

Abstract
Toxic peripheral neuropathies are an important form of acquired polyneuropathy produced by a variety of xenobiotics and different exposure scenarios. Delineating the mechanisms of neurotoxicants and determining the degenerative biological pathways triggered by peripheral neurotoxicants will facilitate the development of sensitive and specific biochemical based methods for identifying neurotoxicants, designing therapeutic interventions and developing structure activity relationships for predicting potential neurotoxicants. This review presents an overview of the general concepts of toxic peripheral neuropathies with the goal of providing insight into why certain agents target the peripheral nervous system and produce their associated lesions. Experimental data and the main hypotheses for the mechanisms of selected agents that produce neuronopathies, axonopathies or myelinopathies including covalent or non-covalent modifications, compromised energy or protein biosynthesis, oxidative injury and disruption of ionic gradients across membranes are presented. The relevance of signaling between the main components of peripheral nerve i.e., glia, neuronal perikaryon, and axon, as a target for neurotoxicants and the contribution of active programmed degenerative pathways to the lesions observed in toxic peripheral neuropathies are also discussed.
Keywords: toxic neuropathy, axonopathy, myelinopathy, neuronopathy, demyelination, axon degeneration, Schwann cell
Introduction
Peripheral neuropathy is an important clinical condition having almost 15% prevalence in the population over age 40 in the United States.1,2 The etiologies of peripheral neuropathies are quite varied including metabolic and infectious diseases, hereditary conditions and nutritional deficiencies. Toxic neuropathies that are produced by xenobiotics targeting components of the peripheral nervous system represent another important cause of acquired polyneuropathy. This review will present an overview of the general concepts of toxic peripheral neuropathies with the goal of providing insight into why certain agents target the peripheral nervous system and produce their associated lesions.
Toxic neuropathies can be environmental, occupational, recreational or iatrogenic; and the prevalence of their cause is governed by geographical and economical factors. In developed countries the most common cause of toxic neuropathy is drug toxicity, particularly that associated with chemotherapy treatments.3 The prevalence of chemotherapy induced peripheral neuropathy was reported to be as high as 68% within the first month of the end of therapy and can be expected to increase as cancer survivorship rates increase.4 In developing countries, environmental and occupational exposures to various agents including arsenic, lead, mercury and organophosphorous compounds are significant peripheral neurotoxicants.5 Additionally, as many manufacturing processes have moved to less developed areas implementing less strict monitoring of occupational exposures, previously recognized peripheral neurotoxicants including hexane, carbon disulfide and newer chlorofluorcarbon replacing agents like 1-bromopropane remain potential risks for peripheral neuropathy.6–8 Two of the most prevalent causes of polyneuropathy, diabetes mellitus and alcoholism, could arguably be considered toxic neuropathies due to the involvement of excess glucose and effects of ethanol. Those neuropathies are beyond the scope of this review and covered in detail elsewhere.9–14
Peripheral neurotoxicants can be categorized in numerous ways including their chemical structure, primary use, or the clinical signs they produce. A commonly used approach for classifying peripheral neuropathies is based upon the primary structural target involved, with those exhibiting initial effects in the neuronal perikaryon, axon or the Schwann cell referred to as neuronopathies, axonopathies or myelinopathies, respectively.15–18 The synapse is also important for the nervous system’s function of transmitting information across space and is the site of action for many pharmacologic and toxic agents. Most of the agents that affect the synapse do not produce structural lesions in the peripheral nervous system and the magnitude of compounds recognized and their mechanisms are beyond the scope of this review.
The specific clinical presentation of a toxic neuropathy will be dependent upon the severity and structural target affected but there are some concepts that generally hold regarding peripheral neuropathies.2,3,19 Typically they are dose-dependent, symmetrical and reversible given adequate time following cessation of exposure. However, for some agents a condition referred to as “coasting’ in which clinical signs continue to progress or persist for a significant period of weeks to months following cessation of exposure has been observed.20–22 The most common presentation of toxic neuropathy involves the largest diameter longest axons associated with numbness, paraesthesia or weakness in a stocking glove distribution.19,23,24 This is a reflection of the fact that the large long axons are ultimately affected whether or not they are the primary critical targets. Accordingly, hind limb grip strength in rodent functional observation batteries (FOB) has been used as a sensitive indicator for toxic neuropathies potentially signaling impairment prior to detectable morphological lesions.25 Many peripheral neurotoxicants not only injure components within the peripheral nervous system but may also have central nervous system involvement, optic neuropathy, rhabdomyopathy (i.e. skeletal muscle lesions) or other systemic involvement. Predisposing factors that can contribute to the risk of toxic neuropathy include diabetes mellitus, certain paraneoplastic conditions, genetics and medical treatments having neurotoxic side effects.
General Concepts of Peripheral Nervous System Structure and Function Relevant to Mechanisms of Toxic Neuropathies
A principal function of the peripheral nervous system is the rapid transmission of signals over a substantial distance. To accomplish this function it has developed unique structural and physiological characteristics. These characteristics present distinct vulnerabilities that are targeted by certain neurotoxicants and contribute to their target organ specificity. Additional factors that can contribute to peripheral nervous system target organ specificity include specific uptake transport systems that can concentrate toxic substances in some types of neurons and the compromised blood nerve barrier at peripheral ganglia.26–29 When investigating mechanisms of peripheral neurotoxicants, the first step is often to examine morphological endpoints to identify and characterize the initial lesion. However caution in interpreting changes must be exercised in that no component can be affected without accompanying changes in the other components. Although an axonopathy or myelinopathy may be observed it is not immediately evident whether these are primary or secondary conditions. Thus an understanding of the temporal and spatial changes, both primary and secondary, is essential to designing follow up analyses to ascertain the initial site of action for a neurotoxicant. Additionally, as our knowledge of intercellular signaling between glia and axons and molecular responses to injury advance, there is promise for the development of sensitive and specific biochemical markers to aid in identifying initial targets of neurotoxicants.
That secondary changes occur in Schwann cells associated with axons experiencing pathological conditions is not surprising when the interdependence of the two is considered. Axons together with Schwann cells achieve their concerted goal through not only their unique structural composition and close physical proximity but also through dynamic communication that regulates the development, maintenance and function of each other.30–34 A variety of molecules has been implicated in signaling between peripheral axons and Schwann cells including myelin-associated glycoprotein (MAG),35 low-affinity nerve growth factor receptor (p75),36 insulin-like growth factor 1 (IGF1),37 and transforming growth factor beta (TGF-β).38 The growth factor neuregulin 1 (NRG1) and the erbB receptors, tyrosine kinases structurally related to the epidermal growth factor receptor (EGFR), are critical regulators in development and maintenance for all of the Schwann cell phenotypes. Schwann cells express erbB2 and erbB3 receptors and multiple NRG1 isoforms (derived from the same gene) are expressed by neurons in the central and peripheral nervous systems that project their axons into the periphery.39–42 Studies support a pivotal role for NRG1 signaling through erbB receptors in promoting expansion and survival of Schwann cells during development and in determining which axons will be myelinated. Homozygous knockout of erbB2 or heterozygous knockout of NRG1 Type III but not Type I results in hypomyelination while overexpression of NRG1 increases myelin thickness suggesting a role of the axon in regulating the thickness of its myelin sheath.43,44 Interruption of axonal NRG1 growth factor expression through primary axonal injury would compromise this signaling and could contribute to the observed alterations in Schwann cell function and structure accompanying axonal injury.
The case for a reciprocal dependence of axons on signaling and support from associated Schwann cells is also strengthening from experimental evidence. 45–47 The neuronal soma has long been considered the most important if not the only source of protein biosynthesis for neurons that is then distributed via axonal transport. The presence of Nissl substance, a large accumulation of rough endoplasmic reticulum, in the soma is a testament to the protein synthetic capacity of the neuron. However, recent studies have supported the ability of protein synthesis to also occur within the axon.31 The protein translated in the axon could contribute to multiple processes including growth, maintenance and responses to injury and it has been shown that transcription factors are produced in developed axons and transported retrograde to the soma possibly to provide information on the status of the axon during injury.48 Two possible sources for the mRNA used to translate proteins in axons have been recognized. One source is perikaryon-derived mRNA which has been detected in axons.49,50 The other source is Schwann cell-derived RNA transferred to axons. This concept was introduced by Singer and Green51 from experiments they performed using the distal ends of transected nerves; and subsequent studies have supported their observations.52,53 The ability of Schwann cells to transfer ribosomes to axons has also been reported.54,55 After injury, transfer of Schwann cell RNA appears concentrated in nodes of Ranvier and Schmidt-Lanterman incisures, the sites of initial myelin structural changes during Wallerian degeneration. That the transfer of RNA and organelles could occur through two plasma membranes is remarkable. Currently it is not understood how this interchange proceeds but it has been suggested that exosomes play a role based upon observations of vesicles with exosomal characteristics in Schwann cells and axons under normal and regenerative conditions.56–58
The interdependence of Schwann cells and axons through signaling not only affords a potential target for neurotoxicants but also suggests that monitoring this interaction may provide a biochemical approach to detecting neuropathy. One marker that has shown promise for assessing the integrity of the Schwann cell axon relationship is p75, which is expressed on Schwann cell precursors and nonmyelinating Schwann cells but not on myelinating Schwann cells. Accordingly, p75 levels are normally low in peripheral nerve but are upregulated in primary demyelinating conditions and as a secondary response to axon injury. This suggests that p75 expression could serve as a marker for neuropathy and upregulation of p75 has been detected in the sciatic nerves of rats administered isoniazid, acrylamide or carbon disulfide at time points prior to the development of morphological or behavioral disturbances.59,60 Additionally, increased levels of p75 protein in serum have been observed in animal models for diabetic neuropathy and nerve crush injury.61 Therefore, analysis of p75 either at the protein or message level could provide a biochemical method for early detection of neuropathy. There is also the potential that analysis of the temporal relationship of p75 expression to other markers of injury could provide insight into the type of neuropathy. For example if myelin specific genes such as myelin protein zero (P0) or myelin basic protein (MBP) were down regulated early prior to changes in axonal markers, it would be consistent with a primary myelinopathy.
Examples are scarce for which a single mechanism is generally accepted to account for all the pathological events observed in a given toxic neuropathy. The major points of contention when considering mechanisms tend to center on the initial critical event and the most relevant molecular target. Typically the more a neurotoxicant is investigated the greater the number of interpretations that exist and for most neurotoxicants there are several potential credible contributing events that are not necessarily mutually exclusive. Ultimately though there are a smaller number of general pathogenic processes through which toxic agents mediate their effects. Among these processes are covalent or non-covalent modifications, compromised energy or protein biosynthesis, oxidative injury and disruption of ionic gradients across membranes. These general mechanisms are common to most toxicants and what bestows selective toxicity for the peripheral nervous system is an ability to target a vital process or structure unique to the peripheral nervous system or some characteristic that favors accumulation of the agent within the peripheral nervous system
Vulnerabilities of Axons to Toxicants
The need to rapidly conduct impulses over relatively great distances presents the nervous system with considerable challenges. The typical cell shape is elongated with a narrow efferent axonal appendage extending from the neuronal perikaryon that in humans may be a meter in length and in larger species even longer. The cylindrical axon extension enables the cell to carry action potentials over distance while minimizing the cytoplasmic volume and cell surface area. However, despite the small axon diameter the axoplasmic volume can be hundreds to thousands of times greater than the cytoplasmic volume within the neuronal cell body due to the axon length. Because the perikaryon is a major source of biosynthesis for components of the axon, there is an essential requirement to distribute these biomolecules that include neurotransmitters, proteins and organelles along the length of the axon via axonal transport.62,63
Transport of cargo synthesized within the neuron proceeds along microtubules mediated by the ATP-dependent microtubule motors kinesin and dynein (Fig. 1).63 These molecules provide mechanochemical force through physical interaction with microtubules and their ATPase activity. Depending on the cargo transported the observed overall rates of movement vary. The fastest component moves about 400 mm/day and is primarily carrying vesicles. An intermediate component of transport moving 50 mm/day is associated with organelles. The slowest component transports cytoskeletal elements and is usually divided into slow component a (SCa) and slow component b (SCb). SCa moves at a rate of 1 mm/day carrying microtubules and neurofilaments with SCb proceeds at a rate of 2–4mm/day carrying microfilaments and microfilament-associated proteins.
Figure 1.
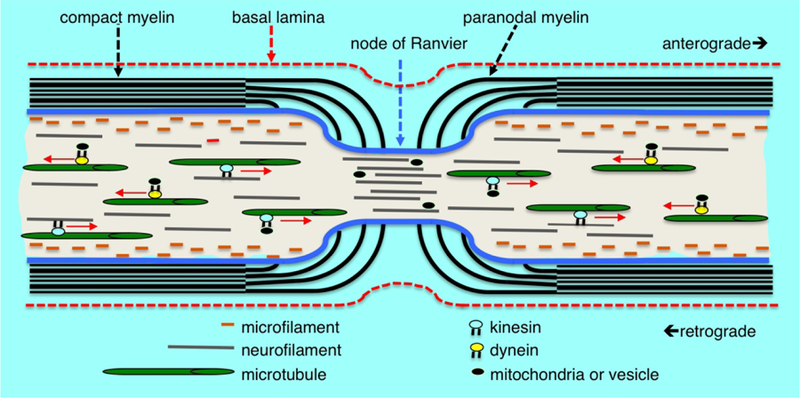
Myelin structure, cytoskeleton arrangement and axonal transport at the node of Ranvier. Microfilaments of polymeric actin align along the axolemma, neurofilaments are distributed throughout the axoplasm and microtubules are arranged more centrally in the axoplasm. Cytoskeletal components, vesicles and mitochondria are transported anterograde and retrograde along microtubule tracts by the mechano-chemical motors kinesin and dynein, respectively. Compact myelin opens into cytoplasmic loops in the paranodal region with microvilli projections contacting the axolemma. There is a rapid decrease in the axonal diameter at the node of Ranvier accompanied by an increased density of neurofilaments having a decreased state of phosphorylation. There is also an increased density of vesicles and other organelles within the axon at the node suggesting an impediment to transport at the nodal constriction.
Anterograde and retrograde transport are both essential with the former providing a vital supply line of biosynthetic components and the latter thought to inform neurons on the status of the distal axon. As demonstrated by experiments, transecting or ligating axons stopping transport results in degeneration of the distal axon. Integral to maintaining normal structure and transport functions are the filamentous proteins of the axonal cytoskeleton.62 These proteins comprise three families categorized based upon their size. The smallest microfilaments of assembled actin filaments are located along the periphery of the axoplasm juxtaposed to the axolemma. The intermediate filament network composed of neurofilament protein polymers extends throughout the axoplasm and plays a role in maintaining axon diameter modulated by associated Schwann cells. Microtubules composed of assembled α/β tubulin dimers arranged in polarized linear arrays more centrally located within the axoplasm are the largest cytoskeleton component. The microtubules play an essential role through providing structural tracts for anterograde and retrograde axonal transport. These cytoskeletal components are all dynamic structures that involve controlled assembly and disassembly to achieve their own transport along the axon and perform their structural functions. Considering that the slowest components migrate on the order of 1 mm/day and that axons can be over a meter in length provides a considerable period of time for cumulative events such as covalent modifications to accumulate on cytoskeletal proteins. Thus collectively the structural components and physiological processes required for axonal transport present unique targets that can account for the ability of some neurotoxicants to produce a primary axonopathy. Chemotherapeutic agents that compromise the dynamics of tubulin elongation and disassembly to disrupt mitotic spindle formation in cancer cells as well as certain environmental chemicals that covalently modify cytoskeletal proteins in a cumulative manner are examples of axonal toxicants that act through this mechanism.62,64,65
Toxic Disorders of Axons
Axonal degeneration is the most commonly observed lesion in toxic peripheral neuropathies. This lesion can be classified as either primary or secondary depending on whether the critical effect is within the axon or another component of the peripheral nervous system, respectively. Using transected nerves in frogs Augustus Waller described a sequence of structural changes in the degenerating axon segment distal to the transection.66 Accordingly the sequence of events he reported is referred to as Wallerian degeneration. Because the structural changes observed in degenerating axons of toxic neuropathies appear to occur through a similar sequence of events it is often referred to as Wallerian-like axonal degeneration.
