

Abstract
Background:
Despite growing evidence of diagnostic yield and clinical utility of whole exome sequencing (WES) in patients with undiagnosed diseases, there remain significant cost and reimbursement barriers limiting access to such testing. The diagnostic yield and resulting clinical actions of WES for patients who previously faced insurance coverage barriers have not yet been explored.
Methods:
We performed a retrospective descriptive analysis of clinical WES outcomes for patients facing insurance coverage barriers prior to clinical WES and who subsequently enrolled in the Undiagnosed Diseases Network (UDN). Clinical WES was completed as a result of participation in the UDN. Payer type, molecular diagnostic yield, and resulting clinical actions were evaluated.
Results:
Sixty-six patients in the UDN faced insurance coverage barriers to WES at the time of enrollment (67% public payer, 26% private payer). Forty-two of 66 (64%) received insurance denial for clinician-ordered WES, 19/66 (29%) had health insurance through a payer known not to cover WES, and 5/66 (8%) had previous payer denial of other genetic tests. Clinical WES results yielded a molecular diagnosis in 23 of 66 patients (35% [78% pediatric, 65% neurologic indication]). Molecular diagnosis resulted in clinical actions in 14 of 23 patients (61%).
Conclusions:
These data demonstrate that a substantial proportion of patients who encountered insurance coverage barriers to WES had a clinically actionable molecular diagnosis, supporting the notion that WES has value as a covered benefit for patients who remain undiagnosed despite objective clinical findings.
Keywords: exome sequencing, diagnostic yield, reimbursement, insurance coverage, rare diseases, undiagnosed diseases, access, genetic testing, policy, public health
Introduction
Undiagnosed diseases can place significant burden on patients and their families. Advances in genomic sequencing technology, including whole exome sequencing (WES), have remodeled the landscape of molecular diagnostic testing in undiagnosed diseases. Many groups have studied the clinical utility of WES, with diagnostic yields of at least 25-28% across various unselected cohorts at genetic testing laboratories (Lee et al. 2014; Retterer et al. 2016; Yang et al. 2013; Yang et al. 2014). Higher diagnostic yields are reported for trio testing (Zhu et al., 2015), pediatric patients (Stark et al., 2016; Tan et al., 2017; Valencia et al., 2015), and patients presenting with neurologic conditions (de Ligt et al., 2012; Srivastava et al., 2014; Thevenon et al., 2016; Vissers et al., 2017).
A growing body of evidence suggests that WES may be an effective first line test for patients with suspected genetic disorders and syndromic presentations (Stark et al., 2016; Tan et al., 2017). There are documented insurance barriers limiting the clinical use of WES for patients with suspected genetic disorders (Bertier, Hetu, & Joly, 2016; Deverka & Dreyfus, 2014; Deverka, Kaufman, & McGuire, 2014; Lennerz et al., 2016). In the early days of clinical WES, many insurance policies either did not cover or did not specifically address coverage of WES (Phillips et al., 2017). As clinical demand for WES increases, private payers have shifted their policies towards coverage of WES, often specifically to pediatric patients with neurodevelopmental disorders (Douglas, Parker, Trosman, Slavotinek, & Phillips, 2019). Coverage of WES may be denied by payers because a patient may not meet specific criteria for coverage, WES may not be mentioned in a policy or explicitly stated as not covered, the reimbursement rate for the test may not be on the fee schedule of payers, or the appropriate preauthorization and appeals may not be pursued by patients or providers. Some insurance policies that exclude WES as a covered benefit qualify this exclusion by stating that the test is not efficacious in making a diagnosis, and is thus “experimental,” or that the test does not have an impact on health outcomes and is thus “not medically necessary” (See Supplemental Table 1). Studies assessing the feasibility of integrating WES into clinical care highlight potential insurance barriers which either dissuade or impede the use of WES in clinically appropriate cases (Atwal et al., 2014; Lazaridis et al., 2016; Shashi et al., 2016). Establishing insurance coverage policies for WES remains difficult when large-scale next-generation sequencing challenges traditional approaches to assess clinical utility and demonstrate medical necessity (Deverka & Dreyfus, 2014).
The proportion of undiagnosed patients encountering insurance coverage barriers to clinical WES is largely unknown, as is the utility of the test in such patients. The Undiagnosed Diseases Network (UDN) cohort provides a unique opportunity to explore the utility of WES in patients who previously faced insurance coverage barriers to WES. To do so, we sought to investigate the diagnostic yield of clinical WES and resulting clinical actions for patients with rare, undiagnosed diseases who had previously faced insurance coverage barriers to WES.
Materials and Methods
Participants
The Undiagnosed Diseases Network (UDN)
The Undiagnosed Diseases Network is a multi-institution research study funded by the NIH Common Fund that seeks to improve the diagnosis and clinical care for patients with undiagnosed diseases (Gahl, Wise, & Ashley, 2015). Through a collaborative research community, UDN team scientists work to understand the etiology and pathogenesis of rare, undiagnosed conditions. Adult and pediatric patients are invited to apply online to the UDN (https://undiagnosed.hms.harvard.edu/). The application must include a study recommendation letter from a healthcare provider which supports the undiagnosed nature of the patient’s objective clinical finding(s). Available clinical diagnostic workup must be exhausted. Applications and full prior medical history are reviewed by interdisciplinary teams within and across seven UDN clinical sites. Applications are prioritized based on their potential to provide a diagnosis (predicted by presence of objective abnormal findings, multiple systems affected, positive family history, etc.) or generate new knowledge about disease mechanisms. Patients are less likely to be accepted if additional workup is deemed unnecessary by expert clinicians on UDN case review committees or if the UDN is unlikely to improve upon the comprehensive workup the applicant already received. Accepted patients in the UDN undergo various genetic, clinical and research evaluations (Reuter et al., 2018), which vary widely depending on the patient.
Participant Sample
We retrospectively reviewed all patients accepted into the Undiagnosed Diseases Network (UDN) meeting eligibility criteria between September 2015 and July 2017. Patients were evaluated at one of seven UDN clinical sites: Baylor College of Medicine, Duke University, Harvard University-affiliated hospitals, National Institutes of Health, Stanford University, University of California Los Angeles, and Vanderbilt University Medical Center. Clinical evaluations, family history, and genetic information for each patient were accessed through the UDN’s central data repository and via site-specific databases. Race, ethnicity, and primary clinical indication are self-reported variables.
