

Abstract
Current therapies for autoimmunity cause significant morbidity and mortality. Adoptive immunotherapy using genetically engineered T cells has led to durable remissions of B cell leukemias and lymphomas, raising the question of whether the approach can be modified to target autoreactive B and T cells to induce durable remissions of autoimmunity. Here we review antigen-specific approaches to modify immune cells to treat autoimmune disease. We focus on recent studies that aim to eliminate or suppress autoimmunity by targeting the disease-causing B or T cells through their B cell receptor or T cell receptor specificities.
Introduction
Autoimmunity results from a mistaken attack by the immune system towards self, resulting in tissue damage. In some cases, organ destruction is the inevitable outcome, requiring lifelong treatments such as insulin or thyroid hormone to replace lost function. In other cases, chronic immunosuppression is required for disease control, which risks life-threatening infections and malignancies. Preferably, therapy for autoimmune diseases would spare protective immunity while specifically targeting pathogenic autoreactive cells.
Over the last decade, the introduction of chimeric antigen receptor (CAR) T cells into clinical practice has revolutionized the treatment of hematologic malignancies [1]. In pivotal trials, T cells isolated from patients with B cell leukemias and lymphomas and genetically modified ex vivo with an anti-CD19 antibody-based immunoreceptor have been shown to activate and expand after re-infusion, causing potent cytolysis of CD19-expressing B cells and long-term remissions of previously refractory cancers [2–6]. The durability of remission has been attributed to the persistence of CAR T cells, which provides ongoing surveillance against cancer recurrence. The ability of CAR T cells to induce durable remission of B cell cancers has spurred interest in engineering T cells to target autoimmune B and T cells, which by analogy holds potential to induce lasting remissions of autoimmunity. In this review, we focus on recent approaches to genetically modify T cells to target pathogenic autoimmune cell populations.
Eliminating autoreactive B cells using autoantigen-based immunoreceptors
Autoreactive lymphocytes are defined by their reactivity against autoantigen, which provides a distinct target for the design of cellular therapies. Our group recently developed chimeric autoantibody receptor (CAAR) T cells that express the autoantigen fragment targeted in the blistering disease pemphigus vulgaris (PV), desmoglein 3 (DSG3), as the extracellular domain of an immunoreceptor, linked to intracellular CD137 and CD3ζ signaling domains. DSG3 CAAR T cells target autoreactive naïve and memory B cells that express anti-DSG3 B cell receptors (BCRs), and indirectly target anti-DSG3 short-lived plasma cells that replenish from the memory B cell pool. The anti-CD20 antibody rituximab can reduce serum autoantibodies to DSG3 to the normal range [7], indicating most if not all of the autoantibodies in PV are produced from short-lived plasma cells. Thus, targeting anti-DSG3 BCRs with a DSG3 CAAR provides a strategy to specifically and potentially durably eliminate DSG3-reactive B cells and their progeny. In preclinical studies, DSG3 CAAR T cells demonstrated specific killing of anti-DSG3 B cells in vitro and induced histologic and serologic remission in a PV mouse hybridoma model [8]. Soluble anti-DSG3 antibodies can either reduce or potentiate CAAR-mediated cytolysis and can also activate DSG3 CAAR T cells to proliferate and produce interferon gamma, suggesting that their presence may partially inhibit but should not prevent DSG3 CAAR T cell function. DSG3 CAAR T cells did not cause cytotoxicity against keratinocytes that express the native desmosomal binding partners of DSG3, either in vitro or in human skin xenografts, which was postulated to be due to low binding affinity and/or suboptimal intercellular distance for cytolysis. Collectively these studies suggested that CAAR T cells can induce antigen-specific B cell depletion and that the CAAR platform could prove to be valuable for other antibody-mediated diseases.
Other antigens have since been used to create CAARs, highlighting the applicability of the approach to both auto- and alloantibody-mediated diseases. T cells expressing a CAAR (termed a B cell antibody receptor (BAR) by the authors) comprised of the A2 or C2 domain of coagulation factor VIII (FVIII) have been engineered [9]; these FVIII BARs target autoreactive B cells that are a major source of morbidity in hemophilia A patients after FVIII replacement therapy due to acquired resistance to FVIII. Expressed in mouse and human T cells, FVIII BARs directed T cell cytolysis against A2- and C2-specific hybridoma cells in vitro and in NSG mice in vivo. Furthermore, they prevented the development of FVIII-reactive IgM B cells after polyclonal stimulation of mouse splenocytes with lipopolysaccharide in vitro and the induction of IgG alloantibodies in FVIII-immunized hemophilic mice.