Degenerating axons can present in a variety of morphologic profiles depending on their stage of degeneration (Fig. 2). In primary axonal degeneration the initial injury occurs within the axon but is accompanied by changes in the associated glial cells and blood-nerve barrier that collectively provide a microenvironment supportive of axonal regeneration. Typically, the first observable structural change is proteolytic digestion of the axolemma and axoplasm leaving only a myelin sheath surrounding a swollen degenerate axon (Fig. 2b). At the light microscopic level this is identifiable as a loss of protein staining within the axoplasm. Axon degeneration proceeds through a programmed pathway of signaling leading to cytoskeletal degeneration. Studies stemming from the discovery of Wallerian Degeneration Slow (Wlds) mice that have a mutation in the enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT) have provided insight into some of the events triggering degeneration. Loss of function of NMNAT2, but not either of the other two isoforms of NMNAT, produces axonal degeneration.67,68 A constant supply of NMNAT2 is required in the axon through axonal transport to prevent degeneration. Therefore disruption of axonal NMNAT2 supply through blocked transport, increased proteosomal breakdown or decreased translation caused by a toxicant is expected to initiate degeneration. Studies have also identified sterile alpha and TIR motif-containing 1 protein (SARM1) as a pro-regulator of degeneration that acts downstream of NMNAT2; and depleting SARM1 in animals that have loss of NMNAT2 function prevents degeneration.68–70 An important consequence of increased SARM1 activity or loss of NMNAT2 function is the activation of dual-leucine-zipper-bearing kinases/mitogen activated protein kinases (DLK/MAPK) signaling that leads to calpain-mediated proteolysis.71,72 Maintenance of myelin-axon contact is essential to supporting intercellular signals across the periaxolemmal space; and if the axon is injured or destroyed the Schwann cell ceases to support the myelin sheath.73,74 The earliest changes in Schwann cells occur in non-compact myelin regions and are present prior to axonal changes observable at the light microscopic level (Fig. 3). These morphological changes include paranodal retraction and dilatation of Schmidt-Lantern incisures.75–80 At the molecular level increased expression of lipolytic enzymes such as phospholipase A2 and demyelinating pathways including the NRG1-erbB2 pathway are activated.81–84 The dilatation of Schmidt-Lanterman incisures results in elevated hydrostatic pressure that displaces compact myelin to form intracellular myelin ovoids (Figure 2c). Studies suggest that actin polymerization and recycling of E-cadherin contribute to these changes in Schmidt-Lanterman clefts and paranodal regions.81 Accompanying the formation of myelin ovoids Schwann cells become hypertrophic, filling with rough endoplasmic reticulum and various vesicular structures and disintegrating compact myelin (Fig. 4a).
Figure 2.


Toluidine blue stained plastic embedded sections of rat sciatic nerve showing stages of axonal degeneration after crush injury. (a) Cross section from a control rat showing the normal axon density and bimodal distribution of large and small myelinated axons. The myelinated axons are surrounded by compact myelin and protein staining of the cytoskeletal and mitochondrial components in the axoplasm is evident. Two Schwann cells (*) have been sectioned at the middle of the internode through their cytoplasm and nucleus and axons sectioned through Schmidt-Lanterman incisures are present (arrows). Clusters of nonmyelinated axons (arrrowheads) are present and a blood vessel (v) is located along the lower boarder of the section. (b) Cross section obtained distal to crush injury. There is a decreased density of axons in the section with very few axons exhibiting normal axoplasm staining. Axons in early stages of axonal degeneration (*) are identifiable by the loss of axoplasm and axolemma staining with the myelin sheath still intact. Many axons are in later stages of axonal degeneration (arrows) demonstrating the progression of myelin collapse to form ovoids. Nucleated cells (arrowheads) present are consistent with dedifferentiated Schwann cells, macrophages or fibroblasts.
Figure 3.
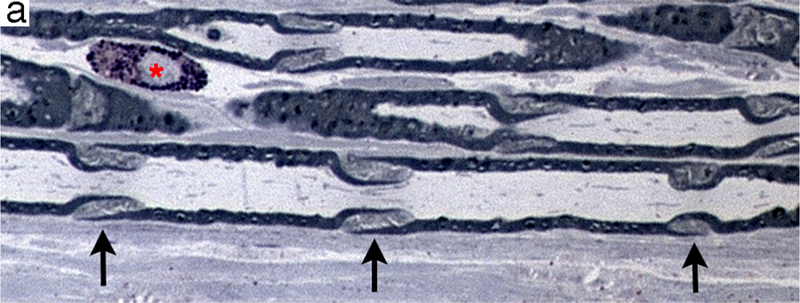

Regions of non-compact myelin that are affected earliest during axonal degeneration. (a) Longitudinal section of plastic-embedded toluidine blue-stained rat sciatic nerve showing Schmidt-Lanterman incisures (arrows) distributed along the internode. These structures consist of non-compact myelin that provide a cytoplasmic pathway connecting the Schwann cell cytoplasm adjacent to the axon to that on the external myelin sheath. A mast cell (*) is present in the endoneurium. (b) Transverse section through the paranodal region of a large myelinated fiber. The axon (ax) is constricted and contains a high density of neuroflaments and organelles. The compact myelin has opened to form folded myelin (fm) and microvilli (mv) that contact the axolemma.
Figure 4.
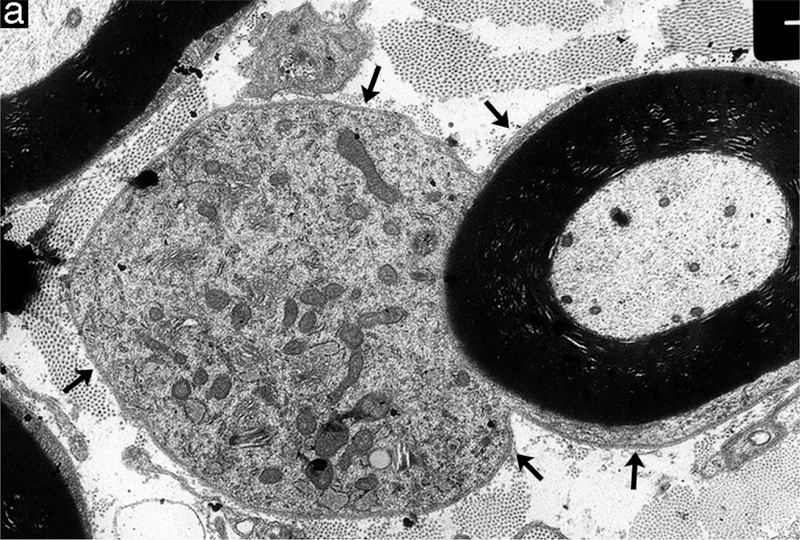
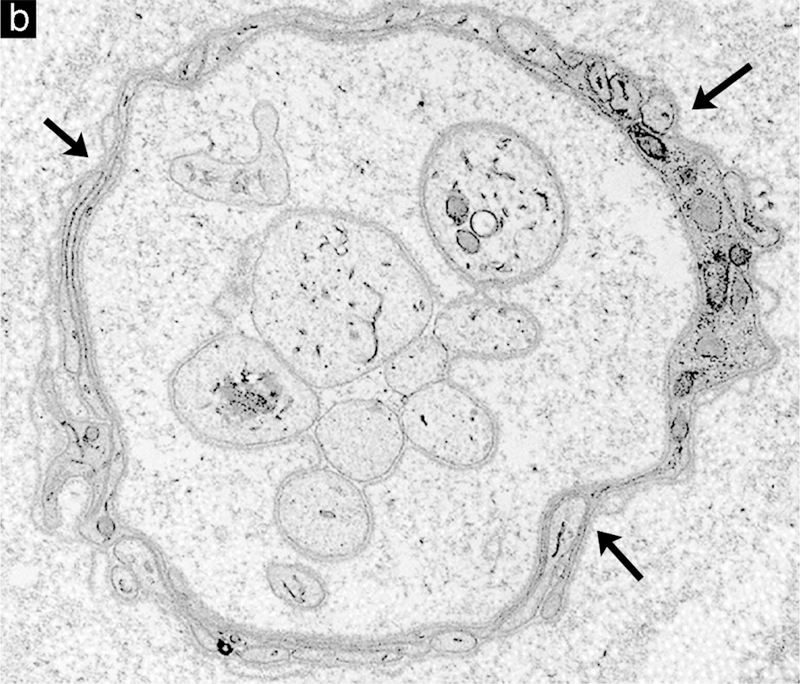

Examples of degenerative and regenerative responses of Schwann cells and axons. (a) Schwann cell hyperplasia exhibiting an increased cytoplasmic volume, enclosed by the extension of basal lamina (arrows), that is filled with rough endoplasmic reticulum and various vesicular structures from disrupted membranous organelles. (b) A cluster of regenerating axonal sprouts that have not yet been myelinated surrounded by persistent basal lamina from previous Schwann cells and the processes of supernumerary Schwann cells (arrows). (c) A regenerated axon (A) ensheathed with thin myelin (M) from its current Schwann cell sectioned through the cell body showing the nucleus (N) and cytoplasm (C) that is surrounded by persistent basal lamina (arrows) of previous Schwann cells and the processes of supernumerary Schwann cells (arrowheads).
Secondary demyelination accompanying Wallerian-like degeneration is not a result of Schwann cell death by necrosis or apoptosis but an active process mediated through reprogramming of myelinating Schwann cells to a demyelinating phenotype. At the molecular level this reprogramming likely recapitulates the events of primary demyelination once the triggering signal from the axon has been transduced. Schwann cells down-regulate lipid and myelin-related proteins and re-express genes that were active in pro-myelinating or previous immature states. However the demyelinating Schwann cell is not just a copy of an immature state and expresses some different genes including sonic hedgehog (SHH) and oligocyte transcription factor 1 (Olig1).85,86 A recognized event in the reprogramming is activation of c-Jun; down-regulating c-Jun delays demyelination after injury and prolongs regeneration.85,87,88 Upregulation of c-Jun appears to be a key factor in reprogramming as it upregulates genes related to neuronal growth and regeneration including N-cadherin, p75 and neuronal cell adhesion molecule (NCAM) and the signaling molecules glial cell-derived neurotrophic factor (GDNF), artemin, SHH, and brain-derived neurotrophic factor BDNF while it downregulates myelin related genes including early growth response protein 2 (Egr2), P0, MBP, periaxin and E-cadherin.85,87,89
There are a number of possible ways Schwann cells can be signaled into the demyelination program and in severed nerves the process starts rapidly within the first hour. However the exact molecular events responsible for initiating the process are not known. One hypothesis is that erbB½ receptors that have been observed to become phosphorylated within minutes of injury contribute to the triggering process.82 Phosphorylation of erbB receptors occurs earliest at Schwann cell microvilli located at the nodes of Ranvier and is then propagated along the plasma membrane. This modification activates Ras-related C3 botulinum substrate GTPase (rac1-GTPase) regulation of actin polymerization, an early event in Schmidt-Lanterman incisure collapse and paranodal myelin retraction. Interestingly Mycobacterium leprae that binds to the alpha-dystroglycan receptor of laminin 2 of Schwann cells produces demyelination and has been observed to activate erbB2.90 This would suggest that the erbB receptors that are activated by NRG1 for proliferation, survival and myelination could also play a role in demyelination.
Removal of myelin debris is more efficient in the peripheral nervous system than the central nervous system and is a concerted effort between demyelinating Schwann cells and macrophages.91,92 Schwann cells appear capable of digesting some of their own myelin sheath but a substantial fraction of the degrading myelin along with parts of the degrading axon are also ejected by the Schwann cell. The ejected myelin is then cleared by macrophages in part by resident macrophages present within the endoneurium and by recruited circulating hematogenous macrophages. Both Schwann cells and macrophages are able to phagocytose extracellular myelin but in addition Schwann cells appear to clear their own intracellular myelin debris through autophagy based upon the presence of autophagosome-like structures and high levels of lysozyme in hypertrophic Schwann cells.93
Additionally, Schwann cells dedifferentiate to a premyelinating mitotic phenotype that align along the basal lamina that continuously encircles the outer Schwann cell plasma membrane over the entire length of each axon including nodes of Ranvier in the peripheral nervous system to form tubular structures referred to as bands of Büngner (Fig. 4b).94,95 Through providing structural guidance and trophic support by expressing several growth factors such as artemin, BDNF and GDNF for the neurites sprouting from the proximal stump these bands enhance the regenerating potential of axons in peripheral nerve.89,96–99 Specialized terminal non-myelinating Schwann cells also guide axon terminals to the neuromuscular junction.100 Once axon regeneration is complete the repair phase Schwann cells stop dividing and differentiate into either the myelinating or nonmyelinating phenotype Schwann cells. The telling signs of remyelination are shorter internodes and a thinner myelin sheath (Fig. 4c).101 Typically the ratio of internode length (L) to myelin outer diameter (D) is ~100 and the ratio of the inner myelin diameter (d) measured between nodes of Ranvier to the outer myelin diameter (D) referred to as the g-ratio is 0.7–0.8.102–104 Thus when L/D is less than 100 and the g-ratio greater than 0.8 it is consistent with remyelination. In contrast axons within the central nervous system do not have an associated basal lamina, proliferating astrocytes form glial scars and the presence of persistent myelin debris all inhibit neurite outgrowth diminishing the potential of axon regeneration. However, even in peripheral nerve, if axon/Schwann cell contact is not reestablished eventually the Schwann cells and bands of Büngner will diminish and be replaced with collagen decreasing the probability of successful regeneration.
Secondary axonal degeneration is either a consequence of neuronal perikaryon death or persistent primary demyelination. In the case of neuronal cell death the associated dendrites and axonal cellular processes are lost without the potential of regeneration. In the case of primary demyelination, the axonal response varies depending on the type and duration of the myelin injury. During incomplete demyelinating conditions such as intramyelinic edema or early stages of complete demyelinating conditions, axonal atrophy is a common finding whereas ongoing complete demyelination can progress to secondary axonal degeneration.
Another type of toxic axonopathy are the neurofilamentous axonopathies that involve accumulations of neurofilaments at various distances from the cell body and in some cases within the perikaryon (Fig. 5).64 Neurofilaments are type IV intermediate filaments that are heteropolymers of non-covalently associated neurofilament protein subunits.62 The subunits include three neuroflament proteins termed low, medium and high molecular weight neurofilament protein based on their rates of migration in gel electrophoresis and lengths of their carboxy terminus. Additionally the filamentous structures also incorporate alpha internexin and peripherin as additional subunits. The neurofilament component of the cytoskeleton is thought to regulate axon diameter and thus plays a role in conduction speed. Schwann cells locally modulate levels of neurofilament phosphorylation providing a potential link between loss of Schwann cell contact and axonal atrophy.105–107 A number of chemicals have been identified through human exposures or experimental animals that produce significant alterations in neurofilament distribution that are accompanied by varying levels of axonal degeneration. These chemicals include carbon disulfide, hexane, methyl-n-butyl ketone, acrylamide and 3,3’-iminodiproprionitrile among others.
Figure 5.


