

Abstract
Gut microbial β-glucuronidase (GUS) enzymes have been suggested to be involved in the estrobolome, the collection of microbial reactions involving estrogens. Furthermore, bacterial GUS enzymes within the gastrointestinal tract have been postulated to be a contributing factor in hormone-driven cancers. However, to date, there has been no experimental evidence to support these hypotheses. Here we provide the first in vitro analysis of the ability of 35 human gut microbial GUS enzymes to reactivate two distinct estrogen glucuronides, estrone-3-glucuronide and estradiol-17-glucuronide, to estrone and estradiol, respectively. We show that certain members within the Loop 1, mini-Loop 1, and FMN-binding classes of gut microbial GUS enzymes can reactivate estrogens from their inactive glucuronides. We provide molecular details of key interactions that facilitate these catalytic processes and present the structures of two novel human gut microbial GUS enzymes related to the estrobolome. Further, we demonstrate that estrogen reactivation by Loop 1 bacterial GUS enzymes can be inhibited both in purified enzymes and in fecal preparations of mixed murine fecal microbiota. Finally, however, despite these in vitro and ex vivo data, we show that a Loop 1 GUS-specific inhibitor is not capable of reducing the development of tumors in the PyMT mouse model of breast cancer. These findings validate that gut microbial GUS enzymes participate in the estrobolome but also suggest that the estrobolome is a multidimensional set of processes on-going within the mammalian gastrointestinal tract that likely involves many enzymes, including several distinct types of GUS proteins.
Keywords: estrogen, structural biology, enzyme structure, breast cancer, X-ray crystallography, beta-glucuronidase, estrobolome, GI microbiota, gut microbiome, hormone reactivation
Introduction
The gastrointestinal (GI)2 microbiome harbors incredible metabolic potential and is intimately connected to human physiology. Possessing 150 times more genes than are found in the human genome, the gut microbiome encodes a vast number of enzymes that function in a variety of metabolic pathways, including the biosynthesis of essential vitamins and the breakdown of complex, nondigestible polysaccharides (1–4). The GI microbiota plays a potentially significant but enigmatic contribution to human health via the estrobolome, the aggregate of the enteric bacterial genes whose products are capable of metabolizing estrogens (5). It has been suggested that a woman's estrobolome plays a key role in a number of hormonal disorders, including breast, endometrial, and ovarian cancers (5–8). Hypothesized to be especially important to metabolism within the estrobolome are bacterial species possessing β-glucuronidase (GUS) enzymes.
During phase II metabolism, UDP-glucuronosyltransferase enzymes (UGTs) append a glucuronic acid moiety to a variety of endo- and xenobiotics, typically inactivating them and marking them for excretion. GUS proteins within the GI tract can intercept this process, cleaving the glucuronic acid, allowing the reactivated compound to be recirculated throughout the body, thereby reversing phase II glucuronidation. In the case of SN-38, the active metabolite of the anticancer drug irinotecan, this process of glucuronidation and deglucuronidation through the action of GUS has been shown to cause severe GI toxicity. By inhibiting the GUS enzymes responsible for this reactivation, the diarrhea associated with administration of SN-38 can be alleviated (9–12). The same has been established for lower GI damage caused by nonsteroidal anti-inflammatory compounds (NSAIDs) (13–17).
Similar to SN-38 and NSAIDs, estrogenic compounds are glucuronidated in the liver during phase II metabolism. Upon entry into the GI tract, they are exposed to GUS enzymes that could, in theory, cleave the sugar moiety, reactivating the parent compound and allowing the unconjugated estrogen to be reabsorbed in the bloodstream and undergo iterative rounds of enterohepatic recirculation (18, 19). This is potentially significant in oncology, as there is now general agreement that the concentrations of unbound estrogens are much higher in plasma and tissues of women with hormone-driven cancers (20–23). Therefore, an estrobolome enriched in β-glucuronidase enzymes that promote estrogen metabolite deconjugation reactions may result in greater reabsorption of free estrogens and a greater risk of hormone receptor–positive (HR +) cancers (Fig. 1).
Figure 1.
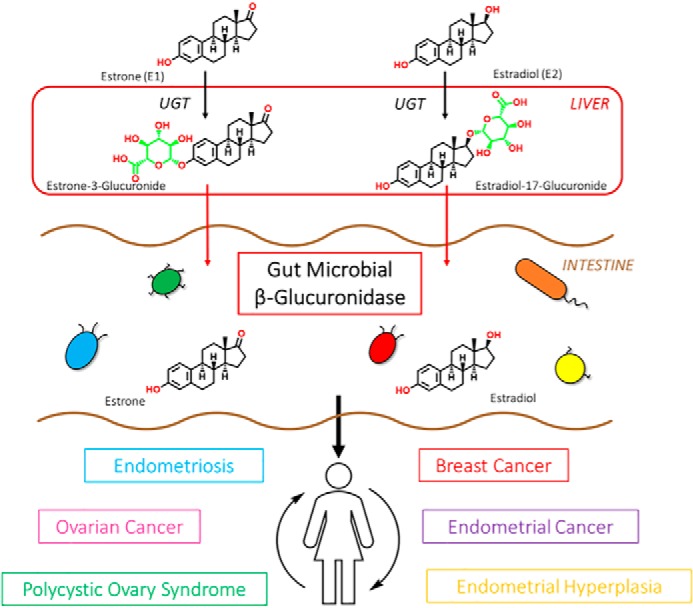
β-Glucuronidase enzymes reactivate estrogens. Gut microbial β-glucuronidase enzymes within the GI deconjugate estrone-3- and estradiol-17-glucuronides to the aglycones estrone and estradiol, respectively. This reactivation allows unbound estrogens to be recirculated through the bloodstream, possibly contributing to a variety of hormonal disorders, including breast cancer and endometriosis.
Although it has been postulated that the GUS enzymes within the estrobolome play an integral role in estrogen reactivation and recirculation, this role has yet to be proven. Here we take a panel of 35 human gut microbial GUS enzymes and the glucuronides of two estrogens, estradiol (E2, dominant during reproductive years) and estrone (E1, dominant after menopause), and assess the ability of these enzymes to process these substrates. With this in vitro panel, we show that 17 of the 35 GUS enzymes tested are capable of reactivating estrogen conjugates. Guided by novel crystal structures of two human gut microbial GUS enzymes and 12 additional structures already in hand, we pinpoint structural features critical to estrogen glucuronide processing. Further, we examine the inhibition of key gut microbial GUS enzymes in vitro, ex vivo, and in vivo and test the estrobolome hypothesis with respect to breast tumor growth in an HR + mouse model. Taken together, our results validate gut microbial GUS enzymes as active members of the estrobolome but highlight the likely complex relationship between the estrobolome and tumorigenesis in vivo.
Results
Identification of estrogen glucuronide–processing gut microbial GUS enzymes
Gut microbial GUS enzymes have been shown to share a common fold but exhibit unique active-site architectures and differential activities with distinct substrates (24, 25). To gain greater insight into the specific sequence-structure-function relationships among GUS proteins, we generated a sequence similarity network (SSN) using sequences of β-glucuronidase enzymes found within the Human Microbiome Project (HMP). The resultant SSN clusters the 279 unique protein sequences based on sequence identity and homology (26).
Of the 279 unique GUS enzymes identified in the HMP, we have cloned, expressed, and purified 35 of these for in vitro study (Fig. 2A (triangles) and Table S1). Enzymes were chosen so that the prevalence of each loop category was comparable with what has been previously reported in the HMP (27). However, an exception to this is the Loop 1 enzymes, which are overrepresented in our panel of 35 proteins, as these have been previously shown to efficiently reactivate small-molecule drug substrates, a key focus of our work (27). Of the 35 enzymes examined, crystal structures have been reported for 18 (Table S1), and these structural data correlate with the family groupings present in the SSN.
Figure 2.