Patients were excluded from the primary analysis if they did not have WES performed as part of their UDN evaluation (i.e., it was planned but pending completion, other sequencing was performed, if clinicians thought a genetic etiology of the disease was unlikely), if they had WES or whole genome sequencing (WGS) prior to acceptance to the UDN, or if data on previous insurance coverage of WES were unavailable.
Procedures
Ascertainment of coverage status and payer
We retrospectively reviewed patient medical records and UDN application materials to determine which patients did not pursue prior WES due to insurance coverage barriers. Because the UDN sites accept applications both from the home institution and from external institutions the format of medical records varies greatly by patient. For patients referred from home institutions, the UDN site had access to their electronic medical record system. For patients that were referred from external institutions, medical records collected may be limited to clinical documentation, labs, procedure notes and imaging. We reviewed all medical records available to the UDN sites for reference to participant payer and insurance coverage information for WES. If the insurance coverage information was not available, the patient was excluded from the primary analysis. The nature of insurance coverage barrier documentation varied by patient, including: clinical documentation in the record that the test was denied, documentation in the UDN referral letter that the test was denied, or insurance denial letter available in the electronic medical record. We did not have access to participants’ primary insurance policies.
Patients facing insurance coverage barriers to WES included three sub-groups that arose via chart review. The first sub-group comprised patients who had documented denial of WES by their insurance, even when pre-authorization was requested prior to acceptance in the UDN. The second sub-group comprised patients whose insurance provider had a universal policy that did not reimburse clinical WES at the time of acceptance to the UDN. For example, during the study period, several state-sponsored insurance plans routinely denied all coverage for WES regardless of indication. The third sub-group comprised patients who had previously been denied coverage for other phenotypically-appropriate genetic tests (such as multi-gene panels). In these cases, the experience of their treating practitioners and their UDN clinicians was that WES would also be denied coverage. For all the sub-groups described, it should be noted that the out-of-pocket cost to patients was prohibitively expensive as evidenced by lack of completion of clinical WES. Payers were considered public if the policy was government-sponsored (i.e., Medicaid, Medicare or TriCare).
Whole exome sequencing and analysis
Clinical WES was performed at a CLIA-certified laboratory as previously described (Supplemental Methods) (Yang et al. 2014). Variant classification and reporting were performed in accordance with American College of Medical Genetics and Genomics recommendations (Richards et al., 2015). All diagnostic variants were confirmed via Sanger sequencing.
Determination of clinical diagnostic yield
Clinical WES reports were reviewed in the context of the medical and family history by UDN clinical personnel. WES was considered diagnostic if: 1) the variant(s) identified on the clinical report was classified as pathogenic or likely pathogenic, 2) clinical features were consistent with disease(s) associated with the gene in which variants have been identified, 3) the patient had sufficient variants to match the mode of inheritance for the given gene, and 4) available evidence suggested that the variant(s) co-segregated with the disease phenotype in the family. The clinical WES report was not considered diagnostic if there was only one pathogenic variant reported in a gene associated with autosomal recessive inheritance, even in cases where there was a second variant in the same gene classified as a variant of uncertain significance (VUS). Patients for whom clinical WES was non-diagnostic were often referred for further evaluation guided by reported variants or other studies, including validation of pathogenicity of VUS via functional assays, research reanalysis of WES data, transcriptomics, metabolome profiling, animal models, or methylation studies.
Clinical actions
Clinical actions resulting from WES were documented by UDN clinical site personnel following participant evaluation. Diagnoses were scored based on medical impact: 1) diagnosis ended the diagnostic odyssey and enabled accurate genetic counseling for recurrence risk; 2) diagnosis confirmed current treatment or medical surveillance; 3) diagnosis initiated a clinically-indicated evaluation or medical surveillance other than medical therapy; 4) diagnosis led to recommendation of a specific therapy; and 5) diagnosis led to patient’s eligibility in research studies or clinical trials. Each diagnosis could have more than one score it if impacted medical management in multiple ways. A diagnosis score of 2, 3, or 4 was considered a resulting clinical action. We were not able to predict or capture downstream evaluations or treatments that were avoided or discontinued as a result of diagnosis.
Data Analysis
As the goal of the study was to describe the outcomes of patients who had any faced insurance coverage barriers to WES, our statistical analysis was primarily descriptive. When performed, statistical tests compared characteristics between sub-groups of patients facing insurance barriers to WES. Categorical variables were compared using two-sided Fisher’s exact tests. A p-value of <0.05 was considered significant. We also performed a sensitivity analysis using only the sub-group of patients with documented denial to calculate diagnostic yield, payer breakdown, and resulting clinical actions. Statistical analyses were performed with SPSS Statistics (IBM, Foster City, CA).
Results
Patient characteristics
The UDN accepted 686 patients between September 16, 2015, and July 31, 2017 (Figure 1). A total of 466 had a molecular genetic evaluation in the UDN. The remaining 220 accepted patients had either not yet completed molecular genetic evaluation or had no genetic testing after UDN investigators determined that a genetic etiology of the patient’s disease was unlikely. For those who had undergone molecular genetic evaluation, WES was completed for 218 of 466 accepted patients. The other 248 patients had WGS (195) or targeted genetic testing (53) and were excluded from this analysis for reasons mentioned above. Of the remaining 218 patients with a molecular genetic evaluation, 48 patients had WES or WGS on a clinical or research basis prior to the UDN evaluation and were excluded. 170 patients had no prior WES or WGS before UDN genetic evaluation and thus WES was completed for the first time as a part of the UDN evaluation. We excluded five patients who were not insured under the United States healthcare system. The remaining 165 patients (45% male, 41% pediatric) were insured under the United States healthcare system prior to UDN evaluation (Figure 1). We reviewed the insurance coverage barriers for this subset of patients.
Figure 1. Identification patients in the UDN undergoing whole exome sequencing (WES) previously facing insurance coverage barriers to WES.