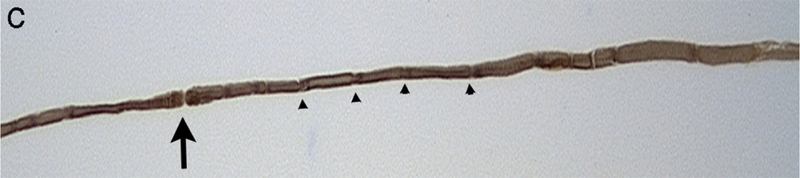

Neurofilamentous axonopathy produced by carbon disulfide released from N,N-diethyldithiocarbamate following oral administration in the muscular branch of the posterior tibial nerve of a rat. (a) Transverse section of large and small myelinated axons obtained from a control animal showing the normal thickness of compact myelin relative to axon diameter and the density and distribution of cytoskeletal components. (b) Transverse section of a myelinated axon present in a treated animal. There is a high density of disorganized neurofilaments in the swollen axon compared to the orderly distribution and alignment in the normal axon. Additionally the myelin is thin and there is sequestering of organelles to the periphery. (c) Teased fiber from a control animal showing a constant diameter and even myelin staining along the internode. A node of Ranvier (arrow) is present and Schmidt-Lanterman incisures (arrowheads) are distributed along the axon. (d) Teased fiber from a treated animal showing the presence of fusiform swellings.
Examples of Agents Producing Peripheral Axonopathies
Taxanes
Paclitaxel is an extract from the Pacific yew tree (Taxus brevifolia) and docetaxol is a semi-synthetic analog of paclitaxel. Both are used as chemotherapeutic agents for several cancers including breast, ovarian and lung cancers. Paclitaxel is more neurotoxic, but both agents can produce dose-dependent neuropathies.4,22,65 The sensory system is more sensitive but motor involvement can also occur. Clinical signs include numbness, neuropathic pain, and cold and mechanical allodynia. The neuropathy is characterized by axonal degeneration with secondary demyelination. An early change is disrupted growth at the distal tips of sensory axons that require ongoing growth to maintain innervation of the epidermis as it turns over.
These compounds cross plasma membranes and bind to the N-terminal region of β-tubulin. This leads to enhanced polymerization and decreased depolymerization.108,109 Integrity of microtubule dynamics is required for axonal transport; and disturbance of microtubule function is thought to interrupt transport of essential cellular components.110 More recently, the observation that paclitaxel decreased levels of some axonal mRNAs but fast transport of organelles was not disrupted suggests this compound may have selective effects on components of transport.111,112 Additional contributing mechanisms that have been proposed include interacting with mitochondria to open the mitochondrial permeability transition pore and activation of calpains.113,114
Vinca Alkaloids
Vincristine and the related vinca alkaloids vinblastine, vinfluine, and vinorelbine are important chemotherapeutics used primarily for hematological cancers. Vincristine is the most neurotoxic and has a high prevalence of peripheral neuropathy associated with treatment.22,65 Neurotoxicity is a common reason to stop treatment; clinical signs may worsen for a period after treatment is stopped. Clinically, sensory signs develop earliest but are often followed by motor and autonomic involvement.
These compounds bind to free tubulin causing the free tubulin to aggregate into non functional polymers, which decreases the free tubulin available to be incorporated into microtubules.115 Similar to taxanes, the disturbance of microtubule function is thought to impair normal axonal transport function.23,110 Vincristine has also been reported to interact with mitochondrial membrane permeability to decrease the uptake and efflux of Ca2+, potentially disturbing intracellular signaling.114
Hexane and Methyl-n-butyl ketone (2-hexanone)
The neurotoxicity of these compounds was recognized by the development of a sensorimotor distal axonopathy in furniture and leather workers and from intentional repeated inhalation of hexane-containing glues.116–119 The ability to reproduce the neuropathy in experimental animals has provided considerable insight into the lesions and molecular mechanisms contributing to the neuropathy.120 Morphologically the neuropathy is characterized by a central peripheral distal neurofilamentous axonopathy. The distal parts of the largest diameter longest myelinated axons are most susceptible and this is reflected in the relative sensitivity of species based on axon size and length. Aggregates of neurofilament protein accumulate in the sub-terminal axon creating axonal swellings accompanied by myelin retraction that often occurs just proximal to the axonal constrictions at nodes of Ranvier. Electron micrographs of these swellings are consistent with an interruption of axonal transport with neurofilamentous aggregates proximal and accumulation of vesicles distal to the nodal constrictions. With continued exposure there is development of axonal atrophy followed by axonal degeneration distal to the swellings.
Hexane is probably one of the most studied toxic neuropathies and there is considerable knowledge regarding the necessary chemical properties of the toxic species and their reactions with proteins. However, there is still debate regarding the most relevant molecular targets and biological processes responsible for axonal degradation.
Investigations in several laboratories have helped to understand the neurotoxic mechanism and based on these studies the neurotoxic conditions are referred to as γ-diketone axonopathies.64,121–123 Both hexane and methyl-n-butylketone are metabolized through ω−1 oxidation to the same neurotoxic species, 2,5-hexanedione, possessing γ spacing of the ketone functions. The requirement for this spacing is well established and has been supported more recently through the use of aromatic analogs such as 1,2-diacetylbenzene.124,125 The γ-diketones are able to react with amino groups of protein lysyl residues to generate 2,5-dimethylpyrrole adducts (Fig 6a). That a subsequent step involving oxidation of the pyrrole adduct to an electrophilic intermediate, that reacts with a protein nucleophile to generate protein cross-links has been supported experimentally. The 2,3-dimethyl analog of 2,5-hexanedione that forms pyrroles and cross-links more rapidly than 2,5-hexanedione was observed to be a more potent neurotoxicant that produced swellings in more proximal locations.126,127 In contrast the 3-acetyl analog of 2,5-hexanedione that forms pyrroles rapidly but does not undergo oxidation and cross-linking did not produce axonal swellings or clinical signs of neurotoxicity.128 Considering their involvement in the axonal swellings, high content of lysyl residues and long biological life time for cumulative derivatization the neurofilament proteins have been proposed as a relevant molecular target. One hypothesis proposes that cross-linking of the neurofilament subunits limits their assembly and disassembly required for transport, particularly at the axonal constrictions occurring at nodes of Ranvier, leading to their accumulation and a blockade of transport.122 However, whether neurofilament accumulation is a critical event to subsequent axonal degeneration has not been clearly established. Studies using crayfish and transgenic mice that did not express neurofilament proteins have exhibited neurotoxicity from 2,5-hexanedione suggesting the involvement of additional mechanisms and that neurofilaments are not a requirement for axonal degeneration.129,130 More recently studies have characterized changes in the transport motors kinesin and dynein and certain microtubule-associated proteins as potential targets contributing to γ-diketone neuropathy.131
Figure 6.
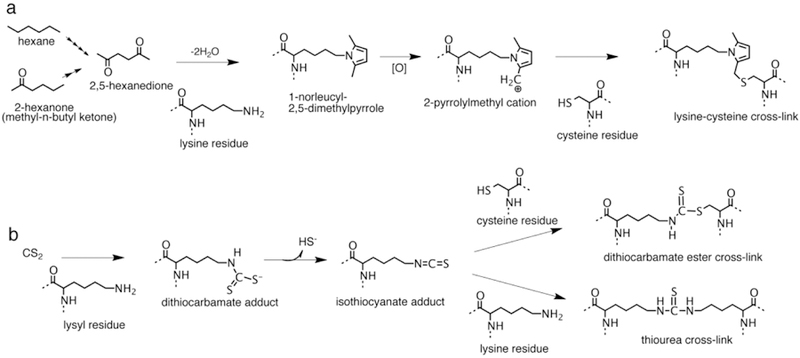
Protein cross-linking pathways for hexane, methyl-n-butyl ketone and carbon disulfide (CS2). (a) Hexane and methyl-n-butyl ketone are metabolized to 2,5-hexanedione through multiple steps of −1 oxidation. 2,5-Hexanedione reacts with protein lysyl residues to generate a 2,5-dimethylpyrrole adduct. In the presence of oxygen the pyrrole adduct oxidizes to a cationic species susceptible to nucleophilic addition by protein amino and sulfhydryl groups producing protein cross-links. Reaction of the oxidized pyrrole adduct with a cysteine residue to produce a possible lysine-cysteine cross-link is shown. (b) Carbon disulfide reacts with protein lysyl residues to form a monoalkyldithiocarbamate adduct. The dithiocarbamate adduct undergoes facile elimination of sulfhydryl ion creating an electrophilic isothiocyanate adduct. The isothiocyanate adduct then undergoes nucleophilic addition by protein sulfhydryl or amino groups to generate dithiocarbamate ester or thiourea protein cross-links.
Carbon Disulfide
The neurotoxicity of this chemical was first recognized during the 19th century associated with the cold vulcanization of rubber in cottage industries and exposure to carbon disulfide is currently a concern in rayon and cellophane production.132 Human cases and experimental models have established that carbon disulfide produces a sensorimotor neurofilamentous distal axonopathy identical to that of hexane (Fig. 5).133
The clinical similarities in these two neuropathies guided mechanistic studies to determine whether a parallel series of protein adduction and cross-linking steps contributed to carbon disulfide neurotoxicity. Using in vitro and in vivo systems it was demonstrated that carbon disulfide can react with amino groups of lysyl residues to generate a monoalkyl dithiocarbamate adduct (Fig. 6b).134–136 The dithiocarbamate adducts undergo a facile elimination of sulfhydryl ion to generate electrophilic isothiocyanate adducts followed by nucleophilic addition of lysyl or sulfhydryl functions to produce thiourea or dithiocarbamate ester cross-links, respectively. Similar to hexane it has been postulated that the stability and long transit time of neurofilament proteins make them the toxicologically relevant target for the development of neurofilamentous swellings in chronic carbon disulfide neuropathy.64,122
1-Bromopropane
Efforts to decrease the production of ozone-depleting solvents such as chlorofluorocarbons has led to the use of new chemicals including 1-bromopropane. Currently 1-bromopropane is used as a solvent vehicle for spray adhesives and for cleaning and degreasing of metal components, electronics, optical instruments and ceramics.137 Clinical reports of human cases and animal studies have demonstrated the neurotoxicity of 1-bromopropane.6,8,138–141 The clinical signs reported included muscle weakness, paresthesias, numbness, urinary incontinence and memory disturbances. Inhalation studies in rats demonstrated a dose-dependent neurotoxicity characterized by decreased forelimb and hind limb grip strengths, decreased motor nerve conduction velocity, increased distal latency, axonal degeneration in the muscular branch of the posterior tibial nerve and axonal swellings in the gracile nucleus.142 The mechanism of 1-bromopropane neurotoxicity has not been delineated but biochemical studies have provided information on the interaction of 1-bromoproane in biological systems. 1-Bromopropane produces S-propyl cysteine adducts on globin in humans and rats and this protein modification has also been identified within the nervous system on neurofilaments of rats exposed to this chemical.143 This alkylating effect could explain the low levels of protein sulfhydryl functions observed in the brains of rats exposed to 1-bromopropane and suggests that similar to the γ-diketones covalent protein modification may be a contributing process to neurotoxicity.144,145 Further investigations will be needed to clarify the role of sulfhydryl modifications to the mechanism of 1-bromopropane.
Vulnerabilities of Schwann Cells and Myelin to Toxicants
Schwann cells ensheath most axons in peripheral nerve. Differentiated Schwann cells can be categorized into several groups based upon their morphology, biochemical composition and the size and location of the axon or axons they associate with. These categories include the myelinating Schwann cells that each associate with a single large motor and certain sensory axons (Fig. 7a), nonmyelinating Schwann cells that each wrap multiple small diameter axons of c-fibers from sensory and postganglionic sympathetic neurons to form Remak bundles (Fig. 7b), paranodal Schwann cells whose compact myelin opens to form myelin villi near nodes of Ranvier (Fig. 3), perisynaptic Schwann cells that are located distally at neuromuscular junctions where they partially wrap around the presynaptic terminal of motor axons, and the satellite cells that associate with neurons in the peripheral nervous system ganglia (Fig. 7c). Our understanding of myelinating Schwann cells and their response to toxicants is the most advanced and will be the primary subject in this discussion.
Figure 7.


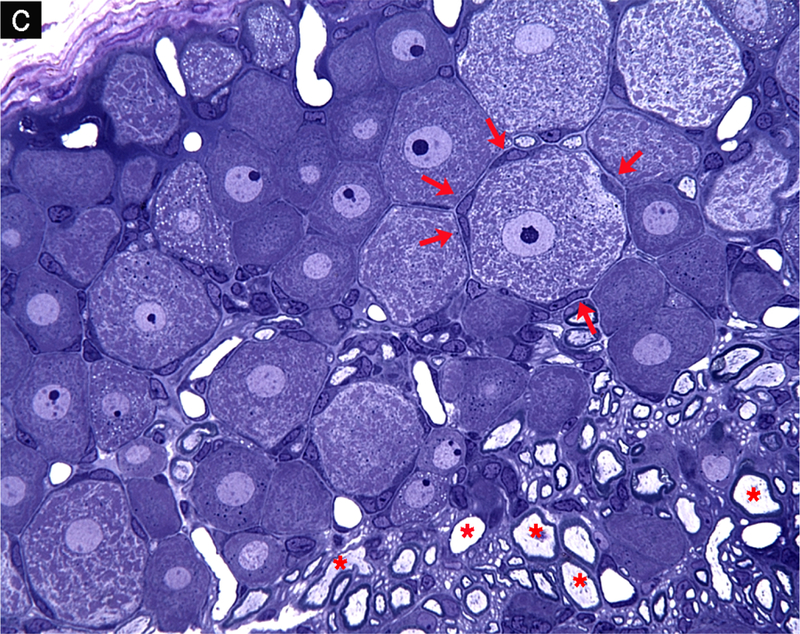
Schwann cell phenotypes. (a) Transverse section through the cell body of a myelinating Schwann cell showing the nucleus (N), cytoplasm (C), compact myelin (M), axon (A) and basal lamina (arrows). (b) Groups of nonmyelinated axons showing the cell bodies of 2 nonmyelinating Schwann cells (*) with their basal lamina wrapped around multiple axons. (c) Plastic-embedded transverse section from a rat dorsal root ganglion showing neurons that have large central nuclei with prominent nucleoli and abundant Nissl substance in the cytoplasm, neurons encircled by multiple satellite cells (arrows) and myelinated axons (*).
Schwann cells associated with larger-diameter axons produce myelin that increases conduction velocity and decreases energy requirements for membrane repolarization.146 They form a lipid-rich wrapping of compact myelin that acts as insulation along axons. This insulation is interrupted at nodes of Ranvier where ion channels are expressed enabling localized depolarization and repolarization that is not perpetuated through the myelin but can depolarize adjacent nodes. The result is saltatory conduction having an increased speed and lower energy requirement than a continuous membrane depolarization along the length of the axon. To perform its insulating function peripheral nerve myelin is ~80% lipid as compared to most plasma membranes that are ~50% lipid.147 The polyunsaturated fatty acids in the myelin plasma membrane are susceptible targets for lipid peroxidation resulting from agents producing free radicals directly or that increase oxidative stress through other mechanisms. The formation and maintenance of compact myelin is dependent upon a number of membrane-associated proteins and the metabolism of specific lipids present within the lipid bilayers (Fig. 8). MBP is a myelin protein thought to stabilize the closely opposed inner cytoplasmic membranes that form the major dense line. P0 is a major protein component of myelin thought to stabilize the junction of the outer plasma membranes through homophilic interactions to form the intraperiod line of compact myelin.148 Additionally in order to maintain the integrity of compact myelin, energy-dependent ion pumps thought to include carbonic anhydrase are required to exclude water from the intraperiod line between the tight wrappings of plasma membrane.149 The metabolic processes and structural features required for compact myelin provide unique targets for toxic agents, particularly those that are hydrophobic that can cross the blood-nerve barrier and accumulate in myelin.