Identification of estrogen-deconjugating β-glucuronidase enzymes. A, sequence similarity network of 279 unique GUS enzymes from the Human Microbiome Project. Each circle represents a unique protein sequence. Of the 279 total proteins shown, 35 enzymes were tested for their ability to reactivate estrone-3-glucuronide and estradiol-17-glucuronide (triangles). Colored triangles represent those enzymes that can reactivate estrogens, gray triangles show those that cannot. Colored squares, the two novel GUS proteins identified here that can reactivate estrogen glucuronides (PDB: 6U7J and 6U7I). B, catalytic efficiencies of 17 GUS enzymes that reactivate estrone-3-glucuronide. Red bars, Loop 1 enzymes; green bars, mini-Loop 1 enzymes; yellow bars, FMN-binding GUS enzymes. F. prausnitzii (Loop 1) through F. prausnitzii L2–6 (FMN) show statistically significant difference at a p value of 0.001 (*) compared with E. eligens and S. agalactiae. Clostridium sp. Marseille-P299 (Loop 1) through F. prausnitzii (mini-Loop 1) show statistically significant difference at a p value of 0.0001 (**) compared with E. eligens (Loop 1) and S. agalactiae (Loop 1). C, catalytic efficiencies of 15 GUS enzymes that reactivate estradiol-17-glucuronide. Red bars, Loop 1 enzymes; green bars, mini-Loop 1 enzymes; yellow bars, FMN-binding GUS enzymes. R. gnavus 3 (FMN) through F. prausnitzii L2–6 (FMN) show statistically significant difference at a p value of 0.001 (*) compared with S. agalactiae (Loop 1) and C. perfringens (Loop 1). Uncultured Clostridium sp. (Loop 1) through F. prausnitzii (mini-Loop 1) show statistically significant difference at a p value of 0.0001 (**) in comparison to S. agalactiae (Loop 1) and C. perfringens (Loop 1). Data are presented as the average of 3 biological replicates ± SEM.
To identify GUS enzymes capable of processing estrogen glucuronides, we employed an assay that couples the formation of glucuronic acid to its utilization by uronate dehydrogenase and subsequent reduction of NAD+ to NADH + H+, to define an end-point cleavage measurement (Fig. S1; see “Experimental procedures”). Of the 35 enzymes tested, 17 were capable of cleaving the glucuronide moiety of estrone (E1-3G), and 15 were capable of cleaving the glucuronide of estradiol (E2-17G). We find that the GUS enzymes capable of cleaving estrogen cluster into three distinct categories: Loop 1 enzymes (red), mini-Loop 1 enzymes (green), and FMN-binding GUS enzymes (yellow). The GUS enzymes that processed glucuronides of estrogen are highlighted on the SSN (Fig. 2A, colored triangles).
These classes have been described in detail previously (27–30); briefly, the Loop 1 and mini-Loop 1 enzymes contain active site–proximal loops that provide favorable interactions for binding smaller substrate-glucuronides. The Loop 1 and mini-Loop 1 enzymes are most frequently associated with the cleavage of small-molecule glucuronides, including drug-glucuronide substrates. The FMN-binding GUS enzymes possess a flavin-mononucleotide cofactor at an allosteric site that aids in structural stability. To date, the exact role of FMN is unclear, but we have found that these enzymes are uniquely capable of small-molecule glucuronide cleavage.
We determined catalytic efficiency (m−1 s−1) values with E1-3G for the 17 enzymes identified. These rates range from 6.40 × 105 to 1.08 × 102 s−1 m−1 (Fig. 2B). The catalytic efficiencies were also determined for the 15 enzymes capable of processing estradiol-17-glucuronide. These rates range from 1.83 × 104 to 1.26 × 102 s−1 m−1 (Fig. 2C). Several of these enzymes have been examined previously with the standard GUS assay substrate 4-nitrophenyl β-d-glucuronide (pNPG), where the catalytic efficiencies range from 9.2 × 105 to 3.6 × 101, comparable with what we observe with the estrogen glucuronides (28). This result suggests that some gut microbial GUS enzymes may have evolved to be able to process estrogen glucuronide substrates efficiently to gain the six-carbon source of energy.
With the estrogen glucuronide substrates, we find that the Loop 1 enzymes are, in general, the fastest processors of both estrone and estradiol glucuronide, with the FMN-binding GUSs the second fastest, and the mini-Loop 1 GUS enzymes as the slowest estrogen processors. In summary, we provide the first detailed in vitro data on the ability of human gut microbial GUS enzymes to process the glucuronides of estrone and estradiol, thus indicating that bacterial GUS proteins are active components of the estrobolome.
Structural rationale for estrogen glucuronide–processing gut microbial GUS enzymes
With concerted modeling and site-directed mutagenesis efforts, we have highlighted the residues involved in catalysis, providing a rationale for rate differences of the three categories. First, we modeled E1-3G and E2-17G into the active site of a tetrameric Loop 1 GUS from Clostridium perfringens (4JKM) (9). Because we know precisely where and how the glucuronide moiety binds (11, 12, 25, 27, 28), and these substrate molecules have a limited number of rotatable bonds, we know with reasonable certainty where these estrogens are within the active site. From these models, we have identified aromatic residues within the Loop 1 architecture that provide potential π-stacking interactions that would facilitate substrate binding to optimize cleavage (Fig. 3A).
Figure 3.

Structural rationale for estrogen glucuronide–processing gut microbial GUS enzymes. Shown in purple and pink are E1-3G and E2-17G, respectively. Catalytic glutamates are highlighted in marine, whereas the N-K glucuronide recognition motif is highlighted in magenta. Statistical differences are annotated with *, **, and *** for p values of 0.001, 0.0001, and <0.0001, respectively. A, active site of Loop 1 GUS from C. perfringens. Shown in cyan are the residues that contribute to the aromatic cage that allow the Loop 1 enzymes to be the fastest processors of estrogen glucuronides. B, activity differences between WT C. perfringens (Loop 1) and mutants. C, active site of mini-Loop 1 GUS from B. fragilis. Shown in cyan are the residues that contribute to an aromatic cage that allow the mini-Loop 1 enzymes to process estrogen glucuronides. D, activity differences between WT B. fragilis (mini-Loop 1) and mutants. E, monomer and active site of FMN-binding GUS from R. gnavus 3. The C-terminal domain (pink) is modeled from a previously resolved structure (5UJ6). F, activity differences between WT R. gnavus GUS (FMN) and mutants. As there are no obvious residues at the active site that contribute to binding, mutants were made to probe steric occlusion of Ser-344 and the role of the C-terminal domain. Data are presented as the average of 3 biological replicates ± SEM.
One of these residues (Phe-363) comes from Loop 1 itself, providing an edge-to-face interaction. The second residue, Phe-368, is donated from the Loop 1 of an adjacent monomer, providing a face-to-face interaction. These residues, coupled with the completely conserved Tyr-472, create an aromatic cage, perhaps explaining why the Loop 1 enzymes exhibit the highest catalytic efficiencies that we measured in vitro (Fig. 3A). To test this hypothesis, mutants of F363A, F368A, and Y472A were created. The Y472A mutation is detrimental to structural stability and as a result is inactive. However, the mutations of F363A and F368A are both stable yet significantly reduce the catalytic efficiency of the molecule. Furthermore, the double mutation, F363A/F368A, eliminates activity of the enzyme entirely (Fig. 3B). These experiments help to confirm our modeling and establish the critical role the aromatic cage in a gut microbial GUS Loop 1 region to the turnover of estrogen glucuronides.
Second, we used the same model of estrogens overlaid with the structure from the tetrameric Bacteroides fragilis β-glucuronidase (3CMG) to identify structural features within the mini-Loop 1 architecture that aid in turnover. Like the Loop 1 GUS enzymes, the mini-Loop 1 possess the conserved tyrosine present in all GUS structures (e.g. Tyr-472 in C. perfringens Loop 1 GUS) and an aromatic residue in the mini-Loop 1, Tyr-389 (Fig. 3C). The tetramer of this GUS is different from the previously resolved Loop 1 GUS enzymes, however, in that the active-site interfaces do not overlap. Indeed, the loop of an adjacent monomer does not extend into the active site like it does in the Loop 1 architecture; thus, it only has the one aromatic residue rather than two, making the aromatic cage notably smaller, perhaps explaining the slower rates of the mini-Loop 1 enzymes compared with the Loop 1 enzymes (Fig. 3C). To test this, we created a Y389A mutation in B. fragilis GUS and found that it reduces the rate of estrone glucuronide cleavage by an order of magnitude, from a rate of 7.75 × 103 to 3.20 × 102 s−1 m−1 (Fig. 3D). Thus, structural modeling and mutagenesis provide a rationale for why the mini-Loop 1 enzymes are less efficient than the Loop 1 enzymes in processing estrone-3-glucuronide.
