

Abstract
Tumor cells adapt to nutrient-limited environments by inducing gene expression that ensures adequate nutrients to sustain metabolic demands. For example, during amino acid limitations, ATF4 in the amino acid response induces expression of asparagine synthetase (ASNS), which provides for asparagine biosynthesis. Acute lymphoblastic leukemia (ALL) cells are sensitive to asparagine depletion, and administration of the asparagine depletion enzyme l-asparaginase is an important therapy option. ASNS expression can counterbalance l-asparaginase treatment by mitigating nutrient stress. Therefore, understanding the mechanisms regulating ASNS expression is important to define the adaptive processes underlying tumor progression and treatment. Here we show that DNA hypermethylation at the ASNS promoter prevents its transcriptional expression following asparagine depletion. Insufficient expression of ASNS leads to asparagine deficiency, which facilitates ATF4-independent induction of CCAAT-enhancer-binding protein homologous protein (CHOP), which triggers apoptosis. We conclude that chromatin accessibility is critical for ATF4 activity at the ASNS promoter, which can switch ALL cells from an ATF4-dependent adaptive response to ATF4-independent apoptosis during asparagine depletion. This work may also help explain why ALL cells are most sensitive to l-asparaginase treatment compared with other cancers.
Keywords: DNA methylation, stress response, tumor metabolism, cell metabolism, amino acid, acute lymphoblastic leukemia, asparagine synthetase, ATF4, CHOP, chromatin modification
Introduction
Regulation of the acquisition and utilization of amino acids is important for supporting tumor cell growth and survival (1, 2). Along this line, asparagine, a nonessential amino acid (NEAA),4 is critical for supporting protein synthesis during tumor progression, when environmental nutrients become limited because of pathophysiological alterations (3–6). The effect of asparagine on protein synthesis is at least partially attributed to its role as a counterion to import essential amino acids (7). Unlike the other 19 proteinogenic amino acids, asparagine is unable to be catabolized in mammalian cells because of a lack of functional asparaginase during evolution (5). Thus, understanding the uptake and de novo biosynthesis of asparagine has been the focus for adaptive mechanisms to nutrient deficiencies in cancer.
The rate-limiting step of de novo biosynthesis of asparagine is regulated by asparagine synthetase (ASNS) (8). It is generally thought that expression of ASNS is low in acute lymphoblastic leukemia (ALL) cells, rendering ALL patients sensitive to treatment with l-asparaginase. l-asparaginase is a purified bacterial enzyme that is administrated by intramuscular injection or intravenous infusion and functions by depleting circulating asparagine (9–11). In ALL cells, a reciprocal correlation between ASNS expression and sensitivity to l-asparaginase treatment has been reported (12, 13), suggesting a role of de novo asparagine biosynthesis in mitigating the nutrient stress caused by depletion of exogenous asparagine. Cells expressing low amounts of ASNS can adapt to l-asparaginase treatment through transcriptional induction of ASNS (14). This process requires activation of general control nonderepressible 2 (GCN2) and the ensuing adaptive amino acid response.
In the amino acid response, GCN2 is activated by uncharged tRNA during amino acid starvation (15, 16). In turn, GCN2 phosphorylates the α subunit of eIF2α, sharply reducing delivery of initiator tRNAs to ribosomes, which culminates in lower global translation (17). Coincidentally, phosphorylation of eIF2α promotes translation of certain stress-responsive mRNAs via ribosome bypass of inhibitory upstream open reading frame (18–20). An important translationally induced gene is ATF4, which activates a transcriptional program that governs genes involved in amino acid biosynthesis, reduction of oxidative stress, and induction of autophagy, which mediate metabolic adaptation (21, 22). ASNS is a direct transcriptional target of ATF4 (23). However, ATF4 can also be activated by other stresses through different eIF2α kinases (24). For example, during endoplasmic reticulum (ER) stress, ATF4 is activated via the eIF2α kinase protein kinase R-like endoplasmic reticulum kinase (PERK), which can culminate in expression of CCAAT-enhancer-binding protein homologous protein (CHOP) and apoptosis (25, 26). It is still uncertain how cell fate decisions are made when both adaptive and apoptotic genes are transcriptionally activated by ATF4, but the duration and amplitude of the stress response are thought to be contributing factors (27).
Here we show that, despite the fact that ATF4 is required for the induction of ASNS in ALL cells following asparagine depletion, activation of ATF4 through the amino acid response pathway was not sufficient to drive transcription of ASNS. Indeed, DNA hypomethylation in the CpG island at the promoter region of ASNS is a prerequisite for ATF4 recruitment and transactivation. In contrast, DNA hypermethylation at the ASNS promoter restricts the accessibility of ATF4 to chromatin, leading to failure of induction of ASNS and an inability to synthesize asparagine de novo when exogenous asparagine is deprived. Furthermore, lack of intracellular asparagine leads to CHOP accumulation and CHOP-dependent apoptosis in an ATF4-independent manner. Using ALL lines as a model, our results provide a comprehensive understanding of the transcriptional regulation of ASNS in tumor cells that inherit distinct chromatin modification status at a specific gene promoter to dictate cell fate decision. These findings will help to facilitate treatment strategies involving l-asparaginase in ALL patients and may accelerate development of therapeutic agents to better target asparagine biosynthesis in cancer.
Results
Expression of ASNS dictates the cellular response to asparagine depletion
We identified a dynamic expression pattern of ASNS protein across a panel of human B- and T-ALL cell lines (Fig. 1A). The amounts of ASNS protein largely correlated with the ability of ALL cells to grow in the absence of exogenous asparagine (Fig. 1B). RS4;11 and DND-41 cells barely expressed ASNS protein and were most sensitive to asparagine depletion–induced growth inhibition and cell death (Fig. 1, B and C). The other ALL lines had no survival defect in asparagine-free medium (data not shown) despite various degrees of growth rate retention (Fig. 1B). To determine whether the expression of ASNS protein is sufficient to support cell growth and drive resistance in the absence of exogenous asparagine, ASNS cDNA was stably expressed in RS4;11 and DND-41 cells through lentiviral vector–mediated transduction. We found that restoring the expression of ASNS in these cells led to rescue of cell proliferation during asparagine depletion (Fig. 1, D and E). Conversely, CRISPR-mediated deletion of the ASNS gene from Nalm-6 cell induced cell death only when exogenous asparagine was deprived (Fig. 1, F and G). These findings support the central role of differential regulation of ASNS expression in ALL cells' sensitivity to asparagine depletion.
Figure 1.

ASNS is both necessary and sufficient for cell growth/survival during asparagine depletion. A, the indicated B- and T-ALL cell lines were cultured in asparagine-replete medium, and protein lysates were analyzed by immunoblot analysis using antibodies specific for ASNS and β-actin. B, relative cell growth of ALL cell lines cultured 4 days in the absence of exogenous asparagine. Results were normalized to cell number collected on day 4 in the presence of exogenous asparagine. C, Nalm-6, RS4;11 and DND-41 cells were cultured in asparagine-replete or -depleted medium for 4 days. Viability was determined by trypan blue staining. D and E, restoration of ASNS expression in RS4;11 and DND-41 cells by cDNA overexpression fully rescued the growth defect during 4-day culture in asparagine-deficient medium. Results are presented as mean ± S.D. of triplicates from a representative experiment. F and G, Nalm-6 cells stably expressing control (Ctrl) guide RNA or two independent guide RNAs targeting ASNS were cultured in asparagine-replete or -depleted medium for 16 h. Protein extracts were subjected to immunoblotting with ASNS antibody. Viability was determined by trypan blue staining on day 4. The data in B–E are presented as mean ± S.D. of triplicates.