Figure 8.
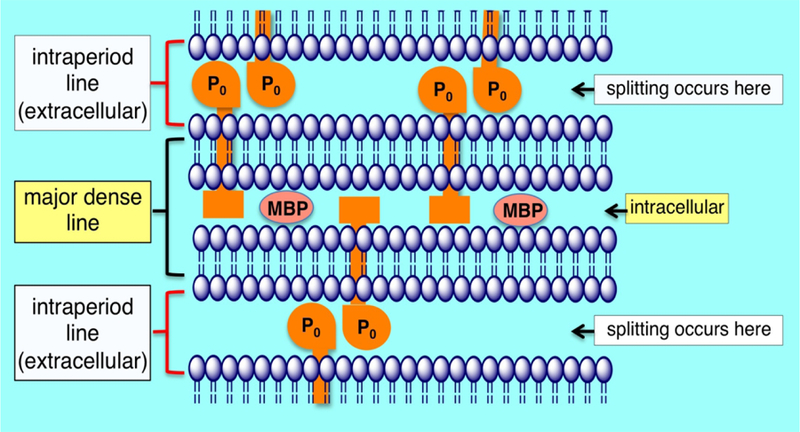
Schematic of peripheral nerve compact myelin. The intraperiod line is formed by the association of the exterior surfaces of the Schwann cell plasma membrane wrapped around the axon. The apposition of the external surfaces is stabilized in part by homophilic interactions of P0 protein. Energy dependent ion pumps thought to include carbonic anhydrase are required to exclude water from the intraperiod line, the typical location that myelin splitting occurs. The major dense line is formed through the exclusion of cytoplasm and close association of the interior surfaces of the Schwann cell plasma membrane. Myelin basic protein is important in stabilizing the apposition of the interior plasma membrane surfaces.
Toxic Disorders of Schwann Cells and Myelin
The high lipid content of myelin introduces unique challenges to fixation and embedding. Therefore when examining the peripheral nervous system for myelin lesions, it is particularly important to discern between lesions and processing artifacts.150 A common artifact that can be misleading is myelin blebbing that results from inadequate dehydration for embedding in plastic. Experimental treatment-related changes can be distinguished from blebbing artifact by the symmetric distribution of the blebs around the axons and the uniform inclusion of all axons within a section. In contrast non-artifact myelin lesions exhibit a range of severity among axons within a section and the lesions of affected axons are typically asymmetric. Other common artifacts include those resulting from crush during handling of the nerve and incomplete penetration of osmium used for the lipid fixation.
As described for axonopathies, myelinopathies can be classified as either primary or secondary. Once demyelination is initiated, the morphological changes including paranodal retraction, collapse of Schmidt-Lanterman incisures, lysosome induction and certain molecular changes are shared suggesting the existence of a common demyelinating phenotype.73 For the case of secondary myelinopathies resulting from axonopathies, similar stages of response are expected for all of the Schwann cells along the entire length of the affected axon section. In contrast individual Schwann cells, typically those maintaining the longest internodes and thickest myelin sheaths of the largest axons, are affected initially in primary demyelination. This can result in segmental demyelination and is thought to result from the greater metabolic demands placed upon this population of Schwann cells (Figs. 9a,b). Significantly, the loss of a single internode can be sufficient to interrupt saltatory conduction.
Figure 9.

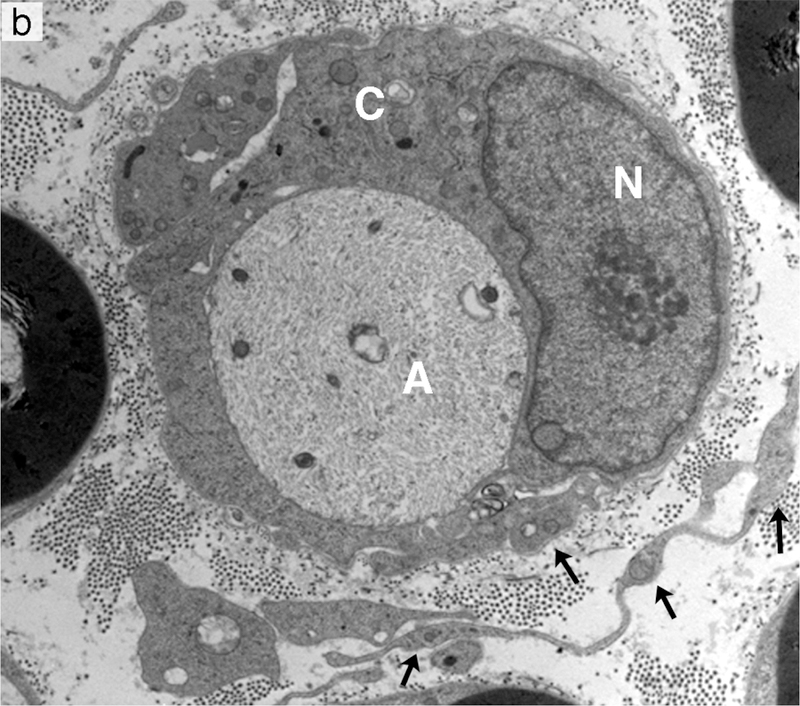
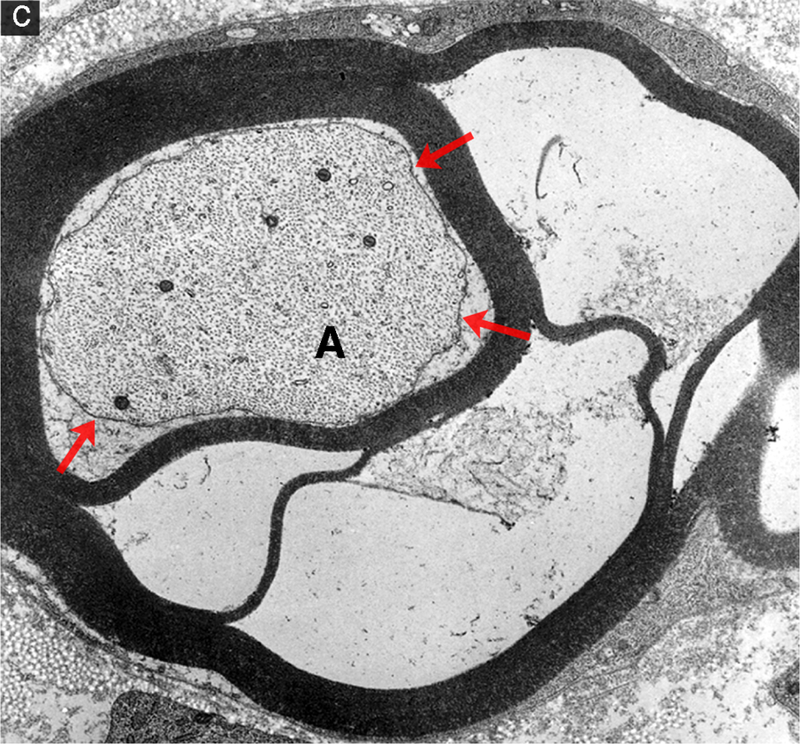
Segmental demyelination and intramyelinic edema in the sciatic nerve of rats administered disulfiram. (a) Teased fiber from an animal exposed to disulfiram showing loss of myelin from an entire internode. An abrupt change to normal myelin thickness is seen on both sides of the demyelinated internode at each node of Ranvier. (b) A demyelinated axon (A) surrounded by a Schwann cell sectioned through the cell body showing the nucleus (N) and increased cytoplasmic volume (C) filled with vesicular structures and debris. Processes of supernumerary Schwann cells are present (arrows). (c) Several layers of the compact myelin lamella have separated and the space created is filled with fluid (intramyelinic edema). The axon (A) has a relatively normal density and distribution of mitochondria and neurofilaments but the axolemma (arrows) has separated from contact with the Schwann cell plasma membrane at the inner layers of compact myelin.
Integral to the structural integrity of compact myelin are the interactions of the associated membrane proteins and the energy-dependent processes required to exclude water. Agents that compromise these processes can produce myelin splitting or intramyelinic edema. This lesion is characterized by extracellular separation of the lamella at the intraperiod line formed by the closely opposed outer membrane protein layers of the Schwann cell membrane wrapped around the axon (Fig. 9c). This type of splitting can be observed in the inner, middle or outer lamella of the compact myelin.
Examples of Agents Producing Peripheral Myelinopathies
Hexachlorophene
The use of hexachlorophene in soaps and detergents as an antimicrobial has resulted in neurotoxicity in newborn humans and animals. This compound is hydrophobic, is absorbed through skin, and enters the nervous system where it accumulates in myelin. The greater susceptibility of newborns is thought to result from the incompletely developed blood-brain barrier during this period. A myelinopathy is produced in both the central and peripheral nervous systems that is characterized by splitting of myelin at the intraperiod lines.151–154 The largest axons appear to be the most susceptible and the optic nerve is particularly vulnerable. Mechanistic studies have supported the ability of hexachlorophene to uncouple oxidative phosphorylation and inhibit carbonic anhydrase.155–157 Both of these effects are thought to interfere with the ability of myelin to exclude water resulting in intramyelinic edema that separates the compact myelin at the intraperiod line.
Triethyltin
Neurotoxicity of triethyltin has been recognized through contamination of medications and as an industrial pollutant. It is most recognized for its central nervous system effects although at high concentrations it also affects the peripheral nervous system.158 Splitting at the intraperiod line with myelin swelling is the characteristic lesion.159 Triethyltin can concentrate in myelin due to its hydrophobicity and the presence of high-affinity binding sites for this compound on myelin.160 Investigations have identified several potential mechanisms to contribute to the myelin lesions. Inhibition of oxidative phosphorylation resulting from inhibition of Mg2+-ATPase has been proposed as one mechanism.161–163 Triethyltin appears to alter ion transport across membranes including the mitochondrial inner membrane anion channel and may directly allow Cl− entry into the intraperiod line.164,165 All of these mechanisms could disrupt the active processes required to exclude water or enhance osmotic pressure to draw an increased volume of water into the intraperiod line.
Tellurium
Toxicity in humans is rare but tellurium provides an experimental model to study primary demyelination. When tellurium is administered to weanling rats, it produces segmental demyelination with the longest axons affected earliest leading to hind limb weakness.166–169 Typically only peripheral nerve is affected and older animals are resistant. The myelin injury results from inhibition of squalene epoxidase, an enzyme involved in cholesterol synthesis.170,171 The susceptible period coincides with a period of rapid myelination in the young animals and a correspondingly high requirement for cholesterol.172 Inhibition of cholesterol synthesis in the rapidly growing peripheral nerve results in decreased expression of myelin-related genes including P0 and MBP.173 The down-regulation occurs in all Schwann cells and can lead to apoptotic cell death.174 Because the exposure to tellurium is systemic, it is interesting that the effects are selective for peripheral nerve myelin. The liver is the major source of peripheral cholesterol and the activity of the hepatic rate-limiting enzyme for cholesterol synthesis, hydroxymethylglutaryl-CoA (HMGCoA) reductase, is upregulated in response to tellurium. As a result the circulating levels of cholesterol are maintained for most organs but access of the circulating cholesterol to the nervous system is prevented by the blood-nerve barrier.175 Because Schwann cells do not upregulate HMGCoA reductase, there is a localized deficiency of cholesterol in the most rapidly developing regions of the nervous system.
Diptheria Toxin
Corynebacterium diphtheria is an example of a microbe that produces a toxin targeting Schwann cells. This bacterium colonizes skin and mucous membranes where it produces localized necrosis and affects the upper respiratory tract initially. Necrosis results from the production of an exotoxin by three biotypes of this bacteria (mitis, intermedius and gravis) that can also spread through the circulatory system to affect other organs including the heart and kidneys.176 The peripheral nervous system can also be affected and the occurrence of polyneuropathy has been reported to be as high as 85% in affected children.177 The first neurologic signs occurring within a week of infection involve sites nearest the infection such as paralysis of the soft palate and pharynx. Subsequently, there can be involvement of cranial nerves and then development of a motor-sensory type of peripheral neuropathy occurring from one week to months afterwards. The exotoxin is thought to not cross the blood-brain barrier but to gain access at peripheral ganglia. The pathology is characterized by paranodal and segmental demyelination with associated slowed and blocked conduction. Typically, axons are spared, but in severe cases secondary axonal degeneration may also occur. The toxin is a typical AB type bacterial exotoxin of 62kDa molecular weight comprised of two peptides joined by a disulfide bond.178 The B segment provides selectivity through binding to the uncleaved precursor form of heparin-binding EGF-like growth factor expressed on Schwann cells.179 The toxin is internalized within an endosome through receptor-mediated endocytosis, and then the two peptides are separated.180 The A fragment catalyzes transfer of an adenosine diphosphoribose from NAD to elongation factor-2 that is essential for the translation of proteins. The toxin does not appear to promote degradation of myelin lipids or protein but produces necrosis in Schwann cells through inhibiting protein synthesis.181,182 Young animals have been reported to be more susceptible to the demyelinating effects possibly due to their greater demand of myelin synthesis.183
Disulfiram and N,N-diethyldithiocarbamate
These compounds find a wide array of applications in agriculture, medicine and industry. The most generally recognized neurotoxicity of these compounds is the dose-dependent neuropathy associated with the use of disulfiram in alcohol aversion therapy.184–187 This compound is also currently being investigated as an adjunct treatment for glioblastoma.188 The therapeutic basis for alcohol aversion therapy is disulfiram’s ability to inhibit acetaldehyde dehydrogenase that leads to elevated levels of acetaldehyde following ingestion of ethanol accompanied by flushing, headache and nausea among other unpleasant symptoms and clinical signs. In animal models, oral disulfiram was observed to produce intramyelinic edema and segmental demyelination with accompanying secondary axonal degneration.189 Disulfiram is reduced to two molecules of N,N-diethyldithiocarbamate in vivo; and mechanistic toxicity studies have mainly focused on this metabolite that also finds considerable application on its own in agriculture and industry. Interestingly N,N-diethyldithiocarbamate has been demonstrated to be capable of producing either a primary myelinopathy190 or primary axonopathy191 depending on the route of exposure. When administered orally, N,N-diethyldithiocarbamate undergoes acid promoted decomposition to carbon disulfide and N,N-diethylamine in the acidic environment of the stomach (Fig. 10).192 The CS2 released can be sufficient to produce a neurofilamentous axonopathy through protein adduction and cross-linking (Fig. 6b).136,193 In contrast, parenteral administration of N,N-diethyldithiocarbamate produced a primary myelinopathy characterized by myelin splitting and segmental demyelination similar to that reported for disulfiram.190 Further investigation determined that the myelinopathy was dependent upon intact N,N-diethyldithiocarbamate forming lipophilic copper complexes that accumulated in peripheral nerve including the myelin compartment. The copper complexes are redox active and promote injury through promoting oxidative injury with lipid peroxidation (Fig. 10).194–196 Analogs of N,N-diethyldithiocarbamate that also complex copper but have hydrophilic nitrogen substituents were not able to accumulate copper in nerve or produce a myelinopathy.197 That intact parent dithiocarbamate is the proximate toxic species was supported by the report that oral administration of acid-stable pyrrolidine dithiocarbamate was able to produce the same myelin lesions as disulfiram.198 Thus, the myelinopathy produced by disulfiram is interpreted to result from the acid stability of disulfiram facilitating systemic delivery of parent compound that subsequently undergoes reduction to N,N-diethyldithiocarbamate.
Figure 10.