Third, we modeled the estrogens within the active site of a dimer from Ruminococcus gnavus 3 GUS (6MVG), an FMN-binding GUS (30). We have previously shown that although these enzymes have an open, planar active site with few aromatic residues, they contain roughly 150 residues that have yet to be resolved in any of the crystal structures we have determined to date (30). However, a model of this domain has been created based on a Loop 2 GUS from Bacteroides uniformis (5UJ6) (27), and this model places the C-terminal domain roughly 15 Å from the active sites of these enzymes. This potentially flexible C-terminal domain could influence catalytic activity of the FMN enzymes (Fig. 3E). Indeed, when this domain is eliminated (mutant: STP641), enzyme activity of R. gnavus 3 GUS is virtually eliminated (Fig. 3F). Thus, the C-terminal domains of the FMN-binding human gut microbial GUS enzymes play critical roles in substrate turnover that allow these FMN-binding GUS proteins to behave akin to Loop 1 GUS enzymes.
Finally, the ratio of the cleavage rate of estrone-3-glucuronide over estradiol-17-glucuronide was calculated for each of the enzymes tested (Table 1). Interestingly, the Loop 1 GUS and mini-Loop 1 GUS orthologs show preference for E1-3G over E2-17G. Furthermore, whereas two of the three mini-Loop 1 GUS proteins tested show some activity with E2-17G, the B. fragilis enzyme does not process E2-17G at all. Two distinctions between these substrates may be involved in these differences. First, E1-3G contains an aromatic ring immediately adjacent to the ether-linked glucuronic acid moiety. The planar feature of this aromatic ring may enhance activity because it can form favorable interactions with the GUS active sites in Loop 1 and some mini-Loop 1 enzymes. In contrast, the more flexible cyclopentane ring in E2-17G cannot form potential π-π stacking interactions at the enzyme active site and thus serves as a less optimal substrate.
Table 1.
Ratio of catalytic efficiencies of E1-3G to E2-17G
Red, green, and yellow labels represent Loop 1, mini-Loop 1, and FMN-binding GUS enzymes, respectively.

Second, the methyl group on E2-17G appears to interfere with optimal positioning of the catalytic glutamates, which would certainly reduce catalytic activity with GUS enzymes (Fig. S2). In contrast, this methyl group in E1-3G is directed away from the active-site glucuronic acid, singular likely allowing these catalytic residues more ready access to the ether linkage to be cleaved in E1-3G compared with E2-17G. Thus, differences in aromatic ring and methyl group positioning between these two estrogen glucuronides likely explain why the 3-glucuronide of E1 is a more efficient substrate for the Loop 1 and mini-Loop 1 GUS enzymes compared with E2-17G.
In contrast, the FMN-binding GUS enzymes that exhibit no preference for the 3- versus 17-positions have more open active sites and may not encounter the same steric hindrance as the Loop 1 and mini-Loop 1 GUS enzymes. Taken together, we provide structure-guided insights into the observed differences in estrogen glucuronide substrate turnover between the three categories of GUS enzymes found to process these compounds.
Novel GUS structures reveal unique active-site architectural motifs
We were particularly intrigued with the observation that although the mini-Loop 1 enzyme from Faecalibacterium prausnitzii and the Loop 1 enzyme from uncultured Clostridium sp. process these estrogenic compounds, they were hundreds of times slower than every other mini-Loop 1 and Loop 1 enzyme examined (Fig. 1, A (squares), B, and C). We reasoned that there may be unique features to the F. prausnitzii and uncultured Clostridium sp. GUS proteins that make them slow with these estrogen glucuronide substrates.
Using a multiple-sequence alignment of all 279 GUS enzymes identified from the HMP, we found that 14 contained a loop insert of roughly 25 residues, like in Eubacterium eligens GUS (6BJW) (29), that were near the active site, adjacent to the Loop 1/mini-Loop 1 site, that could potentially contribute to binding (Fig. S3). F. prausnitzii and Clostridium sp. GUS were both members of this group of 14 enzymes with the hypothesized novel loop. Therefore, we determined the X-ray crystal structures of these two novel tetramers to 2.7 and 2.2 Å resolution, respectively (Table 2), and compared them to the classic architectures observed in several previous structures determined by our laboratory.
Table 2.
Crystallography data collection and refinement statistics
Data were refined using Phenix version 16.1. Values in parentheses are for the highest-resolution shell.
| F. prausnitzii (PDB code 6U7I) | Uncultured Clostridium sp. (PDB code 6U7J) | |
|---|---|---|
| Resolution range (Å) | 39.16–2.702 (2.799–2.702) | 29.31–2.2 (2.279–2.2) |
| Space group | P 21 21 21 | P 1 21 1 |
| Unit cell dimensions a, b, c (Å); α, β, γ (degrees) | 122.597, 129.055, 161.424; 90, 90, 90 | 74.941, 105.655, 189.566; 90, 94.599, 90 |
| Total reflections (F > 0) | 307,596 (25,684) | 398,219 (40,145) |
| Unique reflections | 66,748 (6563) | 146,335 (14,633) |
| Multiplicity | 4.6 (3.9) | 2.7 (2.7) |
| Completeness (%) | 91.80 (81.34) | 97.94 (98.54) |
| Mean I/σ(I) | 7.71 (1.77) | 10.89 (2.57) |
| Wilson B-factor (Å2) | 47.94 | 34.10 |
| Rmerge | 0.1538 (0.6867) | 0.0678 (0.4172) |
| Rmeas | 0.1763 (0.7949) | 0.08378 (0.5126) |
| Rpim (%) | 0.08288 (0.3884) | 0.04852 (0.2942) |
| CC1/2 | 0.969 (0.708) | 0.997 (0.834) |
| Number of non-hydrogen atoms | 18,998 | 20,068 |
| Macromolecules | 18,895 | 18,903 |
| Solvent | 103 | 1165 |
| Protein residues | 2384 | 2277 |
| r.m.s. bond deviations (Å) | 0.010 | 0.008 |
| r.m.s. angle deviations (degrees) | 1.38 | 1.22 |
| Ramachandran favored (%) | 93.77 | 95.52 |
| Ramachandran allowed (%) | 5.98 | 4.44 |
| Ramachandran outliers (%) | 0.25 | 0.04 |
| Rotamer outliers (%) | 0.00 | 0.00 |
| Clashscore | 9.70 | 4.21 |
| Average B-factor (Å2) | 48.47 | 37.64 |
| Macromolecules | 48.48 | 37.45 |
| Solvent | 46.96 | 40.81 |
In the case of the F. prausnitzii GUS (PDB code 6U7I, Fig. 4A), inspection of the active site reveals that this novel loop, adjacent to the mini-Loop 1 site, sterically occludes estrogen binding by this F. prausnitzii GUS. In fact, Phe-153 and Phe-154 (Fig. 4A, inset (purple)) on this loop extend into the active site and may prevent optimal recognition of these estrogenic compounds in this enzyme. Additionally, we observe a unique positioning of a critical aromatic residue situated on this mini-Loop 1 region expected to coordinate estrogen binding (e.g. BfGUS: Tyr-389). We find that this aromatic residue (Tyr-378) is directed away from the active site in this novel structure, 10 Å farther from the active site than previously resolved mini-Loop 1 GUS structures (Fig. 4A, inset). Thus, for F. prausnitzii GUS, steric occlusion by a loop not present in other mini-Loop 1 enzymes likely explains its poor functioning with the two estrogen glucuronides of interest in this report.
Figure 4.