Asparagine is required for tRNA charging and protein synthesis
To determine whether the level of ASNS expression correlates with asparagine biosynthesis, we performed quantitative LC-MS to measure intracellular asparagine. We chose Nalm-6 cell as an example of cells expressing ASNS and RS4;11 cell as an example of ones that barely express ASNS. Asparagine became nearly undetectable in RS4;11 cells following asparagine depletion (Fig. 2A). By comparison, in Nalm-6 cells deprived of asparagine in the medium, there was about 20% of the intracellular asparagine levels compared with cells supplemented with asparagine (Fig. 2A). Furthermore, expressing exogenous ASNS in RS4;11 cells restored 50% intracellular asparagine during asparagine depletion compared with asparagine-replete conditions (Fig. 2B).
Figure 2.
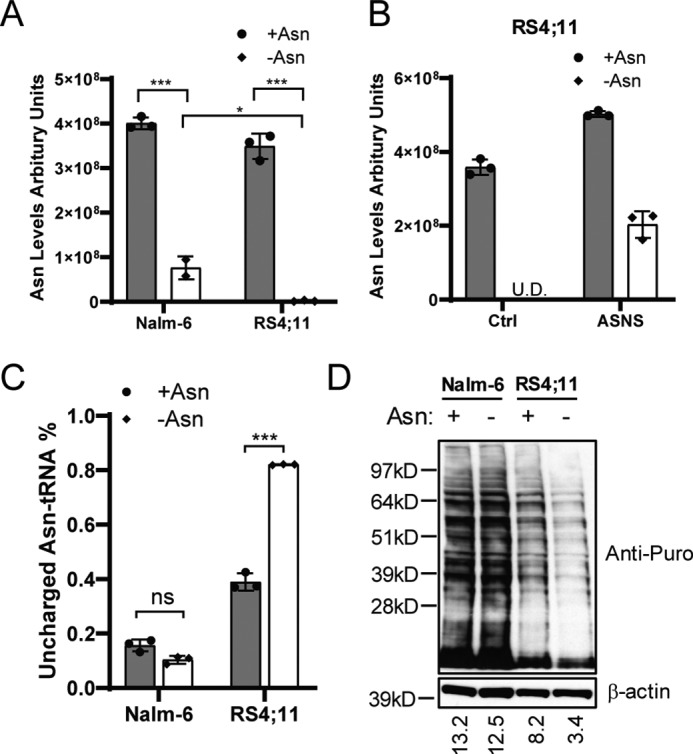
Asparagine production is required for tRNA charging and protein synthesis. A, Nalm-6 and RS4;11 cells were cultured in medium with or without asparagine for 16 h. Intracellular asparagine was measured by LC-MS. B, RS4;11 cells stably expressing a Ctrl vector or ASNS cDNA were cultured in asparagine-replete or -depleted medium for 16 h. Intracellular asparagine was measured by LC-MS. U.D., undetectable. C, Nalm-6 and RS4;11 cells were cultured in asparagine-replete or -depleted conditions for 24 h. The fraction of uncharged tRNAAsn was determined by a tRNA charging assay as described under “Experimental procedures.” D, Nalm-6 and RS4;11 cells were cultured in asparagine-replete or -depleted medium for 24 h. Puromycin (Puro, 90 μm) was added to the culture for 10 min before protein harvest. Immunoblot analysis was performed with an anti-puromycin antibody that recognizes premature puromycin-containing polypeptides. Quantification is presented as signal intensity of the anti-puromycin blot normalized to the anti-β-actin blot (bottom). The results in A and C are presented as mean ± S.D. of triplicates. The p values in A and C were calculated using Student's two-tailed paired t test. *, p < 0.05; ***, p < 0.001; ns, not significant.
We next addressed the levels of aminoacylated tRNAAsn, which would be adversely affected by severe asparagine depletion. Using a covalently linked tRNAAsn charging assay (28), we found that the fraction of uncharged tRNAAsn was appreciable in RS4;11 cells cultured with asparagine supplement, which was sharply increased upon deprivation of asparagine (Fig. 2C). By comparison, there was minimal detectable uncharged tRNAAsn in Nalm-6 cells independent of asparagine addition to the medium. Consistent with these changes in tRNAAsn charging, the global protein synthesis rate was significantly reduced in RS4;11 cells, but not in Nalm-6 cells, following asparagine depletion (Fig. 2D). These results indicate that there is a threshold of asparagine levels in cells that can adversely affect aminoacylation of tRNAAsn, which culminates in lowered global protein synthesis.
The role of the GCN2-ATF4 pathway in induction of ASNS following asparagine deprivation
Because l-asparaginase treatment can induce expression of ASNS in ALL cells in vitro (29), we wondered whether elevated expression of ASNS following asparagine depletion can also contribute to growth restoration in ALL cells that express low to medium levels of ASNS, including Nalm-6, HBP-ALL, and KOPT-K1 cells (Fig. 1, A and B). GCN2 plays an important role in induction of ASNS following l-asparaginase treatment by enhancing the expression of ATF4, which induces transcription of its target genes (14, 30, 31). Indeed, we found that phosphorylation of GCN2 at Thr-899, a measure of activation, and ATF4 protein were induced in response to asparagine depletion in Nalm-6, HPB-ALL, and KOPT-K1 cells, which coincided with induction of ASNS protein (Fig. 3A).
Figure 3.

Activation of ATF4 is not sufficient to drive transcription of the ASNS gene in ALL cells following asparagine depletion. A, Nalm-6, HPB-ALL, and KOPT-K1 cells were cultured in asparagine-replete or -depleted medium for protein harvest. Immunoblot analysis was performed with antibodies specific for phospho-GCN2 (Thr-899), GCN2, ATF4, and ASNS. Protein lysates were collected at 8 h for ATF4 and 24 h for the others. B, Nalm-6, RS4;11, HPB-ALL, and DND-41 cells were cultured in asparagine-replete or -depleted medium for 24 h. Protein lysates were subjected to immunoblot analysis with antibodies for ATF4 and ASNS. C, qPCR analysis of the mRNA of ASNS in Nalm-6, RS4;11, HPB-ALL, and DND-41 cells 24 h after asparagine depletion. The results are presented as mean ± S.D. of triplicates from a representative experiment. D, GESA showed enrichment of amino acid deprivation-induced genes following asparagine depletion (16 h) in RS4;11 cells. E, heatmap of selected amino acid starvation–responsive genes and well-characterized ATF4 target genes. RNAs were collected as triplicates under both asparagine-replete and -depleted conditions. F, proposed working model for transcriptional regulation of ASNS by asparagine deprivation, in which ATF4 may not be able to be recruited to the promoter of ASNS in ALL cells that are unable to turn on ASNS expression.