Disposition and mechanisms of neurotoxicity for disulfiram and N,N-diethyldithiocarbamate. Oral administration of N,N-diethyldithiocarbamate (DEDC) results in acid promoted decomposition to CS2 and diethylamine. The CS2 released produces a neurofilamentous axonopathy through protein cross-linking (see Fig. 6). Disulfiram is more acid-stable and can be absorbed intact following oral administration and then reduced to DEDC in vivo. DEDC derived either from oral disulfiram or parenteral administration of DEDC forms lipophilic redox active complexes with copper that cross the blood-nerve barrier and accumulate in myelin. Redox cycling of the copper complexes in myelin produces free radicals promoting lipid peroxidation that results in demyelination.
Lysolecithin and Ethidium Bromide
Injection or topical application of either of these compounds in the central or peripheral nervous systems produces an acute focal demyelination while sparring axons.199–201 Because total remyelination is usually achieved and the precise location of injury is known these agents have been used as a model to test remyelination therapies and diagnostic methods for demyelinating diseases such as multiple sclerosis or hereditary peripheral nerve diseases. The mechanism for the demyelination produced by ethidium bromide is not known but recent investigations have provided insight into how lysolecithin (lysophosphatidylcholine) produces demyelination.
In addition to the interest of lysolecithin in the experimental setting it is also relevant as a possible contributor in demyelinating disease states. Lysolecithin is an endogenous lysophospholipid produced by phospholipase A2 and is elevated in a number of neuropathological conditions including ischemia,202,203 spinal cord injury,204 amyotrophic lateral sclerosis,205 and multiple sclerosis.206 Additionally, phospholipase A2 is upregulated in the reprogrammed Schwann cell demyelinating phenotype after injury.83 Elevations of lysolecithin in these disease states suggest it may act as an endogenous demyelinating agent.
Using a combination of in vitro and in vivo systems Plemel et al.207 investigated the relative contributions of three demyelinating mechanisms: receptor-mediated toxicity, inflammation-induced injury and nonspecific membrane disruption. From their studies it was concluded that lysolecithin integrates into cellular membranes and when it reaches a critical concentration causes an increase in cell membrane permeability. Thus the data suggest that lysolecithin acts as a non-specific lipid-disrupting agent with the first stage of injury corresponding to the unraveling of the myelin sheath.208 Supporting this interpretation is the ability to reproduce similar myelin lesions using two other lipid-disrupting agents Brij35 and Tween 20.207
Vulnerabilities of Neurons to Toxicants
Certain peripheral neurotoxicants selectively affect the neuronal perikaryon resulting in injury or death, a condition termed neuronopathy. A significant consequence of neuron death is that it is irreversible and accompanied by the loss of all associated cytoplasmic extensions. Thus when significant loss of axons is detected in the peripheral nervous system there exists the potential that it is the result of a primary neuronopathy.
The selectivity of certain agents for neurons typically can be attributed to the ability of the agent to target one of their unique vulnerabilities. Neurons have a high demand for energy primarily derived through aerobic glycolysis that is required for repolarization of the membrane and axonal transport. Also unique to neurons is the magnitude of biosynthetic requirements necessary to support the volume of axoplasm that can be orders of magnitude greater than the neuronal cell body. Selectivity can also result from transport systems associated with neuronal populations that concentrate an agent to toxic levels within that cell type. 6-Hydroxydopamine demonstrates an example of such selective uptake by catecholaminergic neurons in the peripheral nervous system. Neurons in the peripheral nervous system may also demonstrate increased susceptibility due to decreased integrity of the blood nerve barrier at sensory and autonomic ganglia where neurons reside in the periphery.26–29 Support for this concept is provided through the observation that agents producing peripheral neuronopathies produce more widespread toxicity in the nervous system when the blood brain barrier is disrupted experimentally.
Toxic Disorders of the Neuron
Retrograde signaling of injury from the axon results in the chromatolysis response (“axonal reaction”) in the neuronal periykaryon characterized by neuronal enlargement, rounding of the cytoplasmic membrane, eccentric displacement of the nucleus and loss of stainable Nissl substance.209,210 Dissolution of Nissl substance (i.e., free ribosome storage depots associated with rough endoplasmic reticulum) reflects recruitment of ribosomes for accelerated protein synthesis to support cell body and/or cell process repair. There is also increased metabolic activity and transcriptional changes to support regeneration of the injured axon.211,212 However alterations in expression of genes that contribute to cell death and inflammation also occur and it is likely a balance in the overall expression profile that decides the fate of the neuron.213,214 The response of the neurons is signaled to support cells and corresponding gene expression and proliferation occurs in the ganglion satellite cells and macrophages.215,216 Defining the gene expression profiles in the cells of ganglia in response to peripheral nerve injury holds promise for the development of diagnostic biochemical methods of peripheral nerve injury and the design of therapeutic interventions to prevent neuron cell death and facilitate regeneration.
Cell death of the neuron can proceed through necrosis or one of the programmed cell death pathways. Although there are a number of recognized cell death pathways including autophagy and necroptosis in addition to apoptosis the currently recognized agents targeting the neuron appear to work through either necrosis or apoptosis. Biochemical and morphological methods can be used to determine which pathway is involved and have been described in detail in other reviews.217,218 Morphologically necrosis involves cell swelling and disintegration of cellular organelles from lipases, endonucleases, and proteases that are activated by increased intracellular Ca2+ levels that leads to rupture of the nuclear and cell membranes invoking an inflammatory response. Biochemically there is a cessation of protein synthesis and the termination of energy-dependent processes due to the depletion of ATP. Pyridoxamine is an example of an agent targeting dorsal root ganglion (DRG) neurons that produces cell death through necrosis.16 In contrast apoptosis is an active process requiring energy and protein synthesis that proceeds through an orchestrated pathway with specific regulators that can either inhibit or activate the process. Apoptotic cells become shrunken with fragmentation of their chromatin but maintain the structure of their organelles. There is a translocation of phosphatidylserine to the cell surface that enables microglia or macrophages to remove the dead cell with its plasma membrane intact, thus precluding the development of an inflammatory response. The platinum based chemotherapeutic agents produce cell death in DRG neurons through the apoptosis pathway.
Examples of Agents Producing Peripheral Neuronopathies
Chemotherapeutic Platinum Derivatives
Cisplatin, oxaliplatin and carboplatin are platinum based compounds used to treat lung, breast, ovarian and gastrointestinal cancers. In addition to other side effects these chemotherapeutics are associated with a high prevalence of peripheral neuropathy. Large-diameter sensory fibers appear to be the most sensitive resulting in symmetrical glove-stocking distribution of sensory loss including numbness and paresthesias. Neurotoxicity is related to cumulative dose, can persist for months to years and can even worsen for a period following cessation of treatment. In addition, oxaliplatin is recognized to produce an acute non-dose-dependent neuropathy very shortly after infusion exhibited by cold induced paresthesia and dysesthesia of distal extremities These agents target DRG through their ability to accumulate and interact with DNA in this location.219–221 The high density of capillaries27 that are relatively permeable28,29 contribute to an incomplete blood-nerve barrier allowing platinum drugs preferential access to this region of the peripheral nervous system. Uptake by the sensory neurons may also be facilitated by two types of specific transporters, the copper transporter 1 (CTR1) and the organic cation transporter-2 (OCT2).222–224
These agents form platinum intra-strand adducts and inter-strand cross-links. The DNA modifications compromise transcriptional activity in this population of cells noted for their high rate of transcription required to sustain the metabolic demands of maintaining the associated axon. The altered 3-dimensional structure of DNA along with activation of Cyclin D1 and mitogen-activated kinases (MAPKs) are thought to promote entry of these neurons into the cell cycle, which results in a compensatory process of apoptosis in these postmitotic cells.225,226 In addition to nuclear DNA, mitochondrial DNA is also modified. The interactions within mitochondria result in increased oxidative stress, increased p53 tumor suppressor protein (p53) activity, release of cytochrome c and activation of p38 mitogen-activated protein kinases (p38) and extracellular signal-regulated kinases 1 and 2 (ERK½).114,227 Compromised mitochondrial function also leads to a depletion of energy stores vital to this population of cells. Collectively these processes work to activate recognized intrinsic signaling to initiate apoptotic pathways.225,228–230
Oxaliplatin can also exert an effect on the kinetics of the neuron voltage-gated sodium ion channels.231–234 It acts directly at the channel and possibly indirectly through chelation of Ca2+ to increase the duration of the refractory period.231 This effect is thought to contribute to the acute clinical signs of cold paresthesia observed for oxaliplatin from the sensory neurons becoming hyperexcitable. Sensory neurons express transient receptor potential channels that contribute to the neuropathic pain. Oxaliplatin activates the transient receptor potential cation channel subfamily A member 1 (TRPA1) and the transient receptor potential cation channel subfamily M member 8 (TRMP8) providing another mechanism through which this compound may promote cold allodynia.235–237
Methylmercury
Environmental mercury is an important pollutant that exists in both inorganic and organic forms, both of which can produce neurological and developmental effects in humans and wildlife. Methylmercury (organic form) has been responsible for a number of human poisonings and has been the focus of considerable investigation.238 The clinical signs associated with methylmercury intoxication include sensory impairment of the extremities, cerebellar ataxia and visual field constriction.239 In addition to neuron loss in the visual area of the calcarine cortex and granule cells of the cerebellum, methylmercury also produces degeneration of neurons in the DRG along with their associated axons while sparing the peripheral nerve motor axons.239–243
Methylmercury exerts its neurotoxic affects in DRG partly through the ability to accumulate there due to the high density of fenestrated capillaries.26–29 Although the molecular targets and mechanisms of methylmercury are still being delineated, the experimental evidence supports contributions from several processes including disrupted calcium homeostasis, increased oxidative stress and altered glutamate homeostasis leading to excitotoxicity.238,244 Methylmercury alters intracellular calcium levels through disrupting sequestration of intracellular calcium pools and through increasing permeability of the plasma membrane for calcium.245 Elevated oxidative stress may be mediated through the ability of methylmercury to interact with sulfhydryl and selenocysteine functions of antioxidants and antioxidant proteins. Oxidative stress could also be elevated through the accumulation of methylmercury in mitochondria that decreases ATP production and depolarizes the mitochondrial membrane potential.245 Consistent with the affects on mitochondria the activation of signaling pathways involving release of cytochrome c and apoptosis-inducing factor (AIF) leading to both caspase-dependent and caspase-independent apoptotic cell death pathways have been observed in methylmercury-treated cells and apoptosis has been observed in granule cell and DRG cells.244,246 Collectively the number and diversity of significantly altered cellular processes produced by methylmercury suggest the involvement of multiple mechanisms that may act additively or synergistically.
Catecholamines
If not processed correctly through sequestration and storage in a low pH environment, catecholamines can oxidize to form quinones that can via redox cycling produce reactive oxygen species which cause deleterious effects in neurons.247 Dopamine is particularly sensitive to oxidation and an analog of dopamine, 6-hydroxydopamine, has been used to lesion the central and peripheral nervous systems experimentally. When administered peripherally, this compound does not cross the blood-brain barrier and is taken up by transporters at the sympathetic nerve terminals producing a sympathectomy.248 The loss of sympathetic neurons results from retrograde transport of 6-hydroxydopamine to the neuronal perikaryon and increased oxidative stress from the oxidation of 6-hydroxydopamine and subsequent redox cycling similar to that observed with dopamine.249,250 It has also been proposed that 6-hydroxydopamine can inhibit mitochondrial respiration chain complexes I and IV.251 Over expression of copper/zinc (CuZn) superoxide dismutase or glutathione peroxidase has been shown to protect against 6-hydroxydopamine neurotoxicity.252,253
Conclusion
There are many recognized peripheral nervous system toxicants but our understanding of their mechanisms is limited and there are few examples where a single mechanism is generally agreed upon to account for all the pathological events observed in a given toxic neuropathy. For those that have been investigated there are usually a number of potential contributing events recognized. In addition, the diversity of toxicants and the structural complexity and spatial distribution of the peripheral nervous system present a formidable challenge for screening neurotoxicity. However, the capacity of an agent to target the peripheral nervous system can usually be attributed to its ability to circumvent the blood-nerve barrier and compromise one of the vital and unique characteristics the nervous system has evolved to perform its function of rapidly conducting information over distance. Once these requirements have been met there are several general pathogenic processes through which most toxic agents ultimately mediate their effects including covalent or non-covalent modifications, compromised energy or protein biosynthesis, oxidative injury and disruption of ionic gradients across membranes. Because programmed neuronal cell death, demyelination, and axonal degeneration proceed through active programmed pathways, the triggers for these pathways may be the final targets of neurotoxicants that become activated through the shared general pathogenic processes. Thus although the initial critical actions of neurotoxicants may be quite varied and multiple for any given agent there is the potential that common final pathways can be identified to help guide the development of biochemical based methods to complement morphological analyses.
Acknowledgments
I wish to thank Douglas Anthony and Brad Bolon for their help with reviewing electron micrographs and proof reading of the manuscript.