Novel GUS structures reveal unique active-site architectural motifs that impact estrogen glucuronide reactivation rates. In purple and pink are E1-3G and E2-17G, respectively. Catalytic glutamates are highlighted in marine, and the N-K glucuronide recognition motif is highlighted in magenta. A, structure of mini-Loop 1 enzyme from F. prausnitzii. Shown in green is the conserved tyrosine on the mini-Loop 1 region; this residue is 10 Å farther from the active site than previously resolved mini-Loop 1 GUS structures. The active site also reveals a novel loop (purple) that extends into the active site, possibly contributing to its slow rate with these estrogenic substrates. B, structure of Loop 1 enzyme from an uncultured Clostridium sp. revealing that the conserved aromatic residue on the Loop 1 region (Phe-370) is directed away from the active site, toward the solvent (red). In contrast, previously resolved GUS structures of E. eligens (EeGUS), C. perfringens (CpGUS), and B. fragilis (BfGUS) show this aromatic residue directed in the opposite direction, toward the active site.
Second, the 2.2 Å resolution structure of the GUS from the uncultured Clostridium sp. (PDB code 6U7J, Fig. 4B) initially revealed no obvious differences in quaternary or tertiary structure from the other Loop 1 enzymes, as the predicted novel loop contained ordered β-sheets, similar to Escherichia coli GUS (PDB code 3LPF) (11). From E. coli GUS, we know that deletion of this region does not impact substrate turnover with pNPG or diclofenac-glucuronide (28). However, inspection of the active site reveals a unique positioning of a critical aromatic residue situated on the Loop 1 region expected to coordinate estrogen binding in other Loop 1 and mini-Loop 1 GUS enzymes (e.g. for Loop 1 enzymes EeGUS: Phe-374 and CpGUS: Phe-363; for the mini-Loop 1 enzymes BfGUS: Tyr-389).
Whereas these aromatic residues are positioned to contact the steroid scaffold of bound estrogen glucuronides in the standard Loop 1 and mini-Loop 1 GUS structures determined to date, the equivalent residue, Phe-370, in the uncultured Clostridium sp. GUS is directed away from the active site and toward the solvent surface (Fig. 4B, inset). Furthermore, Phe-370 in the uncultured Clostridium sp. GUS is stabilized in this position away from the active site by contacts with Phe-379, His-419, and Trp-149 in this GUS structure and thus is not likely to be capable of shifting in position into the active site to facilitate the binding of estrogen glucuronides. Taken together, these novel human gut microbial GUS crystal structures provide molecular explanations for their relatively poor activity with the estrogen glucuronides examined.
In vitro inhibition of gut microbial GUS enzymes that process estrogen glucuronides
We have developed a series of chemotypes that are selective for and potently and nonlethally inhibit human gut microbial GUS enzymes (9–12, 29). To date, these compounds are selective for the Loop 1 group of microbial GUS enzymes (28, 29). Of the compounds examined, one inhibitor, UNC10201652, is particularly potent, as it contains a piperazine ring that intercepts the catalytic cycle of Loop 1 microbial GUS enzymes and is capable of forming a long-lived inhibitor-glucuronide moiety that remains bound at the enzyme's active site (30). Thus, we chose to examine UNC10201652 (Fig. S4) for its ability to inhibit estrogen glucuronide processing by a range of Loop 1 and non-Loop 1 gut microbial GUS enzymes.
As expected, UNC10201652 potently inhibits E1-3G and E2-17G processing by several “standard” Loop 1 gut microbial GUS enzymes, particularly those that showed efficient activities with these substrates in vitro, such as E. eligens and C. perfringens (Fig. 5A). UNC10201652 does not inhibit the activities of the Loop 1 GUS enzymes from F. prausnitzii or Lactobacillus rhamnosus, however, which corroborates previous observations regarding these enzymes (28). Furthermore, it does not affect the activities of the unique gut microbial GUS enzymes from F. prausnitzii or the uncultured Clostridium sp. examined above. Variability in UNC10201652 potency may be a result of differences in the amino acid sequences at the Loop 1 region, as previously hypothesized (28). In addition, UNC10201652 may be ineffective against F. prausnitzii (mini-Loop 1) because of the novel loop identified here.
Figure 5.

Inhibition by UNC10201652. A, in vitro inhibition of all 17 estrogen-reactivating enzymes identified in this study. UNC10201652 is only effective toward the Loop 1 GUS architecture. B, EC50 plot of WT and Δgus E. coli K-12 MG1655 cells incubated with UNC10201652. As expected, the knockout strain shows no activity with estrogen glucuronide substrates. WT cells exhibit sub-micromolar EC50 values. L1, Loop 1; mL1, mini-Loop 1. Data are presented as the average of 3 biological replicates ± SEM.
We also tested UNC10201652's effect on the processing of estrogen glucuronide substrates by non-Loop 1 human gut microbial GUS enzymes in vitro. As expected, because UNC10201652 has been shown to be specific for Loop 1 GUS enzymes with standard small-molecule GUS substrates and glucuronidated drugs (28, 29), this compound had no effect on the non-Loop 1 enzymes capable of processing estrogen glucuronides in our in vitro studies (Fig. 5A). Thus, UNC10201652 is a potent and effective inhibitor of estrogen glucuronide processing by a targeted subset of the gut microbial Loop 1 GUS enzymes examined.
Finally, we tested the ability of UNC10201652 to inhibit estrogen glucuronide conversion by living E. coli cells. We have previously constructed a variant of E. coli K-12 MG1655 cells in which the gus gene was truncated to remove the amino acids between the two conserved catalytic glutamates (GUSΔ413–504). As expected, in this knockout strain, we see no GUS activity in cultured cells (Fig. 5B). We find with WT E. coli K-12 MG1655 cells that UNC10201652 shows EC50 values of 155 ± 7 and 148 ± 8 nm toward E1-3G and E2-17G processing, respectively (Fig. 5B), similar to what has been observed previously with this inhibitor and other substrates (29). Thus, taken together, we have demonstrated that our most potent gut microbial GUS inhibitor to date is effective in vitro and in cells against several key estrogen glucuronide–processing Loop 1 GUS enzymes, but does not inhibit all of the β-glucuronidases that would appear to participate in the complete estrobolome.
In fimo evaluation of estrobolome modulation
Next we sought to examine the reactivation of estrogen glucuronides by gut microbial GUS enzymes present in mammalian intestinal contents. We use the Latin term in fimo to describe studies on fecal samples obtained ex vivo. In this in fimo study, we obtained fecal sample preparations from 11 BALB/c mice (6 female, 5 male) and tested their ability to deconjugate E1-3G and E2-17G in fimo. Fecal samples were homogenized, sonicated, and then subjected to centrifugation, after which the resulting supernatant was used to quantify GUS activity.
After incubation of the estrogen conjugate and the in fimo specimen for 1 h, we find that every fecal sample is capable of processing these estrogen glucuronides to varying degrees to produce the parent compounds, estrone and estradiol. This indicates that GUS enzymes are indeed active members of the estrobolome (Fig. 6, A and B). Further, we tested the ability of UNC10201652 to inhibit estrogen glucuronide conversion by these in fimo preparations. We find that upon the addition of 10 μm UNC10201652, we can inhibit in fimo the formation of estrone and estradiol from E1-3G and E2-17G, respectively. All samples tested showed a trend toward reduction in these reaction rates. The variability in response to UNC10201652 likely arises from the differential levels of Loop 1 GUS enzymes present in each sample. Taken together, these data support the conclusion that gut microbial GUS enzymes are active components of the estrobolome and that such enzymes may be amenable to control using targeted small-molecule inhibitors.
Figure 6.

Ex vivo and in vivo application of UNC10201652. Statistical differences are annotated as follows: *, p = 0.001. A, fecal samples from 11 mice were incubated with E1-3G in the presence and absence of UNC10201652. Fecal samples incubated with inhibitor show decreased activity. B, fecal samples from 11 mice were incubated with E2-17G in the presence and absence of UNC10201652. Fecal samples incubated with inhibitor show decreased activity. C, lesion size of PyMT-treated mouse model of breast cancer. Mice treated with UNC10201652 show no difference in lesion size compared with control mice. Data are presented as the average of 3 biological replicates ± SEM.
In vivo model of the estrobolome hypothesis
We have previously shown for both irinotecan and NSAIDs that targeted Loop 1 GUS inhibitors effectively alleviate GI toxicity associated with these drugs (11, 13, 14). This is despite the fact that we have also shown that other GUS orthologs, including the mini-Loop 1 and FMN GUS enzymes, also reactivate these glucuronides (28). As such, we hypothesized that our inhibitor could similarly be used to prevent tumor growth in an HR + breast cancer model.
