


Abstract
Spatially resolved visualization of RNA processing and structures is important for better studying single-cell RNA function and landscape. However, currently available RNA imaging methods are limited to sequence analysis, and not capable of identifying RNA processing events and structures. Here, we developed click-encoded rolling FISH (ClickerFISH) for visualizing RNA polyadenylation and structures in single cells. In ClickerFISH, RNA 3′ polyadenylation tails, single-stranded and duplex regions are chemically labeled with different clickable DNA barcodes. These barcodes then initiate DNA rolling amplification, generating repetitive templates for FISH to image their subcellular distributions. Combined with single-molecule FISH, the proposed strategy can also obtain quantitative information of RNA of interest. Finally, we found that RNA poly(A) tailing and higher-order structures are spatially organized in a cell type-specific style with cell-to-cell heterogeneity. We also explored their spatiotemporal patterns during cell cycle stages, and revealed the highly dynamic organization especially in S phase. This method will help clarify the spatiotemporal architecture of RNA polyadenylation and structures.
INTRODUCTION
RNA plays diverse biological roles in coding, decoding, expression and regulation of genes. Its synthesis, processing and higher-order structures underlie fundamental biological processes and critical functions (1,2). Especially, 3′-end polyadenylation processing, also known as poly(A) tailing, influences RNA stability and translation (3,4), and is related to cancer activation and metastasis (5). Ascribed to conformational flexibility, RNA species can fold into stem-loop or other structures, which is essential for their information-carrying, regulatory and catalytic roles (6–9). Therefore, determining RNA polyadenylation and structures is crucial to expanding our understanding of RNA function and networks. Visualizing these RNA features in single cells can reveal their subcellular distribution pattern and cell-to-cell variation, yet it remains scarcely explored.
As we know, many methods have been established for imaging specific RNA sequence (10–17) and protein or enzyme activity (18–21). Among them, these RNA imaging included mRNA, microRNA and even base modification analysis, and provided the spatial location information in single cells. Additionally, some in situ imaging technologies such as single-molecule fluorescence in situ hybridization (smFISH) (22,23) and enzymatic DNA amplification-assisted FISH (24–29) have achieved single-molecule sensitivity based on signal amplification, and determined spatially resolved RNA copies in single cells. Despite the effectiveness, these methods can only detect RNA sequence of interest due to hybridization-assisted target recognition. Thus they are not capable of labeling RNA polyadenylation and identifying structured RNA regions. On the other hand, visualizing protein synthesis (30,31) and post-translational processing (19,32,33) in single cells have been demonstrated by coupling metabolic or chemical labeling with high-efficiency DNA amplification. Nevertheless, neither the chemical labels nor amplification methods used in protein imaging are suitable for RNA analysis. Therefore, to develop amplified imaging strategy for visualizing single-cell RNA polyadenylation and structures is an urgent demand and a formidable challenge.
We herein developed click-encoded rolling FISH (Clicker-FISH) as a general and scalable approach for translating cellular RNA features into chemical barcodes and spatially resolved images. As shown in Figure 1, RNA poly(A) tails and single-stranded/duplex regions are labeled with different chemical tags by metabolic labeling in living cells or post modification in fixed cells. They are encoded with clickable DNA barcodes, and their subcellular distributions can be visualized by enzymatic amplification-assisted FISH. Notably, the fluorescent DNA probe is hybridized to the DNA amplicon with repetitive sequence, rather than to target RNA as that in typical FISH. As the DNA amplicon is chemically linked to target RNA, we still call the proposed strategy FISH.
Figure 1.

Overview of ClickerFISH visualizing RNA processing and structures. (A) The workflow for visualizing RNA polyadenylation and single-stranded/duplex regions. (B) The detailed chemical reactions are illustrated, respectively. DNA1, DNA2 and DNA3 indicate the clickable DNA barcodes initiating rolling amplification.
MATERIALS AND METHODS
Materials
2-Ethynyl-adenosine (2-EA) and 2-Ethynyl-ATP (2-EATP) were purchased from Jena Bioscience (Jena, Germany). Azide-modified 2-methylnicotinic acid imidazolide (NAI-N3) from MedChem Express (Shanghai, China). Succinimidyl-[4-(psoralen-8-yloxy)]butyrate (SPB), DBCO-cy5 and azide-FAM were obtained from Sigma Aldrich. Trans-Cyclooctene-amine (TCO-amine) and methyltetrazine-NHS ester were obtained from Click Chemistry Tools (Scottsdale, USA). Tetrazine-cy3 (Tz-cy3) and azide-cy5 (N3-cy5) were purchased from Lumiprobe (Maryland, USA). All chemicals were used as received without further purification. The reactions were performed in RNase-free water and buffers. The oligonucleotides used in this work (Supplementary Table S1) were synthesized by Sangon Biological Co. Ltd (Shanghai, China). DNA marker, T4 DNA ligase, Exonuclease I, Exonuclease III, RiboLock RNase Inhibitor and Trizol Reagent were obtained from Takara Biotechnology Co. Ltd (Dalian, China). Poly(U) polymerase, phi29 DNA polymerase and shrimp alkaline phosphatase (rSAP) were purchased from New England Biolabs Ltd (Beijing, China) and RNase I from ThermoFisher Scientific.
