylation-induced topological switch is reversible upon dephosphorylation. First we tested the reversibility of the phosphorylation-induced topological switch at steady state in proteoliposomes composed of WT E. coli lipid composition and containing LacY exhibiting one engineered PDK1 phosphorylation site, a change in the net charge of EMDs C2/C4/C6 to −2/0/−2, and a diagnostic cysteine in EMD C6 of LacY lacking all other cysteine residues. To test the effect of dephosphorylation on EMD C6 topology, the λ protein phosphatase was encapsulated in the proteoliposomes. Phosphorylation of LacY was triggered by addition of PDK1 to the outside of the proteoliposomes. Kinase activity was inhibited after 30-min incubation at 20 °C by addition of 10 mm GSK 2334470 (Chemical Abstracts Service (CAS) 1227911-45-6), a PDK1-specific inhibitor. Assessment of the LacY phosphorylation state allowed us to determine that the phosphorylation levels of LacY are lower in the presence of encapsulated phosphatase (Fig. 2A, lane 3) and that they can be entirely abrogated upon simultaneous inhibition of PDK1 (Fig. 2A, lane 4).
Figure 2.
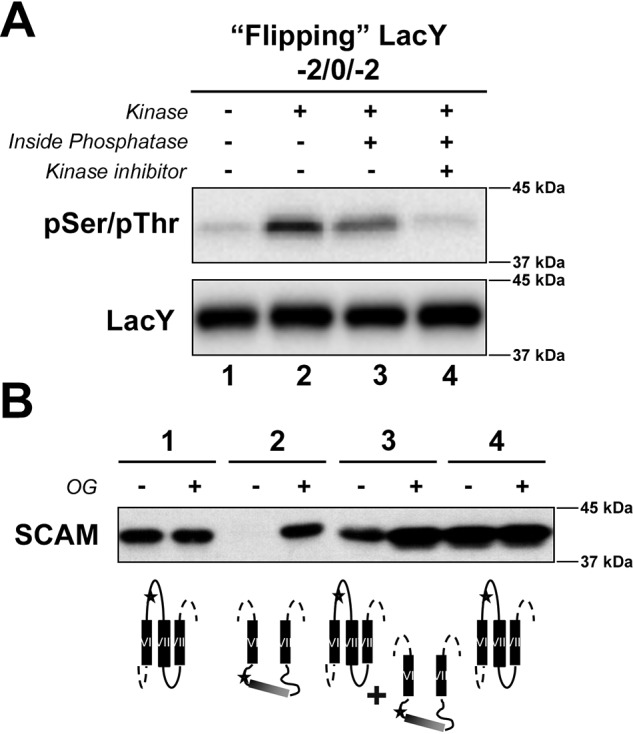
Determination of the reversibility of phosphorylation-induced topological switch of a membrane protein upon dephosphorylation in the proteoliposomes at steady state. A, determination of the phosphorylation state of the LacY template −2/0/−2 containing a single engineered PDK1 sequence in EMD C6 in the presence or absence of external kinase and its inhibitor, with and without encapsulated phosphatase, as indicated. LacY was reconstituted in proteoliposomes made of E. coli native lipids. The phosphorylation state of LacY was visualized by Western blotting using an anti-pSer/pThr antibody after quenching in Laemmli buffer. B, determination of the orientation of EMD C6 of LacY with altered EMD net charge and containing a single cysteine replacement using SCAMTMD under the various conditions indicated in A. Proteoliposomes were exposed to MBP before or after addition of OG. The deduced orientation of EMD C6 is also shown. The orientation of LacY TMDs VI–VIII is summarized for LacY in proteoliposomes. Stars indicate the position of single Cys replacements in EMD C6 (H205C), used to determine topological orientation.
We validated that addition of 10 mm GSK 2334470 leads to full inhibition of PDK1, as evidenced by the absence of LacY phosphorylation in the presence of GSK 2334470 (Fig. S1A). Additionally, we verified that the reversibility of LacY phosphorylation is solely due to the action of the encapsulated phosphatase. We conducted the same experiments as described above using the “nonflipping” −2/+2/+2 LacY template, which has been shown to maintain its correct topological orientation upon addition of one phosphate group (27). No changes in LacY phosphorylation state were observed in the presence or absence of encapsulated phosphatase or upon PDK1 inhibition (Fig. S2A), indicating the absence of “leakage” of the phosphatase activity or spontaneous dephosphorylation. As expected, no change in LacY topology was observed under the various conditions tested (Fig. S2B).
Next we tested whether phosphorylation impacted the overall topological orientation of LacY. SCAMTMD assessment of EMD C6 topology at steady state (Fig. 2B) further revealed that dephosphorylation of LacY impacted its overall topological orientation. Indeed, full LacY topological inversion was observed in the presence of kinase only (Fig. 2B, lane 2), whereas mixed topologies were observed with encapsulated phosphatase (Fig. 2B, lane 3). Phosphorylation of LacY followed by its full dephosphorylation led to complete reversal to correct the topology (Fig. 2B, lane 4). Together, our results indicate that the level of dephosphorylation correlates with the amount of correct EMD C6 topology, with the majority of the topological switch observed upon complete phosphorylation of LacY. This finding is of particular importance for proteins undergoing multisite phosphorylation events, where an increasing local phosphate content could tip the scale toward topological switch. Because these multiple phosphorylation sites tend to cluster around the same region in a protein (30, 31), in silico prediction of the potential target protein could be achieved.