MMTV-PyMT is a transgenic mouse model in which the expression of the oncogene is driven by the mouse mammary tumor virus promoter, resulting in widespread transformation of the mammary epithelium and development of multifocal mammary adenocarcinomas and metastatic lesions in the lung and lymph nodes (31–33). Additionally, in this model, mice exhibit a gradual loss of steroid hormone receptors (estrogen and progesterone) similar to human breast cancer. Because the MMTV-PyMT mouse model is characterized by short latency, high penetrance, and a high incidence of lung metastasis, it was chosen to test the role of β-glucuronidase enzymes within the GI as they contribute to total estrogen burden and breast cancer risk.
PyMT females were gavaged beginning at 4 weeks old with UNC10201652 (or saline for the control group) for 9 weeks every day except weekends (20 μg/mouse/day). On the final day, animals were sacrificed, and histology analysis of breast tissue, liver, and kidneys was performed. We hypothesized that the group given UNC10201652 would have fewer and smaller tumors by the end of the treatment period, as the Loop 1 GUS enzymes were shown to be fastest in vitro at reactivating estrogens from their glucuronides. In fact, we see no difference in lesion size or quantity in mammary pads between the control and treated mice (Fig. 6B, Fig. S5 and Tables S2 and S3). This result may be because only one inhibitor chemotype was examined, at a single dose, with one dosing regimen; at this point, however, we only have potent gut microbial GUS inhibitors that target Loop 1 GUS enzymes. Therefore, such a compound may be insufficient to block the reactivation of estrogens in the gut that potentially participate in tumor growth in this model.
Discussion
The estrobolome, first defined in 2011 as the aggregate of all enteric bacteria capable of metabolizing estrogen, is predicted to impact endogenous estrogen metabolism by modulating the enterohepatic circulation of estrogens, thus affecting plasma estrogen levels (5). This initial review and subsequent contributions to the literature suggest that gut microbial β-glucuronidase enzymes are especially important in total estrogen circulation (5–8). Further, it has been hypothesized that an estrobolome rich in deconjugating GUS enzymes would be a contributing factor in breast cancer.
Here, we probe the potential roles of gut microbial GUS proteins in the estrobolome by testing a panel of 35 human gut microbial GUS enzymes with two endogenous small-molecule estrogens: estradiol-17-glucuronide and estrone-3-glucuronide. Guided by the structures of 14 GUS enzymes, including novel structures reported here, we have identified characteristics of GUS enzymes that contribute to deconjugation. In general, specific members of three subtypes of GUS enzymes are able to process these glucuronides: Loop 1 GUS, mini-Loop 1 GUS, and FMN-binding GUS. In addition to defining key residues involved in estrogen glucuronide processing, we demonstrated that we can inhibit estrogen reactivation by Loop 1 GUS enzymes using a targeted microbial GUS inhibitor, UNC10201652. Despite these promising preliminary data, UNC10201652 alone was not able to impact tumor development in the PyMT mouse model of breast cancer.
We have previously demonstrated that GUS orthologs other than the Loop 1 enzymes are capable of processing irinotecan and NSAIDs, yet our targeted Loop 1 inhibitors effectively alleviate gut toxicity associated with these drugs (11, 13, 14, 28). As such, we hypothesized that the same paradigm could apply to inhibiting hormone reactivation in the GI and possibly impact tumor growth in the PyMT mouse model of HR + breast cancer. However, we see no difference in mammary lesion size between mice treated with vehicle or UNC10201652.
Lack of impact on breast tumor lesions may be because only a single inhibitor chemotype, at a single dose, with one dosing regimen was examined. This inhibition may not be sufficient to disrupt estrogen regeneration in the GI tract that has been hypothesized to be reabsorbed and trafficked to the developing tumors in breast tissues. Future studies could focus on variabilities in dosing and regimen to answer this question more completely. However, ultimately, the inability to combat breast tumor formation in the PyMT model is most likely the consequence of the complexity of breast tumor formation.
There is increasing evidence from epidemiological, animal, and in vitro studies that endogenous estrogens are involved in breast carcinogenesis (34, 35). As such, steroid hormone biosynthetic pathways have been under investigation for decades, and there is abundant information on their metabolism in humans. Cytochromes P450, UGTs, sulfotransferases, and catechol-O-methyltransferases are just a few of the major enzymatic families that metabolize estrone, for example (37–41). Some of the major metabolites of estrone are highlighted in Fig. 7. It is possible that estrone and estradiol are first glucuronidated in the liver and, after being transported to the gut, are immediately acted on by GUS enzymes. These GUS enzymes then reactivate the parent estrogens and allow them to be further metabolized into methoxy-estrogens, semiquinones, quinones, etc. Such considerations might explain why targeting GUS has no impact on tumorigenesis in the PyMT model. However, it must be noted that sulfonation and hydroxylation play much larger roles in human metabolism than glucuronidation does. The action of sulfatase enzymes is perhaps more likely the preferred deconjugation step of estrogenic metabolites in the gut. Future studies will be required to evaluate the role of sulfatases in gut estrogen metabolism.
Figure 7.

Estrone metabolism. Although many metabolites are possible, this study focuses on the deconjugation of estrone-3-glucuronide to estrone. In addition to being glucuronidated, estrone can be sulfonated, hydroxylated, reduced, etc. to form metabolites that can be used at distal sites.
Further, the metabolites outlined in Fig. 7 do not include the incredible, diverse, and surprising amount of novel chemistry that occurs in the gastrointestinal tract. For example, the sterol scaffold–related bile acids are a good example of our growing understanding of the biotransformations that occur during mammalian-microbial symbiosis. Bile acids can be conjugated, deconjugated, hydrolyzed, epimerized, oxidized, methylated, etc., to play roles in absorption and digestion (42–45). Therefore, like bile acids, it may be that endogenous estrogens are transformed and repurposed for use in other areas of the body, like distal mucosal or receptor sites. We therefore propose that the estrobolome acts as an estrogen reservoir in the gut and is capable of creating estrogenic metabolites for local and nonlocal functions, rather than only simple reactivation. Future studies with other model systems and deeper bioanalytical investigations will be required to unravel how gut estrogen metabolism may affect hormone-dependent tumor growth in vivo. Taken together, these observations support the conclusion that the estrobolome's effects on breast cancer development are likely complex and multivariant.
Conclusion
Guided by initial data with 35 gut microbial GUS enzymes and then by the subsequent detailed analysis of 14 GUS proteins, including the resolution of relevant novel crystal structures, we have pinpointed molecular characteristics that contribute to estrogen deconjugation. These are key initial data to validate the fact that gut microbial GUS enzymes are functional members of the estrobolome. Furthermore, we postulate that the gut may serve as a reservoir for estrogenic metabolites capable of acting locally and perhaps distantly in systemic homeostasis and the development of disease.
Experimental procedures
Gene synthesis, expression, and purification of GUSs
All genes were codon-optimized for heterologous expression in E. coli, synthesized by BioBasic, and incorporated into a pLIC-His vector via ligation-independent cloning, and resultant plasmids were transformed into BL21-G E. coli cells. Glycerol stocks were made from overnight cultures and snap-frozen and stored at −80 °C. Verification of successful transformation and sample integrity were determined by DNA sequencing.
Cultures of 100 ml of LB with ampicillin were inoculated with glycerol stock and incubated overnight at 37 °C with shaking at 225 rpm. For protein expression, 50 ml of the overnight culture, ∼40 μl of Antifoam 204, and 750 μl of 2000× ampicillin were added to 1.5 liter of LB in a 2.5-liter Erlenmeyer flask and incubated at 37 °C at 225 rpm. At an optical density of ∼0.6, the temperature was reduced to 18 °C, and the culture was induced with isopropyl 1-thio-β-d-galactopyranoside (100 μm) and incubated overnight with shaking at 225 rpm. Cultures were spun down in a Sorvall Instruments RC-3B centrifuge at 4500 × g for 25 min in 1-liter round, flat-bottom plastic bottles. Cultures were resuspended in LB and transferred to a 50-ml Falcon tube and spun down in a Thermo Scientific Sorvall ST 40R centrifuge for 15 min at 5000 × g. Supernatant was discarded, and cell pellets were stored at −80 °C until purification.