Footnotes
Declaration of Conflicting Interests
The author declares no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population ≥40 years of age with and without diabetes: 1999–2000 national health and nutrition examination survey. Diabetes Care 2004;27(7):1591–1597. [DOI] [PubMed] [Google Scholar]
- 2.Barrell K, Smith AG. Peripheral Neuropathy. Med Clin North Am 2019;103(2):383–397. [DOI] [PubMed] [Google Scholar]
- 3.Karam C, Dyck P. Toxic Neuropathies. Semin Neurol 2015;35(04):448–457. [DOI] [PubMed] [Google Scholar]
- 4.Seretny M, Currie GL, Sena ES, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014;155(12):2461–2470. [DOI] [PubMed] [Google Scholar]
- 5.Trivedi S, Pandit A, Ganguly G, Das SK. Epidemiology of peripheral neuropathy: an Indian perspective. Ann Indian Acad Neurol 2017;20(3):173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samukawa M, Ichihara G, Oka N, Kusunoki S. A case of severe neurotoxicity associated with exposure to 1-bromopropane, an alternative to ozone-depleting or global-warming solvents. Arch Intern Med 2012;172(16):1257–1260. [DOI] [PubMed] [Google Scholar]
- 7.Meyer-Baron M, Kim EA, Nuwayhid I, Ichihara G, Kang S-K. Occupational exposure to neurotoxic substances in Asian countries - challenges and approaches. Neurotoxicology 2012;33(4):853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichihara G, Li W, Shibata E, et al. Neurologic abnormalities in workers of a 1-bromopropane factory. Environ Health Perspect 2004;112(13):1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi M, Zochodne DW. Diabetic neuropathy and the sensory neuron: New aspects of pathogenesis and their treatment implications. J Diabetes Investig 2018;9(6):1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iqbal Z, Azmi S, Yadav R, et al. Diabetic Peripheral Neuropathy: Epidemiology, Diagnosis, and Pharmacotherapy. Clin Ther 2018;40(6):828–849. [DOI] [PubMed] [Google Scholar]
- 11.Azmi S, Petropoulos IN, Ferdousi M, Ponirakis G, Alam U, Malik RA. An update on the diagnosis and treatment of diabetic somatic and autonomic neuropathy. F1000Res 2019;8:186. doi: 10.12688/f1000research.17118.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azmi S, Ferdousi M, Kalteniece A, et al. Diagnosing and managing diabetic somatic and autonomic neuropathy. Ther Adv Endocrinol Metab 2019;10:204201881982689. doi: 10.1177/2042018819826890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chopra K, Tiwari V. Alcoholic neuropathy: possible mechanisms and future treatment possibilities. Brit J Clin Pharmacol 2012;73(3):348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellion M, Gilchrist JM, La Monte De S. Alcohol-related peripheral neuropathy: Nutritional, toxic, or both? Muscle Nerve 2011;43(3):309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barohn RJ, Amato AA. Pattern-recognition approach to neuropathy and neuronopathy. Neurol Clin 2013;31(2):343–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jortner BS. Common structural lesions of the peripheral nervous system. Toxicol Pathol 2019;97:019262331982606. doi: 10.1177/0192623319826068. [DOI] [PubMed] [Google Scholar]
- 17.Jortner BS. Mechanisms of toxic injury in the peripheral nervous system: neuropathologic considerations. Toxicol Pathol 2000;28(1):54–69. [DOI] [PubMed] [Google Scholar]
- 18.Rao DB, Jortner BS, Sills RC. Animal models of peripheral neuropathy due to environmental toxicants. ILAR J 2014;54(3):315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cashman CR, Höke A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci Lett 2015;596:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanat O, Ertas H, Caner B. Platinum-induced neurotoxicity: A review of possible mechanisms. World J Clin Oncol 2017;8(4):329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiorazzi A, Semperboni S, Marmiroli P. Current view in platinum drug mechanisms of peripheral neurotoxicity. Toxics 2015;3(3):304–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: An unresolved issue. Neurología 2010;25(2):116–131. [PubMed] [Google Scholar]
- 23.Fukuda Y, Li Y, Segal RA. A mechanistic understanding of axon degeneration in chemotherapy-induced peripheral neuropathy. Front Neurosci 2017;11:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas PK. Peripheral neuropathy. Br Med J 1970;1(5692):349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moser VC, Phillips PM, Morgan DL, Sills RC. Carbon disulfide neurotoxicity in rats: VII. Behavioral evaluations using a functional observational battery. Neurotoxicology 1998;19(1):147–157. [PubMed] [Google Scholar]
- 26.Maurer LL, Philbert MA. The mechanisms of neurotoxicity and the selective vulnerability of nervous system sites. Handb of Clin Neurol 2015;131(1):61–70. [DOI] [PubMed] [Google Scholar]
- 27.Jimenez-Andrade JM, Herrera MB, Ghilardi JR, Vardanyan M, Melemedjian OK, Mantyh PW. Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: implications for chemical-induced peripheral sensory neuropathies. Mol Pain 2008;4(1):10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirakawa H, Okajima S, Nagaoka T, Kubo T, Takamatsu T, Oyamada M. Regional differences in blood-nerve barrier function and tight-junction protein expression within the rat dorsal root ganglion. Neuroreport 2004;15(3):405–408. [DOI] [PubMed] [Google Scholar]
- 29.Abram SE, Yi J, Fuchs A, Hogan QH. Permeability of injured and intact peripheral nerves and dorsal root ganglia. Anesthesiology 2006;105(1):146–153. [DOI] [PubMed] [Google Scholar]
- 30.Court FA, Alvarez J. Schwann Cell and Axon: An interlaced unit—from action potential to phenotype expression. In: von Bernhardi R ed. Glial Cells in Health and Disease of the CNS. Advances in Experimental Medicine and Biology Switzerland; Springer Nature AG; 2016;194:183–201. [DOI] [PubMed] [Google Scholar]
- 31.López-Leal R, Alvarez J, Court FA. Origin of axonal proteins: Is the axon-Schwann cell unit a functional syncytium? Cytoskeleton 2016;73(10):629–639. [DOI] [PubMed] [Google Scholar]
- 32.Boerboom A, Dion V, Chariot A, Franzen R. Molecular mechanisms involved in Schwann cell plasticity. Front Mol Neurosci 2017;10(Pt 5):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corfas G. Mechanisms and roles of axon-Schwann cell interactions. J Neurosci 2004;24(42):9250–9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong K, Babetto E, Beirowski B. Axon degeneration: make the Schwann cell great again. Neural Regen Res 2017;12(4):518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin X, Crawford TO, Griffin JW, et al. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 1998;18(6):1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 2002;298(5596):1245–1248. [DOI] [PubMed] [Google Scholar]
- 37.Syroid DE, Maycox PR, Burrola PG, et al. Cell death in the Schwann cell lineage and its regulation by neuregulin. Proc Natl Acad Sci USA 1996;93(17):9229–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guénard V, Gwynn LA, Wood PM. Transforming growth factor-beta blocks myelination but not ensheathment of axons by Schwann cells in vitro. J Neurosci 1995;15(1):419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corfas G, Rosen KM, Aratake H, Krauss R, Fischbach GD. Differential expression of ARIA isoforms in the rat brain. Neuron 1995;14(1):103–115. [DOI] [PubMed] [Google Scholar]
- 40.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature 1995;378(6555):386–390. [DOI] [PubMed] [Google Scholar]
- 41.Adlkofer K, Lai C. Role of neuregulins in glial cell development. Glia 2000;29(2):104–111. [DOI] [PubMed] [Google Scholar]
- 42.Birchmeier C, Nave K-A. Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia 2008;56(14):1491–1497. [DOI] [PubMed] [Google Scholar]
- 43.Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C. A dual role of erbB2 in myelination and in expansion of the Schwann cell precursor pool. The J Cell Biol 2000;148(5):1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michailov GV, Sereda MW, Brinkmann BG, et al. Axonal neuregulin-1 regulates myelin sheath thickness. Science 2004;304(5671):700–703. [DOI] [PubMed] [Google Scholar]
- 45.Sotelo JR, Canclini L, Kun A, et al. Glia to axon RNA transfer. Devel Neurobio 2013;74(3):292–302. [DOI] [PubMed] [Google Scholar]
- 46.Nave K-A. Myelination and support of axonal integrity by glia. Nature 2010;468(7321):244–252. [DOI] [PubMed] [Google Scholar]
- 47.Nave K-A. Myelination and the trophic support of long axons. Nat Rev Neurosci 2010;11(4):275–283. [DOI] [PubMed] [Google Scholar]
- 48.Ben-Yaakov K, Dagan SY, Segal-Ruder Y, et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J 2012;31(6):1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogelaar CF, Gervasi NM, Gumy LF, et al. Axonal mRNAs: Characterisation and role in the growth and regeneration of dorsal root ganglion axons and growth cones. Mol Cell Neurosci 2009;42(2):102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor AM, Wu J, Tai HC, Schuman EM. Axonal translation of -Catenin regulates synaptic vesicle dynamics. J Neurosci 2013;33(13):5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singer M, Green MR. Autoradiographic studies of uridine incorporation in peripheral nerve of the newt, Triturus. J Morphol 1968;124(3):321–344. [DOI] [PubMed] [Google Scholar]
- 52.Gambetti P, Autilio-Gambetti L, Shafer B, Pfaff LD. Quantitative autoradiographic study of labeled RNA in rabbit optic nerve after intraocular injection of (3H)uridine. J Cell Biol 1973;59(3):677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benech C, Sotelo JRJ, Menendez J, Correa-Luna R. Autoradiographic study of RNA and protein synthesis in sectioned peripheral nerves. Exp Neurol 1982;76(1):72–82. [DOI] [PubMed] [Google Scholar]
- 54.Court FA, Midha R, Cisterna BA, et al. Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia 2011;59(10):1529–1539. [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Verrilli MA, Court FA. Transfer of vesicles from Schwann cells to axons: a novel mechanism of communication in the peripheral nervous system. Front Physiol 2012;3:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kun A, Otero L, Sotelo-Silveira JR, Sotelo JR. Ribosomal distributions in axons of mammalian myelinated fibers. J Neurosci Res 2007;85(10):2087–2098. [DOI] [PubMed] [Google Scholar]
- 57.Li Y-C, Li Y-N, Cheng C-X, et al. Subsurface cisterna-lined axonal invaginations and double-walled vesicles at the axonal-myelin sheath interface. Neurosci Res 2005;53(3):298–303. [DOI] [PubMed] [Google Scholar]
- 58.Li Y-C, Cheng C-X, Li Y-N, Shimada O, Atsumi S. Beyond the initial axon segment of the spinal motor axon: fasciculated microtubules and polyribosomal clusters. J Anat 2005;206(6):535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toews AD, Harry GJ, Lowrey KB, Morgan DL, Sills RC. Carbon disulfide neurotoxicity in rats: IV. Increased mRNA expression of low-affinity nerve growth factor receptor--a sensitive and early indicator of PNS damage. Neurotoxicology 1998;19(1):109–116. [PubMed] [Google Scholar]
- 60.Roberson MD, Toews AD, Bouldin TW, Weaver J, Goines ND, Morell P. NGFR-mRNA expression in sciatic nerve: a sensitive indicator of early stages of axonopathy. Brain Res Mol Brain Res 1995;28(2):231–238. [DOI] [PubMed] [Google Scholar]
- 61.Chilton L, Middlemas A, Gardiner N, Tomlinson DR. The p75 neurotrophin receptor appears in plasma in diabetic rats-characterisation of a potential early test for neuropathy. Diabetologia 2004;47(11):1924–1930. [DOI] [PubMed] [Google Scholar]
- 62.Pyle S, Reuhl KR. Cytoskeletal Elements in Neurotoxicity. In: Philbert M, ed. Comprehensive Toxicology Vol 13 2nd ed. Kidlington, United Kingdom: Elsevier Ltd; 2010:71–87. [Google Scholar]
- 63.Lasek RJ, Garner JA, Brady ST. Axonal transport of the cytoplasmic matrix. J Cell Biol 1984;99(1 Pt 2):212s–221s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Llorens J Toxic neurofilamentous axonopathies– accumulation of neurofilaments and axonal degeneration. J Intern Med 2013;273(5):478–489. [DOI] [PubMed] [Google Scholar]
- 65.Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro Oncol 2012;14(suppl_4):iv45–iv54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waller A Experiments on the section of the glosso-pharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Edinb Med Surg J 1851;76(189):369–376. [PMC free article] [PubMed] [Google Scholar]
- 67.Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 2010;8(1):e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci 2013;33(33):13410–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Osterloh JM, Yang J, Rooney TM, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012;337(6093):481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015;348(6233):453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J, Wu Z, Renier N, et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 2015;160(1–2):161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 2016;89(3):449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park HT, Kim JK, Tricaud N. The conceptual introduction of the “demyelinating Schwann cell” in peripheral demyelinating neuropathies. Glia 2018;67(4):571–581. [DOI] [PubMed] [Google Scholar]
- 74.Tricaud N, Park HT. Wallerian demyelination: chronicle of a cellular cataclysm. CMLS, Cell Mol Life Sci 2017;74(22):4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams PL, Hall SM. Chronic Wallerian degeneration--an in vivo and ultrastructural study. J Anat 1971;109(Pt 3):487–503. [PMC free article] [PubMed] [Google Scholar]
- 76.Causey G, Palmer E. The centrifugal spread of structural change at the nodes in degenerating mammalian nerves. J Anat 1953;87(2):185–191. [PMC free article] [PubMed] [Google Scholar]
- 77.Ghabriel MN, Allt G. The role of Schmidt-Lanterman incisures in Wallerian degeneration. I. A quantitative teased fibre study. Acta Neuropathol 1979;48(2):93–93. [PubMed] [Google Scholar]
- 78.Ghabriel MN, Allt G. The role of Schmidt-Lanterman incisures in Wallerian degeneration. II. An electron microscopic study. Acta Neuropathol 1979;48(2):95–103. [DOI] [PubMed] [Google Scholar]
- 79.Webster HF. The relationship between Schmidt-Lantermann incisures and myelin segmentation during Wallerian degeneration. Ann N Y Acad Sci 1965;122:29–38. [DOI] [PubMed] [Google Scholar]
- 80.Griffin JW, Drucker N, Gold BG, et al. Schwann cell proliferation and migration during paranodal demyelination. J Neurosci 1987;7(3):682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jung J, Cai W, Lee HK, et al. Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J Neurosci 2011;31(6):2009–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guertin AD. Microanatomy of axon/glial signaling during Wallerian degeneration. J Neurosci 2005;25(13):3478–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De S, Trigueros MA, Kalyvas A, David S. Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol Cell Neurosci 2003;24(3):753–765. [DOI] [PubMed] [Google Scholar]
- 84.Paul JA, Gregson NA. An immunohistochemical study of phospholipase A2 in peripheral nerve during Wallerian degeneration. J Neuroimmunol 1992;39(1–2):31–47. [DOI] [PubMed] [Google Scholar]
- 85.Arthur-Farraj PJ, Latouche M, Wilton DK, et al. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012;75(4):633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma KH, Hung HA, Srinivasan R, Xie H, Orkin SH, Svaren J. Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex 2. J Neurosci 2015;35(22):8640–8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parkinson DB, Bhaskaran A, Arthur-Farraj P, et al. c-Jun is a negative regulator of myelination. J Cell Biol 2008;181(4):625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee HJ, Shin YK, Park HT. Mitogen activated protein kinase family proteins and c-jun signaling in injury-induced Schwann cell plasticity. Exp Neurobiol 2014;23(2):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fontana X, Hristova M, Da Costa C, et al. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol 2012;198(1):127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tapinos N, Ohnishi M, Rambukkana A. ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat Med 2006;12(8):961–966. [DOI] [PubMed] [Google Scholar]