In addition, La/SSB CAARs have been described [10]; expressed in the natural killer cell line NK92MI, these immunoreceptors were active in vitro against Jurkat cells expressing anti-La BCRs and against La-reactive human B cells in vitro. La/SSB autoantibodies are associated with Sjogren’s syndrome and systemic lupus erythematosus (SLE) and cause neonatal lupus pathology in mice after placental transfer [11]. Although La/SSB autoantibodies do not account for the full spectrum of symptoms observed in Sjogren’s or SLE and are rarely found in the absence of the main pathogenic autoantibodies targeting the Ro/SSA autoantigen in neonatal lupus, elimination of La-reactive B cells could represent part of a therapeutic approach for neonatal lupus.
Targeting autoreactive T cells with p:MHCI immunoreceptors
Autoreactive T cells are the major mediators of disease in several autoimmune conditions, either through direct cytotoxic damage or the production of cytokines that cause inflammation and activate downstream immune responses. Autoreactive T cells are activated through their T cell receptor (TCR) by cognate peptide bound to MHC (MHCI and MHCII for CD8 and CD4 T cells, respectively). MHCI is expressed on nearly all nucleated mammalian cells, whereas MHCII is primarily expressed by specialized antigen-presenting cells (APCs).
In human type 1 diabetes (T1D), CD8 T cells recognize peptides derived from a variety of pancreatic β cell antigens, including proinsulin, GAD65, IA-2, IGRP, ZNT8, and others [12], many of which are also targeted by cytotoxic lymphocytes (CTLs) in the non-obese diabetic (NOD) mouse. In NOD mice, CTLs against proinsulin appear early and are required for expansion of IGRP-reactive CTLs and subsequent diabetes onset [13,14]. Transgenic mice expressing a chimeric immunoreceptor consisting of an insulin peptide linked to mouse β2-microglobulin and CD3ζ (InsCD3ζ) developed diabetes at similar rates as nontransgenic mice [15]. However, activation of InsCD3ζ T cells ex vivo followed by infusion into NOD mice resulted in lower incidence and delayed onset of diabetes [16]. Subsequently, immunoreceptors encoding a chimera of insulin or IGRP peptide linked to human β2-microglobulin and the mouse CD3ζ cytoplasmic domain were engineered to force presentation of linked peptides in the context of murine H-2Kd MHCI upon its assembly with β2-microglobulin. Gene-engineered mouse T cells expressed the chimeric peptide-β2-microglobulin receptor in the context of MHCI (p:MHCI) after RNA electroporation and demonstrated in vitro killing of insulin and IGRP-reactive CTLs. In NOD mice, reduced incidence and delayed onset of diabetes was induced by insulin but not IGRP p:MHCI T cells [17]. Although the diversity of self-peptides recognized by autoreactive T cells in human patients may complicate the implementation of such a therapeutic approach, these experiments provide proof of concept for antigen-specific targeting of autoreactive CD8 T cells and suggest that preventive adoptive immunotherapy could protect high-risk individuals from developing diabetes.
Engineering antibody-based immunoreceptors against p:MHCII to target antigen presenting cells
Whereas CD8 cytotoxic T cells directly cause tissue destruction in diseases such as T1D, autoreactive CD4 T cells can promote autoimmunity by secreting cytokines that can promote inflammation and activate humoral immune responses. Because CD4 T cells are activated by peptide bound to MHCII (p:MHCII) on APCs, depletion of APCs through an antibody-based receptor targeting p:MHCII is a strategy to abrogate CD4 T cell-mediated autoimmunity [18]. A monoclonal antibody (mAb287) against the insulin B chain 9–23 peptide in the context of I-Ag7 MHCII has been cloned and characterized. This pre-pro-insulin peptide is critical for development of diabetes in NOD mice, as a single mutation in position 16 protects mice from diabetes onset [19]. MAb287 CAR T cells caused specific cytolysis of APCs presenting the insulin self-peptide in vitro. A single infusion of mAb287 CAR T cells significantly delayed the onset of diabetes in NOD mice, but CAR T cells did not persist past 10–15 weeks in vivo, resulting in a similar overall incidence of diabetes by 30 weeks compared to control mice. Future work in the field would need to focus on cloning and characterizing antibodies specific for the peptide-binding pocket of the MHCII bound to self-peptides, which can be technically challenging [20,21] due to the diversity of self-peptides and MHC haplotypes involved in disease.