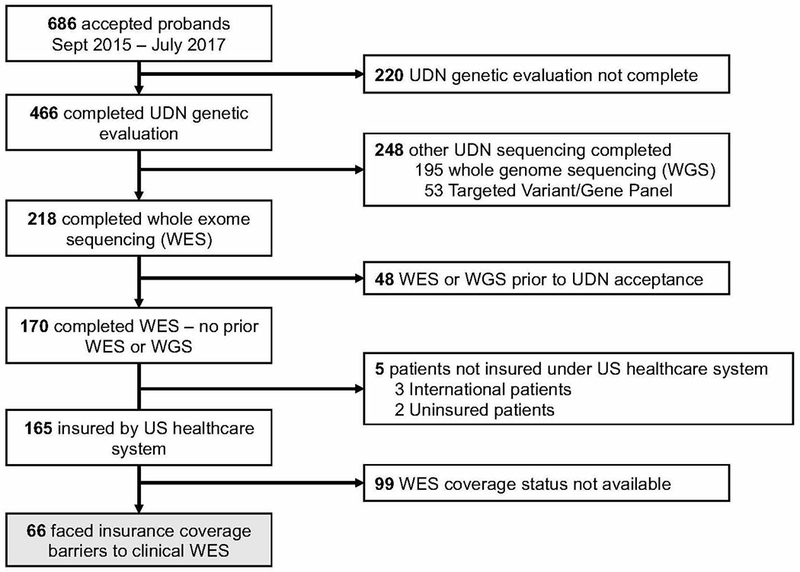
Sixty-six of 165 (40%) patients undergoing WES had no documented insurance coverage for WES prior to the UDN evaluation. WGS – whole genome sequencing; WES – whole exome sequencing; UDN – Undiagnosed Diseases Network
Identification of patients facing insurance barriers to WES
We identified 66 patients (40%) who had previously faced insurance coverage barriers limiting access to clinical WES. There were 99 patients for which WES insurance coverage data were unavailable. Demographic information and clinical indications for the 66 patients are described in Table 1. The majority of patients had public insurance (44/66, 67%) (Figure 2a) and documented denial of WES when requested clinically prior to UDN acceptance (42/66, 64%) (Figure 2b).
Table 1.
Patient Characteristics
| Variable | Total w/Insurance Coverage Barriers for WES N/66 (%) | Diagnostic WES N/23 (%) | Non-diagnostic WES N/43 (%) | |
|---|---|---|---|---|
| Male sex | 33 (50) | 9 (39) | 24 (56) | |
| Age (years) | 14.6 (1 - 56)a | 12.3 (2 – 48)a | 15.8 (1 – 56)a | |
| Pediatric (<18yo) | 48 (73) | 18 (78) | 30 (70) | |
| Race | ||||
| White | 43 (65) | 18 (78) | 25 (58) | |
| Asian | 4 (6) | 1 (4) | 3 (7) | |
| Black/African American | 7 (11) | 1 (4) | 6 (14) | |
| Other | 10 (15) | 2 (9) | 8 19) | |
| Not reported | 2 (3) | 1 (4) | 1 (2) | |
| Hispanic/Latino | 17 (26) | 6 (26) | 11 (26) | |
| Primary clinical indication | ||||
| Neurology | 30 (46) | 15 (65) | 15 (35) | |
| Other | 8 (12) | 3 (13) | 5 (12) | |
| Musculoskeletal/Orthopedic | 10 (15) | 3 (13) | 7 (16) | |
| Gastroenterology | 4 (6) | 1 (4) | 3 (7) | |
| Endocrinology | 3 (5) | 1 (4) | 2 (4) | |
| Cardiology and vascular | 1 (2) | -- | 1 (2) | |
| Allergy/Immunology | 2 (3) | -- | 2 (5) | |
| Dentistry/Craniofacial | 2 (3) | -- | 2 (5) | |
| Rheumatology | 2 (3) | -- | 2 (5) | |
| Hematology | 1 (2) | -- | 1 (2) | |
| Nephrology | 1 (2) | -- | 1 (2) | |
| Ophthalmology | 1 (2) | -- | 1 (2) | |
| N/A | 1 (2) | -- | 1 (2) | |
| Type of WES | ||||
| Proband-only | 4 (6) | 1 (4) | 3 (7) | |
| Duo | 10 (15) | 2 (9) | 8 (19) | |
| Trio | 40 (61) | 15 (65) | 25 (58) | |
| Quad | 8 (12) | 5 (22) | 3 (7) | |
| Other | 4 (6) | -- | 4 (9) | |
mean (range)
Figure 2. Insurance coverage barriers to clinical WES.
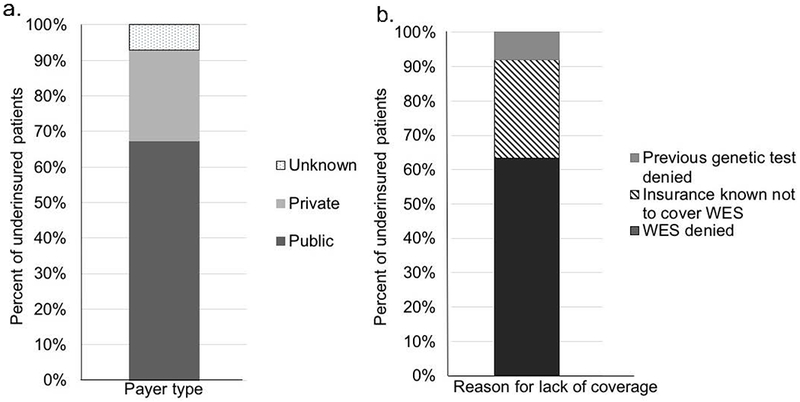
(a) Payer types for patients with insurance coverage barriers to WES. The majority had a public insurance policy (44/66, 67%). The remainder had a private insurance policy (17/66, 26%). The identity of the payer was unknown for a minority (5/66, 8%).
(b) Nature of insurance coverage barrier for patients without prior WES. The majority of patients (42/66, 64%) had documented denial of WES. The remainder either had an insurance type with a policy known not to cover WES (19/66, 29%) or had previous denial of a genetic test such as multi-gene panel (5/66, 8%).
WES diagnostic outcomes
We focused our analysis on the group of 66 patients with who faced insurance coverage barriers to WES to address the questions of diagnostic yield and clinical actionability of WES. Review of clinical WES performed as a part of UDN evaluation revealed 23/66 (35%) of patients received a molecular diagnosis. Most patients diagnosed by clinical WES were pediatric (18/23, 78%) or were patients whose primary phenotype was neurologic (15/23, 65%) (Table 1). Patients with neurological phenotypes were more likely to receive a molecular diagnosis from WES compared to those with a different primary clinical indication (50% vs. 29%; p=0.02). Fully 22/23 (96%) of patients who were diagnosed by clinical WES had at least one other family member sequenced to assist in genetic analysis (duo: 2/23; trio: 15/23; quad 5/23). When additional family members were included for WES analysis, the diagnostic yield was higher overall but not statistically significant, compared to proband-only WES (24% vs. 13%; p = 0.19). Diagnostic yields across age group, race, and ethnicity were not significantly different.