Preparation of circularized padlocks and amplicons in vitro
The circularized padlock probes were prepared by a multi-step reaction of hybridization, ligation and digestion. 10 μM padlock probes were hybridized with 30 μM linker prones in 10 μl 1 × T4 DNA ligase reaction buffer at 55°C for 2 h. Then, 3 μl T4 DNA ligase (350 U/μl) was added and the solution was incubated at 37°C for 2 h and then at 65°C for 30 min. After that, 5 μl Exonuclease I (5 U /μl) and 2 μl Exonuclease III (200 U /μl) were added to digest linear DNA. The enzymes were inactivated by heating at 80°C for 30 min. The resulting probes were stored at –20°C. In a typical amplification reaction, 10 μl of 1× phi29 DNA polymerase reaction buffer containing 1 μl of circularized padlock probes, 2 μl dNTPs (2 mM) and 2 μl of DNA barcode (2 μM) and 0.5 μl phi29 DNA polymerase (10 U/μl) were incubated at 37°C for 1 h and then at 65°C for 10 min to stop the reaction. The amplicons were characterization by gel electrophoresis and atomic force microscopy (AFM). These single-stranded amplicons can be assembled as monodisperse nanoparticles driven by liquid crystallization and dense packaging rather than Watson-Crick base pairing (27). AFM tips (model ScanAsyst-Air; Bruker multimode 8) are used in this work. A freshly cleaved mica surface for sample mounting is firstly prepared. 10 mM Ni2+ solution is added to the center of the mica surface for 3 min. After rinsing the mica surface vigorously with Milli-Q water followed by its dry with nitrogen gas, 2 μl solution of amplicons is pipetted onto the center of the mica surface for incubation of 10 min. Then, 10 μl of annealing buffer is added. The image is operated in ScanAsyst-air model under ambient air.
Gel electrophoresis and mass spectrometry analysis
The in vitro polyadenylation products were analyzed by 15% denaturing PAGE in 1× TBE buffer at a 165 V constant voltage for 60 min. The isolated RNA samples and rolling amplicons were analyzed by 1% agarose gel electrophoresis in 1× TBE buffer. The gels were visualized with UV or fluorescence channel by ChampChemI imaging system (Sagecreation, Beijing, China). For MS analysis, the samples were analyzed on a Waters I-Class Vion IMS Qtof using a reverse phase column (Waters BEH C18, 1.7 μm, 2.1 × 50 mm) detected by electrospray ionization (positive ion mode). Liquid chromatography was performed by a gradient analysis: flow rate: 0.4 ml/min; eluent A: H2O in 0.1% (v/v) formic acid; eluent B: MeOH; gradient: 0 min-7 min, 95% A, 5% B, curve: initial; 7 min-9 min, 0% A, 100% B, curve: 6; 9 min-11 min, 0% A, 100% B, curve: 6; 11 min-12 min, 95% A, 5% B, curve: 6; 12 min-13 min, 95% A, 5% B, curve: 6.
Characterization of RNA chemical labels
Firstly, the polyadenylation of both a 22-nt RNA oligonucleotide with 2-EATP in vitro and cellular RNA with 2-EA in living cells were performed, respectively. In a typical in vitro polyadenylation reaction (20 μl), 1 μg 22-nt RNA substrate, 1 mM 2-EATP, 9 mM ATP and 6 units of Poly(U) polymerase supplemented with its buffer were used. The reaction was performed at 37°C for 2 h. The polyadenylation product was clicked with N3-cy5 and then purified and precipitated with 50% isopropanol. Gel electrophoresis was used to analyze these products. For the analysis of cellular RNA polyadenylation, 2 × 106 cultured cells were treated with 2 μM ActD for 1 h and then with 100 μM 2-EA for 1 h metabolic labeling. The RNA was directly isolated and then digested into nucleosides by RNase I and rSAP for mass spectrometry analysis (Thermo Scientific Q Exactive Focus Orbitrap LC–MS/MS system). In the experiments of RNase H/oligo(dT)-catalyzed degradation of poly(A) tails, these treated cells were fixed and labeled with N3-cy5 before RNA isolation. Then, the acylation of NTP and total RNA by NAI-N3 were demonstrated. In experiments testing NTP acylation, 10 μM each NTP (ATP, UTP, GTP or CTP) and 1 mM NAI-N3 were incubated in the buffer (100 mM HEPES, pH 8.0, 6 mM MgCl2 and 100 mM NaCl) at 37°C for 1 h. The products were dephosphorylated by rSAP to nucleosides for mass spectrometry analysis. For the acylation of total RNA, 25 μg RNA was incubated with 200 mM NAI-N3 at 37°C for 30 min. After purification, the sample was treated with 100 μM DBCO-cy5 at 37°C for 1 h. A second purification was used to remove excess DBCO-cy5 for the gel electrophoresis analysis. The synthesis of trans-cyclooctene-functionalized psoralen derivative (TCO-Pso) was carried out by incubating SPB with TCO-amine. The product was tested by mass spectrometry.
Cell cultures and RNA isolation
Mammalian cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% of heat-inactivated fetal bovine serum and 1% antibiotics penicillin/streptomycin (100 U/ml) in a humidified incubator containing CO2 (5%) at 37°C. Lovastatin, hydroxyurea and colchicine were used to synchronize cells in G1, S and G2 phase, respectively. In general, RNA was harvested using Trizol Reagent according to the manufacturer's instructions. For the isolation of RNA in 2-EA-treated cells that were fixed before N3-cy5 labeling, 2 × 106 cells seeded in a six-well plate was treated with Proteinase K (2 mg/ml) for 1 h at 50°C. Then, the cell lysate was subjected to the protocol of Trizol Reagent.