Directing regulatory T cells with antigen-specific immunoreceptors to suppress autoreactive B and/or T cells
While the aforementioned strategies rely on cytotoxic T cells to eliminate autoreactive target cells, similar approaches have been employed using regulatory T cells (Tregs) to suppress autoreactive immune responses. Tregs prevent autoimmunity in healthy individuals through a variety of mechanisms, including downregulation of costimulatory molecules on APCs by CTLA-4 or secretion of inhibitory mediators such as IL-10, TGFβ, or adenosine, among others [22,23]. Because they do not lyse target cells, Tregs should pose less risk for off-target cytotoxicity compared to gene-engineered conventional T cells, but could cause immunosuppression more broadly than intended due to secretion of soluble mediators.
Localizing Tregs with an antibody-based CAR to an antigen restricted to a certain anatomic compartment creates a spatially-confined immunosuppressive environment that could be beneficial in several autoimmune diseases. Tregs to suppress alloimmune responses to factor replacement therapy and in transplanted organs (via FVIII or HLA-A2) have previously been reviewed and will not be covered here [24–27]. CAR Tregs targeting carcinoembryonic antigen (CEA) were shown to accumulate in the colon and were able to suppress colitis in mouse models [28]. Intriguingly, they also prevented the development of colon carcinomas, likely by prevention of chronic inflammation. Similarly, CAR Tregs targeting myelin oligodendrocyte glycoprotein (MOG) migrated to the brain after intranasal delivery in mice, reduced neuroinflammation and disease symptoms in a mouse model of multiple sclerosis, and prevented subsequent disease flares upon re-challenge with autoantigen [29]. Recently, CAR Tregs targeting citrullinated vimentin, which is localized to the inflamed synovium of rheumatoid arthritis patients, have been engineered and pre-clinical studies are underway to test their efficacy in rheumatoid arthritis (C. Raffin et al., J Immunol May 1, 2018, 200 (1 Supplement) 176.17).
TCR-based Tregs with defined specificity have also been engineered. Retrovirally transduced Tregs expressing TCRs specific for ovalbumin (OTII Tregs) were evaluated in an animal model for antigen-induced arthritis, in which one joint was injected with methylated bovine serum albumin (mBSA) to induce joint inflammation, and the other joint was injected with mBSA and ovalbumin. OTII Tregs suppressed inflammation in joints where ovalbumin was present but not in mBSA-injected joints, indicating that spatially localized activation of Treg function can suppress immune reactions throughout the region where the antigen is present [30]. TCR-modified human Tregs for T1D have also been generated using human TCRs isolated from islet-specific T cell clones [31]. CD4 and CD8 T cell proliferation were suppressed in vitro even in the absence of cognate peptide, indicating non-specific immunosuppressive effects, but antigen-specific Treg suppression was enhanced after stimulation with cognate peptide. Future studies will need to identify a diversity of TCRs with appropriate affinity and MHC restriction, as well as ablate the endogenous TCR to prevent aberrant pairing of native and engineered TCR chains.
Challenges in translation to human clinical trials
In addition to technology-specific limitations described in each of the aforementioned sections, there may be general challenges associated with the development of cell therapies for the treatment of autoimmune diseases that warrants further discussion. If efficacious, a key consideration will be the durability of the response and associated safety of the therapy. For cell therapies involving both T cells in oncology [2–6] and Tregs in autoimmune disease [32–33], the association between cell persistence and durability of response is variable and may depend on the pre-conditioning regimen, target cell burden, antigen density, cell phenotype and homing in ways that are disease-specific. Administration of autologous T cells engineered to express surface autoantigen may cause a disease flare, if an unintended immune-stimulatory effect exceeds the expected suppressive or cytotoxic activity of the engineered cells. If observed in clinical testing, some solutions to these issues could include incorporating a “suicide switch” or strategies to induce selective receptor expression and/or activation, disrupting the expression of the endogenous TCR, and/or novel pre-conditioning regimens that may be less toxic than those currently used in oncology CAR T cell treatment protocols.
Conclusions
Recent advances in antigen-specific targeting of cellular immunotherapies have resulted in several novel approaches to eliminate or suppress autoreactive lymphocytes (summarized in Figure 1), strategies that may prevent autoimmune disease onset or avoid the morbidity and mortality associated with globally immunosuppressive regimens. Genetically-engineered immunotherapies to redirect T lymphocytes hold promise to achieve these therapeutic ideals, and may induce durable remissions of autoimmunity similar to results observed with B cell leukemias and lymphomas. Clinical trials are being designed to bring these technologies from bench to bedside, which will mark a new era in the development of autoimmune disease therapies.