Next we tested whether the rate of the topological switch is influenced by the rate of LacY dephosphorylation and/or compartmentalization of kinase and phosphatase. To do so, we measured the rate of phosphorylation-induced topological switch using proteoliposomes made of WT E. coli lipids. We used a LacY template exhibiting an engineered PDK1 phosphorylation site, a change in the net charge of EMD C2/C4/C6 to −2/0/−2, and a Trp/IAEDANS FRET pair allowing real-time monitoring of EMD C6 topology. In all cases, phosphorylation of LacY was triggered by addition of PDK1 to the solution. Real-time measurements were conducted under two conditions: both kinase and phosphatase are located in the same compartment (outside of proteoliposomes), or kinase and phosphatase are located on opposite sides of the membrane (phosphatase encapsulated in the proteoliposomes). By varying the phosphatase:kinase ratio, we were able to modulate the rate of phosphorylation/dephosphorylation.
Our data (Fig. 3) demonstrated that the nature of the phosphorylation/dephosphorylation cycle influences the phosphorylation-induced topological rearrangement of LacY. One-phase exponential decay kinetics (Fig. 3B) are observed when kinase and phosphatase are located in different compartments (phosphorylation and dephosphorylation occurring on two different LacY topomers), whereas more complex two/three-phase decay kinetics (Fig. 3A) are observed when kinase and phosphatase are in the same compartment (phosphorylation and dephosphorylation occurring on the same LacY topomer before its topological switch). One possible explanation for these differences lies in the sequestration effects occurring when enzyme concentrations (kinase and phosphatase) become equal and/or higher than that of the substrate (LacY) (32). Under these conditions, LacY may be bound to phosphatase more often when phosphatase content increases, therefore preventing its topological inversion and changing the dynamics of the phosphorylation-induced topological switch.
Figure 3.

Determination of the effects of dephosphorylation rates on the dephosphorylation-induced topological switch of a membrane protein. LacY template −2/0/−2 with a single Trp replacement in EMD C6, V331C labeled with IAEDANS and containing a single engineered PDK1 sequence in EMD C6 was reconstituted in proteoliposomes made of E. coli native lipids. A and B, real-time FRET measurements monitoring phosphorylation-induced topological switch of LacY in the presence of various concentrations of phosphatase located either outside of proteoliposomes (A) or encapsulated in proteoliposomes (B). Molar kinase:phosphatase ratios are indicated next to the measured kinetics. Data represent the averaged normalized fluorescence expressed as the ratio FMeas/F0. Schematics of the measured events are shown, with the varied rates of dephosphorylation highlighted. C, the flipping rates, kF1, determined from linear regression fits of the first 30 s of the data shown in A and B. In all cases, the data represent mean values ± S.D. from four experimental replicates.
Additionally, we determined that the rate of the phosphorylation-induced topological switch is inversely correlated with the dephosphorylation rate, as evidenced by decreasing rates of inversion when increasing amounts of phosphatase are present (Fig. 3C). As expected, LacY topological flipping rates decrease when both kinase and phosphatase are located in the same compartment (Fig. 3A). In this case, the LacY topological switch is in direct competition with LacY dephosphorylation (as depicted in the diagram in Fig. 3A). When kUP1 < kF1, then LacY flipping is favored, and decreases in FRET follow a one-phase (ratio 1:0, no phosphatase) or two-phase decay (ratios 1:1 and 1:2). When kUP1 > kF1 (ratios 1:5 to 1:50), then phosphorylation/dephosphorylation cycles are occurring faster than LacY flipping. However, after LacY has flipped, it becomes trapped in its inverted topology because of the absence of phosphatase in the proteoliposomes, thus explaining the progressive shift of this equilibrium toward inverted LacY over time, independent of kinase:phosphatase molar ratios. These results indicate that the phosphorylation-induced topological switch of a membrane protein is favored under conditions where kinase activity/concentration is higher than phosphatase activity/concentration and when both enzymes are present in the same subcellular location. The probability of this inversion decreases significantly with increasing phosphatase activity/concentration.
The determination that topological flipping rates are also influenced by the presence of different content of encapsulated phosphatase (Fig. 3B) prompted us to investigate the real-time reversibility of the phosphorylation-induced topological switch upon dephosphorylation. To do so, we used the same experimental conditions as in Fig. 3B (outside kinase, encapsulated phosphatase) with a 1:1 kinase:phosphatase molar ratio. Real-time measurements of the LacY topological switch were conducted with and without addition of kinase inhibitor (GSK 2334470). The results presented in Fig. 4A indicate that, upon inhibition of LacY phosphorylation (200 s after triggering LacY phosphorylation by addition of external PDK1), the topological switch can be reversed (i.e. return to LacY correct topology), as shown previously in steady-state topology determination (Fig. 2), with the rate of reverse topological switch (kF2 as depicted in Fig. 4B, 0.00394 ± 0.00014 s−1) close to the rate of topological switch in the presence of kinase only (kF1 as depicted in Fig. 4B, 0.00462 ± 0.00007 s−1).
Figure 4.