- 91.Hirata K, Kawabuchi M. Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech 2002;57(6):541–547. [DOI] [PubMed] [Google Scholar]
- 92.Barrette B, Hebert MA, Filali M, et al. Requirement of myeloid cells for axon regeneration. J Neurosci 2008;28(38):9363–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 2015;210(1):153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weinberg HJ, Spencer PS. The fate of Schwann cells isolated from axonal contact. J Neurocytol 1978;7(5):555–569. [DOI] [PubMed] [Google Scholar]
- 95.Stoll G, Muller HW. Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol 1999;9(2):313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cattin A-L, Burden JJ, Van Emmenis L, et al. Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 2015;162(5):1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fernandez-Valle C, Bunge RP, Bunge MB. Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. J Neurocytol 1995;24(9):667–679. [DOI] [PubMed] [Google Scholar]
- 98.Kim HA, Mindos T, Parkinson DB. Plastic fantastic: Schwann cells and repair of the peripheral nervous system. Stem Cells Transl Med 2013;2(8):553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boyd JG, Gordon T. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp Neurol 2003;183(2):610–619. [DOI] [PubMed] [Google Scholar]
- 100.Kang H, Tian L, Thompson WJ. Schwann cell guidance of nerve growth between synaptic sites explains changes in the pattern of muscle innervation and remodeling of synaptic sites following peripheral nerve injuries. J Comp Neurol 2019;527(8):1388–1400. [DOI] [PubMed] [Google Scholar]
- 101.Schroder JM. Altered ratio between axon diameter and myelin sheath thickness in regenerated nerve fibers. Brain Res 1972;45(1):49–65. [DOI] [PubMed] [Google Scholar]
- 102.Ford MC, Alexandrova O, Cossell L, et al. Tuning of Ranvier node and internode properties in myelinated axons to adjust action potential timing. Nat Commun 2015;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peters A, Palay SL, Webster HD. The Fine Structure of the Nervous System : Neurons and Their Supporting Cells 3rd ed. New York: Oxford University Press; 1991. [Google Scholar]
- 104.Rushton WAH. A theory of the effects of fibre size in medullated nerve. J Physiol 1951;115(1):101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 1992;68(3):451–463. [DOI] [PubMed] [Google Scholar]
- 106.Mata M, Kupina N, Fink DJ. Phosphorylation-dependent neurofilament epitopes are reduced at the node of Ranvier. J Neurocytol 1992;21(3):199–210. [DOI] [PubMed] [Google Scholar]
- 107.Hsieh ST, Kidd GJ, Crawford TO, et al. Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci 1994;14(11 Pt 1):6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shemesh OA, Spira ME. Paclitaxel induces axonal microtubules polar reconfiguration and impaired organelle transport: implications for the pathogenesis of paclitaxel-induced polyneuropathy. Acta Neuropathol 2010;119(2):235–248. [DOI] [PubMed] [Google Scholar]
- 109.Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW. An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res 2007;1168:46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 2013;37:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bobylev I, Joshi AR, Barham M, et al. Paclitaxel inhibits mRNA transport in axons. Neurobiology of Disease 2015;82:321–331. [DOI] [PubMed] [Google Scholar]
- 112.Gornstein EL, Schwarz TL. Neurotoxic mechanisms of paclitaxel are local to the distal axon and independent of transport defects. Exp Neurol 2017;288:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang MS, Davis AA, Culver DG, Wang Q, Powers JC, Glass JD. Calpain inhibition protects against taxol-induced sensory neuropathy. Brain 2004;127(Pt 3):671–679. [DOI] [PubMed] [Google Scholar]
- 114.Canta A, Pozzi E, Carozzi V. Mitochondrial dysfunction in chemotherapy-induced peripheral neuropathy (CIPN). Toxics 2015;3(2):198–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sahenk Z, Brady ST, Mendell JR. Studies on the pathogenesis of vincristine-induced neuropathy. Muscle Nerve 1987;10(1):80–84. [DOI] [PubMed] [Google Scholar]
- 116.Herskowitz A, Ishii N, Schaumburg H. n-Hexane neuropathy. A syndrome occurring as a result of industrial exposure. N Engl J Med 1971;285(2):82–85. [DOI] [PubMed] [Google Scholar]
- 117.Rizzuto N, De Grandis D, Di Trapani G, Pasinato E. n-Hexane polyneuropathy. An occupational disease of shoemakers. Eur Neurol 1980;19(5):308–315. [DOI] [PubMed] [Google Scholar]
- 118.Chang YC. Patients with n-hexane induced polyneuropathy: a clinical follow up. Br J Ind Med 1990;47(7):485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang C-C. Polyneuropathy induced by n-hexane intoxication in Taiwan. Acta Neurol Taiwan 2008;17(1):3–10. [PubMed] [Google Scholar]
- 120.Krasavage WJ, O’Donoghue JL, DiVincenzo GD, Terhaar CJ. The relative neurotoxicity of methyl-n-butyl ketone, n-hexane and their metabolites. Toxicol Appl Pharmacol 1980;52(3):433–441. [DOI] [PubMed] [Google Scholar]
- 121.Graham DG. Neurotoxicants and the cytoskeleton. Curr Opin Neurol 1999;12(6):733–737. [DOI] [PubMed] [Google Scholar]
- 122.Graham DG, Amarnath V, Valentine WM, Pyle SJ, Anthony DC. Pathogenetic studies of hexane and carbon disulfide neurotoxicity. Crit Rev Toxicol 2008;25(2):91–112. [DOI] [PubMed] [Google Scholar]
- 123.LoPachin RM. Protein Adduct Formation as a Molecular Mechanism in Neurotoxicity. Toxicol Sci 2005;86(2):214–225. [DOI] [PubMed] [Google Scholar]
- 124.Kim MS, Sabri MI, Miller VH, Kayton RJ, Dixon DA, Spencer PS. 1,2-diacetylbenzene, the neurotoxic metabolite of a chromogenic aromatic solvent, induces proximal axonopathy. Toxicol Appl Pharmacol 2001;177(2):121–131. [DOI] [PubMed] [Google Scholar]
- 125.Kim M-S, Hashemi SB, Spencer PS, Sabri MI. Amino acid and protein targets of 1,2-diacetylbenzene, a potent aromatic gamma-diketone that induces proximal neurofilamentous axonopathy. Toxicol Appl Pharmacol 2002;183(1):55–65. [DOI] [PubMed] [Google Scholar]
- 126.Anthony DC, Boekelheide K, Graham DG. The effect of 3,4-dimethyl substitution on the neurotoxicity of 2,5-hexanedione. I. Accelerated clinical neuropathy is accompanied by more proximal axonal swellings. Toxicol Appl Pharmacol 1983;71(3):362–371. [DOI] [PubMed] [Google Scholar]
- 127.Anthony DC, Boekelheide K, Anderson CW, Graham DG. The effect of 3,4-dimethyl substitution on the neurotoxicity of 2,5-hexanedione. II. Dimethyl substitution accelerates pyrrole formation and protein crosslinking. Toxicol Appl Pharmacol 1983;71(3):372–382. [DOI] [PubMed] [Google Scholar]
- 128.St Clair MBG, Amarnath V, Moody MA, Anthony DC, Anderson CW, Graham DG. Pyrrole oxidation and protein cross-linking as necessary steps in the development of gamma-diketone neuropathy. Chem Res Toxicol 1988;1(3):179–185. [DOI] [PubMed] [Google Scholar]
- 129.Stone JD, Peterson AP, Eyer J, Oblak TG, Sickles DW. Neurofilaments are nonessential to the pathogenesis of toxicant-induced axonal degeneration. J Neurosci 2001;21(7):2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sickles DW, Pearson JK, Beall A, Testino A. Toxic axonal degeneration occurs independent of neurofilament accumulation. J Neurosci Res 1994;39(3):347–354. [DOI] [PubMed] [Google Scholar]
- 131.Sabri MI, Hashemi SB, Lasarev MR, Spencer PS. Axonopathy-inducing 1,2-diacetylbenzene forms adducts with motor and cytoskeletal proteins required for axonal transport. Neurochem Res 2007;32(12):2152–2159. [DOI] [PubMed] [Google Scholar]
- 132.Beauchamp ROJ, Bus JS, Popp JA, Boreiko CJ, Goldberg L. A critical review of the literature on carbon disulfide toxicity. Crit Rev Toxicol 1983;11(3):169–278. [DOI] [PubMed] [Google Scholar]
- 133.Sills RC, Harry GJ, Morgan DL, Valentine WM, Graham DG. Carbon disulfide neurotoxicity in rats: V. Morphology of axonal swelling in the muscular branch of the posterior tibial nerve and spinal cord. Neurotoxicology 1998;19(1):117–127. [PubMed] [Google Scholar]
- 134.Valentine W, Amarnath V. Covalent cross-linking of proteins by carbon disulfide. Chem Res Toxicol 1992;5(2):254–62. [DOI] [PubMed] [Google Scholar]
- 135.Valentine W, Amarnath V, Graham D. CS2-mediated cross-linking of erythrocyte spectrin and neurofilament protein: Dose response and temporal relationship to the formation of axonal swellings. Toxicol Appl Pharmacol 1997;142(1):95–105. [DOI] [PubMed] [Google Scholar]
- 136.Erve JCL, Amarnath V, Graham DG, Sills RC, Morgan AL, Valentine WM. Carbon disulfide and N, N-diethyldithiocarbamate generate thiourea cross-links on erythrocyte spectrin in vivo. Chem Res Toxicol 1998;11(5):544–549. [DOI] [PubMed] [Google Scholar]
- 137.Ichihara G, Kitoh J, Li W, Ding X, Ichihara S, Takeuchi Y. Neurotoxicity of 1-bromopropane: Evidence from animal experiments and human studies. J Adv Res 2012;3(2):91–98. [Google Scholar]
- 138.Yu X, Ichihara G, Kitoh J, et al. Neurotoxicity of 2-bromopropane and 1-bromopropane, alternative solvents for chlorofluorocarbons. Environ Res 2001;85(1):48–52. [DOI] [PubMed] [Google Scholar]
- 139.Majersik JJ, Caravati EM, Steffens JD. Severe neurotoxicity associated with exposure to the solvent 1-bromopropane (n-propyl bromide). Clin Toxicol 2007;45(3):270–276. [DOI] [PubMed] [Google Scholar]
- 140.Li W, Shibata E, Zhou Z, et al. Dose-dependent neurologic abnormalities in workers exposed to 1-bromopropane. J Occup Environ Med 2010;52(8):769–777. [DOI] [PubMed] [Google Scholar]
- 141.Wang T-H, Wu M-L, Wu Y-H, et al. Neurotoxicity associated with exposure to 1-bromopropane in golf-club cleansing workers. Clin Toxicol 2015;53(8):823–826. [DOI] [PubMed] [Google Scholar]
- 142.Ichihara G, Kitoh J, Yu X, et al. 1-Bromopropane, an alternative to ozone layer depleting solvents, is dose-dependently neurotoxic to rats in long-term inhalation exposure. Toxicol Sci 2000;55(1):116–123. [DOI] [PubMed] [Google Scholar]
- 143.Valentine H, Amarnath K, Amarnath V, et al. Globin s-propyl cysteine and urinary N-acetyl-S-propylcysteine as internal biomarkers of 1-bromopropane exposure. Toxicol Sci 2007;98(2):427–435. [DOI] [PubMed] [Google Scholar]
- 144.Wang H, Ichihara G, Ito H, et al. Biochemical changes in the central nervous system of rats exposed to 1-bromopropane for seven days. Toxicol Sci 2002;67(1):114–120. [DOI] [PubMed] [Google Scholar]
- 145.Wang H, Ichihara G, Ito H, et al. Dose-dependent biochemical changes in rat central nervous system after 12-week exposure to 1-bromopropane. Neurotoxicology 2003;24(2):199–206. [DOI] [PubMed] [Google Scholar]
- 146.Rasband MN, Macklin WB. Myelin Structure and Biochemistry. In: Brady ST, Siegel GJ, Albers RW, Price DL, eds. Basic Neurochemistry 8th ed. New York: Academic Press; 2012:180–199. [Google Scholar]
- 147.Aschner M, Toews AD. Myelin and Myelination. In: Philbert M, ed. Comprehensive Toxicology Vol 13 2nd ed. Kidlington, United Kingdom: Elsevier Ltd; 2010:181–198. [Google Scholar]
- 148.Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell 1985;40(3):501–508. [DOI] [PubMed] [Google Scholar]
- 149.Reiss DS, Lees MB, Sapirstein VS. Is Na + ATPase a myelin-associated enzyme? J Neurochem 1981;36(4):1418–1426. [DOI] [PubMed] [Google Scholar]
- 150.Valentine WM. Fixation and processing of the peripheral nervous system. In: Aminoff MJ, Daroff RB eds. Encyclopedia of the Neurological Sciences 2nd ed. Oxford: Academic Press; 2014:317–323. [Google Scholar]
- 151.Towfighi J, Gonatas NK, McCree L. Hexachlorophene-induced changes in central and peripheral myelinated axons of developing and adult rats. Lab Invest 1974;31(6):712–721. [PubMed] [Google Scholar]
- 152.Towfighi J, Gonatas NK, McCree L. Hexachlorophene neuropathy in rats. Lab Invest 1973;29(4):428–436. [PubMed] [Google Scholar]
- 153.Atkins CE, Johnson RK. Clinical toxicities of cats. Vet Clin North Am 1975;5(4):623–652. [DOI] [PubMed] [Google Scholar]
- 154.Tripier MF, Berard M, Toga M, Martin-Bouyer G, Le Breton R, Garat J. Hexachlorophene and the central nervous system. Toxic effects in mice and baboons. Acta Neuropathol 1981;53(1):65–74. [DOI] [PubMed] [Google Scholar]
- 155.Cammer W, Fredman T, Rose AL, Norton WT. Brain carbonic anhydrase: activity in isolated myelin and the effect of hexachlorophene. J Neurochem 1976;27(1):165–171. [DOI] [PubMed] [Google Scholar]
- 156.Rose AL, Wisniewski HM, Cammer W. Neurotoxicity of hexachlorophene: new pathological and biochemical observations. J Neurol Sci 1975;24(4):425–435. [DOI] [PubMed] [Google Scholar]
- 157.Pleasure D, Towfighi J, Silberberg D, Parris J. The pathogenesis of hexachlorophene neuropathy: in vivo and in vitro studies. Neurology 1974;24(11):1068–1075. [DOI] [PubMed] [Google Scholar]
- 158.Graham DI, de Jesus PV, Pleasure DE, Gonatas NK. Triethyltin sulfate-induced neuropathy in rats. Electrophysiologic, morphologic, and biochemical studies. Arch Neurol 1976;33(1):40–48. [DOI] [PubMed] [Google Scholar]
- 159.Aleu FP, Katzman R, Terry RD. Fine structure and electrolyte analyses of cerebral edema induced by alkyl tin intoxication. J Neuropathol Exp Neurol 1963;22(3):403–413. [DOI] [PubMed] [Google Scholar]
- 160.Lock EA, Aldridge WN. The binding of triethyltin to rat brain myelin. J Neurochem 1975;25(6):871–876. [DOI] [PubMed] [Google Scholar]
- 161.Aldridge WN, Cremer JE. The biochemistry of organo-tin compounds; diethyltin dichloride and triethyltin sulphate. Biochem J 1955;61(3):406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lock EA. The action of triethyltin on the respiration of rat brain cortex slices. J Neurochem 1976;26(5):887–892. [DOI] [PubMed] [Google Scholar]
- 163.Wassenaar JS, Kroon AM. Effects of triethyltin on different ATPases, 5’-nucleotidase and phosphodiesterases in grey and white matter of rabbit brain and their relation with brain edema. Eur Neurol 1973;10(6):349–370. doi: 10.1159/000114294. [DOI] [PubMed] [Google Scholar]
- 164.Powers MF, Beavis AD. Triorganotins inhibit the mitochondrial inner membrane anion channel. J Biol Chem 1991;266(26):17250–17256. [PubMed] [Google Scholar]
- 165.Selwyn MJ, Dawson AP, Stockdale M, Gains N. Chloride-hydroxide exchange across mitochondrial, erythrocyte and artificial lipid membranes mediated by trialkyl- and triphenyltin compounds. Eur J Biochem 1970;14(1):120–126. [DOI] [PubMed] [Google Scholar]
- 166.Duckett S, Said G, Streletz LG, White RG, Galle P. Tellurium-induced neuropathy: correlative physiological, morphological and electron microprobe studies. Neuropathol Appl Neurobiol 1979;5(4):265–278. [DOI] [PubMed] [Google Scholar]
- 167.Lampert P, Garro F, Pentschew A. Tellurium neuropathy. Acta Neuropathol 1970;15(4):308–317. [DOI] [PubMed] [Google Scholar]
- 168.Said G, Duckett S. Tellurium-induced myelinopathy in adult rats. Muscle Nerve 1981;4(4):319–325. [DOI] [PubMed] [Google Scholar]
- 169.Bouldin TW, Samsa G, Earnhardt TS, Krigman MR. Schwann cell vulnerability to demyelination is associated with internodal length in tellurium neuropathy. J Neuropathol Exp Neurol 1988;47(1):41–47. [DOI] [PubMed] [Google Scholar]
- 170.Harry GJ, Goodrum JF, Bouldin TW, Wagner-Recio M, Toews AD, Morell P. Tellurium-induced neuropathy: metabolic alterations associated with demyelination and remyelination in rat sciatic nerve. J Neurochem 1989;52(3):938–945. [DOI] [PubMed] [Google Scholar]
- 171.Wagner-Recio M, Toews AD, Morell P. Tellurium blocks cholesterol synthesis by inhibiting squalene metabolism: preferential vulnerability to this metabolic block leads to peripheral nervous system demyelination. J Neurochem 1991;57(6):1891–1901. [DOI] [PubMed] [Google Scholar]