These findings led us to think that loss of GCN2/ATF4 signaling may explain the diminished ASNS expression of RS4;11 and DND-41 cells and their heightened sensitivity to asparagine depletion (Fig. 1B). Of interest, we found that ATF4 accumulation following asparagine depletion is not defective in RS4;11 and DND-41 cells, whereas there was minimal induction of ASNS protein and mRNA (Fig. 3, B and C). To further address whether there is a defect in the general amino acid response or ATF4-directed gene expression, we performed RNA-Seq analysis of RS4;11 cells subjected to asparagine depletion. Using gene set enrichment analysis (GSEA), we found that a collection of amino acid starvation–responsive genes was induced by asparagine depletion, with the exception of ASNS (Fig. 3D). A heatmap illustrates the induction patterns for a collection of ATF4 target genes involved in amino acid and protein homeostasis (Fig. 3E). These results suggest that although the GCN2/ATF4 pathway functions appropriately in ALL cells that are sensitive to asparagine depletion, there is an intrinsic defect in transcriptional expression of ASNS (Fig. 3F).
DNA methylation status within the CpG island region of the ASNS promoter dictates ATF4-dependent induction of ASNS following asparagine depletion
DNA methylation within the CpG island of the gene promoter is a central mechanism of gene silencing (32). Because DNA methylation within the promotor of the ASNS gene has been reported (33), we hypothesized that the chromatin structure around the promoter of the ASNS gene may restrict ATF4 accessibility to drive gene transcription in leukemic cells that are deficient for induced ASNS expression following asparagine depletion (Fig. 3F). To test this, we performed bisulfite sequencing analyses in the CpG island region of the ASNS promoter. Nearly all predicted CpG islands were methylated in RS4;11 cells that are sensitive to asparagine depletion (Fig. 4A, bottom). By comparison, the majority of the predicted CpG islands were unmethylated in Nalm-6 cells, which efficiently adapts to depletion of asparagine (Fig. 4A, top).
Figure 4.

Promoter hypermethylation restricts ATF4-dependent induction of ASNS following asparagine depletion. A, bisulfite sequencing analyses revealed that the ASNS promoter is hypermethylated in RS4;11 cells. As a control, the same region is hypomethylated in Nalm-6 cells. Each dot represents a predicted CpG site. Each line represents one sequencing result from a bulk population of cells. B, RS4;11 cells were maintained in medium containing low levels of exogenous asparagine (20 μm, 20% of complete LCM) for 4 weeks. The resulting cells were then plated on 1% methylcellulose prepared in asparagine-free LCM for another 4 weeks to establish the RS4;11/R line, which was maintained in asparagine-deficient LCM. C, growth curve of RS4;11 and RS4;11/R cells cultured in in asparagine-replete or -depleted medium for 4 days. Results are presented as mean ± S.D. of triplicates from a representative experiment. D, RS4;11 and RS4;11/R cells were cultured in asparagine-replete or -depleted medium for 24 h. Protein lysates were analyzed by immunoblot with antibodies specific for p-GCN2 (Thr-899), GCN2, ATF4, and ASNS. E, RS4;11 and RS4;11/R cells were cultured under the same condition as in D. RNA was extracted and subjected to qPCR analysis. The result is normalized to 18S rRNA as an internal control. Results are presented as mean ± S.D. of triplicates from a representative experiment. F, bisulfite sequencing results of the ASNS promoter from a bulk population of RS4;11/R cells. G, RS4;11 and RS4;11/R cells were cultured in asparagine-replete or -depleted medium for 24 h. A ChIP assay was performed using an ATF4-specific antibody or an isotype IgG control antibody. Promoter-specific qPCR amplification was done with primer pairs specific for the promoter region of ASNS (left) or SESN2 (right). Results were normalized to total input DNA prior to antibody enrichment. Data are presented as mean ± S.D. of triplicates from a representative experiment. The p values were calculated using Student's two-tailed paired t test. **, p < 0.01; ***, p < 0.001; ns, not significant. H, RS4;11/R cells stably expressing control guide RNA (sgCtrl) or ATF4-specific guide RNA were cultured in asparagine-replete or -depleted medium for 24 h. Protein lysates were analyzed by immunoblot analyses for ATF4 and ASNS.
To determine the functional consequence of the promoter methylation status on ASNS expression and the response of leukemic cells to asparagine depletion, we established an RS4;11 line that is resistant to asparagine depletion by subjecting the parental RS4;11 cells to rounds of culture in low levels of asparagine in the medium, followed by culturing in the absence of asparagine supplement (Fig. 4B). The resulting resistant cells, designated RS4;11/R, displayed the ability to proliferate in asparagine-free medium (Fig. 4C). Consistently, we observed restoration of ASNS expression at both the protein and mRNA levels (Fig. 4, D and E). The results from bisulfite sequencing showed that a significant portion of the DNA in RS4;11/R cells was now unmethylated at nearly all predicted CpG islands in the promoter region of ASNS (Fig. 4F). These results suggest that the DNA methylation status of the ASNS promoter is an important reason for the differences in ASNS expression and the cell sensitivity to asparagine depletion.
Next we addressed whether the methylation status at the ASNS promoter dictates ATF4 recruitment in ALL cells in response to asparagine depletion. We performed a ChIP assay with an ATF4 antibody followed by promoter-specific PCR amplification. ATF4 occupancy at the ASNS promoter was blocked in RS4;11 cells independent of the presence or absence of asparagine in the medium (Fig. 4G, left panel). However, ATF4 occupancy was restored in RS4;11/R cells even under asparagine-replete conditions, and ATF4 binding was further enhanced when exogenous asparagine was deprived (Fig. 4G, left panel). As a control, there was induced recruitment of ATF4 to the promoter region of another target gene, SESN2 (Fig. 3E), following asparagine depletion in both the RS4;11 and RS4;11/R cells (Fig. 4G, right panel). Of importance is that ATF4 was still required for ASNS expression in RS4;11/R cells. CRISPR was used to selectively delete the ATF4 gene, leading to abolishment of ASNS expression independent of the availability of asparagine in the medium (Fig. 4H). Of interest, readdition of asparagine to RS4;11/R cells for 8 days did not significantly alter ASNS expression (Fig. S1), suggesting the involvement of additional regulatory mechanisms to control the expression of ASNS when the cells are fully adapted to culturing conditions without any exogenous asparagine.
Asparagine deficiency leads to CHOP-dependent apoptosis
l-asparaginase treatment in mice induces a profound amino acid starvation response in normal tissues, with up-regulation of many known ATF4 target genes, including ASNS and CHOP (34). Our results suggest that DNA hypermethylation at the promoter region of ASNS genes specifically blocked ATF4-dependent adaptation to asparagine depletion by inhibiting transcription of the ASNS gene while promoting ATF4-dependent induction of apoptotic genes to dictate cell fate decision. Because CHOP is also a direct transcriptional target of ATF4 (26), which plays a critical role in programmed cell death during ER/unfolded protein response stress (35, 36), we sought to determine whether CHOP was induced in ALL lines that were unable to induce ASNS expression because of promoter hypermethylation. We found that CHOP mRNA and protein were induced by asparagine depletion in RS4;11 and DND-41 cells but not in Nalm-6 and HPB-ALL cells (Fig. 5, A and B). Induction of CHOP was accompanied by increased caspase-3 cleavage, a marker of apoptosis (Fig. 5A). Furthermore, Q-VD, a pan-caspase inhibitor, blocked apoptosis in RS4;11 and DND-41 cells following asparagine depletion (Fig. 5C), supporting the idea that caspase activity facilitates cell death. To determine whether CHOP is critical for apoptosis in response to asparagine depletion, we used CRISPR to delete the CHOP gene in the RS4;11 cells. Loss of the CHOP gene enhanced ALL cell survival following asparagine depletion (Fig. 5, D and E). We conclude that induction of CHOP expression is critical for apoptosis induced in ALL cells subjected to asparagine depletion.