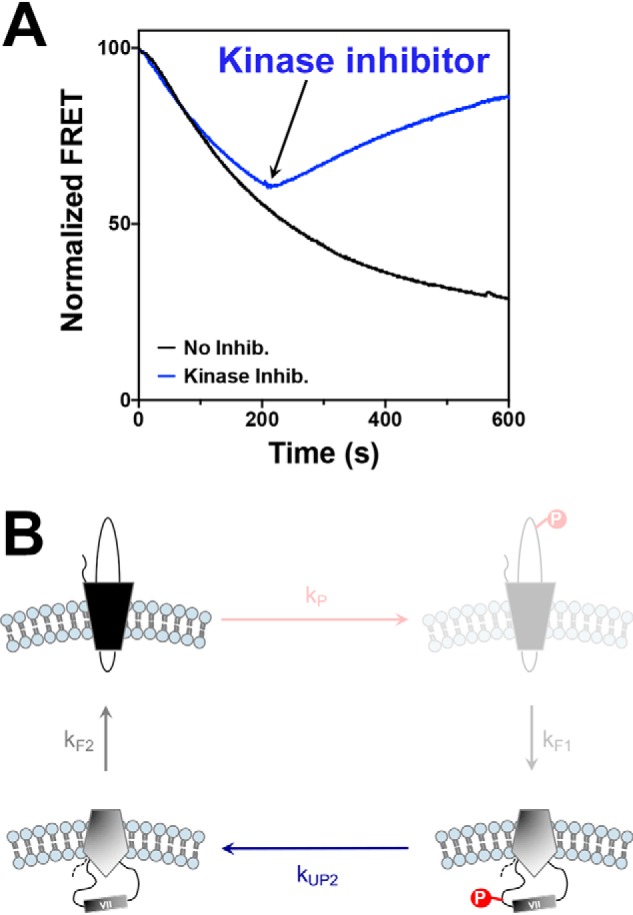
Real-time determination of the reversibility of phosphorylation-induced topological switch of a membrane protein upon dephosphorylation in proteoliposomes. A, real-time FRET measurements monitoring the topological switch of EMD C6 in LacY in the presence of encapsulated phosphatase with or without addition of kinase inhibitor (Inhib) GSK 2334470. LacY template −2/0/−2 containing a single engineered PDK1 sequence in EMD C6 was reconstituted in proteoliposomes made of E. coli native lipids. Data represent the averaged normalized fluorescence expressed as the ratio FMeas/F0. Experiments were repeated three to five times, and the data represent mean values. B, schematics of the measured events, with the inhibited steps (phosphorylation and topological inversion) shaded.
To further investigate the relationship between dephosphorylation rate and topological switch reversal, we modulated the level of phosphatase enzymatic activity by coencapsulating λ protein phosphatase with various amounts of one of its known inhibitors, ATP (Fig. 5). Because modulation of phosphatase activity in the lumen of the proteoliposomes leads to different topological steady-state equilibria (the amount of inverted LacY at steady state inversely correlates with the phosphatase activity), we first triggered phosphorylation of LacY to induce its topological rearrangement (Fig. 5A) and then let the system equilibrate for at least 30 min at 20 °C (as evidenced by a stable FRET signal). When equilibrium was reached, the kinase inhibitor was added to the solution, and real-time FRET was monitored (Fig. 5B). Our results indicate that the rate of topological switch reversal positively correlates with phosphatase activity, as evidenced by slower FRET recovery for increasing ATP concentrations (Fig. 5C). This approach allowed us to independently determine the rates of topological switch reversal (kF2) without interference from kP or kF1 and solely assess their dependence on dephosphorylation rates (kUP2). From this determination, we can conclude that the rate of topological reversal (flipping from inverted to correct, kF2) is slower than the rate of topological switch (flipping from correct to inverted, kF1) in membranes exhibiting the same lipid composition (E. coli total lipids), prompting us to test this observation in the context of other lipid compositions.
Figure 5.

Determination of the rate of dephosphorylation-induced topological switch of a membrane protein after inhibition of kinase activity. A and B, real-time FRET measurements monitoring phosphorylation-induced topological switch of EMD C6 in the LacY −2/0/−2 template reconstituted in proteoliposomes made of E. coli total lipids before (A) and after (B) addition of kinase inhibitor. LacY proteoliposomes with encapsulated phosphatase were incubated at 20 °C for 30 min after addition of excess PDK1. When a fluorescence steady state was reached, the PDK1 inhibitor GSK 2334470 was added. The various traces depict the topological switch of EMD C6 with various amounts of ATP encapsulated in the proteoliposomes, allowing the determination of kF2. Data represent the averaged normalized fluorescence expressed as the ratio FMeas/F0. C, the flipping rates, kF2, determined from single exponential fit of the data shown in B. In all cases, the data represent mean values ± S.D. from four experimental replicates.