Visualizing RNA polyadenylation and structures
In a typical experiment, 6000 cells were seeded on a collagen A-coated coverglass enclosed in a PDMS chamber (4 mm in diameter) at 37°C for overnight. When the cells reached the desired confluency, they were cultured with 2 μM ActD for 1 h to inhibit transcription. The drugs were kept in the medium after the addition of 100 μM 2-EA for 1 h incubation. After that, they were fixed in 4% (w/v) paraformaldehyde for 10 min at room temperature.
The cells were then permeabilized with 0.5% (v/v) Triton X-100 for 5 min at room temperature and then briefly washed three times with PBS. Cells were reacted with 2 mM NAI-N3 and 0.5 μl RiboLock RNase Inhibitor (40 U/ml) at 37°C for 30 min. After three washes with PBS, a copper-free click reaction was carried out at 37°C for 60 min in the presence of 0.1 μM DBCO-barcode and 2.5 μg/ml yeast tRNA. The cell were washed three times in wash buffer (2x SSC, 300 mM NaCl, 30 mM Sodium Citrate, pH 7), each consisting of at least 5 minutes. Then, we performed the copper(I)-catalyzed click reaction between 2-EA and N3. A solution containing N3-barcode, 2.5 μg/ml yeast tRNA, 1 mM CuSO4 and 100 mM sodium ascorbate (made fresh and added to solution immediately before use) was added to the samples for 45 min incubation at room temperature. The samples were then rinsed with wash buffer, and then the nuclear DNA was stained and blocked by 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 20 min at room temperature.
After that, these cells were incubated with 0.4 mM TCO-Pso in PBS for 10 min and then were irradiated with 365 nm UV radiation for 20 min. The third click reaction was performed by incubating cells with freshly synthesized 1 μM Tz-barcode and 2.5 μg/ml yeast tRNA in PBS for 1 h at 37°C in the dark. Tz-barcode was prepared by mixing NH2-barcode and methyltetrazine-NHS ester dissolved in DMSO at room temperature for 3 h. The samples were then rinsed with wash buffer.
For enzymatic rolling amplification, 20 μl of 1× SSC hybridization mixture containing three circularized padlocks (200 nM each), 0.5 μl of RiboLock RNase Inhibitor (40 U/ml) and 1μl of yeast tRNA (50 μg/ml) were added to the fixed cells for 3 h incubation at 37°C. After washing three times, the cell samples were incubated with 20 μl 1× phi29 DNA polymerase buffer containing 5 units phi29 DNA polymerase, 0.5 μl RiboLock RNase Inhibitor (40 U/ml), 2.5 mM dNTPs and 0.5 μl BSA (10 mg/ml) at 37°C for 2 h.
After washing twice, the cell samples were incubated with 200 nM fluorophore-labeled DNA probes in 2× SSC and 20% formamide buffer for 30 min at 37°C. The samples were washed three times. The nucleus and mitochondria were stained by DAPI and MitoTracker® Red CMTMRos (Invitrogen), respectively. The cells were washed three times before imaging. All fluorescence images were acquired using a laser scanning confocal microscopy (TCS SP8 STED 3X, Leica). The cellular images were acquired using a 63× water objective (NA 1.2).
Imaging processing and data extraction
Cells were randomly selected, and over ∼25 cells were chosen for cell samples. The fluorescence intensity correlation of two marks (ssRNA and dsRNA, RNA polyadenylation and mitochondria) was calculated by LAS X Version: 3.1.5.16308. Intensity distributions of RNA polyadenylation and DAPI signals were measured as a function of radial distance from the nuclear center toward the cell membrane. The average intensity within a series of annuli is extracted and calculated by MATLAB. Each channel was normalized to its mean intensity per cell. The codes and commands in MATLAB are shown in the Supplementary information. Pearson correlations between polyadenylation and DAPI signals were measured on a per-pixel basis within the nucleus. Correlation data was fit to a linear regression on a cell population basis. The RGB intensity information of each channel were extracted by MATLAB.
RESULTS
Overview and characterization of ClickerFISH
In this work, we use an adenosine analog 2-ethynyl adenosine (2-EA, appending an alkyne moiety to the C2 atom) to label poly(A) tails during cellular polyadenylation process (4). Azide-modified 2-methylnicotinic acid imidazolide (NAI-N3) enables high 2′-hydroxyl acylation of nucleotides in flexible or single-stranded RNA regions instead of base-paired duplex regions (34). On the other hand, RNA duplex regions can be tagged by trans-cyclooctene-functionalized psoralen derivative (TCO-Pso) that is prepared by crosslinking succinimidyl-[4-(psoralen-8-yloxy)]butyrate (SPB) and TCO-amine. It is well known that psoralen and its derivatives intercalate into base-paired nucleotides, preferentially crosslinking juxtaposed pyrimidines (especially thymine and uracil) upon 365 nm UV irradiation (35). Three sets of click reactions, including copper(I)-catalyzed N3-alkyne and copper-free N3-dibenzocyclooxtyne (DBCO) and alkene-tetrazine (Tz), are utilized here. To avoid cross interference, we performed copper-free click reactions before the copper(I)-catalyzed one.