Figure 1. Genetically engineered T cell therapeutic strategies for autoimmune disease.
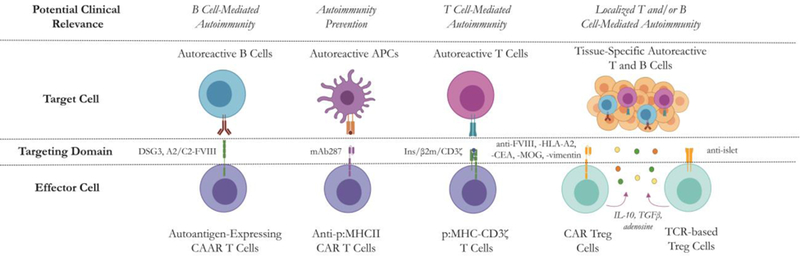
Chimeric autoantibody receptor (CAAR) T cells for antigen-specific B cell depletion, anti-peptide:MHC CAR T cells for autoreactive APC depletion, peptide:MHC immunoreceptor T cells for autoreactive T cell depletion and CAR- or TCR-Tregs for autoimmune B or T cell suppression are depicted. Figure created using BioRender.
Highlights.
T cells can be genetically modified to ablate or suppress autoreactive immune cells
Autoantigen-engineered T cells eliminate antigen-specific B cells
Peptide-MHCI directs T cell cytolysis toward autoreactive CD8 T cells
Antibodies to peptide-MHCII target autoreactive CD4 T cell responses
Regulatory T cells inhibit autoimmune B and T cell responses
Acknowledgements
This work was supported by R01-AR068288 (ASP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest statement
ASP: co-founder, equity, compensation, and grant funding from Cabaletta Bio, focused on targeted cellular immunotherapy of autoimmune diseases including pemphigus. Inventor on patents licensed by Cabaletta Bio and Novartis for cellular immunotherapy of autoimmune diseases. CTE: founding equity in Cabaletta Bio, inventor on patents licensed by Cabaletta Bio and Novartis. DKL: consultant, Cabaletta Bio. Co-founder with equity, NanoXCell Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.June CH, Sadelain M: Chimeric Antigen Receptor Therapy. N Engl J Med 2018, 379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jager U, Jaglowski S, Andreadis C, Westin JR, et al. : Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019, 380:45–56. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et al. : Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 2018, 378:449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. : Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018, 378:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. : Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017, 377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, Lin Y, Braunschweig I, Hill BT, Timmerman JM, et al. : Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 2019, 20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, Caillot F, Golinski ML, Labeille B, Picard-Dahan C, et al. : First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet 2017, 389:2031–2040. [DOI] [PubMed] [Google Scholar]
- **8.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, et al. : Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]; Engineering Dsg3 CAAR T cells, the authors demonstrate that these cells specifically eliminate Dsg3-specific B cell hybridomas in vitro and in vivo. This is the first publication to report proof of concept that autoantigen-based immunoreceptors can specifically eliminate autoreactive B cells as a potential therapeutic strategy for B cell-mediated autoimmune disease.
- **9.Parvathaneni K, Scott DW: Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv 2018, 2:2332–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]; Generating separate FVIII A2-BAR and C2-BAR T cells, the authors show in vitro and in vivo activity in both a hybridoma mouse model and active immune mouse model of hemophilia A inhibitors. This is the first published study to extend the concept of CAAR T cells to an alloimmune setting.