Our previous work on the phosphorylation-induced topological switch of a membrane protein indicated substantial importance of the membrane properties (lipid composition and membrane fluidity) regarding both the rates and steady-state levels of LacY inversion (27). Therefore, we further examined the influence of membrane lipid composition on the rate of topological switch reversibility using proteoliposomes made of lipids mimicking the membrane composition of eukaryotic organelles (endoplasmic reticulum, Golgi, endosomes, and plasma membrane, with compositions described previously (27)). These proteoliposomes contained a LacY template exhibiting an engineered PDK1 phosphorylation site, a change in the net charge of EMDs C2/C4/C6 to −2/0/−2, a Trp/IAEDANS FRET pair allowing real-time monitoring of EMD C6 topology, and where the λ protein phosphatase was encapsulated with various ATP concentrations. The addition of external PDK1 first triggered the LacY topological switch. Then the system was allowed to equilibrate and reach a steady-state stable FRET signal. Phosphorylation was stopped by addition of a PDK1-specific inhibitor (GSK 2334470), and real-time FRET increases were monitored (Fig. 6). Exponential fits of the data allowed determination of flipping rates (kF2) for the various lipid compositions and the various ATP contents (Fig. 6, histograms). The results illustrate that the rate of topological switch reversal, kF2, is influenced by the membrane lipid composition, similar to what was observed for the rate of the phosphorylation-induced topological switch (27).
Figure 6.

Dephosphorylation-induced topological switch of a membrane protein is influenced by the organelle lipid environment. A–D, real-time FRET measurements monitoring phosphorylation-induced topological switch of EMD C6 in the LacY −2/0/−2 template reconstituted in proteoliposomes mimicking the lipid composition of endosomes (A), the Golgi (B), the endoplasmic reticulum (C), and the plasma membrane (D). Kinase inhibitor addition is indicated by arrows. The traces depict the topological switch of EMD C6 with various amounts of ATP encapsulated in proteoliposomes, with increasing amounts correlating with decreasing phosphatase activities. The bar graphs display the flipping rates, kF2, determined from single exponential fit of the data shown above. In all cases, the data represent mean values ± S.D. from four experimental replicates.
It should be noted that, in the absence of ATP (0 mm), determined kF2 values for a given lipid composition are lower than previously determined kF1 values, indicating slower topological reorientation from inverted to correct LacY. One possible explanation is that the energy penalty for inserting a transmembrane peptide back into a lipid membrane is higher than the one associated with releasing TMD VII from the membrane, the latter being driven by the N-terminal bundle (TMDs I–VI). Also, the rate of dephosphorylation may be slower than the rate of phosphorylation and/or the phosphatase interacts longer with LacY than with the kinase, as these two enzymes might present different catalytic characteristics.
Our results indicate that the rates of topological reorientation of a membrane protein induced either by posttranslational phosphorylation or dephosphorylation are dependent on the membrane lipid composition following trends similar to our previous work (27). Topological inversion/reversal happens fastest in the plasma membrane but is significantly slower in endosomes. Also, a gradual increase in dephosphorylation-dependent topological inversion is observed along the secretory pathway (endoplasmic reticulum < Golgi < plasma membrane), where increases in sphingomyelin and cholesterol contents and decreases in anionic lipid content occur. Together, our findings point toward a possible new regulatory mechanism for membrane protein activity/localization (cycles of posttranslational phosphorylation/dephosphorylation) and highlight the importance of the localization and activity of the regulatory enzymes (kinase and phosphatase) regarding the lifetime of the posttranslational modification and possible downstream signal transduction.
Discussion
Phosphorylation and dephosphorylation of proteins are widely recognized as key regulatory elements of cellular functions, affecting a diversity of proteins and mechanisms (3). Protein phosphorylation and dephosphorylation can regulate signaling in multiple ways, including activation/inhibition through conformational changes, creation of binding site(s) for proteins containing specific domains (i.e. complex formation), and control of cellular localization (23, 26). In the case of membrane proteins, phosphorylation-induced structural changes have been shown to modulate the functions of ion channels (33, 34), aquaporins (35), the ligand specificity of CD36 (36), the structure of tight junctions (37), and mitochondrial function (38) as well as alter their trafficking/targeting (39).
Our study adds to this long list of regulatory roles the possibility to regulate the topological orientation of a membrane protein through phosphorylation/dephosphorylation of its EMDs. This is of particular interest for surface membrane proteins because our results demonstrate that the rates of topological inversion are faster at the plasma membrane than they are in endosomes or at the Golgi apparatus. Receptor tyrosine kinases (RTKs) are single-pass transmembrane receptors located at the surface of various cell types. They are key constituents of signal transduction pathways involved in cellular growth, differentiation, metabolism, and motility (40). One very well studied example is the epidermal growth factor receptor, a member of the ErbB family of RTKs, which is up-regulated in many cancer cell types, including breast cancer (41). Genomic analyses of single-nucleotide variants revealed over 44,000 deposited variations in the Single Nucleotide Polymorphism Database from the National Center for Biotechnology Information (NCBI), with 15 of them listed as pathogenic. Three pathogenic epidermal growth factor receptor SNPs result in increasing the negative charge content of the cytoplasmic domain (rs121913230, where the Gly residue is mutated to a phosphorylatable Ser, and rs121913428, where a Gly residue is mutated to a negatively charged Asp) and decreasing the positive charge content in the extracellular domain (rs139429793, where a Glu residue is mutated to a positively charged Lys). Positively charged residues are dominant in orienting EMDs toward the cytoplasm (42), which makes any mutation leading to charge alteration or creation of a phosphorylation site in a cytoplasmic domain a potential means to alter the topology of a membrane protein.