Figure 5.

Asparagine deficiency leads to CHOP-dependent apoptosis. A, Nalm-6, RS4;11, HPB-ALL, and DND-41 cells were cultured in asparagine-replete or -depleted medium for 48 h. Protein lysates were subjected to immunoblot analyses for CHOP and caspase-3. Arrows, cleaved active forms of caspase-3. B, the leukemic cells in A were cultured under the same condition as in A for 24 h. RNAs were extracted for qPCR analyses of CHOP normalized to 18S rRNA. C, RS4;11 and DND-41 cells were cultured in asparagine-replete or -depleted medium or asparagine-depleted medium containing 20 μm Q-VD for 96 h. Apoptosis was quantified by Annexin V and propidium iodide staining followed by flow cytometry analyses. Results are presented as one representative of three independent experiments. D, RS4;11 cells stably expressing a Ctrl guide RNA or CHOP guide RNA were subjected to asparagine depletion for 24 h. Protein lysates were analyzed by immunoblot for CHOP. E, the same cells as in D were cultured in asparagine-replete or -depleted medium for 96 h. The cell death percentage was quantified by trypan blue staining. Results are presented as mean ± S.D. of triplicates. The p value was calculated using Student's two-tailed paired t test. **, p < 0.01.
CHOP induction under asparagine deficiency is ATF4-independent
Because CHOP is a well-defined ER stress response gene and a target of ATF4, we next sought to determine whether ATF4 is required for CHOP induction following asparagine depletion. Although deletion of ATF4 by CRISPR in Nalm-6 cells ablated induced ASNS expression in response to asparagine depletion, there was more robust induction of CHOP protein and mRNA expression with loss of ATF4 (Fig. 6, A and B). The induction of CHOP was accompanied by growth/survival defects in ATF4-deficient Nalm-6 cells following asparagine depletion (Fig. 6C). Furthermore, mouse embryonic fibroblasts (MEFs) deleted for ATF4 did not express ASNS, which correlated with strong induction of CHOP following asparagine depletion (Fig. 6, D and E). These findings suggest that CHOP is an important trigger of apoptosis in ALL cells that are unable to adapt to asparagine depletion. Of importance, induced CHOP expression can occur independent of ATF4.
Figure 6.
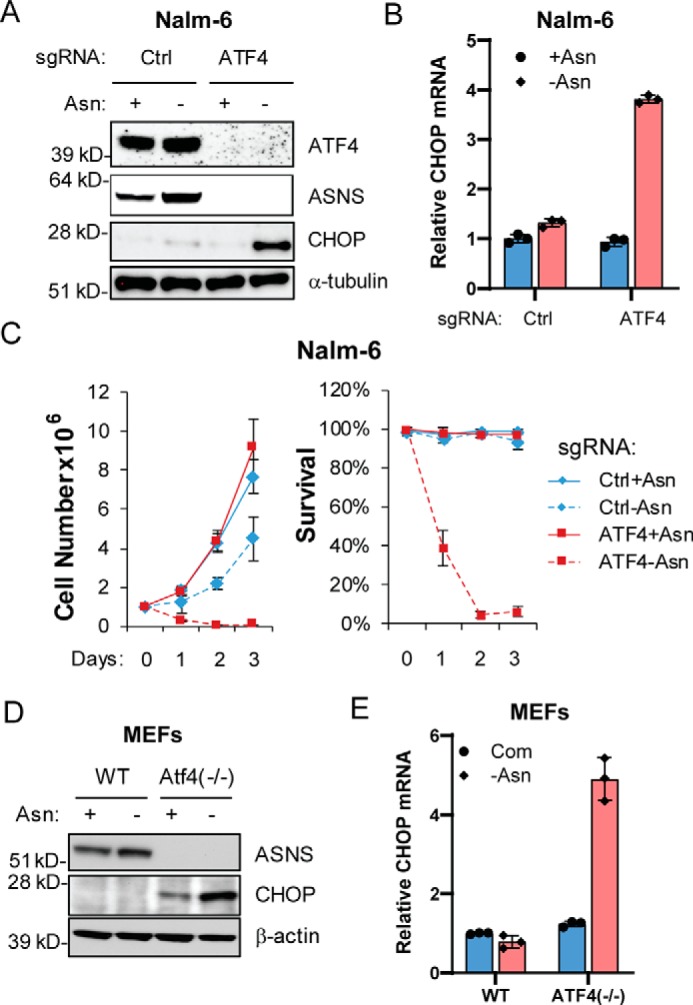
CHOP induction under asparagine deficiency is ATF4-independent. A and B, Nalm-6 cells stably expressing a Ctrl guide RNA or ATF4 guide RNA were subjected to asparagine depletion for 24 h. Protein lysates were analyzed by immunoblot for ATF4, ASNS, and CHOP. RNAs were subjected to qPCR analysis for CHOP, and the results are presented as mean ± S.D. of triplicates from a representative experiment. C, the same cells as in A were cultured in asparagine-replete or -depleted medium for 4 days. Viable cell numbers and the percentage of viable cells were recorded. Results are presented as mean ± S.D. of triplicates from a representative experiment. D and E, WT or Atf4−/− MEFs were cultured in asparagine-replete (Com) or -depleted (-Asn) medium for 24 h. Protein lysates were analyzed by immunoblot for Asns and Chop. RNAs were subjected to qPCR analysis for CHOP, and the results are presented as mean ± S.D. of triplicates from a representative experiment.
Discussion
In this study, we found that a critical molecular feature dictating the ability of ALL cells to induce the expression of ASNS is the DNA methylation status at the promoter region of ASNS. As a result, the ability to induce expression of ASNS is critical in driving resistance to asparagine depletion. When the ASNS promoter is hypomethylated, transcription of ASNS is induced by ATF4 through a GCN2-dependent adaptive amino acid response. However, when the ASNS promoter is hypermethylated, ATF4 is not able to be recruited to the cis-regulatory element for transactivation (Fig. 4G). In addition, we provided an example of the hierarchy between the methylation status of the gene promoter and key transcription factor. Using a standard ALL cell line (Nalm-6) and an experimentally derived asparagine-insensitive ALL line (RS4;11/R), we showed that hypomethylation at the ASNS promoter is necessary, but not sufficient, to drive transcription of ASNS without ATF4 in ALL cells (Figs. 6A and 4H). Recently, DNA methylation at the promoter region of ASNS has been found to be a feature of TLX1-positive T-ALL patient samples, which correlates with a better prognosis (37). To further elucidate the genetic components that dictate promoter methylation status in ALL cells may provide better management of ALL patients in the future.