We first investigated whether 2-ethynyl ATP (2-EATP) can work as a substrate like ATP in polyadenylation reactions. The in vitro 3′ tailing of a 22-nt RNA oligonucleotide was performed. The products were further clicked with N3-modified fluorescence, and then analyzed by denaturing polyacrylamide gel electrophoresis (PAGE). According to the UV and fluorescence images (left panel of Figure 2A), poly(A) tails were generated by reactions containing 2-EATP. To confirm specific incorporation of 2-EA throughout poly(A) tails during cellular RNA polyadenylation processes, we incubated MCF-10A cells with 2-EA in the presence of a transcription inhibitor actinomycin D (ActD). As we know, ActD prevents RNA elongation by binding to DNA at dG residues. The total RNA was isolated and clicked with fluorescence signal. We later digested the poly(A) tails of RNA samples by the oligo(dT)-RNase H treatment. As shown in the middle of Figure 2A, the 2-EA signal in clicked RNA from ActD-treated cells was lost. We further found 2-EA in isolated RNA from ActD-treated cells by mass spectrometry (Figure 2A). These data indicate that 2-EA can be specifically incorporated into poly(A) tails during polyadenylation in living cells. Then, we investigated the acylation of NTP and total RNA by NAI-N3. The mass spectrometry results confirmed the acylation of all four nucleotide at both 2′ and 3′ hydroxyl (Figure 2B and Supplementary Figure S1–S4). The gel electrophoresis visualized the efficient acylation of total RNA (Figure 2B). Finally, the TCO-Pso tag was synthesized and characterized (Figure 2C).
Figure 2.

Characterization of RNA chemical labels. (A) 2-EA is incorporated into poly(A) chains in vitro (left panel) and in cells (middle panel); right panel is the mass spectrum of 2-EA from RNase I/rSAP-treated total RNA (calculated mass of [C12H14N5O4]+ = 292.1040, found: 292.1048). (B) NAI-N3 can label ATP at both 2′-OH and 3′-OH (left panel, mass spectrum, ATP-2NAI-N3, calculated mass of [C24H22N13O6]+ = 588.1811, found: 588.1791), and total RNA (right panel). (C) The synthesis of TCO-Pso for tagging RNA duplex regions. Left, LC-MS extracted ion chromatograms of TCO (black), SPB (blue) and TCO-Pso (red); right, the mass spectrum of TCO-Pso (calculated mass of [C27H33N2O7]+ = 497.2282, found: 497.2269).
The enzymatic amplification in ClickerFISH is based on rolling circle replication of short DNA circles (24,27,28,36). Three sets of clickable barcode primers and corresponding padlock probes (Supplementary Table S1) were designed to minimize non-specific complementarity using the NCBI’s BLAST Human transcripts program. The resulting amplicons were characterized as monodisperse particles of 50–150 nm in diameter by AFM (Figure 3A). And they own tens of kb length and hundreds of a periodically repeated sequences according to agarose gel image, allowing high-efficiency amplification of FISH signals. These probe sets all executed independent DNA amplification without cross interference, indicating the sequence orthogonality. Notably, the length and size of amplicons can be easily regulated by reaction conditions (27,37). We then used ClickerFISH to visualize RNA polyadenylation in ActD/2-EA-treated cells. The non-amplifying system that uses clickable fluorescent dyes was designed as a control. As shown in Figure 3B and Supplementary Figure S5A, no nonspecific signals are induced by other two unmatched barcodes. And ClickerFISH achieves much higher and more fluorescence signals than the non-amplifying control, which allows visualizing RNA molecules with limited labeling sites. Additionally, many bright spots with various sizes and fluorescence intensities are observed. The different staining patterns depend on the distribution and density of RNA labeling site, and non-uniform RCA efficiency in the complex intracellular environment including crowded proteins and other biomacromolecules. We also analyzed cell samples treated with RNase I or oligo(dT)-RNase H prior to ClickerFISH reactions. The fluorescence signals nearly disappeared (Supplementary Figure S5B), validating the ClickerFISH signals in cells.
Figure 3.

Amplification and imaging performance of ClickerFISH. (A) Characterization of enzymatic amplification. Left, AFM image of amplicons, scale bar = 300 nm; right, gel electrophoresis image of click-encoded amplicons. (B) Representative cell images (green, polyadenylation; blue, DAPI). The scale bars of all cell images in this work is 10 μm. The upper inset illustrates a typical RNA polyadenylation and corresponding imaging; The AAUAAA is the Poly(A) Signal (PAS) sequence.