Moreover, these RTKs undergo controlled turnover at the cell surface, constantly recycling between the plasma membrane and endosomes (43). Upon activation by a specific ligand and RTK autophosphorylation, increased turnover of RTKs is observed, leading to internal trafficking through endosomes toward proteolytic degradation. The existence of a phosphoprotein gradient from the plasma membrane (high kinase, low phosphatase) to the nucleus (low kinase, high phosphatase) has been verified both theoretically (44, 45) and experimentally (46, 47). Theoretical studies have shown that levels of phosphatase activity play a critical role in signal propagation (48), with lower phosphatase activity linked to further propagation of phosphorylation-induced signaling through the cell. We showed that lower phosphatase activity is linked to slower rates of topological reversal upon dephosphorylation. For the same phosphatase activity, slower rates of inversion are observed in endosomes compared with the plasma membrane, indicating that phosphorylation-induced topological inversion of a membrane protein at the plasma membrane will occur faster than its dephosphorylation-induced topological reversion in endosomes. This could constitute a new regulatory mechanism controlling the lifetime of a phosphoprotein in the cell, possibly prolonging the signaling properties of this given protein, shifting the fine balance of cellular homeostasis.
Studies of the structure and function of several bacterial secondary transporters have established that orientation of a membrane protein TMD in a lipid bilayer depends on the net positive charge of EMDs facing the cytoplasm and the net negative charge density of the membrane surface, determined by anionic phospholipid content (42, 49, 50). We established that lipid-dependent membrane protein organization is dynamic during and after membrane protein assembly in vivo and independent of other cellular factors in vitro (28, 51–53). Dependence of TMD topology solely on the intrinsic properties of a membrane protein and its lipid environment indicates a thermodynamically driven process that can occur in any cell membrane at any time.
Further studies are required to address the thermodynamics of individual TMD movement in and out of a particular bilayer context upon phosphorylation/dephosphorylation. Specifically, expanding previous work on the “biological” hydrophobicity scale (54) to membrane lipid compositions mimicking the biological membranes encountered by membrane proteins throughout their lifespan (biosynthesis at the endoplasmic reticulum, trafficking along the secretory pathway, functional site, trafficking toward degradation) and specific peptides mimicking the target TMDs would allow us to address the generality of the events uncovered in this study.
Experimental procedures
Reagents
All lipids were purchased from Avanti Polar Lipids. Except for E. coli–mimicking membranes where phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL) from E. coli were used, care was taken to use phospholipids from similar eukaryotic sources (brain for PE, phosphatidylcholine, phosphatidylserine, and sphingomyelin; liver for phosphatidylinositol; heart for CL; and soy for PG) to limit fatty acid heterogeneity among phospholipid classes. Anti-phosphoserine/threonine antibody (catalog no. ab17464) was purchased from Abcam. PDK1 kinase was purchased from Sigma. IAEDANS and 3-(N-maleimidopropionyl) biocytin (MPB) were purchased from Molecular Probes. HiTrap columns, the PD10 desalting column, and Vivaspin concentrators (50,000 molecular weight cutoff) were purchased from GE Healthcare. The ECL kit, Imperial protein stain solution, HRP-labeled secondary antibody, micro-BCA protein reagent assay, Slide-A-Lyzer G2 dialysis cassettes, and avidin-HRP were purchased from Thermo Pierce. Octyl glucoside (OG) and dodecyl maltoside were purchased from Anatrace. Complete protease inhibitor was purchased from Roche Molecular Biochemicals. SpinOUT columns were purchased from G Biosciences. DNase and all other reagents were purchased from Sigma.
Bacterial strains, plasmids, growth condition, and LacY purification
Strain AL95 (pss93::kanR lacY::Tn9 (a kanamycin resistance gene inserted into the gene encoding phosphatidylserine synthase and a tetracycline resistance gene inserted into the gene encoding LacY, respectively)) with (WT lipid composition) or without (lacking PE and containing mostly PG and CL) plasmid pDD72GM (pssA+ genR (a gene encoding phosphatidylserine synthase and a gene encoding gentamycin resistance, respectively) on plasmid pSC101 with a temperature-sensitive replicon (52) were used as hosts for expression of plasmids encoding LacY derivatives. Plasmid pT7–5/C-less LacY (ampR, ColE1 replicon), LacY in which Cys residues are replaced with Ser, was used to construct plasmids expressing LacY derivatives under OPlac regulation and containing multiple amino acid replacements in a derivative of LacY (−2/+2/+2 and −2/0/−2 in EMDs C2/C4/C6) encoding a single Cys replacement at His-205 in otherwise Cys-less LacY (52). The LacY charge templates −2/0/−2 (which contains the mutations K69E, K74E, K131E, K211E, and K218E) and −2/+2/+2 (which contains the mutations K69E and K74E) have been described previously (52). The latter plasmids were used to generate the templates to engineer phosphorylation sites in EMD C6 to be used for SCAMTMD assays. The amino acid sequence FAAFSY was inserted between amino acids 195 and 196 to engineer the PDK1 phosphorylation site. Plasmid pT7–5/C-less LacY/V331C encoding a single Cys replacement at Val-331 in otherwise Cys-less LacY, where Trp replacement was made in EMD C6 at position 205 (28), was used to generate the templates containing the PDK1 engineered phosphorylation site in EMD C6 to be used for real-time monitoring of TMD flipping by FRET. LacY was engineered with a His6 tag at the C terminus to facilitate purification and was expressed under control of the LacY operator/promoter by growth of cells in the presence of 1 mm isopropyl 1-thio-β-d-galactopyranoside. Cells were grown in Luria-Bertani (LB)-rich medium containing ampicillin (100 μg/ml) to an A600 of 0.6, induced by addition of isopropyl 1-thio-β-d-galactopyranoside, and grown until cell arrest occurred. Purification of LacY was carried out at 4 °C or on ice as described previously (53). Protein content during purification and the concentration of LacY in proteoliposomes were determined by the micro-BCA protein assay according to the manufacturer's instructions.