- 172.Rawlins FA, Smith ME. Myelin synthesis in vitro: a comparative study of central and peripheral nervous tissue. J Neurochem 1971;18(10):1861–1870. [DOI] [PubMed] [Google Scholar]
- 173.Toews AD, Lee SY, Popko B, Morell P. Tellurium-induced neuropathy: a model for reversible reductions in myelin protein gene expression. J Neurosci Res 1990;26(4):501–507. [DOI] [PubMed] [Google Scholar]
- 174.Berciano MT, Calle E, Fernandez R, Lafarga M. Regulation of Schwann cell numbers in tellurium-induced neuropathy: apoptosis, supernumerary cells and internodal shortening. Acta Neuropathol 1998;95(3):269–279. [DOI] [PubMed] [Google Scholar]
- 175.Jurevics H, Morell P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem 1995;64(2):895–901. [DOI] [PubMed] [Google Scholar]
- 176.McAuley JH, Fearnley J, Laurence A, Ball JA. Diphtheritic polyneuropathy. J Neurol, Neurosurg Psychiatry 1999;67(6):825–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kanwal SK, Yadav D, Chhapola V, Kumar V. Post-diphtheritic neuropathy: a clinical study in paediatric intensive care unit of a developing country. Trop Doct 2012;42(4):195–197. [DOI] [PubMed] [Google Scholar]
- 178.Falnes PO, Sandvig K. Penetration of protein toxins into cells. Curr Opin Cell Biol 2000;12(4):407–413. [DOI] [PubMed] [Google Scholar]
- 179.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell 1992;69(6):1051–1061. [DOI] [PubMed] [Google Scholar]
- 180.Skretting G, Torgersen ML, van Deurs B, Sandvig K. Endocytic mechanisms responsible for uptake of GPI-linked diphtheria toxin receptor. J Cell Sci 1999;112 ( Pt 22):3899–3909. [DOI] [PubMed] [Google Scholar]
- 181.Pleasure DE, Feldmann B, Prockop DJ. Diphtheria toxin inhibits the synthesis of myelin proteolipid and basic proteins by peripheral nerve in vitro. J Neurochem 1973;20(1):81–90. [DOI] [PubMed] [Google Scholar]
- 182.Jolly RD. The pathogenic action of the exotoxin of Corynebacterium ovis. J Comp Pathol 1965;75(4):417–431. [DOI] [PubMed] [Google Scholar]
- 183.Matheson DF, Cavanagh JB. Protein synthesis in peripheral nerve and susceptibility to diphtheritic neuropathy. Nature 1967. 214(5089):721–2. [DOI] [PubMed] [Google Scholar]
- 184.Frisoni GB, Di Monda V. Disulfiram neuropathy: a review (1971–1988) and report of a case. Alcohol Alcohol 1989;24(5):429–437. [PubMed] [Google Scholar]
- 185.Behan C, Lane A, Clarke M. Disulfiram induced peripheral neuropathy: between the devil and the deep blue sea. Ir J Psychol Med 2007;24(3):115–116. [DOI] [PubMed] [Google Scholar]
- 186.Tran AT, Rison RA, Beydoun SR. Disulfiram neuropathy: two case reports. J Med Case Rep 2016; 10(72):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Santos T, Martins Campos A, Morais H. Sensory-motor axonal polyneuropathy involving cranial nerves: An uncommon manifestation of disulfiram toxicity. Clin Neurol Neurosurg 2017;152:12–15. [DOI] [PubMed] [Google Scholar]
- 188.Huang J, Campian JL, Gujar AD, et al. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J Neurooncol 2016;128(2):259–266. [DOI] [PubMed] [Google Scholar]
- 189.Tonkin EG, ERVE JCL, Valentine WM. Disulfiram produces a non-carbon disulfide-dependent schwannopathy in the rat. J Neuropathol Exp Neurol 2000;59(9):786–797. [DOI] [PubMed] [Google Scholar]
- 190.Tonkin EG, Valentine HL, Zimmerman LJ, Valentine WM. Parenteral N,N-diethyldithiocarbamate produces segmental demyelination in the rat that is not dependent on cysteine carbamylation. Toxicol Appl Pharmacol 2003;189(2):139–150. [DOI] [PubMed] [Google Scholar]
- 191.Johnson DJ, Graham DG, Amarnath V, Amarnath K, VAlentine WM. Release of carbon disulfide is a contributing mechanism in the axonopathy produced by N,N-diethyldithiocarbamate. Toxicol Appl Pharmacol 1998; 142(2):288–296. [DOI] [PubMed] [Google Scholar]
- 192.Johnson DJ, Graham DG, Amarnath V, Amarnath K, Valentine WM. The measurement of 2-thiothiazolidine-4-carboxylic acid as an Index of the in vivo release of CS2 by dithiocarbamates. Chem Res Toxicol 1996;9(5):910–916. [DOI] [PubMed] [Google Scholar]
- 193.Erve JCL, Amarnath V, Sills RC, Morgan DL, Valentine WM. Characterization of a valine–lysine thiourea cross-link on rat globin produced by carbon disulfide or N,N-diethyldithiocarbamate in vivo. Chem Res Toxicol 1998;11(10):1128–1136. [DOI] [PubMed] [Google Scholar]
- 194.Viquez OM, Lai B, Ahn JH, Does MD, Valentine HL, Valentine WM. N,N-diethyldithiocarbamate promotes oxidative stress prior to myelin structural changes and increases myelin copper content. Toxicol Appl Pharmacol 2009;239(1):71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Viquez OM, Valentine HL, Friedman DB, Olson SJ, Valentine WM. Peripheral nerve protein expression and carbonyl content in N,N-diethlydithiocarbamate myelinopathy. Chem Res Toxicol 2007;20(3):370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Viquez OM, Valentine HL, Amarnath K, Milatovic D, Valentine WM. Copper accumulation and lipid oxidation precede inflammation and myelin lesions in N,N-diethyldithiocarbamate peripheral myelinopathy. Toxicol Appl Pharmacol 2008;229(1):77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Valentine HL, Viquez OM, Amarnath K, et al. Nitrogen substituent polarity influences dithiocarbamate-mediated lipid oxidation, nerve copper accumulation, and myelin injury. Chem Res Toxicol 2009;22(1):218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Valentine HL, Amarnath K, Amarnath V, Valentine WM. Dietary copper enhances the peripheral myelinopathy produced by oral pyrrolidine dithiocarbamate. Toxicol Sci 2006;89(2):485–494. [DOI] [PubMed] [Google Scholar]
- 199.Riet-Correa G, Fernandes CG, Pereira LAV, Graça DL. Ethidium bromide-induced demyelination of the sciatic nerve of adult Wistar rats. Braz J Med Biol Res 2002;35(1):99–104. [DOI] [PubMed] [Google Scholar]
- 200.Hollis ER II, Ishiko N, Tolentino K, et al. A novel and robust conditioning lesion induced by ethidium bromide. Experimental Neurology 2015;265(C):30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wallace VCJ, Cottrell DF, Brophy PJ, Fleetwood-Walker SM. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci 2003;23(8):3221–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Koizumi S, Yamamoto S, Hayasaka T, et al. Imaging mass spectrometry revealed the production of lyso-phosphatidylcholine in the injured ischemic rat brain. Neuroscience 2010;168(1):219–225. [DOI] [PubMed] [Google Scholar]
- 203.Shanta SR, Choi CS, Lee JH, et al. Global changes in phospholipids identified by MALDI MS in rats with focal cerebral ischemia. J Lipid Res 2012;53(9):1823–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hanada M, Sugiura Y, Shinjo R, et al. Spatiotemporal alteration of phospholipids and prostaglandins in a rat model of spinal cord injury. Anal Bioanal Chem 2012;403(7):1873–1884. [DOI] [PubMed] [Google Scholar]
- 205.Hanrieder J, Ewing AG. Spatial elucidation of spinal cord lipid- and metabolite-regulations in amyotrophic lateral sclerosis. Sci Rep 2014;4:5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pieragostino D, D’Alessandro M, di Ioia M, et al. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol Biosyst 2015;11(6):1563–1572. [DOI] [PubMed] [Google Scholar]
- 207.Plemel JR, Michaels NJ, Weishaupt N, et al. Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia 2018;66(2):327–347. [DOI] [PubMed] [Google Scholar]
- 208.Hall SM, Gregson NA. The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibres of the adult mouse. J Cell Sci 1971;9(3):769–789. [DOI] [PubMed] [Google Scholar]
- 209.Hanz S, Fainzilber M. Retrograde signaling in injured nerve--the axon reaction revisited. J Neurochem 2006;99(1):13–19. [DOI] [PubMed] [Google Scholar]
- 210.Scheib J, Höke A. Advances in peripheral nerve regeneration. Nat Rev Neurol 2013;9(12):668–676). [DOI] [PubMed] [Google Scholar]
- 211.Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol 2008;18(3):276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Martin SL, Reid AJ, Verkhratsky A, Magnaghi V, Faroni A. Gene expression changes in dorsal root ganglia following peripheral nerve injury: roles in inflammation, cell death and nociception. Neural Regen Res 2019;14(6):939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li S, Xue C, Yuan Y, et al. The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci Rep 2015:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chandran V, Coppola G, Nawabi H, et al. A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 2016;89(5):956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ohara PT, Vit J-P, Bhargava A, et al. Gliopathic pain: when satellite glial cells go bad. Neuroscientist 2009;15(5):450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kwon MJ, Kim J, Shin H, et al. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J Neurosci 2013;33(38):15095–15108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Elmore S Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dennis KE, Valentine WM. Degenerative and Regenerative Events in the Central and Peripheral Nervous System. In: Philbert M, ed. Comprehensive Toxicology Vol 6 3rd ed. Oxford: New York, New York: Elsevier, Inc.; 2018:50–69. [Google Scholar]
- 219.Carozzi VA, Canta A, Chiorazzi A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci Lett 2015;596:90–107. [DOI] [PubMed] [Google Scholar]
- 220.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther 2009;8(1):10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology 2006;27(6):992–1002. [DOI] [PubMed] [Google Scholar]
- 222.Cavaletti G, Ceresa C, Nicolini G, Marmiroli P. Neuronal drug transporters in platinum drugs-induced peripheral neurotoxicity. Anticancer Res 2014;34(1):483–486. [PubMed] [Google Scholar]
- 223.Ciarimboli G Membrane transporters as mediators of cisplatin effects and side effects. Scientifica 2012;2012(9):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sprowl JA, Ciarimboli G, Lancaster CS, et al. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. P Natl Acad Sci Usa 2013;110(27):11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest 1998;101(12):2842–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Scuteri A, Galimberti A, Maggioni D, et al. Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology 2009;30(2):312–319. [DOI] [PubMed] [Google Scholar]
- 227.Podratz JL, Knight AM, Ta LE, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiology of Disease 2011;41(3):661–668. doi: 10.1016/j.nbd.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.McDonald ES, Windebank AJ. Cisplatin-induced apoptosis of DRG neurons involves bax redistribution and cytochrome c release but not fas receptor signaling. Neurobiol Dis 2002;9(2):220–233. [DOI] [PubMed] [Google Scholar]
- 229.Alaedini A, Xiang Z, Kim H, Sung Y-J, Latov N. Up-regulation of apoptosis and regeneration genes in the dorsal root ganglia during cisplatin treatment. Exp Neurol 2008;210(2):368–374. doi: 10.1016/j.expneurol.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 230.Cavaletti G, Tredici G, Petruccioli MG, et al. Effects of different schedules of oxaliplatin treatment on the peripheral nervous system of the rat. Eur J Cancer 2001;37(18):2457–2463. [DOI] [PubMed] [Google Scholar]
- 231.Adelsberger H, Quasthoff S, Grosskreutz J, Lepier A, Eckel F, Lersch C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur J Pharmacol 2000;406(1):25–32. [DOI] [PubMed] [Google Scholar]
- 232.Park SB, Lin CS-Y, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Dose effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS ONE 2011;6(4):e18469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wu S-N, Chen B-S, Wu Y-H, Peng H, Chen L-T. The mechanism of the actions of oxaliplatin on ion currents and action potentials in differentiated NG108–15 neuronal cells. Neurotoxicology 2009;30(4):677–685. [DOI] [PubMed] [Google Scholar]
- 234.Webster RG, Brain KL, Wilson RH, Grem JL, Vincent A. Oxaliplatin induces hyperexcitability at motor and autonomic neuromuscular junctions through effects on voltage-gated sodium channels. Brit J Pharmacol 2005;146(7):1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Nassini R, Gees M, Harrison S, et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011;152(7):1621–1631. [DOI] [PubMed] [Google Scholar]
- 236.Zhao M, Isami K, Nakamura S, Shirakawa H, Nakagawa T, Kaneko S. Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol Pain 2012;8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Park J-H, Chae J, Roh K, et al. Oxaliplatin-Induced Peripheral Neuropathy via TRPA1 Stimulation in Mice Dorsal Root Ganglion Is Correlated with Aluminum Accumulation. PLoS ONE 2015;10(4):e0124875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Farina M, Rocha JBT, Aschner M. Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies. Life Sci 2011;89(15–16):555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Nagashima K A review of experimental methylmercury toxicity in rats: neuropathology and evidence for apoptosis. Toxicol Pathol 1997;25(6):624–631. [DOI] [PubMed] [Google Scholar]
- 240.Hunter D, Russel DS. Focal cerebral and cerebellar atrophy in a human subject due to organic mercury compounds. J Neurol, Neurosurg Psychiatry 1954;17(4):235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Irukayama K, Kai F, Fujiki M, Kondo T. Studies on the origin of the causative agent of Minamata disease. III. Industrial wastes containing mercury compounds from Minamata Factory. Kumamoto Med J 1962;15:57–68. [PubMed] [Google Scholar]
- 242.Takeuchi T Pathology of Minamata disease. With special reference to its pathogenesis. Acta Pathol Jpn 1982;32 Suppl 1:73–99. [PubMed] [Google Scholar]
- 243.Shinoda Y, Ehara S, Tatsumi S, et al. Methylmercury-induced neural degeneration in rat dorsal root ganglion is associated with the accumulation of microglia/macrophages and the proliferation of Schwann cells. J Toxicol Sci 2019;44(3):191–199. [DOI] [PubMed] [Google Scholar]
- 244.Ceccatelli S, Dare E, Moors M. Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact 2010;188(2):301–308. [DOI] [PubMed] [Google Scholar]
- 245.Atchison WD, Hare MF. Mechanisms of methylmercury-induced neurotoxicity. FASEB J 1994;8(9):622–629. [DOI] [PubMed] [Google Scholar]
- 246.Wilke RA, Kolbert CP, Rahimi RA, Windebank AJ. Methylmercury induces apoptosis in cultured rat dorsal root ganglion neurons. Neurotoxicology 2003;24(3):369–378. [DOI] [PubMed] [Google Scholar]
- 247.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol 1978;14(4):633–643. [PubMed] [Google Scholar]
- 248.Thoenen H, Tranzer JP. Chemical sympathectomy by selective destruction of adrenergic nerve endings with 6-Hydroxydopamine. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol 1968;261(3):271–288. [DOI] [PubMed] [Google Scholar]
- 249.Tiffany-Castiglioni E, Saneto RP, Proctor PH, Perez-Polo JR. Participation of active oxygen species in 6-hydroxydopamine toxicity to a human neuroblastoma cell line. Biochem Pharmacol 1982;31(2):181–188. [DOI] [PubMed] [Google Scholar]
- 250.Storch A, Kaftan A, Burkhardt K, Schwarz J. 6-Hydroxydopamine toxicity towards human SH-SY5Y dopaminergic neuroblastoma cells: independent of mitochondrial energy metabolism. J Neural Transm 2000;107(3):281–293. [DOI] [PubMed] [Google Scholar]
- 251.Glinka Y, Gassen M, Youdim MB. Mechanism of 6-hydroxydopamine neurotoxicity. J Neural Transm Suppl 1997;50:55–66. [DOI] [PubMed] [Google Scholar]
- 252.Asanuma M, Hirata H, Cadet JL. Attenuation of 6-hydroxydopamine-induced dopaminergic nigrostriatal lesions in superoxide dismutase transgenic mice. Neuroscience 1998;85(3):907–917. [DOI] [PubMed] [Google Scholar]
- 253.Bensadoun JC, Mirochnitchenko O, Inouye M, Aebischer P, Zurn AD. Attenuation of 6-OHDA-induced neurotoxicity in glutathione peroxidase transgenic mice. Eur J Neurosci 1998;10(10):3231–3236. [DOI] [PubMed] [Google Scholar]