Visualizing single-cell RNA polyadenylation and structures of different cell types
Then we profiled global RNA polyadenylation and structures in three human breast cell types (epithelial MCF-10A, non-invasive MCF-7 and invasive MBA-MD-231), revealing cell type-specific spatial organization of RNA processing and structures with significant single-cell variation. The poly(A) tail is added at the end of transcription in nucleus and poly(A)-binding protein promotes the export of RNA from nucleus into cytoplasm. We thus hypothesized that poly(A) tailing signal would be accumulated and more prominent in cytoplasm of individual cells. In MCF-10A cells, this signal intensity gradually increased from the nucleus center to the outside of the nucleus periphery (Figure 4A). They lacked clusters of polyadenylation fluorescence in the nucleus, and there is no significant correlation (r = 0.086) between polyadenylation and DAPI signals. However, analysis of MCF-7 and MBA-MD-231 cells showed exceptions to this simplistic pattern. In these two cells, prominent nuclear clusters of polyadenylation foci were present, and they exhibited a positive correlation (r = 0.224 and 0.213) between polyadenylation and DAPI signals. The distribution of the polyadenylation fluorescence varied dramatically from cell to cell in all three cell types. The notable distinction of RNA polyadenylation in these cell types may imply the post-transcriptional regulation of oncogene or suppressor gene without genetic alteration (5). We also developed polyadenylation labeling with MitoTracker imaging to study mitochondrial RNA processing. Three-color image combining MitoTracker® Red CMTMRos, DAPI and 2-EA signal clearly delineated a high overlap between Mito and poly(A) labeling signal outside of the nucleus (Supplementary Figure S6).
Figure 4.

Cell type-specific organization of RNA polyadenylation and structures. (A) RNA polyadenylation analysis. Each cell type has three columns; each contains: (left) a representative cell image (blue, DAPI; green, polyadenylation fluorescence); (middle) signal intensity of DAPI and polyadenylation as a function of distance from nuclear center to cell membrane. Each line represents one cell and the bold lines indicate the means. n is the number of cells; (right) Pearson correlation of polyadenylation and DAPI signal intensity. (B) RNA structures analysis. Left, representative cell images from different cell types. right, cell type-specific organization of RNA structures. The violin plot represents of the correlation coefficients between RNA flexible signal and duplex signal per cell in different cell types. **** (P < 0.0001), ** (P < 0.005), Student's t-test.
The spatial distribution fashion of RNA structures in single cells was then investigated. Due to psoralen crosslinking both duplex RNA and DNA, fixed cells were firstly incubated with DAPI that binds strongly to duplex DNA with a preference for adenine-thymine rich regions. We found that DAPI occupied binding sites (juxtaposed pyrimidines) and blocked following psoralen crosslinking of DNA (Supplementary Figure S7). Strong colocalization of RNA flexible (single-stranded) signal with duplex signal are observed (Figure 4B and Supplementary Figure S8), indicating transcripts are organized into higher-level architectures to impact gene regulation. These structured RNAs are heterogeneously distributed in the cytoplasm and nucleus, and the spatial maps are quite distinctive in different cell lines (3). Notably, the correlation coefficient in cancer cell lines (MCF-7 and MBA-MD-231) is significantly higher than that in MCF-10A. These results imply that RNA functions in a structure-dependent manner by interacting with RNA-binding proteins or microRNAs (38). Combined with smFISH, the proposed strategy can also obtain quantitative information of RNA sequence of interest. The nucleoporin 43 (Nup43) mRNA is used as an example (39). The imaging result in Supplementary Figure S9 reveals varied structure organization of the same mRNA copies at different subcellular locations. Collectively, all these observations revealed that different cell types have a diverse spatial distribution and organization of RNA processing and structures.
Cell cycle-specific organization of RNA polyadenylation and structures
Finally, we simultaneously imaged RNA polyadenylation, flexible regions and duplex structures in the cell cycle using three-barcode ClickerFISH. It is well known that RNA is transcribed rapidly in G1 phase and G2 phase. And DNA replication occurs in S phase, whereas the rate of RNA transcription is very low during this phase. An interesting question is how transcripts are organized during the cell cycle. As shown in Figure 5, Supplementary Figures S10 and S11, RNA polyadenylation and structures are spatially organized in a cell cycle-specific fashion. In both MCF-10A and MDA-MB-231 cells, the ClickerFISH signal decreased at the cell-cycle transition from G1 (2N DNA content) to S phase and then increased at S/G2 (4N DNA content) transition. However, analysis of MCF-7 cells showed exception to this fashion. These results explored the spatiotemporal patterns of RNA polyadenylation and structures, and revealed their dynamic organization especially in S phase (40).
Figure 5.