For all 23 patients diagnosed, the reported genetic variants fit the clinical phenotype and familial co-segregation supported the expected inheritance pattern for the respective gene (Table 2). Almost all (21/23, 91%) of the molecular diagnoses were made on the basis of pathogenic or likely pathogenic variants in genes with established disease-associations. The remaining two genes (USP7; CAMK2B) (Hao et al., 2015; Küry et al., 2017) had emerging evidence implicating them as causing human disease at the time of sequencing (Table 2).
Table 2.
Molecular diagnoses and impact of diagnosis on clinical actions for patients facing insurance coverage barriers to WES)
| Gene (Inheritance Pattern) | Diagnosis (OMIM #) | Diagnosis scorea | Resulting clinical actions | Primary clinical indication | Age group | WES type |
|---|---|---|---|---|---|---|
| Diagnoses with resulting clinical actions | ||||||
| SDHA (AR) | Mitochondrial respiratory chain complex II deficiency (OMIM 252011) | 1, 4 | Initiation of mitochondrial cocktail recommended; advised medications to avoid | Neurology | Peds | Trio |
| RPS6KA3 (XL) | Coffin-Lowry syndrome (OMIM 303600) | 1, 3, 4 | Audiology assessment recommended; clarify prognosis to prevent further unnecessary diagnostic testing or treatment; guide psychiatric treatment | Neurology | Adult | Trio |
| MAGEL2 (AD) | Schaaf-Young syndrome (OMIM 615547) | 1, 3, 5 | Referrals to endocrinology, developmental pediatrician, dietician; sleep study for surveillance of sleep apnea; monitor for excessive eating, scoliosis with aging; connection with research group studying the disorder | Neurology | Peds | Trio |
| RARS2 (AR) | Pontocerebellar Hypoplasia, type 6 (OMIM 611523) | 1, 3, 5 | Facilitated participation in gene-specific drug trial; surveillance of other disease-related features | Neurology | Peds | Quad |
| TRIP11 (AR) | Achondrogenesis, type IA (OMIM 200600) | 1, 2, 3 | Facilitated participation in gene-specific drug trial; surveillance of other disease-related features | Musculoskeletal | Peds | Trio |
| FBN1 (AD); TRPS1 (AD) | Marfan syndrome OMIM 154700); Tricho-rhino-phalangeal syndrome (OMIM 190350) | 1, 3 | Referral to cardiologist to establish cardiac surveillance | Musculoskeletal | Peds | Trio |
| DDX3X (XL) | Mental retardation, 102 (OMIM 300958) | 1, 3 | Testing for mitochondrial dysfunction; clarify prognosis to prevent further unnecessary diagnostic testing or treatment | Other | Adult | Proband only |
| ARID1B (AD) | Coffin-Siris Syndrome 1 (OMIM 135900) | 1, 3 | Referral to establish care with gastroenterologist, ophthalmologist, audiologist; tumor surveillance | Neurology | Peds | Quad |
| TANGO2 (AR) | Recurrent metabolic encephalomyopathic crisis, rhabdomyolysis, cardiac arrhythmias, and neurodegeneration (OMIM 616878) | 1, 3 | Referral to cardiologist for ongoing arrhythmia surveillance | Other | Peds | Trio |
| SYNE1 (AR) | Spinocerebellar ataxia, 8 (OMIM 610743) | 1, 3 | Pulmonary function testing; clarify prognosis to prevent further unnecessary diagnostic testing or treatment | Neurology | Adult | Duo |
| FOXG1 (AD) | FOXG1 syndrome; Rett syndrome, congential variant (OMIM 613454) | 1, 3 | Referral to movement disorder specialist | Neurology | Peds | Trio |
| DYNC1H1 (AD) | Mental retardation, 13 (OMIM 614563) | 1, 3 | Referred to specialists based on molecular diagnosis | Neurology | Peds | Trio |
| SRCAP (AD) | Floating-Harbor Syndrome (OMIM 136140) | 1, 2 | Continuation of endocrine surveillance per diagnosis | Endocrinology | Peds | Trio |
| ROGDI (AR) | Floating-Harbor Syndrome (OMIM 136140) | 1, 2 | Validation of previous management recommendations | Neurology | Adult | Quad |
| Eligibility for research studies or clinical trials only | ||||||
| ADNP (AD) | Helsmoortel-van der Aa syndrome (OMIM 615873) | 1, 5 | Referral to relevant clinical trial for neuroprotective drug | Gastroenterology | Peds | Trio |
| NAGLU (AR) | Mucopolysaccharidosis type IIIB (Sanfilippo B Syndrome) (OMIM 252920) | 1, 5 | Eligibility for gene therapy clinical trials | Neurology | Adult | Trio |
| ALG13 (XL) | Epileptic encephalopathy, early infantile, 36 (OMIM 300884) | 1, 5 | Referred to additional research studies | Neurology | Peds | Quad |
| No impact on clinical actions | ||||||
| CAMK2Bb (AD) | CAMK2B-related neurodevelopmental disorder25 | 1 | End of diagnostic odyssey | Other | Peds | Trio |
| GNAO1 (AD) | Epileptic encephalopathy, early infantile, 17 (OMIM 615473) | 1 | End of diagnostic odyssey | Neurology | Peds | Trio |
| USP7b (AD) | USP7-related neurodevelopmental disorder26 | 1 | End of diagnostic odyssey | Neurology | Peds | Trio |
| EEF1A2 (AD) | Mental Retardation, 38 (OMIM 616393); Epileptic encephalopathy, early infantile 33 (OMIM 616409) | 1 | End of diagnostic odyssey | Neurology | Peds | Trio |
| ASXL3 (AD) | Bainbridge-Ropers syndrome (OMIM 615485) | 1 | End of diagnostic odyssey | Musculoskeletal | Peds | Duo |
| GRIN2A (AD) | Epilepsy, focal, with speech disorder and with or without intellectual disability (OMIM 245570) | 1 | End of diagnostic odyssey | Neurology | Peds | Quad |
AR - autosomal recessive; AD - autosomal dominant; XL - X-linked; Peds - pediatric
1 - diagnosis resulted in the end of the diagnostic odyssey and enabled accurate genetic counseling for recurrence risk; 2 - diagnosis confirmed current treatment and medical surveillance; 3 - diagnosis initiated a clinically-indicated evaluation or medical surveillance other than medical therapy ; 4 - diagnosis led to recommendation of a specific therapy; 5 - diagnosis led to patient’s eligibility in research studies and/or clinical trials.