IAEDANS labeling
Trp replacement derivatives of LacY/V331C were purified from PE-containing and PE-lacking cells. Labeling of LacY with IAEDANS was carried out as described previously (28). The labeling ratio was quantified by comparing the absorption spectrum of labeled protein with unlabeled protein using extinction coefficient of ϵ334 = 5,700 M−1 cm−1 for IAEDANS. Typical labeling efficiency was above 85%.
Preparation of proteoliposomes
Proteoliposomes were formed by reconstitution of protein into small unilamellar liposomes of various lipid compositions as described previously (53) by incorporation of LacY into preformed liposomes. For encapsulation of λ protein phosphatase with and without various concentrations of ATP, proteoliposome reconstitution was conducted in the presence of the indicated concentration of phosphatase (at 1 μm for most assays, ranging from 1–50 μm when conducting assays with phosphatase located outside of or in proteoliposomes) and/or the indicated concentration of ATP (0, 1, 2, 4, 5, or 10 mm). Liposomes (containing the specified lipid composition and 1.5% OG) were mixed with purified LacY in a 500:1 lipid:protein ratio in the presence of λ protein phosphatase and/or ATP (at the indicated concentrations), incubated at 30 °C with gentle agitation for 10 min, and then placed on ice for 20 min. To remove OG, the mixture was diluted at least 30-fold into 50 mm KPi (pH 7.5), 50 mm MgCl2, and 2 mm β-mercaptoethanol and centrifuged at 190,000 × gav for 45 min to recover proteoliposomes. Proteoliposomes were suspended in 50 mm Tris-HCl (pH 7.5) and 100 mm NaCl, and unincorporated λ protein phosphatase and/or ATP was removed by size exclusion chromatography on a PD10 column equilibrated with 50 mm Tris-HCl (pH 7.5) and 100 mm NaCl.
Chemical protein topology mapping
Topological determination (SCAMTMD) of TMDs is based on the controlled membrane permeability of the thiol-specific reagent MPB and its reactivity with diagnostic cysteine residues in EMDs of LacY as described previously (53). Using LacY with a single cysteine engineered in EMD C6, the degree of mixed or dual topologies coexisting within the same proteoliposome membrane was assessed with the standard biotinylating procedure with MPB and quantification by densitometry.
Topological assessment by FRET
The various fluorescence measurements were conducted using a QuantaMaster model QM3-SS (Photon Technology International), a cuvette-based fluorescence spectrometer. All measurements were conducted in degassed 50 mm Tris-HCl (pH 7.5)/100 mm NaCl. Using a Peltier TE temperature controller, the sample was held constant at 20 °C. Data were collected and analyzed using Felix 32 software. Real-time protein flipping was monitored at an excitation wavelength of 295 nm (for Trp) and an emission wavelength of 475 nm (for IAEDANS), both with a 1-nm bandpass filter. The proteoliposomes (containing 1 μm IAEDANS-labeled LacY at a lipid to protein ratio (w/w) of 500) were first equilibrated at 20 °C. Phosphorylation was then triggered by addition of 1 μm PDK1 under constant stirring at 20 °C. We used the FRET values observed before phosphorylation (F0) to normalize the FRET values (FMeas) using Equation 1.
| (Eq. 1) |
In all cases, before starting kinetics measurements, care was taken to measure and adjust Trp fluorescence before phosphorylation to start with a similar fluorescence signal from the donor.
Data analysis and fitting
All data analyses and fitting were conducted using Prism 8 software (GraphPad Software Inc.). In all figures, solid lines indicate fits, and error bars mark standard deviation for the measured parameters.
Author contributions
H. V. conceptualization; H. V. and V. K. P. S. M. formal analysis; H. V. methodology; H. V. and W. D. writing-original draft; H. V. writing-review and editing; V. K. P. S. M. and W. D. investigation; W. D. resources; W. D. supervision; W. D. funding acquisition; W. D. project administration.
Supplementary Material
Acknowledgment
We thank H. R. Kaback for providing plasmids and reagents.
This work was supported by NIGMS, National Institutes of Health Grants R37 GM20478 and R01 GM121493 and the John S. Dunn Research Foundation (to W. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- EMD
- extramembrane domain
- NT
- N-terminal
- IAEDANS
- 5-(2-iodoacetyl)-amino-ethyl-amino-naphthalene-1-sulfonic acid
- TMD
- transmembrane domain
- SCAMTMD
- substituted cysteine accessibility method for determining TMD orientation
- PE
- phosphatidylethanolamine
- RTK
- receptor tyrosine kinase
- PG
- phosphatidylglycerol
- CL
- cardiolipin
- MPB
- 3-(N-maleimidopropionyl) biocytin
- OG
- octyl glucoside.