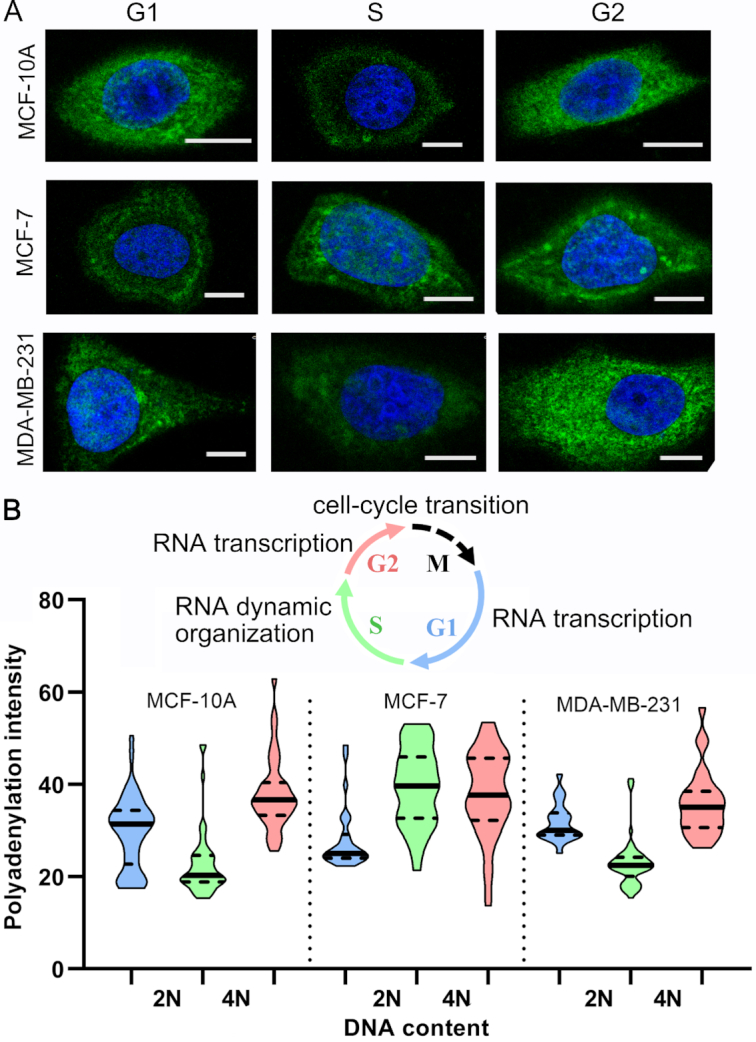
Cell cycle-specific RNA polyadenylation. (A) Representative cell images (green, polyadenylation fluorescence; blue, DAPI; scale bar, 10 μm). (B) The violin plot (G1, blue; S, green; G2, pink) depicting RNA polyadenylation intensity distribution. The cell numbers are 43, 49, 39, 30, 33, 50, 27, 57 and 28, respectively (from left to right). The inset diagram indicates RNA features during cell cycle transitions. M represents the Mitosis phase.
DISCUSSION
Our goal here was to develop a general strategy to visualize RNA polyadenylation and structures in single cells. As far as we know, many methods have been established for detecting expression level of specific RNA in fixed cells (22,23,25,27–29) or live cells (10–13). And some of them achieved single-molecule sensitivity and determined RNA copy numbers based on signal amplification. However, these methods are dependent on sequence hybridization for the recognition of RNA of interest, and not able to label polyadenylation events and identify structured RNA regions. On the other hand, metabolic and chemical labeling approaches have been employed to tag and visualize protein synthesis and post-translational modification (30–32,41). Inspired by these results, we speculate that RNA polyadenylation and structures can also be visualized by coupling related chemical labeling with amplified imaging methods. Thus, we chose three representative clickable chemicals to each label RNA polyadenylation events (4), single-stranded regions (34) and double-stranded regions (35,38). They have been demonstrated to specifically react with these RNA features rather than other biomolecules such as proteins or DNA in cells. We also design clickable DNA barcode probes to distinguish these chemicals and initiate DNA amplification-assisted FISH imaging.
For the first time, our strategy ClickerFISH described the subcellular distribution of these RNA features which are indispensable for better studying single-cell RNA function and landscape. This method uses three kinds of click chemistry reactions to encode these RNA features into amplified fluorescence signals. We reveals cell type-specific spatial organization of RNA polyadenylation and structures with single-cell variation. We also explored their spatiotemporal patterns during different cell cycle stages, and found the highly dynamic organization especially in S phase. Additionally, implementing barcoded DNA probes enabled multiplexed readout. Except these RNA features, ClickerFISH can be expanded for detecting RNA modifications such as N6-methyladenosine and pseudouridine. Simultaneous visualization of RNA and protein processing events or features may also be feasible with more sets of barcoded DNA probes. In addition, two methods, nuclear magnetic resonance (NMR) spectroscopy (42) and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) (43,44), have been well established for the analysis of RNA structures. The NMR method requires isotopically labeled RNAs by chemical synthesis or in vitro transcription, and is limited to short RNAs. SHAPE analyzes base flexibility in RNAs of various lengths based on structure-sensitive acylation reagents and modest extensions of acylated RNAs. Both NMR and SHAPE determine the RNA structure with single nucleotide resolution, yet they can’t provide subcellular distribution information.
Combined with sequence-specific smFISH, ClickerFISH achieved quantitative analysis of RNA of interest. In ClickerFISH, the amplicon is visualized as a bright fluorescent spot after the hybridization of fluorescence DNA probes. The intensity of these spots represents the number of label site of each RNA molecule. To obtain these RNA features of multiplexed and even transcriptome sequences, it is required to integrate ClickerFISH with single-cell transcriptomics imaging methods based on sequential hybridization (45–47). Simultaneously visualizing RNA polyadenylation or structures and sequence of single-cell transcriptome will be a challenging and highly significant work.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Miss Lu and Miss Hao at Instrument Analysis Center of Xi’an Jiaotong University for their assistance with MS and fluorescence imaging analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation of China [31671013, 21705124, 21874105]; China Postdoctoral Science Foundation [2017M613102, 2018T111032]; Fundamental Research Funds for the Central Universities and ‘Young Talent Support Plan’ of Xi’an Jiaotong University. Funding for open access charge: National Science Foundation of China [21874105].
Conflict of interest statement. None declared.