Because perturbing amino acid metabolism has been proposed to be a potential strategy to treat cancer (38), it is critical to understand how tumor cells make decisions between adaptation versus cell death during metabolic stress. The fact that ALL cells respond differentially to asparagine depletion provides a compelling example to interrogate this question. Both adaptive and apoptotic responses can be induced by ATF4 during amino acid starvation (27). Whether this relies on selective expression of ATF4 target genes or involves ATF4-independent mechanism is still unclear. Here we showed that CHOP was induced only in ALL cells that are unable to turn on the expression of ASNS following asparagine depletion to trigger apoptosis (Fig. 5, A–E). Of interest is that induction of CHOP did not require the presence of ATF4 but correlated with a lack of intracellular asparagine (Fig. 6, A–E). It has been shown that ATF6 may compensate for the loss of ATF4 to drive CHOP transcription during ER/UPR stress (39). Whether amino acid starvation shares a similar machinery or employs a different context-specific molecular component to drive the induction of CHOP warrants further investigation. Therefore, we propose a model where promoter hypermethylation of ASNS prevents ATF4-dependent adaptation by blocking transcription of ASNS, which consequently leads to ATF4-independent induction of CHOP and apoptosis through intracellular depletion of asparagine (Fig. 7).
Figure 7.
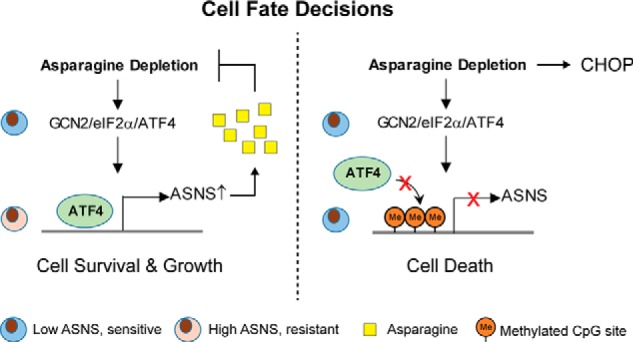
Proposed model of cell fate decisions following asparagine depletion. Asparagine depletion induces ATF4 through a GCN2-dependent amino acid response. When the ASNS promoter is hypomethylated, ATF4 is recruited to the promoter region to induce gene expression. As a result, increased expression of ASNS drives de novo biosynthesis of asparagine to support cell survival and growth (left). When the ASNS promoter is hypermethylated, ATF4 recruitment is blocked. Consequently, failure to induce ASNS expression leads to intracellular deficiency, which triggers CHOP induction and apoptosis in an ATF4-independent manner (right).
Asparagine has recently emerged as a critical NEAA to support tumor cell growth/survival in a variety of settings where nutrient limitation becomes a barrier for further tumor progression (3–5, 40, 41). Thus, understanding the acquisition and utilization of asparagine in tumor cells becomes an important topic. Like other NEAAs, asparagine acquisition can be achieved by direct uptake from the environment via a cell surface transporter or de novo biosynthesis in the cell. However, the preferential route of asparagine acquisition is context-dependent. For example, in breast tumors, loss of function of de novo biosynthesis through genetic inhibition of ASNS significantly reduces the incidence of lung metastases without perturbing tumor growth at primary sites (4), suggesting that limitation of exogenous asparagine in the lungs or the process of metastasis can create dependence on de novo biosynthesis during tumor progression. In addition, KRAS mutant non-small-cell lung cancer cells up-regulate ASNS to support cell survival and protein synthesis through ATF4 regulation, suggesting a direct connection between oncogenic signaling and the adaptive amino acid response pathway (3). Furthermore, stroma cells in the tumor environment can engage ATF4-dependent ASNS induction and asparagine biosynthesis to feed tumor cell asparagine during glutamine limitation (41). Thus, elucidating the molecular mechanism that governs the expression of ASNS is essential for understanding tumor cell adaptation to nutrient limitation during tumor progression.
On the other hand, l-asparaginase has activity only in ALL and certain cases of T/nature killer cell lymphoma, suggesting that their route of asparagine acquisition is different from other cancers (9). It is thought that ALL cells inherit intrinsic properties to limit their capacity to synthesize asparagine de novo. However, despite their overall sensitivity to asparagine depletion, we found that differential expression of ASNS following asparagine depletion is central for dictating an adaptive or apoptotic response in ALL cells. Although the GCN2/ATF4 pathway plays an important role in induction of ASNS following asparagine depletion, it is not sufficient to drive ASNS expression unless the promoter region of ASNS is hypomethylated. Together, our results elucidate a comprehensive program involving an adaptive amino acid response and chromatin modification component to regulate ASNS expression and de novo asparagine biosynthesis. Failure of this process switches cells from programmed adaptation to programmed cell death following deprivation of exogenous asparagine, which can broadly affect therapeutic interventions that aim to targeting asparagine bioavailability in cancer.
Experimental procedures
Cell culture
All human T-ALL and B-ALL cell lines were cultured at 37 °C in 5% CO2 in lymphocyte culture medium (LCM). The base of LCM is a 1:1 ratio mixture of DMEM and Iscove's modified Dulbecco's medium. For practical reasons and quality control purposes, we can consistently generate LCM in the laboratory by supplementing high-glucose DMEM (11965092, Thermo Fisher Scientific) with 0.14 mm l-alanine, 0.17 mm l-proline, 5.3 × 10−5 mm biotin, 9.59 × 10−5 mm vitamin B12, 9.8 × 10−5 mm sodium selenite, 10 mm HEPES, 55 μm β-mercaptoethanol, 100 units/ml penicillin/streptomycin, 2 mm l-glutamine, 10% FBS, and 0.1 mm l-asparagine. MEFs were cultured in DMEM (11965092, Thermo Fisher Scientific) supplemented with 10% FBS; 100 units/ml penicillin/streptomycin; 2 mm l-glutamine; a mixture of nonessential amino acids containing 0.1 mm glycine, l-alanine, l-asparagine, l-aspartic acid, l-glutamic acid, l-proline, and l-serine (11140050, Thermo Fisher Scientific); and 55 μm β-mercaptoethanol. For cell growth experiments, cell viability and numbers were recorded in triplicates using the Vi-CellXR cell viability analyzer (Beckman Coulter).
Asparagine deprivation experiments
Leukemic cells were centrifuged, and the supernatant was removed by aspiration. Cell pellets were resuspended in asparagine-free LCM. Asparagine-free LCM was made from high-glucose DMEM as described above, with the exception of using 10% dialyzed FBS and not adding l-asparagine. MEFs were rinsed with PBS once and then cultured with DMEM excluding l-asparagine.
Puromycin labeling assay
Cells were cultured in the presence or absence of asparagine for 24 h. Puromycin (90 μm) was added to the cultured cells for 10 min before protein harvest. Puromycin-incorporated polypeptides were resolved by BisTris gel and detected by immunoblotting with an anti-puromycin antibody. The rate of global protein synthesis was defined by the ratio between the anti-puromycin signal and anti-β-actin signal per sample using ImageJ software.