References
- 1. Hunter T. (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 10.1016/0092-8674(95)90405-0 [DOI] [PubMed] [Google Scholar]
- 2. Cori C. F., Schmidt G., and Cori G. T. (1939) The synthesis of a polysaccharide from glucose-1-phosphate in muscle extract. Science 89, 464–465 10.1126/science.89.2316.464 [DOI] [PubMed] [Google Scholar]
- 3. Rapundalo S. T. (1998) Cardiac protein phosphorylation: functional and pathophysiological correlates. Cardiovasc. Res. 38, 559–588 10.1016/S0008-6363(98)00063-7 [DOI] [PubMed] [Google Scholar]
- 4. Bononi A., Agnoletto C., De Marchi E., Marchi S., Patergnani S., Bonora M., Giorgi C., Missiroli S., Poletti F., Rimessi A., and Pinton P. (2011) Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011, 329098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen P. (2000) The regulation of protein function by multisite phosphorylation: a 25-year update. Trends Biochem. Sci. 25, 596–601 10.1016/S0968-0004(00)01712-6 [DOI] [PubMed] [Google Scholar]
- 6. Humphrey S. J., James D. E., and Mann M. (2015) Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 26, 676–687 10.1016/j.tem.2015.09.013 [DOI] [PubMed] [Google Scholar]
- 7. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 10.1038/nrm2039 [DOI] [PubMed] [Google Scholar]
- 8. Easty D., Gallagher W., and Bennett D. C. (2006) Protein tyrosine phosphatases, new targets for cancer therapy. Curr. Cancer Drug Targets 6, 519–532 10.2174/156800906778194603 [DOI] [PubMed] [Google Scholar]
- 9. Gee C. E., and Mansuy I. M. (2005) Protein phosphatases and their potential implications in neuroprotective processes. Cell. Mol. Life Sci. 62, 1120–1130 10.1007/s00018-005-5008-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 [DOI] [PubMed] [Google Scholar]
- 11. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Wiśniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 10.1016/j.celrep.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 12. Wu R., Haas W., Dephoure N., Huttlin E. L., Zhai B., Sowa M. E., and Gygi S. P. (2011) A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods. 8, 677–683 10.1038/nmeth.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ardito F., Giuliani M., Perrone D., Troiano G., and Lo Muzio L. (2017) The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int. J. Mol. Med. 40, 271–280 10.3892/ijmm.2017.3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sacco F., Perfetto L., Castagnoli L., and Cesareni G. (2012) The human phosphatase interactome: an intricate family portrait. FEBS Lett. 586, 2732–2739 10.1016/j.febslet.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forester C. M., Maddox J., Louis J. V., Goris J., and Virshup D. M. (2007) Control of mitotic exit by PP2A regulation of Cdc25C and Cdk1. Proc. Natl. Acad. Sci. U.S.A. 104, 19867–19872 10.1073/pnas.0709879104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sim A. T., and Ludowyke R. I. (2002) The complex nature of protein phosphatases. IUBMB Life 53, 283–286 10.1080/15216540213462 [DOI] [PubMed] [Google Scholar]
- 17. Yadav L., Tamene F., Göös H., van Drogen A., Katainen R., Aebersold R., Gstaiger M., and Varjosalo M. (2017) Systematic analysis of human protein phosphatase interactions and dynamics. Cell Syst. 4, 430–444.e5 10.1016/j.cels.2017.02.011 [DOI] [PubMed] [Google Scholar]
- 18. Bauman A. L., and Scott J. D. (2002) Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat. Cell Biol. 4, E203–E206 10.1038/ncb0802-e203 [DOI] [PubMed] [Google Scholar]
- 19. Cohen P., and Cohen P. T. (1989) Protein phosphatases come of age. J. Biol. Chem. 264, 21435–21438 [PubMed] [Google Scholar]
- 20. Gold M. G., Lygren B., Dokurno P., Hoshi N., McConnachie G., Taskén K., Carlson C. R., Scott J. D., and Barford D. (2006) Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell. 24, 383–395 10.1016/j.molcel.2006.09.006 [DOI] [PubMed] [Google Scholar]
- 21. Kinderman F. S., Kim C., von Daake S., Ma Y., Pham B. Q., Spraggon G., Xuong N. H., Jennings P. A., and Taylor S. S. (2006) A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell 24, 397–408 10.1016/j.molcel.2006.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kholodenko B. N. (2006) Cell-signaling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7, 165–176 10.1038/nrm1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Day E. K., Sosale N. G., and Lazzara M. J. (2016) Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 40, 185–192 10.1016/j.copbio.2016.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Purvis J. E., and Lahav G. (2013) Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956 10.1016/j.cell.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scott J. D., and Pawson T. (2009) Cell signaling in space and time: where proteins come together and when they're apart. Science 326, 1220–1224 10.1126/science.1175668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishi H., Shaytan A., and Panchenko A. R. (2014) Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 5, 270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vitrac H., MacLean D. M., Karlstaedt A., Taegtmeyer H., Jayaraman V., Bogdanov M., and Dowhan W. (2017) Dynamic lipid-dependent modulation of protein topology by posttranslational phosphorylation. J. Biol. Chem. 292, 1613–1624 10.1074/jbc.M116.765719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vitrac H., MacLean D. M., Jayaraman V., Bogdanov M., and Dowhan W. (2015) Dynamic membrane protein topological switching upon changes in phospholipid environment. Proc. Natl. Acad. Sci. U.S.A. 112, 13874–13879 10.1073/pnas.1512994112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bogdanov M., Heacock P. N., and Dowhan W. (2002) A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 21, 2107–2116 10.1093/emboj/21.9.2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schweiger R., and Linial M. (2010) Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct 5, 6 10.1186/1745-6150-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freschi L., Osseni M., and Landry C. R. (2014) Functional divergence and evolutionary turnover in mammalian phosphoproteomes. PLoS Genet. 