REFERENCES
- 1. Goodarzi H., Nguyen H.C.B., Zhang S., Dill B.D., Molina H., Tavazoie S.F.. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016; 165:1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fehrmann R.S.N., Karjalainen J.M., Krajewska M., Westra H.J., Maloney D., Simeonov A., Pers T.H., Hirschhorn J.N., Jansen R.C., Schultes E.A. et al.. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015; 47:115–125. [DOI] [PubMed] [Google Scholar]
- 3. Wu X., Bartel D.P.. Widespread Influence of 3′-End structures on mammalian mRNA processing and stability. Cell. 2017; 169:905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Curanovic D., Cohen M., Singh I., Slagle C.E., Leslie C.S., Jaffrey S.R.. Global profiling of stimulus-induced polyadenylation in cells using a poly(A) trap. Nat. Chem. Biol. 2013; 9:671–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayr C., Bartel D.P.. Widespread Shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009; 138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ziv O., Gabryelska M.M., Lun A.T.L., Gebert L.F.R., Sheu-Gruttadauria J., Meredith L.W., Liu Z.Y., Kwok C.K., Qin C.F., MacRae I.J. et al.. COMRADES determines in vivo RNA structures and interactions. Nat. Methods. 2018; 15:785–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramani V., Qiu R., Shendure J.. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 2015; 33:980–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rouskin S., Zubradt M., Washietl S., Kellis M., Weissman J.S.. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2013; 505:701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li M., Zheng M., Wu S., Tian C., Liu D., Weizmann Y., Jiang W., Wang G., Mao C.. In vivo production of RNA nanostructures via programmed folding of single-stranded RNAs. Nat. Commun. 2018; 9:2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo X., Xue B., Feng G., Zhang J., Lin B., Zeng P., Li H., Yi H., Zhang X.L., Zhu H. et al.. Lighting up the native viral RNA genome with a fluorogenic probe for the live-cell visualization of virus infection. J. Am. Chem. Soc. 2019; 141:5182–5191. [DOI] [PubMed] [Google Scholar]
- 11. Ying Z.M., Wu Z., Tu B., Tan W., Jiang J.H.. Genetically encoded fluorescent RNA sensor for ratiometric imaging of MicroRNA in living tumor cells. J. Am. Chem. Soc. 2017; 139:9779–9782. [DOI] [PubMed] [Google Scholar]
- 12. He X., Zeng T., Li Z., Wang G., Ma N.. Catalytic molecular imaging of MicroRNA in living cells by DNA-Programmed nanoparticle disassembly. Angew. Chem. Int. Ed. 2016; 55:3073–3076. [DOI] [PubMed] [Google Scholar]
- 13. Wu C., Cansiz S., Zhang L., Teng I.T., Qiu L., Li J., Liu Y., Zhou C., Hu R., Zhang T. et al.. A nonenzymatic hairpin DNA cascade reaction provides high signal gain of mRNA imaging inside live cells. J. Am. Chem. Soc. 2015; 137:4900–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu L., Gao Y., Kuang H., Liz-Marzán L.M., Xu C.. MicroRNA-Directed intracellular Self-Assembly of chiral nanorod dimers. Angew. Chem. Int. Ed. 2018; 57:10544–10548. [DOI] [PubMed] [Google Scholar]
- 15. Wu Z., Liu G.-Q., Yang X.L., Jiang J.H.. Electrostatic nucleic acid nanoassembly enables hybridization chain reaction in living cells for ultrasensitive mRNA imaging. J. Am. Chem. Soc. 2015; 137:6829–6836. [DOI] [PubMed] [Google Scholar]
- 16. Zhang P., He Z., Wang C., Chen J., Zhao J., Zhu X., Li C.-Z., Min Q., Zhu J.J.. In situ amplification of intracellular microRNA with MNAzyme nanodevices for multiplexed imaging, logic operation, and controlled drug release. ACS Nano. 2014; 9:789–798. [DOI] [PubMed] [Google Scholar]
- 17. Ranasinghe R.T., Challand M.R., Ganzinger K.A., Lewis B.W., Softley C., Schmied W.H., Horrocks M.H., Shivji N., Chin J.W., Spencer J.. Detecting RNA base methylations in single cells by in situ hybridization. Nat. Commun. 2018; 9:655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian R., Ding L., Yan L., Lin M., Ju H.. A robust probe for lighting up intracellular telomerase via primer extension to open a nicked molecular beacon. J. Am. Chem. Soc. 2014; 136:8205–8208. [DOI] [PubMed] [Google Scholar]
- 19. Li S., Liu Y., Liu L., Feng Y., Ding L., Ju H.. A Hierarchical Coding Strategy for Live Cell Imaging of Protein-Specific Glycoform. Angew. Chem. Int. Ed. 2018; 130:12183–12187. [DOI] [PubMed] [Google Scholar]
- 20. Wang L.J., Ma F., Tang B., Zhang C.Y.. Sensing telomerase: From in vitro detection to in vivo imaging. Chem. Sci. 2017; 8:2495–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang H., Chen S., Li P., Wang L., Li J., Li J., Yang H.H., Tan W.. Nongenetic approach for imaging protein dimerization by aptamer recognition and proximity-induced DNA assembly. J. Am. Chem. Soc. 2018; 140:4186–4190. [DOI] [PubMed] [Google Scholar]
- 22. Battich N., Stoeger T., Pelkmans L.. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods. 2013; 10:1127. [DOI] [PubMed] [Google Scholar]