Mass spectrometry analysis of metabolite asparagine
Ten million cells were collected by centrifugation. The supernatant was aspirated, and the pellets were washed thoroughly once with ice-cold 1× Hanks' Balanced Salt solution (14025092, Life Technology). The cellular metabolite was extracted with 80% methanol on ice. The supernatant was collected and dried with a SpeedVac (SPD111V, Thermo Fisher Scientific) connected to a refrigerated vapor trap (RVT5105, Thermo Fisher Scientific) at room temperature. Dried samples were resuspended in 80:20 acetonitrile:water and analyzed using a Thermo Q-Exactive mass spectrometer coupled to a Vanquish Horizon Ultra-High-Performance Liquid Chromatography. Metabolites were separated on a 150 × 2.1 mm SeQuant Polyether ether ketone (PEEK) HPLC column with ZIC-pHILIC (5 μm) polymeric beads (Millipore Sigma). Samples were run with a gradient of solvent A (95% acetonitrile and 5% water) and solvent B (10 mm NH4Ac (pH = 5.5)) as follows: 0 min, 5% B; 2 min, 5% B; 18 min, 60% B; 19 min 90% B; 24 min, 90% B; and 25.5 min, 5% B. Data were collected on full scan positive mode. The settings for the ion source were as follows: 12 auxiliary gas flow rate, 40 sheath gas flow rate, 1 sweep gas flow rate, 3.5 kV spray voltage, 340 °C capillary temperature, and 250 °C heater temperature. Asparagine was identified based on the exact m/z and retention time of an asparagine chemical standard. Data were analyzed with Maven (42, 43), normalized to the internal standard of [13C4,15N2]asparagine (1 pmol/sample), and then to the packed cell volume of each sample.
tRNA charging assay
We adopted an assay to measure the percentage of asparagine-charged tRNA based on the literature (28). In brief, total cellular RNA was extracted with TRIzol (15596026, Life Technologies). 2 μg of RNA was incubated with 12.5 mm NaIO4 or 12.5 mm NaCl (as a nonoxidization control) in acidic buffer (sodium acetate buffer (pH = 4.5)) in the dark and then quenched with 0.3 m glucose. Each sample was spiked with 7.3 ng of yeast phenylalanine tRNA (R4018, Sigma) and then subjected to desalination through a MicroSpin G-25 column (27532501, GE Healthcare). Desalted RNA was precipitated with cold ethanol and then subjected to deacylation in 50 mm Tris-HCl (pH 9.0). 200∼400 ng of tRNA was used to ligate with a 5′-adenylated adaptor (5′-/5rApp/TGGAATTCTCGGGTGCCAAGG/3ddC/-3′) using T4 RNA ligase 2 truncated KQ (M0373S, New England Biolabs). Then a single-strand oligo (5′-GCCTTGGCACCCGAGAATTCCA-3′) complementary to the adaptor was used to generated cDNA using the SuperScript RT IV First-Strand Synthesis System (18091050, Life Technologies). cDNA was diluted and then subjected to quantitative PCR to detect amino acid–specific tRNA with corresponding primer pairs: yeast phenylalanine, GCGGAYTTAGCTCAGTTGGGAGAG (forward) and GAGAATTCCATGGTGCGAAYTCTGTGG (reverse); human asparagine, GTCTCTGTGGCGCAATCGGT (forward) and GAGAATTCCATGGCGTCCCTGG (reverse). The results were normalized to yeast phenylalanine first, and then the uncharged tRNA fraction was calculated by subtracting the charged fraction (NaIO4-treated) from the total (NaCl-treated).
Molecular cloning and virus production
Mouse ASNS cDNA was ordered from Dharmacon and then cloned into the LeGO-iG2 vector backbone (Addgene, 27341). Guide RNAs were designed using the Feng Zhang laboratory CRISPR design resource (http://crispor.tefor.net/ (47))5 and cloned into the pLentiCRISPRv2-Puro vector (Addgene, catalog no. 98290). The calcium phosphate method (44) was used to produce the lentivirus. We used pMD2.G (Addgene, catalog no. 12259) and psPAX2 (Addgene, catalog no.12260) as packaging plasmids.
Western blotting
Protein was extracted by using 1× radioimmune precipitation assay buffer (diluted from 10× radioimmune precipitation assay lysis buffer, Millipore, catalog no. 20-188) with protease inhibitors (Thermo Scientific, catalog no. 1860932) and phosphatase inhibitors (Thermo Scientific, catalog no. 78428). Total proteins of an equal amount (20 μg) were separated on NuPAGE BisTris gels (Invitrogen, catalog no. NP0322BOX) and then transferred to nitrocellulose membranes (Bio-Rad, catalog no. 1620115). Membranes were blocked in 5% milk and then incubated with corresponding primary antibodies overnight at 4 °C. Membranes were washed with 1× Tris-buffered saline with Tween 20 (TBST; diluted from 20× TBST, Santa Cruz Biotechnology, catalog no. 362311) and then incubated with HRP-conjugated secondary antibody (ECL anti-rabbit IgG, Sigma, catalog no. NA934V; ECL anti-mouse IgG, Sigma, catalog no. NA931V; 1:5000 dilution). Membranes were washed with 1× TBST and subjected to chemiluminescent Western ECL detection. We used Pierce ECL Western blot substrate (Thermo Scientific, catalog no. 32106) to detect α-tubulin and β-actin signals and SuperSignal West Pico Plus chemiluminescent substrate (Thermo Scientific, catalog no. 34578) to detect other protein signals. The blots were stripped with Restore Western blot stripping buffer (Thermo Scientific, catalog no. 21059), washed with 1× TBST, and then reprobed with the appropriate primary antibodies for signal detection. Primary antibodies were as follows: ASNS (14681-1-AP, ProteinTech), puromycin (MABE343, EMD Millipore), phospho-GCN2(Thr-899) (Ab75836, Abcam), CHOP/DDIT3 (Ab11419, Abcam), GCN2 (3302, Cell Signaling), caspase-3 (9662, Cell Signaling), β-actin (A2228, Sigma), α-tubulin (T9026, Sigma), and ATF4 (45).
mRNA quantification
Cells were collected 24 h after starvation. Total RNA was then isolated with TRIzol (15596026, Life Technologies) according to the manufacturer's instructions. 500 μg of total RNA was processed for cDNA synthesis with random hexamer primers using EasyScript Plus RTase from the EasyScript Plus cDNA Synthesis Kit (Lamda Biotech, catalog no. G235). The synthesized cDNA was then subjected to qPCR amplification with designed PCR primers for human ASNS, CHOP, and 18S rRNA.
RNA-Seq and GSEA
We used STAR 2.4 to align the RNA-Seq samples to the reference genome (hg19) and count the number of reads mapping to each gene in the ensemble GRCh37 gene model. Differential expression between the different groups was performed through the use of DESeq 1.22.1. GSEA analysis (46) was applied in weighted mode against the gene set collection in MSigDB (v5.1). Gene sets with a size over 5000 genes or smaller than 10 genes were excluded from further analysis. Each gene set was permuted 1000 times to calculate the p value and false discovery rate (FDR) values. The heatmap was generated with Multi Experiment Viewer (http://mev.tm4.org/#/welcome)5 with gene/row adjustment by root mean square.