- 10.Meng H, Sun X, Song Y, Zou J, An G, Jin Z, Yang L: La/SSB chimeric autoantibody receptor modified NK92MI cells for targeted therapy of autoimmune disease. Clin Immunol 2018, 192:40–49. [DOI] [PubMed] [Google Scholar]
- 11.Tran HB, Macardle PJ, Hiscock J, Cavill D, Bradley J, Buyon JP, Gordon TP: Anti-La/SSB antibodies transported across the placenta bind apoptotic cells in fetal organs targeted in neonatal lupus. Arthritis Rheum 2002, 46:1572–1579. [DOI] [PubMed] [Google Scholar]
- 12.Pugliese A: Autoreactive T cells in type 1 diabetes. J Clin Invest 2017, 127:2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, Colman PG, Harrison LC, Lew AM, Thomas HE, et al. : Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006, 116:3258–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnamurthy B, Mariana L, Gellert SA, Colman PG, Harrison LC, Lew AM, Santamaria P, Thomas HE, Kay TW: Autoimmunity to both proinsulin and IGRP is required for diabetes in nonobese diabetic 8.3 TCR transgenic mice. J Immunol 2008, 180:4458–4464. [DOI] [PubMed] [Google Scholar]
- 15.Scott GS, Fishman S, Margalit A, Siew LK, Chapman S, Wen L, Gross G, Wong FS: Developing a novel model system to target insulin-reactive CD8 T cells. Ann N Y Acad Sci 2008, 1150:54–58. [DOI] [PubMed] [Google Scholar]
- 16.Scott GS, Fishman S, Khai Siew L, Margalit A, Chapman S, Chervonsky AV, Wen L, Gross G, Wong FS: Immunotargeting of insulin reactive CD8 T cells to prevent diabetes. J Autoimmun 2010, 35:390–397. [DOI] [PubMed] [Google Scholar]
- *17.Fishman S, Lewis MD, Siew LK, De Leenheer E, Kakabadse D, Davies J, Ziv D, Margalit A, Karin N, Gross G, et al. : Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol Ther 2017, 25:456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using electroporation of mRNA, the authors reprogrammed T cells with p:MHCI linked to CD3ζ, which specifically killed diabetogenic T cells in vitro and delayed onset of disease in a mouse model of diabetes. Although proof of concept for this approach was previously reported by the same group, this study is the first to leverage methods of genetic engineering ex vivo with partially humanized immunoreceptors to further demonstrate the feasibility of this therapeutic strategy.
- *18.Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, Miao D, Yu L, Pietropaolo M, Davidson HW: Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J Autoimmun 2019, 96:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]; Previous studies from the same group characterized a monoclonal antibody that is specific for insulin peptide in the context of MHCII. Converting these properties into a CAR construct that has the potential to specifically and durably target diabetogenic APCs better than the antibody alone, the authors demonstrate that mAb287 CAR T cells delay the onset of disease in a mouse model of diabetes.
- 19.Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, et al. : Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005, 435:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahan R, Reiter Y: T-cell-receptor-like antibodies - generation, function and applications. Expert Rev Mol Med 2012, 14:e6. [DOI] [PubMed] [Google Scholar]
- 21.Spanier JA, Frederick DR, Taylor JJ, Heffernan JR, Kotov DI, Martinov T, Osum KC, Ruggiero JL, Rust BJ, Landry SJ, et al. : Efficient generation of monoclonal antibodies against peptide in the context of MHCII using magnetic enrichment. Nat Commun 2016, 7:11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC: Regulatory T cells in the treatment of disease. Nat Rev Drug Discov 2018. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt A, Oberle N, Krammer PH: Molecular mechanisms of treg-mediated T cell suppression. Front Immunol 2012, 3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, Pratt KP, Shevach EM, Scott DW: Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood 2015, 125:1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang AH, Yoon J, Kim YC, Scott DW: Targeting Antigen-Specific B Cells Using Antigen-Expressing Transduced Regulatory T Cells. J Immunol 2018, 201:1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, Levings MK: Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest 2016, 126:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JP, Payne AS: Advances in Targeting CAR-T Therapy for Immune-Mediated Diseases. Cell & Gene Therapy Insights 2018, 4:255–265. [Google Scholar]
- *28.Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z: Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther 2014, 22:1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors engineered CAR Tregs specific for CEA that suppressed colitis in mouse models and decreased colon cancer tumor burden. After several reports demonstrating the promise of antigen-redirected CAR Treg in both allo- and autoimmune models, this is further evidence in a different disease supporting the potentially broad applicability of the therapeutic hypothesis.
- 29.Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS: CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation 2012, 9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright GP, Notley CA, Xue SA, Bendle GM, Holler A, Schumacher TN, Ehrenstein MR, Stauss HJ: Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci U S A 2009, 106:19078–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, Maher J, Tree TI: Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun 2017, 79:63–73. [DOI] [PubMed] [Google Scholar]; Isolating TCRs from human islet-specific T cell clones, the authors engineered TCR-based Tregs for type 1 diabetes that show antigen-specific suppression in vitro and compare favorably to polyclonal Tregs. Polyclonal Treg cell therapy is currently in clinical trials for the treatment of diabetes, but this and other reports have shown that antigen-specific Tregs have higher specific potency, furthering the promise of antigen-specific Treg therapy.
- 32.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long AS, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q: Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science Translational Medicine 2015, 315:315–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, DeFor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE: Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 2011, 117: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]