Cell pellets were lysed in 30 ml of Nickel A buffer (20 mm KH2PO4, 500 mm NaCl, 50 mm imidazole (pH 7.4)) with DNase, lysozyme, and a Roche EDTA-free protease inhibitor tablet. The resultant cell slurry was sonicated on a Fisher sonic dismembrator model 500 twice with 1-s pulses for 1.5 min. The resultant lysate was subsequently spun down on a Beckman Coulter J2-HC centrifuge for 1 h at 17,000 rpm. The supernatant was subject to filtration with a 0.22-μm filter prior to purification.
Protein was first purified with an Aktaxpress FPLC (Amersham Bioscience) via a nickel-nitrilotriacetic acid column. Protein was eluted in one step using Nickel B buffer (20 mm KH2PO4, 500 mm NaCl, 500 mm imidazole (pH 7.4)). The eluent was then subject to size-exclusion chromatography on a HiLoadTM 16/60 Superdex 200 gel filtration column. Size exclusion buffer was utilized for elution (20 mm Hepes, 50 mm NaCl (pH 7.4)). Fractions were collected, and an SDS-polyacrylamide gel was performed to assess purity and stability of the enzyme. Protein concentration was determined on an ND-1000 spectrophotometer, and then cultures were snap-frozen in liquid nitrogen and stored at −80 °C.
Site-directed mutagenesis of GUSs
All mutants were created via site-directed mutagenesis. Mutagenesis primers were synthesized by Integrated DNA Technologies. Mutant plasmids were sequenced by Eton Bioscience to confirm successful mutagenesis (Table S4).
SSN construction
The sequence similarity network diagram of GUS enzyme sequences was generated using the Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST) online tool (26). The sequences obtained from the GUS rubric were used in combination with the EFI-EST “FASTA” tool to create a sequence with 279 nodes. Each node represents sequences bearing ≥90% sequence identity to each other. A BLAST E-value of 1 × 10−220 was employed.
In vitro estrogen-processing coupled assay
E1-3G and E2-17G were purchased as solids (Toronto Research Chemicals, North York, Ontario, Canada) and resuspended in DMSO to a concentration of 10 mm. In vitro assays were conducted in a 50-μl total volume. Reactions consisted of 5 μl of uronate dehydrogenase (1 μm final), 5 μl of enzyme (30 nm final), 10 μl of NAD+ (2 mm final), and 30 μl of E1-3G or E2-17G (500 μm final), all diluted in assay buffer (50 mm HEPES, 50 mm NaCl, various pH) (Fig. S1) (47, 48). The pH of each reaction was chosen based on the optimal pH determined for each GUS with pNPG (28). Reactions were incubated at 37 °C for 30 min, after which reactions were quenched with an equivalent volume of sodium carbonate. Final absorbance was monitored at 340 nm in a BMG Labtech PHERAstar plate reader. Enzyme was considered to process substrate if the final absorbance monitored was at least twice that of the minus enzyme control. Data were calculated from the average absorbance of three biological replicates ± S.E.
Manual docking of estrogens in PyMOL
Estrone and estradiol were accessed from the PDB in previously solved crystal structures. These were then imported into PyMOL and manually aligned to the GlcA-bound BuGUS-1 structure (PDB code 6DW6) with the three-button editing tool. After manual alignment of the sugar monosaccharides, structures of the Loop 1–, mini-Loop 1–, and FMN-binding GUS enzymes were aligned to the monomer of BuGUS-1. Visual inspection and final figures after alignment were generated in PyMOL.
In vitro HPLC estrogen-processing assay
E1-3G and E2-17G were purchased as solids (Toronto Research Chemicals) and resuspended in DMSO to a concentration of 10 mm. In vitro assays were conducted at 37 °C in a 50-μl total volume. Reactions consisted of 10 μl of assay buffer (50 mm HEPES, 50 mm NaCl, various pH), 10 μl of enzyme (various concentrations), and 30 μl of E1-3G or E2-17G (various concentrations) diluted in assay buffer. The pH of each reaction was chosen based on the optimal pH determined for each GUS with pNPG (28). Reactions were quenched at six time intervals with 50 μl of 25% trichloroacetic acid. After centrifugation at 13,000 × g for 10 min, the resultant supernatant was subjected to HPLC analysis. The concentration of E1-3G or E2-17G remaining at each time point was quantified on an Agilent 1260 Infinity II liquid chromatograph system. Samples were separated on an Agilent InfinityLab Poroshell 120 C18 column (4.6 × 100 mm, 2.7-μm particle size) at 38 °C. The flow rate was 0.9 ml/min, and the injection volume was 10 μl. LC conditions were set at 98% water with 0.1% formic acid (A) for 2 min and then ramped linearly over 10 min to 98% acetonitrile with 0.1% formic acid (B) and held until 14 min. At 15 min, the gradient was switched back to 100% A and allowed to re-equilibrate until 17 min. E1 and E2 were monitored at 280 nm. The concentration of E1 and E2 were determined from a standard curve (0–500 μm E1-3G/E2-17G in assay buffer). Resultant progress curves were fit by a custom linear regression analysis program in MATLAB. Initial velocities were then plotted against substrate concentration and fit with linear regression in Microsoft Excel to determine catalytic efficiency (kcat/Km). Control reactions were performed in which enzyme was substituted with buffer. Background hydrolysis was not observed at each pH tested. Reactions were performed in triplicate for each enzyme. Data were calculated from the average absorbance of three biological replicates ± S.E.
Crystallization and structure determination
Both structures were derived from crystals grown at 20 °C via the sitting-drop method in Hampton Research 3-well Midi Crystallization Plates (Swissci) by an Art Robbins Instruments Crystal Phoenix robot with the following drop conditions. For F. prausnitzii GUS, 100 nl of 10 mg/ml protein were added to 100 nl of 0.1 m BisTris-HCl, pH 8.5, and 1.8 m magnesium sulfate. For uncultured Clostridium sp. GUS, 100 nl of 12 mg/ml protein were added to 100 nl of 0.2 m calcium chloride, 0.1 m Tris-HCl, pH 8.5, and 20% (w/v) PEG 4000.
Crystal specimens were cryo-protected in the crystallization conditions as described above with the addition of 20% glycerol, and diffraction data were collected at 100 K on APS Beamline 23-ID-D. The data were processed with XDS, and both structures were solved via molecular replacement in Phenix (46) using the C. perfringens GUS structure (PDB code 4JKM) as a search model. The LLG and TFZ scores were 4147 and 47.1, respectively, for F. prausnitzii GUS. The LLG and TFZ scores were 6724 and 67.1, respectively, for the uncultured Clostridium sp. GUS. The resulting starting model and maps from molecular replacement were then employed in the AutoBuild function of Phenix. Structures were refined in Phenix and visually inspected and manually built using COOT (36). Final PDB coordinates for all structures have been deposited to the RCSB Protein Data Bank with corresponding PDB codes 6U7I and 6U7J for F. prausnitzii and uncultured Clostridium sp., respectively.
In vitro inhibition assay
Reactions consisted of 10 μl of GUS (15 nm final), 5 μl of inhibitor (1 μm final for inhibitor UNC10201652 and 10 μm final for all other inhibitors), 30 μl of estrone-3-glucuronide or estradiol-17-glucuronide (900 μm final), and 10 μl of assay buffer (25 mm NaCl, 25 mm HEPES, pH 7.4 final). Reactions were initiated by the addition of pNPG and then incubated for 1 h, after which the end-point absorbance was determined. Final percentage inhibition was determined via HPLC as described above.
In-cell inhibition assay
WT E. coli K-12 MG1655 was grown overnight in 10 ml of LB, and a 100-μl portion was subcultured the following morning in 5 ml of fresh LB. Cells were grown to an optical density of ∼0.6 and used for the cell-based assay. Reactions were carried out in Costar 96-well black clear-bottom plates. Reaction volumes consisted of 90 μl of cells premixed with 700 μm estrone-3-glucuronide or estradiol-17-glucuronide and various concentrations of 10 μl of inhibitor. This reaction was incubated for 24 h at 37 °C with a low evaporation lid. Incubations were quenched by the addition of 50 μl of 0.2 m sodium carbonate. Absorbance values were measured at 410 nm in a BMG Labtech PHERAstar plate reader. Percentage inhibition and EC50 values were determined as described previously (29).