Bisulfite sequencing
Genomic DNA was isolated from 10 million cells using the Blood & Cell Culture DNA Midi Kit (Qiagen, catalog no. 13343) following the manufacturer's instructions. Genomic DNA was isolated from fresh cultured suspension cells using the Blood & Cell Culture DNA Midi Kit (Qiagen, catalog no. 13343) and then subjected to CT conversion using the EZ DNA Methylation Kit (Zymo Research, catalog no. D5001) following the manufacturer's instructions. Genomic DNA was amplified from the CT-converted samples with primers specific for the CpG island of human asparagine synthetase and then cloned into the pGEM-T Easy vector (Promega, catalog no. A1360). T7 primers were used for the following Sanger sequencing.
ChIP assay
2.5 × 107 cells were resuspended in 25 ml of PBS and fixed with 1% formaldehyde (final concentration) on a platform rocker at room temperature for 10 min. 1.4 ml of 2.5 m glycine was added and incubated for another 5 min on a platform rocker to quench the cross-linking reaction. Cells were washed and lysed in 2 ml of cell lysis buffer A (20 mm Tris-HCl (pH 8.0), 85 mm KCl, and 0.5% NP-40) and incubated on ice for 10 min. Nuclei were pelleted by centrifugation at 1350 × g for 5 min at 4 °C. The nuclei were then resuspended in 750 μl of lysis buffer B (50 mm Tris-HCl (pH 8.0), 10 mm EDTA, 1% SDS, and protease inhibitor mixture) and sonicated until the majority of DNA fragments were between 200 and 500 bp in size. The sonicated materials were then centrifuged at 20,000 × g for 10 min at 4 °C to collect the supernatant. 10% of the supernatant (75 μl) was used as input, and 300 μl of the supernatant was used for each immunoprecipitation (IP) reaction.
The 300-μl supernatant sample was diluted 5-fold by adding 1.2 ml of IP dilution buffer (1.25% Triton X-100, 187.5 mm NaCl, 20 mm Tris-HCl (pH 8.0), and protease inhibitor mixture) and incubated overnight at 4 °C by rotating with 30 μl of protein G Dynabeads (Life Technologies) preincubated with ATF4 antibody (Cell Signaling, catalog no. 11815) or normal rabbit IgG control. The beads were washed consecutively with 1 ml of low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.0), and 150 mm NaCl, twice), high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.0), and 500 mm NaCl, once), LiCl wash buffer (0.25 m LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl (pH 8.0), once) and TE wash buffer (50 mm NaCl, 10 mm Tris (pH 8.0), and 1 mm EDTA, once) and incubated with 125 μl of elution buffer (1% SDS and 0.1 M sodium bicarbonate) for 15 min on a thermomixer at 1000 rpm and 65 °C. The supernatant was collected by magnet separation from the beads. 5 μl of 5 m NaCl was added to each eluate and incubated at 65 °C overnight. 30 μl of input samples were incubated with 95 μl of elution buffer, and 5 μl of 5 m NaCl was used for reverse cross-linking. 2 μl of RNase A (0.5 mg/ml) was added to each IP and input sample for 30 min of incubation at 37 °C. 2 μl of proteinase K (20 mg/ml) was then added to each sample and incubated for 2 h at 55 °C. A PCR purification kit (Qiagen) was used to recover DNA from each sample in 40 μl of elution buffer. 10 μl of DNA was used for qPCR amplification to determine enrichment of regions of the ASNS promoter.
Apoptosis assessment experiment
Cells were washed twice with cold PBS and then resuspended in Annexin V binding buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, and 2.5 mm CaCl2) at a density of 5 × 106 cells/ml. 5 μl of FITC-Annexin V (Biolegend, catalog no. 640905) and 0.1 μg of propidium iodide solution (Thermo Fisher Scientific) were added to 100 μl of cell suspension for 15-min incubation at room temperature (25 °C) in the dark. 400 μl of Annexin V binding buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, and 2.5 mm CaCl2) was then added to each tube, followed by flow cytometry analysis on the FL1 and FL3 channels.
Author contributions
J. J., S. S., G. S., M. Z., J. F., and J. Z. data curation; J. J., S. S., G. S., B. K., L. Z., C. Z., J. F., and J. Z. formal analysis; J. J., S. S., and J. Z. validation; J. J. and J. Z. investigation; J. J. visualization; J. J., N. N. P., and J. Z. methodology; J. J. and J. Z. writing-original draft; J. J., S. S., R. C. W., and J. Z. writing-review and editing; H. F., R. K., and R. C. W. resources; J. Z. conceptualization; J. Z. supervision; J. Z. funding acquisition.
Supplementary Material
Acknowledgment
We thank the Flow Cytometry Core at the Indiana University Simon Cancer Center for cell sorting services.
This work was supported by Children's Cancer Research Fund Grant 567830 and St. Baldrick's Foundation Grant 578621 (to J. Z.) and Riley Children's Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
The RNA-Seq results were deposited in the GEO Repository with accession number GSE135420.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- NEAA
- nonessential amino acid
- ASNS
- asparagine synthetase
- ALL
- acute lymphoblastic leukemia
- ER
- endoplasmic reticulum
- cDNA
- complementary DNA
- GSEA
- gene set enrichment analysis
- MEF
- mouse embryonic fibroblast
- LCM
- lymphocyte culture medium
- qPCR
- quantitative PCR
- IP
- immunoprecipitation
- Ctrl
- control.