- 23. Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S.. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008; 5:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue C., Zhang S.X., Ouyang C.H., Chang D., Salena B.J., Li Y., Wu Z.S.. Target-Induced catalytic assembly of Y-Shaped DNA and its application for in situ imaging of MicroRNAs. Angew. Chem. Int. Ed. 2018; 130:9887–9891. [DOI] [PubMed] [Google Scholar]
- 25. Deng R., Zhang K., Wang L., Ren X., Sun Y., Li J.. DNA-Sequence-Encoded rolling circle amplicon for Single-Cell RNA imaging. Chem. 2018; 4:1373–1386. [Google Scholar]
- 26. Wu Z., Fan H., Satyavolu N.S.R., Wang W., Lake R., Jiang J.H., Lu Y.. Imaging endogenous metal ions in living cells using a DNAzyme–catalytic hairpin assembly probe. Angew. Chem. Int. Ed. 2017; 129:8847–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu G., Hu R., Zhao Z., Chen Z., Zhang X., Tan W.. Noncanonical Self-Assembly of multifunctional DNA nanoflowers for biomedical applications. J. Am. Chem. Soc. 2013; 135:16438–16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jia H., Li Z., Liu C., Cheng Y.. Ultrasensitive detection of microRNAs by exponential isothermal amplification. Angew. Chem. Int. Ed. 2010; 49:5498–5501. [DOI] [PubMed] [Google Scholar]
- 29. Choi H.M.T., Chang J.Y., Trinh L.A., Padilla J.E., Fraser S.E., Pierce N.A.. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 2010; 28:1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang J., Xue J., Yan Z., Zhang S., Qiao J., Zhang X.. Photoluminescence lifetime imaging of synthesized proteins in living cells using an Iridium–Alkyne probe. Angew. Chem. Int. Ed. 2017; 56:14928–14932. [DOI] [PubMed] [Google Scholar]
- 31. Tom Dieck S., Kochen L., Hanus C., Heumüller M., Bartnik I., Nassim-Assir B., Merk K., Mosler T., Garg S., Bunse S.. Direct visualization of newly synthesized target proteins in situ. Nat. Methods. 2015; 12:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao X., Hannoush R.N.. Single-cell imaging of Wnt palmitoylation by the acyltransferase porcupine. Nat. Chem. Biol. 2014; 10:61–66. [DOI] [PubMed] [Google Scholar]
- 33. Hui J., Bao L., Li S., Zhang Y., Feng Y., Ding L., Ju H.. Localized chemical remodeling for live cell imaging of Protein‐Specific glycoform. Angew. Chem. Int. Ed. 2017; 56:8139–8143. [DOI] [PubMed] [Google Scholar]
- 34. Spitale R.C., Flynn R.A., Zhang Q.C., Crisalli P., Lee B., Jung J.-W., Kuchelmeister H.Y., Batista P.J., Torre E.A., Kool E.T. et al.. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015; 519:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sharma E., Sterne-Weiler T., O’Hanlon D., Blencowe B.J.. Global mapping of human RNA-RNA interactions. Mol. Cell. 2016; 62:618–626. [DOI] [PubMed] [Google Scholar]
- 36. Lin C., Wang X., Liu Y., Seeman N.C., Yan H.. Rolling circle enzymatic replication of a complex Multi-Crossover DNA nanostructure. J. Am. Chem. Soc. 2007; 129:14475–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu R., Zhang X., Zhao Z., Zhu G., Chen T., Fu T., Tan W.. DNA Nanoflowers for multiplexed cellular imaging and traceable targeted drug delivery. Angew. Chem. Int. Ed. 2014; 53:5821–5826. [DOI] [PubMed] [Google Scholar]
- 38. Lu Z., Zhang Q.C., Lee B., Flynn R.A., Smith M.A., Robinson J.T., Davidovich C., Gooding A.R., Goodrich K.J., Mattick J.S. et al.. RNA Duplex map in living cells reveals Higher-Order transcriptome structure. Cell. 2016; 165:1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mellis I.A., Gupte R., Raj A., Rouhanifard S.H.. Visualizing adenosine-to-inosine RNA editing in single mammalian cells. Nat. Methods. 2017; 14:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park J.E., Yi H., Kim Y., Chang H., Kim V.N.. Regulation of Poly(A) tail and translation during the somatic cell cycle. Mol. Cell. 2016; 62:462–471. [DOI] [PubMed] [Google Scholar]
- 41. Li G., Montgomery J.E., Eckert M.A., Chang J.W., Tienda S.M., Lengyel E., Moellering R.E.. An activity-dependent proximity ligation platform for spatially resolved quantification of active enzymes in single cells. Nat. Commun. 2017; 8:1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Varani G., Aboul-ela F., Allain F.H. T.. NMR investigation of RNA structure. Prog. Nucl. Mag. Res. Sp. 1996; 29:51–127. [Google Scholar]
- 43. Merino E.J., Wilkinson K.A., Coughlan J.L., Weeks K.M.. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005; 127:4223–4231. [DOI] [PubMed] [Google Scholar]
- 44. Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y.. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013; 9:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coskun A.F., Cai L.. Dense transcript profiling in single cells by image correlation decoding. Nat. Methods. 2016; 13:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X.. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015; 348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lubeck E., Coskun A.F., Zhiyentayev T., Ahmad M., Cai L.. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods. 2014; 11:360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.