recently described gene-disease association
Impact of diagnosis on clinical actions for diagnostic WES
The diagnosis scores and resulting clinical actions for each of the 23 patients who received a molecular diagnosis are represented in Table 2. Not including new eligibility for clinical trials or enablement of accurate recurrence risk counseling, 61% (14/23) of patients diagnosed by WES had resulting clinical actions. This included patients for whom the diagnosis confirmed current treatment or medical surveillance (3/23), initiated a clinically-indicated evaluation or medical surveillance (11/23), including referrals to specialist providers for ongoing surveillance of disease-related manifestations (i.e., tumor surveillance), or initiated a specific medical therapy (2/23). For example, one patient diagnosed with a TANGO2-related disorder (OMIM 616878), a disease characterized by recurrent metabolic encephalomyopathic crises, rhabdomyolysis, childhood-onset lethal cardiac arrhythmias, and neurodegeneration, established ongoing care with a cardiologist to monitor for arrhythmias and was recommended to avoid QT prolongating medications.
Sensitivity analysis
As a sensitivity analysis, we calculated our main outcome measures with only the 42 patients in the documented denial sub-group. Payer breakdown, diagnostic yield and patients with clinical actions are represented in Supplemental Tables 3 and 4. Overall diagnostic yield and clinical action outcomes are consistent with the primary results. More patients in the documented denial sub-group had private insurance.
Non-diagnostic WES
We investigated the outcomes of the 43 patients whose WES was considered non-diagnostic. Of these, 15/43 (35%) of non-diagnostic WES cases had a pathogenic variant reported that was not sufficient to make a molecular diagnosis due to mismatch of inheritance pattern or lack of genotype-phenotype correlation (Figure 3). Patients with pathogenic variants are considered non-diagnostic if they lacked a second pathogenic variant (in the case of autosomal recessive inheritance) or if the variant did not fit the clinical phenotype. Additionally, 19/43 (44%) of all non-diagnostic WES without a pathogenic or likely pathogenic variant reported at least one VUS in a gene related to the patient’s clinical phenotype (Figure 3). The remainder only had research variants not meeting American College of Medical Genetics and Genomics reporting standards but which were included in an addendum document issued by the clinical laboratory and used by the UDN clinical sites for research purposes.
Figure 3. Variant types reported for patients with non-diagnostic WES.
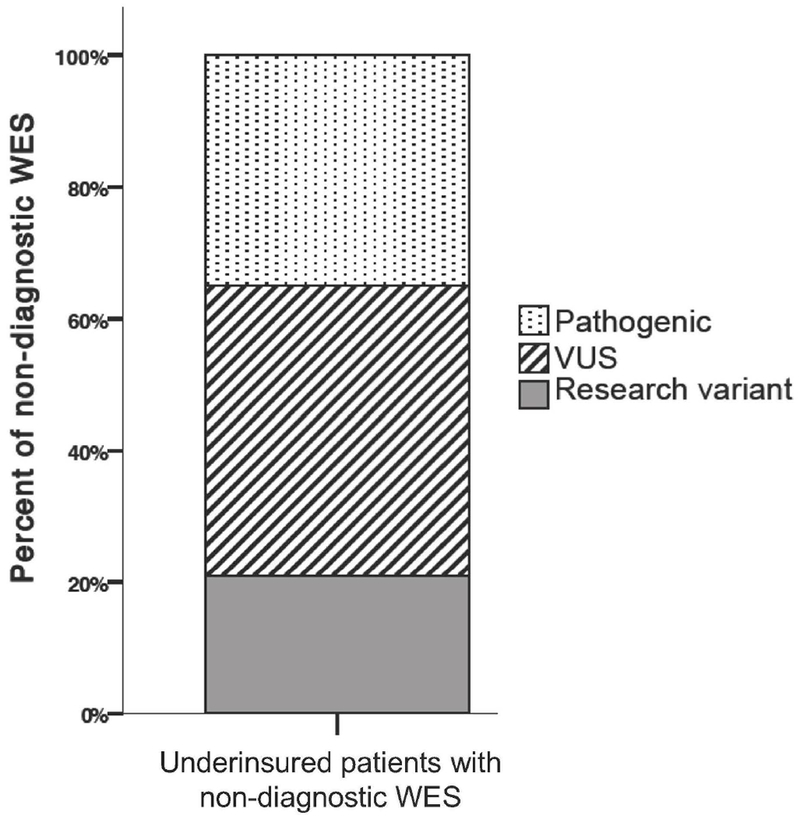
15/43 (35%) had a pathogenic variant reported, 19/43 (44%) of patients without a pathogenic or likely pathogenic variant reported at least one VUS in a gene related to the patient’s clinical phenotype, and 9/43 (21%) only had a research variant.
Despite a non-diagnostic clinical WES, 8/43 patients (19%) received a diagnosis through the UDN, either as a part of the UDN clinical evaluation or research studies beyond the clinical WES report (Supplemental Table 2). All of these additional studies and diagnoses were guided by variants reported on WES, even when WES on initial analysis was non-diagnostic. If these consequent diagnoses are included, the diagnostic rate across all patients facing insurance coverage barriers is 47% (31/66).
Discussion
We retrospectively investigated the diagnostic yield of clinical WES in a cohort of patients with undiagnosed diseases who encountered insurance coverage barriers that prevented completion of WES on a clinical basis. We also captured resulting clinical actions following molecular diagnosis. Prior to this study, the clinical characteristics of patients with insurance barriers to clinical WES were not known. Also unknown was the diagnostic yield in such patients.
A substantial proportion of patients without prior WES (40%) did not complete the test on a clinical basis due to insurance coverage barriers, and the majority of patients facing such barriers had public insurance (67%). Few studies have systematically measured the proportion of individuals facing insurance barriers to WES (Lazaridis et al., 2016). As previously described, not all payer policies mention WES (Phillips et al., 2017). Thus, coverage policies may be unknown by the clinical team if the test was never requested on a clinical basis or if the test was denied in the absence of a pre-authorization process. Even if a payer does include WES as a covered benefit, the patient may not meet specific criteria permitting coverage or the patient may still have a prohibitively large out-of-pocket cost. Genetic testing laboratories may be a valuable source of these data, and, in fact, one clinical laboratory estimated half of WES tests ordered were denied coverage (Deverka et al., 2014). However, there are no data on the outcome of clinical WES in patients facing insurance barriers since clinical WES was likely never completed. Prior studies of research exome testing have not been guided by ACMG recommendations (Richards et al., 2015) and rigorous CLIA protocol, as was done in this study. Thus, our study represents a unique cohort to interrogate outcomes of clinical WES in patients facing insurance barriers.