In fimo estrogen deconjugation and inhibition
All animal studies were approved by the University of North Carolina Institutional Animal Care and Use Committee, in accordance with the Care and Use of Laboratory Animals guidelines set by the National Institutes of Health. BALB/c mice were individually housed in specific pathogen-free conditions with sterile ventilator cages containing corn bedding, with ad libitum access to chow and water. Fecal pellets were collected from each mouse shortly by gentle abdominal palpation and snap-frozen in sterile microcentrifuge tubes. To perform the assay, frozen fecal samples were rehydrated in 15× assay buffer (w/v; 20 mm HEPES, 50 mm NaCl, pH 6.5, 1× Complete® Protease inhibitor mixture (Roche Applied Science). Bacterial cells were lysed using a Tissuelyzer II (Qiagen) for 2 min at 30 Hz. Homogenate was sonicated for 4 min and then clarified by centrifugation for 5 min at 13,000 × g. All experimental manipulation until this point occurred at 4 °C. 0.1 mg/ml protein from the fecal slurry supernatant was used to initiate the hydrolysis reaction of 1 mm estrone-3-glucuronide or estradiol-17-glucuronide resuspended in the same buffer. Parallel reactions containing 10 μm inhibitor were used to determine percentage inhibition; additionally, only estrogen or only buffer/fecal slurry were used as negative controls. An aliquot of each sample was heat-inactivated at 95 °C and used in the assay for further background establishment. Each sample was assayed using two technical replicates with the HPLC protocol above. The total amount of estrogen produced was calculated from a standard curve and then normalized to the total fecal protein content calculated using a standard Bradford assay.
UNC10201652 GUS inhibitor administration in PyMT mouse model
All animal studies were completed in accordance with the Care and Use of Laboratory Animals guidelines set by the National Institutes of Health. PyMT males, a gift from the Jeffery Pollard laboratory, and FVB females were put in one cage for genotyping pups. Males are kept for continuous breeding with FVB females. PyMT females were used for UNC10201652 assays. Two groups of 11 mice each were individually housed in a specific pathogen–free vivarium maintained with a 12-h/12-h light/dark cycle, in specific pathogen–free conditions with sterile ventilator cages containing corn bedding, with ad libitum access to chow and water. Beginning at 4 weeks old, female PyMT mice were gavaged with UNC10201652 for a final concentration of 100 μl/20 μg/mouse (final DMSO 0.67%) every day except weekends for 9 weeks. On the last day, animals were deeply anesthetized with CO2 followed by manual cervical dislocation to collect samples for histology analysis and evaluation of hyperplasia and adenoma in breast, liver, lung, and kidney tissue.
Author contributions
S. M. E. and H. L. data curation; S. M. E. formal analysis; S. M. E. investigation; S. M. E., H. L., and L. L. methodology; S. M. E. writing-original draft; L. R. R., X. L., and S. M. funding acquisition; S. M. E. supervision; S. M. E. and M. R. R. project administration; M. R. R. conceptualization; M. R. R. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the following core services for assistance in generating the data: Cancer Center Grant P30CA013330 (Principal Investigator: David Goldman), the Institute for Animal Studies (Officers: Sunder Shrestha, Jorge Larocca, Linda Jelicks, and Lawrence Herbst), and the Histology and Comparative Pathology Facility of the Albert Einstein College of Medicine (Director: Amanda P. Beck) (Bronx, NY).
This work was supported by National Institutes of Health Grants CA098468 and CA207416 (to M. R. R.), the Merck Exploratory Science Center (to M. R. R.), and National Science Foundation Graduate Research Fellowship Program Grant DGS-1650116 (to S. M. E.). This animal study was fully supported by the Department of Defense, United States Army Medical Research and Materiel Command Congressionally Directed Medical Research Programs, 2016 Breast Cancer Research Program Breakthrough Award–Funding Level 1 (CDMRP Log BC161093; GRANT12158914; Award W81XWH-17-1-0023) (Principal Investigator S. M.). Matthew R. Redinbo is a Founder of Symberix, Inc., which is developing microbiome-targeted therapeutics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S4 and Figs. S1–S5.
The atomic coordinates and structure factors (codes 6U7I and 6U7J) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GI
- gastrointestinal
- GUS
- β-glucuronidase
- SSN
- sequence similarity network
- HMP
- human microbiome project
- UGT
- UDP-glucuronosyltransferase
- E1-3G
- estrone-3-glucuronide
- E2-17G
- estradiol-17-glucuronide
- pNPG
- 4-nitrophenyl β-d-glucuronide
- HR +
- hormone receptor–positive
- LB
- lysogeny broth
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- NSAID
- nonsteroidal anti-inflammatory drug.
References
- 1. Flint H. J., Scott K. P., Duncan S. H., Louis P., and Forano E. (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306 10.4161/gmic.19897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flint H. J., Duncan S. H., Scott K. P., and Louis P. (2007) Interactions and competition within the microbial community of the human colon: links between diet and health. Environ. Microbiol. 9, 1101–1111 10.1111/j.1462-2920.2007.01281.x [DOI] [PubMed] [Google Scholar]
- 3. LeBlanc J. G., Milani C., de Giori G. S., Sesma F., van Sinderen D., and Ventura M. (2013) Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr. Opin. Biotechnol. 24, 160–168 10.1016/j.copbio.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 4. Koh A., De Vadder F., Kovatcheva-Datchary P., and Bäckhed F. (2016) From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 10.1016/j.cell.2016.05.041 [DOI] [PubMed] [Google Scholar]
- 5. Plottel C. S., and Blaser M. J. (2011) Microbiome and malignancy. Cell Host Microbe 10, 324–335 10.1016/j.chom.2011.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adams S. (2016) Estrobolome disparities may lead to developing bimoarkers that could mitigate cancer risk. J. Natl. Cancer Inst. 108, djw130 10.1093/jnci/djw130 [DOI] [Google Scholar]
- 7. Baker J. M., Al-Nakkash L., and Herbst-Kralovetz M. M. (2017) Estrogen–gut microbiome axis: physiological and clinical implications. Maturitas 103, 45–53 10.1016/j.maturitas.2017.06.025 [DOI] [PubMed] [Google Scholar]
- 8. Chen K. L., and Madak-Erdogan Z. (2016) Estrogen and microbiota crosstalk: should we pay attention? Trends Endocrinol. Metab. 27, 752–755 10.1016/j.tem.2016.08.001 [DOI] [PubMed] [Google Scholar]
- 9. Wallace B. D., Roberts A. B., Pollet R. M., Ingle J. D., Biernat K. A., Pellock S. J., Venkatesh M. K., Guthrie L., O'Neal S. K., Robinson S. J., Dollinger M., Figueroa E., McShane S. R., Cohen R. D., Jin J., et al. (2015) Structure and inhibition of microbiome β-glucuronidases essential to the alleviation of cancer drug toxicity. Chem. Biol. 22, 1238–1249 10.1016/j.chembiol.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts A. B., Wallace B. D., Venkatesh M. K., Mani S., and Redinbo M. R. (2013) Molecular insights into microbial β-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 84, 208–217 10.1124/mol.113.085852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wallace B. D., Wang H., Lane K. T., Scott J. E., Orans J., Koo J. S., Venkatesh M., Jobin C., Yeh L. A., Mani S., and Redinbo M. R. (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 10.1126/science.1191175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takasuna K., Hagiwara T., Hirohashi M., Kato M., Nomura M., Nagai E., Yokoi T., and Kamataki T. (1996) Involvement of β-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 56, 3752–3757 [PubMed] [Google Scholar]