10, e1004062 10.1371/journal.pgen.1004062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blüthgen N., Bruggeman F. J., Legewie S., Herzel H., Westerhoff H. V., and Kholodenko B. N. (2006) Effects of sequestration on signal transduction cascades. FEBS J. 273, 895–906 10.1111/j.1742-4658.2006.05105.x [DOI] [PubMed] [Google Scholar]
- 33. Levitan I. B. (1994) Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu. Rev. Physiol. 56, 193–212 10.1146/annurev.ph.56.030194.001205 [DOI] [PubMed] [Google Scholar]
- 34. Lee S. C., Lan W. Z., Kim B. G., Li L., Cheong Y. H., Pandey G. K., Lu G., Buchanan B. B., and Luan S. (2007) A protein phosphorylation/dephosphorylation network regulates a plant potassium channel. Proc. Natl. Acad. Sci. U.S.A. 104, 15959–15964 10.1073/pnas.0707912104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nesverova V., and Törnroth-Horsefield S. (2019) Phosphorylation-dependent regulation of mammalian aquaporins. Cells 8, E82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asch A. S., Liu I., Briccetti F. M., Barnwell J. W., Kwakye-Berko F., Dokun A., Goldberger J., and Pernambuco M. (1993) Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science 262, 1436–1440 10.1126/science.7504322 [DOI] [PubMed] [Google Scholar]
- 37. Dörfel M. J., and Huber O. (2012) Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J. Biomed. Biotechnol. 2012, 807356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim S., Smith K. R., Lim S. T., Tian R., Lu J., and Tan M. (2016) Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 6, 25 10.1186/s13578-016-0089-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Offringa R., and Huang F. (2013) Phosphorylation-dependent trafficking of plasma membrane proteins in animal and plant cells. J. Integr. Plant Biol. 55, 789–808 10.1111/jipb.12096 [DOI] [PubMed] [Google Scholar]
- 40. Hubbard S. R., and Miller W. T. (2007) Receptor tyrosine kinases: mechanisms of activation and signaling. Curr. Opin. Cell Biol. 19, 117–123 10.1016/j.ceb.2007.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gullick W. J. (1990) The role of the epidermal growth factor receptor and the c-erbB-2 protein in breast cancer. Int. J. Cancer Suppl. 5, 55–61 [DOI] [PubMed] [Google Scholar]
- 42. Bogdanov M., Dowhan W., and Vitrac H. (2014) Lipids and topological rules governing membrane protein assembly. Biochim. Biophys. Acta. 1843, 1475–1488 10.1016/j.bbamcr.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goh L. K., and Sorkin A. (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 5, a017459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brown G. C., and Kholodenko B. N. (1999) Spatial gradients of cellular phospho-proteins. FEBS Lett. 457, 452–454 10.1016/S0014-5793(99)01058-3 [DOI] [PubMed] [Google Scholar]
- 45. Lipkow K., Andrews S. S., and Bray D. (2005) Simulated diffusion of phosphorylated CheY through the cytoplasm of Escherichia coli. J. Bacteriol. 187, 45–53 10.1128/JB.187.1.45-53.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niethammer P., Bastiaens P., and Karsenti E. (2004) Stathmin-tubulin interaction gradients in motile and mitotic cells. Science 303, 1862–1866 10.1126/science.1094108 [DOI] [PubMed] [Google Scholar]
- 47. Kalab P., Weis K., and Heald R. (2002) Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science 295, 2452–2456 10.1126/science.1068798 [DOI] [PubMed] [Google Scholar]
- 48. Markevich N. I., Tsyganov M. A., Hoek J. B., and Kholodenko B. N. (2006) Long-range signaling by phosphoprotein waves arising from bistability in protein kinase cascades. Mol. Syst. Biol. 2, 61 10.1038/msb4100108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dowhan W., Vitrac H., and Bogdanov M. (2019) Lipid-assisted membrane protein folding and topogenesis. Protein J. 38, 274–288 10.1007/s10930-019-09826-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dowhan W., and Bogdanov M. (2009) Lipid-dependent membrane protein topogenesis. Annu. Rev. Biochem. 78, 515–540 10.1146/annurev.biochem.77.060806.091251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bogdanov M., and Dowhan W. (2012) Lipid-dependent generation of dual topology for a membrane protein. J. Biol. Chem. 287, 37939–37948 10.1074/jbc.M112.404103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bogdanov M., Xie J., Heacock P., and Dowhan W. (2008) To flip or not to flip: lipid-protein charge interactions are a determinant of final membrane protein topology. J. Cell Biol. 182, 925–935 10.1083/jcb.200803097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vitrac H., Bogdanov M., and Dowhan W. (2013) In vitro reconstitution of lipid-dependent dual topology and postassembly topological switching of a membrane protein. Proc. Natl. Acad. Sci. U.S.A. 110, 9338–9343 10.1073/pnas.1304375110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 10.1038/nature03216 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.