Our outcomes of diagnostic yield and resulting clinical actions aim to measure the clinical utility of WES for our population of patients. Our finding of a diagnostic yield by WES of 35% for UDN patients previously facing insurance barriers to WES indicates that a substantial proportion of this subset of patients could have received a molecular diagnosis earlier had clinical WES been financially feasible. Furthermore, our data show that those patients who did receive a diagnosis by WES are mostly pediatric (78%) and have a primary neurologic finding (65%). Most patients who received a diagnosis had family member comparator exomes included in genetic analysis (96%). In this study, the diagnostic yield of WES in patients facing insurance barriers is similar to that of other WES cohorts described in the literature, in which the majority of patients had WES covered to some degree as evidenced by the fact that it was completed (de Ligt et al., 2012; Srivastava et al., 2014; Stark et al., 2016; Tan et al., 2017; Thevenon et al., 2016; Valencia et al., 2015; Vissers et al., 2017).
Both commercial and public payers have denied coverage for WES for patients in our cohort on the basis that it is experimental or investigational, in some cases due to sensitivity and specificity measures (Deverka & Dreyfus, 2014). Our diagnostic yield of 35% does not include those patients who were diagnosed via research-based methods. In our study, clinical WES provided diagnoses to patients facing insurance barriers for the test, with 91% of diagnostic cases involving established genes with known disease associations. The diagnostic yield suggests that at least a subset of patients facing insurance barriers would have received an established molecular diagnosis had WES been completed before their application the UDN.
An important consideration for payers in developing coverage policies for and providing authorization of WES is its medical necessity and impact on clinical decision-making. In order to address these issues, our secondary outcome measured clinical actions resulting from molecular diagnoses delivered by WES. While our study did not collect longitudinal data on health outcomes, we found that the molecular diagnoses provided by WES guided clinical decision-making for the patients in the short term. The majority of our patients (61%) facing insurance barriers who ultimately reached a molecular diagnosis had at least one clinical action item as a result of WES, beyond enabling genetic counseling for accurate recurrence risk counseling and not including eligibility for clinical trials. The most frequent impact of diagnosis was the initiation of clinically-indicated evaluation or medical surveillance. For some patients, new treatment options became available as a result of molecular diagnosis via WES.
The challenges of integrating new genomic technology into medicine are not unique to WES. In many ways the incorporation of WES into clinical care mimics the challenges faced during the early days of chromosomal microarray testing. When microarray testing first emerged, clinicians reported frequent insurance denials due to the test being deemed experimental, not covered, or not medically necessary (Riggs et al., 2014), despite demonstration of its diagnostic potential and impact on medical management (Coulter et al., 2011; Ellison et al., 2012). The diagnoses made by WES are no less medically necessary than the diagnoses that are made by chromosomal microarray. Both are untargeted tests that have the potential to diagnose a wide variety of genetic syndromes. Both chromosomal microarray and WES can reveal genetic diagnoses and enable clinical actions like those reported in our study.
Further complicating the development of nuanced and appropriate policies on coverage for WES is the lack of data regarding its cost-effectiveness. Existing data supports the use of WES in patients with suspected genetic disorders early in the diagnostic process in order to shorten the diagnostic odyssey and improve efficiency of evaluations, especially in patients with neurologic conditions (Stark et al., 2016; Tan et al., 2017; Vissers et al., 2017; Walsh et al., 2017). However, such literature on WES utilization lacks clear presentation of cost data, is limited by small cohort sizes, and use outcome measures that are difficult to translate into health economic policy (Schwarze, Buchanan, Taylor, & Wordsworth, 2018). Although a cost-effectiveness analysis was not feasible nor a goal in this study, patients were subjected to non-diagnostic clinical evaluations, laboratory testing, and molecular testing prior to their diagnoses that would likely have been unnecessary had clinical WES been available.
Nineteen percent of patients with non-diagnostic WES achieved diagnosis through subsequent UDN clinical and research evaluations. Similarly, all patients with non-diagnostic WES had genetic variants found on WES considered for additional investigation through reanalysis. While the clinical WES report alone did not yield a diagnosis for this subset of our patients, WES data guided both clinical and research workup and create opportunity for future reanalysis of patient genetic data. It is estimated that 250 new gene-disease associations are curated yearly (Wenger, Guturu, Bernstein, & Bejerano, 2017), which has contributed to recent work establishing the value of periodic reanalysis of clinical WES data. These studies report that WES reanalysis results in new diagnoses in 10-36% of cases (Eldomery et al., 2017; Wenger et al., 2017). As more patients undergo testing, further phenotype-genotype correlations will be established, advances are made in bioinformatics tools, and knowledge about the genetic basis of disease expands. This further suggests that WES, although non-diagnostic for some at its initial interpretation, may eventually lead to clinical diagnosis in the future.
Study Limitations
Our study is limited by the small sample size and to patients covered by the United States Healthcare system, which might not be generalizable to settings with universal health care. We relied on secondary sources for data on insurance denial and lack of coverage since review of patient insurance policy was not feasible or documented consistently in patients’ medical records by the ordering clinical team. We did not have further information about whether all payers were denying all requests for WES at that time, or whether it was a case-by-case basis due to the retrospective nature of the study. We acknowledge that those patients undergoing WES in our study had a higher a priori likelihood of identifying a molecular diagnosis compared to all-comers of clinical WES for several reasons. First, a clinical provider prior to the UDN had to have recommended WES in order to learn that it was not a covered benefit, indicating that there was a reasonable suspicion for an underlying genetic etiology. Second, the UDN applications are reviewed for acceptance in part for likelihood of identifying a genetic diagnosis, so the accepted patients in the UDN are likely enriched for those with underlying molecular diagnoses. Third, we may not be capturing those patients and families who face insurance coverage barriers to WES but who are less motivated to find a diagnosis and, thus, do not apply to the UDN. Finally, our cohort of patients undergoing WES was enriched for neurologic indications.
Practice Implications
Insurance coverage policies for genetic testing are evolving. Our study adds to a body of literature supporting the clinical utility of WES, specifically for underinsured patients. As the evidence of the clinical utility of WES increases and the cost of testing decreases, payers may have a larger body of literature to assess when developing their nuanced coverage policies. Until then, there remain a population of patients without access to clinically appropriate WES. Our study demonstrates that a substantial proportion of patients facing insurance coverage barriers to WES are denied the opportunity for molecular genetic diagnosis, particularly neurology patients. Genetic counselors, who are often at the front line of insurance coverage disparities for genetic testing, can leverage this data to continue to advocate for patients’ access to WES.