- 13. LoGuidice A., Wallace B. D., Bendel L., Redinbo M. R., and Boelsterli U. A. (2012) Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J. Pharmacol. Exp. Ther. 341, 447–454 10.1124/jpet.111.191122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saitta K. S., Zhang C., Lee K. K., Fujimoto K., Redinbo M. R., and Boelsterli U. A. (2014) Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44, 28–35 10.3109/00498254.2013.811314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boelsterli U. A., Redinbo M. R., and Saitta K. S. (2013) Multiple NSAID-induced hits injure the small intestine: underlying mechanisms and novel strategies. Toxicol. Sci. 131, 654–667 10.1093/toxsci/kfs310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boelsterli U. A., and Ramirez-Alcantara V. (2011) NSAID acyl glucuronides and enteropathy. Curr. Drug Metab. 12, 245–252 10.2174/138920011795101877 [DOI] [PubMed] [Google Scholar]
- 17. Allison M. C., Howatson A. G., Torrance C. J., Lee F. D., and Russell R. I. (1992) Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 327, 749–754 10.1056/NEJM199209103271101 [DOI] [PubMed] [Google Scholar]
- 18. Sher A., and Rahman M. A. (2000) Enterohepatic recycling of estrogen and its relevance with female fertility. Arch. Pharmacol. Res. 23, 513–517 10.1007/BF02976582 [DOI] [PubMed] [Google Scholar]
- 19. Vree T. B., and Timmer C. J. (1998) Enterohepatic cycling and pharmacokinetics of oestradiol in postmenopausal women. J. Pharm. Pharmacol. 50, 857–864 10.1111/j.2042-7158.1998.tb04000.x [DOI] [PubMed] [Google Scholar]
- 20. Travis R. C., and Key T. J. (2003) Oestrogen exposure and breast cancer risk. Breast Cancer Res. 5, 239–247 10.1186/bcr628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Key T. J., and Pike M. C. (1988) The role of oestrogens and progestagens in the epidemiology and prevention of breast cancer. Eur. J. Cancer Clin. Oncol. 24, 29–43 10.1016/0277-5379(88)90173-3 [DOI] [PubMed] [Google Scholar]
- 22. Roe F. J. (1979) Aetiology of breast cancer: a brief review. Invest. Cell Pathol. 2, 45–53 [PubMed] [Google Scholar]
- 23. Kaaks R., Rinaldi S., Key T. J., Berrino F., Peeters P. H. M., Biessy C., Dossus L., Lukanova A., Bingham S., Khaw K. T., Allen N. E., Bueno-de-Mesquita H. B., van Gils C. H., Grobbee D., Boeing H., et al. (2005) Postmenopausal serum androgens, oestrogens and breast cancer risk: the European prospective investigation into cancer and nutrition. Endocr. Relat. Cancer 12, 1071–1082 10.1677/erc.1.01038 [DOI] [PubMed] [Google Scholar]
- 24. Little M. S., Pellock S. J., Walton W. G., Tripathy A., and Redinbo M. R. (2018) Structural basis for the regulation of β-glucuronidase expression by human gut Enterobacteriaceae. Proc. Natl. Acad. Sci. U.S.A. 115, E152–E161 10.1073/pnas.1716241115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pellock S. J., Walton W. G., Biernat K. A., Torres-Rivera D., Creekmore B. C., Xu Y., Liu J., Tripathy A., Stewart L. J., and Redinbo M. R. (2018) Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biol. Chem. 293, 18559–18573 10.1074/jbc.RA118.005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerlt J. A., Bouvier J. T., Davidson D. B., Imker H. J., Sadkhin B., Slater D. R., and Whalen K. L. (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 10.1016/j.bbapap.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pollet R. M., D'Agostino E. H., Walton W. G., Xu Y., Little M. S., Biernat K. A., Pellock S. J., Patterson L. M., Creekmore B. C., Isenberg H. N., Bahethi R. R., Bhatt A. P., Liu J., Gharaibeh R. Z., and Redinbo M. R. (2017) An atlas of β-glucuronidases in the human intestinal microbiome. Structure 25, 967–977.e5 10.1016/j.str.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biernat K. A., Pellock S. J., Bhatt A. P., Bivins M. M., Walton W. G., Tran B. N. T., Wei L., Snider M. C., Cesmat A. P., Tripathy A., Erie D. A., and Redinbo M. R. (2019) Structure, function, and inhibition of drug reactivating human gut microbial β-glucuronidases. Sci. Rep. 9, 825 10.1038/s41598-018-36069-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pellock S. J., Creekmore B. C., Walton W. G., Mehta N., Biernat K. A., Cesmat A. P., Ariyarathna Y., Dunn Z. D., Li B., Jin J., James L. I., and Redinbo M. R. (2018) Gut microbial β-glucuronidase inhibition via catalytic cycle interception. ACS Cent. Sci. 4, 868–879 10.1021/acscentsci.8b00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pellock S. J., Walton W. G., Ervin S. M., Torres-Rivera D., Creekmore B. C., Bergan G., Dunn Z. D., Li B., Tripathy A., and Redinbo M. R. (2019) Discovery and characterization of FMN-binding β-glucuronidases in the human gut microbiome. J. Mol. Biol. 431, 970–980 10.1016/j.jmb.2019.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fantozzi A., and Christofori G. (2006) Mouse models of breast cancer metastasis. Breast Cancer Res. 8, 212 10.1186/bcr1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weigelt B., Peterse J. L., and van 't Veer L. J. (2005). Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591–602 10.1038/nrc1670 [DOI] [PubMed] [Google Scholar]
- 33. Lin E. Y., Jones J. G., Li P., Zhu L., Whitney K. D., Muller W. J., and Pollard J. W. (2003) Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 163, 2113–2126 10.1016/S0002-9440(10)63568-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kwa M., Plottel C. S., Blaser M. J., and Adams S. (2016) The intestinal microbiome and estrogen receptor–positive female breast cancer. J. Natl. Cancer Inst. 108, 10.1093/jnci/djw029 10.1093/jnci/djw029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Samavat H., and Kurzer M. S. (2015) Estrogen metabolism and breast cancer. Cancer Lett. 356, 231–243 10.1016/j.canlet.2014.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 37. Abel E. L., Lyon R. P., Bammler T. K., Verlinde C. L. M. J., Lau S. S., Monks T. J., and Eaton D. L. (2004) Estradiol metabolites as isoform-specific inhibitors of human glutathione S-transferases. Chem. Biol. Interact. 151, 21–32 10.1016/j.cbi.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 38. Rogan E. G., Badawi A. F., Devanesan P. D., Meza J. L., Edney J. A., West W. W., Higginbotham S. M., and Cavalieri E. L. (2003) Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: potential biomarkers of susceptibility to cancer. Carcinogenesis 24, 697–702 10.1093/carcin/bgg004 [DOI] [PubMed] [Google Scholar]
- 39. Raftogianis R., Creveling C., Weinshilboum R., and Weisz J. (2000) Chapter 6: Estrogen metabolism by conjugation. J. Natl. Cancer Inst. Monogr. 2000, 113–124 10.1093/oxfordjournals.jncimonographs.a024234 [DOI] [PubMed] [Google Scholar]
- 40. Martucci C. P., and Fishman J. (1993) P450 enzymes of estrogen metabolism. Pharmacol. Ther. 57, 237–257 10.1016/0163-7258(93)90057-K [DOI] [PubMed] [Google Scholar]
- 41. Zhu B. T., and Conney A. H. (1998) Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis 19, 1–27 10.1093/carcin/19.1.1 [DOI] [PubMed] [Google Scholar]
- 42. Chiang J. Y. L. (2013) Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212 10.1002/cphy.c120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Molinaro A., Wahlström A., and Marschall H.-U. (2018) Role of bile acids in metabolic control. Trends Endocrinol. Metab. 29, 31–41 10.1016/j.tem.2017.11.002 [DOI] [PubMed] [Google Scholar]
- 44. Hofmann A. F., and Hagey L. R. (2008) Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65, 2461–2483 10.1007/s00018-008-7568-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chiang J. Y. L. (2009) Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966 10.1194/jlr.R900010-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 47. Little M. S., Ervin S. M., Walton W. G., Tripathy A., Xu Y., Liu J., and Redinbo M. R. (2018). Active site flexibility revealed in crystal structures of Parabacteroides merdae β-glucuronidase from the human gut microbiome. Protein Sci. 27, 2010–2022 10.1002/pro.3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pellock S. J., Walton W. G., and Redinbo M. R. (2019). Selecting a Single Stereocenter: The Molecular Nuances That Differentiate β-Hexuronidases in the Human Gut Microbiome. Biochemistry. 58, 1311–1317 10.1021/acs.biochem.8b01285 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.