References
- 1. Vander Heiden M. G., and DeBerardinis R. J. (2017) Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pavlova N. N., and Thompson C. B. (2016) The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gwinn D. M., Lee A. G., Briones-Martin-Del-Campo M., Conn C. S., Simpson D. R., Scott A. I., Le A., Cowan T. M., Ruggero D., and Sweet-Cordero E. A. (2018) Oncogenic KRAS Regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to l-asparaginase. Cancer Cell 33, 91–107.e6 10.1016/j.ccell.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knott S. R. V., Wagenblast E., Khan S., Kim S. Y., Soto M., Wagner M., Turgeon M. O., Fish L., Erard N., Gable A. L., Maceli A. R., Dickopf S., Papachristou E. K., D'Santos C. S., Carey L. A., et al. (2018) Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381 10.1038/nature25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pavlova N. N., Hui S., Ghergurovich J. M., Fan J., Intlekofer A. M., White R. M., Rabinowitz J. D., Thompson C. B., and Zhang J. (2018) As Extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428–438.e5 10.1016/j.cmet.2017.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ye J., Kumanova M., Hart L. S., Sloane K., Zhang H., De Panis D. N., Bobrovnikova-Marjon E., Diehl J. A., Ron D., and Koumenis C. (2010) The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 29, 2082–2096 10.1038/emboj.2010.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krall A. S., Xu S., Graeber T. G., Braas D., and Christofk H. R. (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 10.1038/ncomms11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lomelino C. L., Andring J. T., McKenna R., and Kilberg M. S. (2017) Asparagine synthetase: function, structure, and role in disease. J. Biol. Chem. 292, 19952–19958 10.1074/jbc.R117.819060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van den Berg H. (2011) Asparaginase revisited. Leuk. Lymphoma 52, 168–178 10.3109/10428194.2010.537796 [DOI] [PubMed] [Google Scholar]
- 10. Avramis V. I. (2012) Asparaginases: biochemical pharmacology and modes of drug resistance. Anticancer Res. 32, 2423–2437 [PubMed] [Google Scholar]
- 11. Balasubramanian M. N., Butterworth E. A., and Kilberg M. S. (2013) Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am. J. Physiol. Endocrinol. Metab. 304, E789–E799 10.1152/ajpendo.00015.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holleman A., Cheok M. H., den Boer M. L., Yang W., Veerman A. J., Kazemier K. M., Pei D., Cheng C., Pui C. H., Relling M. V., Janka-Schaub G. E., Pieters R., and Evans W. E. (2004) Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med. 351, 533–542 10.1056/NEJMoa033513 [DOI] [PubMed] [Google Scholar]
- 13. Su N., Pan Y. X., Zhou M., Harvey R. C., Hunger S. P., and Kilberg M. S. (2008) Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr. Blood Cancer 50, 274–279 10.1002/pbc.21213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura A., Nambu T., Ebara S., Hasegawa Y., Toyoshima K., Tsuchiya Y., Tomita D., Fujimoto J., Kurasawa O., Takahara C., Ando A., Nishigaki R., Satomi Y., Hata A., and Hara T. (2018) Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. U.S.A. 115, E7776–E7785 10.1073/pnas.1805523115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wek S. A., Zhu S., and Wek R. C. (1995) The histidyl-tRNA synthetase-related sequence in the eIF-2α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 15, 4497–4506 10.1128/MCB.15.8.4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong J., Qiu H., Garcia-Barrio M., Anderson J., and Hinnebusch A. G. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279 10.1016/S1097-2765(00)00028-9 [DOI] [PubMed] [Google Scholar]
- 17. Kilberg M. S., Shan J., and Su N. (2009) ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 20, 436–443 10.1016/j.tem.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vattem K. M., and Wek R. C. (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274 10.1073/pnas.0400541101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Young S. K., Palam L. R., Wu C., Sachs M. S., and Wek R. C. (2016) Ribosome elongation stall directs gene-specific translation in the integrated stress response. J. Biol. Chem. 291, 6546–6558 10.1074/jbc.M115.705640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., and Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 21. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., and Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 22. B'chir W., Maurin A. C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., and Bruhat A. (2013) The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 10.1093/nar/gkt563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen H., Pan Y. X., Dudenhausen E. E., and Kilberg M. S. (2004) Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J. Biol. Chem. 279, 50829–50839 10.1074/jbc.M409173200 [DOI] [PubMed] [Google Scholar]
- 24. Wek R. C., Jiang H. Y., and Anthony T. G. (2006) Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 25. Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., and Kaufman R. J. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma Y., Brewer J. W., Diehl J. A., and Hendershot L. M. (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351–1365 10.1016/S0022-2836(02)00234-6 [DOI] [PubMed] [Google Scholar]
- 27. Wortel I. M. N., van der Meer L. T., Kilberg M. S., and van Leeuwen F. N. (2017) Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 28, 794–806 10.1016/j.tem.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loayza-Puch F., Rooijers K., Buil L. C., Zijlstra J., Oude Vrielink J. F., Lopes R., Ugalde A. P., van Breugel P., Hofland I., Wesseling J., van Tellingen O., Bex A., and Agami R. (2016) Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494 10.1038/nature16982 [DOI] [PubMed] [Google Scholar]
- 29. Fine B. M., Kaspers G. J., Ho M., Loonen A. H., and Boxer L. M. (2005) A genome-wide view of the in vitro response to l-asparaginase in acute lymphoblastic leukemia. Cancer Res. 65, 291–299 [PubMed] [Google Scholar]
- 30. Richards N. G., and Kilberg M. S. (2006) Asparagine synthetase chemotherapy. Annu. Rev. Biochem. 75, 629–654 10.1146/annurev.biochem.75.103004.142520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bunpo P., Dudley A., Cundiff J. K., Cavener D. R., Wek R. C., and Anthony T. G. (2009) GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti-cancer agent l-asparaginase. J. Biol. Chem. 284, 32742–32749 10.1074/jbc.M109.047910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deaton A. M., and Bird A. (2011) CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren Y., Roy S., Ding Y., Iqbal J., and Broome J. D. (2004) Methylation of the asparagine synthetase promoter in human leukemic cell lines is associated with a specific methyl binding protein. Oncogene 23, 3953–3961 10.1038/sj.onc.1207498 [DOI] [PubMed] [Google Scholar]
- 34. Reinert R. B., Oberle L. M., Wek S. A., Bunpo P., Wang X. P., Mileva I., Goodwin L. O., Aldrich C. J., Durden D. L., McNurlan M. A., Wek R. C., and Anthony T. G. (2006) Role of glutamine depletion in directing tissue-specific nutrient stress responses to l-asparaginase. J. Biol. Chem. 281, 31222–31233 10.1074/jbc.M604511200 [DOI] [PubMed] [Google Scholar]
- 35. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., and Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 10.1101/gad.12.7.982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., and Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Touzart A., Lengliné E., Latiri M., Belhocine M., Smith C., Thomas X., Spicuglia S., Puthier D., Pflumio F., Leguay T., Graux C., Chalandon Y., Huguet F., Leprêtre S., Ifrah N., et al. (2019) Epigenetic silencing affects l-asparaginase sensitivity and predicts outcome in T-ALL. Clin. Cancer Res. 25, 2483–2493 10.1158/1078-0432.CCR-18-1844 [DOI] [PubMed] [Google Scholar]
- 38. Mirzaei H., Suarez J. A., and Longo V. D. (2014) Protein and amino acid restriction, aging and disease: from yeast to humans. Trends Endocrinol. Metab. 25, 558–566 10.1016/j.tem.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fusakio M. E., Willy J. A., Wang Y., Mirek E. T., Al Baghdadi R. J., Adams C. M., Anthony T. G., and Wek R. C. (2016) Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 27, 1536–1551 10.1091/mbc.E16-01-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang J., Fan J., Venneti S., Cross J. R., Takagi T., Bhinder B., Djaballah H., Kanai M., Cheng E. H., Judkins A. R., Pawel B., Baggs J., Cherry S., Rabinowitz J. D., and Thompson C. B. (2014) Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 10.1016/j.molcel.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Linares J. F., Cordes T., Duran A., Reina-Campos M., Valencia T., Ahn C. S., Castilla E. A., Moscat J., Metallo C. M., and Diaz-Meco M. T. (2017) ATF4-induced metabolic reprograming is a synthetic vulnerability of the p62-deficient tumor stroma. Cell Metab. 26, 817–829.e6 10.1016/j.cmet.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melamud E., Vastag L., and Rabinowitz J. D. (2010) Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 82, 9818–9826 10.1002/0471250953.bi1411s37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clasquin M. F., Melamud E., and Rabinowitz J. D. (2012) LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinformatics Chapter 14, Unit 14.11 10.1021/ac1021166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen C., Liu Y., Lu C., Cross J. R., Morris J. P. 4th, Shroff A. S., Ward P. S., Bradner J. E., Thompson C., and Lowe S. W. (2013) Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 27, 1974–1985 10.1101/gad.226613.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang H. Y., Wek S. A., McGrath B. C., Lu D., Hai T., Harding H. P., Wang X., Ron D., Cavener D. R., and Wek R. C. (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24, 1365–1377 10.1128/MCB.24.3.1365-1377.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J. S., and Concordet J. P. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.