Research Recommendations
Future studies could assess insurance coverage and diagnostic relevant diagnostic outcomes for all-comers of WES, including review of primary sources of insurance policies and denials. As the genetic testing market expands to include clinical sequencing of the entire genome, it may be valuable to describe the landscape of insurance policies for such testing. Collaboration between clinicians and payers could facilitate appropriate study design and review of available evidence to inform healthcare policies.
Supplementary Material
Acknowledgements:
Patients and families of the Undiagnosed Diseases Network; Members of the Undiagnosed Diseases Network: David R. Adams, Aaron Aday, Mercedes E. Alejandro, Patrick Allard, Mahshid S. Azamian, Carlos A. Bacino, Ashok Balasubramanyam, Hayk Barseghyan, Gabriel F. Batzli, Alan H. Beggs, Babak Behnam, Hugo J. Bellen, Anna Bican, David P. Bick, Camille L. Birch, Braden E. Boone, Bret L. Bostwick, Lauren C. Briere, Donna M. Brown, Matthew Brush, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Shan Chen, Gary D. Clark Terra R. Coakley, Joy D. Cogan, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Charles Curnin, Precilla D’Souza, Mariska Davids, Jyoti G. Dayal, Esteban C. Dell’Angelica, Shweta U. Dhar, Katrina M. Dipple, Laurel A. Donnell-Fink, Naghmeh Dorrani, Daniel C. Dorset, Emilie D. Douine, David D. Draper, David J. Eckstein, Lisa T. Emrick, Gregory M. Enns, Ascia Eskin, Cecilia Esteves, Tyra Estwick, Carlos Ferreira, Brent L. Fogel, Noah D. Friedman, William A. Gahl, Emily Glanton, Rena A. Godfrey, Alica M. Goldman, David B. Goldstein, Sarah E. Gould, Jean-Philippe F. Gourdine, Catherine A. Groden, Andrea L. Gropman, Melissa Haendel, Rizwan Hamid, Neil A. Hanchard, Lori H. Handley, Francis High, Ingrid A. Holm, Jason Hom, Ellen M. Howerton, Yong Huang, Fariha Jamal, Yong-hui Jiang, Jean M. Johnston, Angela L. Jones, Lefkothea Karaviti, David M. Koeller, Isaac S. Kohane, Donna M. Krasnewich, Susan Korrick, Elizabeth L. Krieg, Jennifer E. Kyle, Seema R. Lalani, C. Christopher Lau, Jozef Lazar, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Shawn E. Levy, Richard A. Lewis, Sharyn A. Lincoln, Sandra K. Loo, Richard L. Maas, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Thomas C. Markello, Ronit Marom, Martin G. Martin, Julian A. Martínez-Agosto, Thomas May, Allyn McConkie-Rosell, Colleen E. McCormack, Alexa T. McCray, Jason D. Merker, Thomas O. Metz, Matthew Might, Paolo M. Moretti, Marie Morimoto, David R. Murdock, Jennifer L. Murphy, Donna M. Muzny, Michele E. Nehrebecky, Stan F. Nelson, J. Scott Newberry, John H. Newman, Sarah K. Nicholas, Donna Novacic, Jordan S. Orange, James P. Orengo, J. Carl Pallais, Jeanette C. Papp, Neil H. Parker, Loren DM. Pena, Jennifer E. Posey, John H. Postlethwait, Lorraine Potocki, Barbara N. Pusey, Amy K. Robertson, Lance H. Rodan, Jacinda B. Sampson, Susan L. Samson, Molly C. Schroeder, Daryl A. Scott, Prashant Sharma, Edwin K. Silverman, Janet S. Sinsheimer, Kevin S. Smith, Nicholas Stong, Jennifer A. Sullivan, Queenie K.-G. Tan, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Tiina K. Urv, Zaheer M. Valivullah, Eric Vilain, Tiphanie P. Vogel, Daryl M. Waggott, Colleen E. Wahl, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jijun Wan, Michael F. Wangler, Patricia A. Ward, Katrina M. Waters, Bobbie-Jo M. Webb-Robertson, Monte Westerfield, Anastasia L. Wise, Lynne A. Wolfe, Elizabeth A. Worthey, Shinya Yamamoto, Amanda J. Yoon, Guoyun Yu, Chunli Zhao, Allison Zheng
Dr. Melanie Myers served as Action Editor on the manuscript review process and publication decision.
Funding sources: Research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Numbers listed below. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest Statement
Euan A. Ashley is a Founder of Personalis Inc., Deep Cell Inc, and advisor to Genome Medical and SequenceBio. Matthew T. Wheeler has modest ownership interest in Personalis Inc. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the clinical exome sequencing offered in the Medical Genetics Laboratory and Whole Genome Laboratory and the authors who are faculty members are indicated in the affiliation section on the title page. No other authors have relevant conflicts of interest to disclose.
Clinical trial registration: . Registered 19 May 2015.
Clinical sites
U01HG007709 (Baylor College of Medicine)
U01HG007672 (Duke University)
U01HG007690 (Harvard Teaching Hospitals)
U01HG007708 (Stanford Medicine)
U01HG007703 (University of California Los Angeles)
U01HG007674 (Vanderbilt University)
Coordinating Center
U01HG007530 (Harvard Medical School)
Sequencing Core
U01HG007942 (Baylor College of Medicine)
Ethics statement: The clinical trial () was approved by the Institutional Review Board of NHGRI (NIH Study Reference Number 15-HG-0130), and written informed consent and assent was obtained from the patient and family members.
References
- Atwal PS, Brennan M-L, Cox R, Niaki M, Platt J, Homeyer M, … Hudgins L (2014). Clinical whole-exome sequencing: are we there yet? Genetics in Medicine, 16(9), 717–9. 10.1038/gim.2014.10 [DOI] [PubMed] [Google Scholar]
- Bertier G, Hetu M, & Joly Y (2016). Unsolved challenges of clinical whole-exome sequencing: A systematic literature review of end-users’ views. BMC Medical Genomics, 9(52). 10.1186/s12920-016-0213-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter ME, Miller DT, Harris DJ, Hawley P, Picker J, Roberts AE, … Irons M (2011). Chromosomal microarray testing influences medical management. Genetics in Medicine, 13(9), 770–776. 10.1097/GIM.0b013e31821dd54a [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, … Vissers LELM (2012). Diagnostic exome sequencing in persons with severe intellectual disability. New England Journal of Medicine, 367(20), 1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Deverka PA, & Dreyfus JC (2014). Clinical integration of next generation sequencing: Coverage and reimbursement challenges. Journal of Law, Medicine & Ethics, (Fall), 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverka PA, Kaufman D, & McGuire AL (2014). Overcoming the reimbursement barriers for clinical sequencing. JAMA : The Journal of the American Medical Association, 312(18), 1857–1858. 10.1001/jama.2014.14915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas MP, Parker SL, Trosman JR, Slavotinek AM, & Phillips KA (2019). Private payer coverage policies for exome sequencing ( ES ) in pediatric patients : trends over time and analysis of evidence cited. Genetics in Medicine, 21(1), 152–160. 10.1038/s41436-018-0043-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, … Lupski JR (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Medicine, 9(26). 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison JW, Ravnan JB, Rosenfeld JA, Morton SA, Neill NJ, Williams MS, … Shaffer LG (2012). Clinical Utility of Chromosomal Microarray Analysis. Pediatrics, 130(5), e1085–e1095. 10.1542/peds.2012-0568 [DOI] [PubMed] [Google Scholar]
- Gahl WA, Wise AL, & Ashley EA (2015). The Undiagnosed Diseases Network of the National Institutes of Health: A National Extension. Journal of the American Medical Association, 314(17), 1797–1798. 10.1001/jama.2015.12249.Conflict [DOI] [PubMed] [Google Scholar]
- Hao Y-H, Fountain MD, Tacer KF, Xia F, Bi W, Kang S-HL, … Moran Roc, R. P (2015). USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder Article USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in. Molecular Cell, 59, 956–969. 10.1016/j.molcel.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küry S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, … Mercier S (2017). De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. American Journal of Human Genetics, 101(5), 768–788. 10.1016/j.ajhg.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis K, Schahl K, Cousin M, Babovic-vuksanovic D, Riegert-johnson DL, Gavrilova RH, … Ferber MJ (2016). Outcome of whole exome sequencing for diagnostic odyssey cases of an individualized medicine clinic: The Mayo Clinic experience. Mayo Clinic Proceedings, 91(3), 297–307. 10.1016/j.mayocp.2015.12.018 [DOI] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-rivera F, … Nelson SF (2014). Clinical exome sequencing for genetic identification of rare mendelian disorders. Journal of the American Medical Association, 312(18), 1880–1887. 10.1001/jama.2014.14604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennerz JK, McLaughlin HM, Baron JM, Rasmussen D, Sumbada Shin M, Berners-Lee N, … Iafrate AJ (2016). Health care infrastructure for financially sustainable clinical genomics. The Journal of Molecular Diagnostics, 18(5), 697–706. 10.1016/j.jmoldx.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KA, Deverka PA, Trosman JR, Douglas MP, Chambers JD, Weldon CB, & Dervan AP (2017). Payer coverage policies for multigene tests. Nature Biotechnology, 35(7), 614–617. 10.1038/nbt.3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, … Bale S (2016). Clinical application of whole-exome sequencing across clinical indications. Genetics in Medicine, 18(7), 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Reuter CM, Brimble E, DeFilippo C, Dries AM, Enns GM, Ashley EA, … Wheeler MT (2018). A New Approach to Rare Diseases of Children: The Undiagnosed Diseases Network. Journal of Pediatrics, 196, 291–297.e2. 10.1016/j.jpeds.2017.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Wain KE, Riethmaier D, Smith-Packard B, Faucett WA, Hoppman N, … Miller DT (2014). Chromosomal microarray impacts clinical management. Clinical Genetics, 85(2), 147–153. 10.1111/cge.12107 [DOI] [PubMed] [Google Scholar]
- Schwarze K, Buchanan J, Taylor JC, & Wordsworth S (2018). Are whole-exome and whole-genome sequencing approaches cost-effective ? A systematic review of the literature. Genetics in Medicine, 0(August 2017). 10.1038/gim.2017.247 [DOI] [PubMed] [Google Scholar]
- Shashi V, McConkie-Rosell A, Schoch K, Kasturi V, Rehder C, Jiang YH, … McDonald MT (2016). Practical considerations in the clinical application of whole-exome sequencing. Clinical Genetics, 89(2), 173–181. 10.1111/cge.12569 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, … Fatemi A (2014). Clinical whole exome sequencing in child neurology practice. Annals of Neurology, 76(4), 473–483. 10.1002/ana.24251 [DOI] [PubMed] [Google Scholar]
- Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, … White SM (2016). A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genetics in Medicine, 18(11), 1090–1096. 10.1038/gim.2016.1 [DOI] [PubMed] [Google Scholar]
- Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, … White SM (2017). Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatrics, 171(9), 855–862. 10.1001/jamapediatrics.2017.1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, … Rivière JB (2016). Diagnostic odyssey in severe neurodevelopmental disorders: Toward clinical whole-exome sequencing as a first-line diagnostic test. Clinical Genetics, 89(6), 700–707. 10.1111/cge.12732 [DOI] [PubMed] [Google Scholar]
- Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, … Zhang K (2015). Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: A pediatric center’s experience. Frontiers in Pediatrics, 3(August), 1–16. 10.3389/fped.2015.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, van Nimwegen KJM, Schieving JH, Kamsteeg E-J, Kleefstra T, Yntema HG, … Willemsen MAAP (2017). A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genetics in Medicine, 19(9), 1055–1063. 10.1038/gim.2017.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M, Bell KM, Chong B, Creed E, Brett GR, Pope K, … Ryan MM (2017). Diagnostic and cost utility of whole exome sequencing in peripheral neuropathy. Annals of Clinical and Translational Neurology, 4(5), 318–325. 10.1002/acn3.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Guturu H, Bernstein JA, & Bejerano G (2017). Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genetics in Medicine, 19(2), 209–214. 10.1038/gim.2016.88 [DOI] [PubMed] [Google Scholar]
- Yang Y, Bainbridge MN, Ph D, Willis A, Ph D, Ward PA, … Eng CM (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. New England Journal of Medicine, 369(16), 1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, … Boerwinkle E (2014). Molecular findings among patients referred for clinical whole-exome sequencing. Journal of the American Medical Association, 312(18), 1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu Y-F, McSweeney KM, … Goldstein DB (2015). Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genetics in Medicine, 17(10), 774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.