

Abstract
An unique catalytic strategy was recently reported for the glmS ribozyme [Bingaman et al., Nat. Chem. Biol. 2017, 13, 439−445] that involves promotion of productive hydrogen bonding of the O2′ nucleophile to facilitate its activation. We provide broad evidence of this strategy in the hammerhead, pistol, and VS ribozymes and 8-17 DNAzyme, enabled by a functionally important divalent metal ion that interacts with the scissile phosphate and disrupts nonproductive competitive hydrogen bonding with the O2′ nucleophile. This strategy, designated tertiary gamma (3°γ) catalysis, illustrates an additional role for divalent ions in ribozyme catalysis.
Keywords: RNA catalysis, gamma catalysis, catalytic metal, metalloenzymes, computational, free energy simulations
Much of our understanding of catalytic RNAs is gleaned from detailed studies of small self-cleaving nucleolytic ribozymes that perform site-specific phosphodiester bond cleavage reactions and share structural and mechanistic features.1,2 The general strategies for catalyzing phosphodiester bond cleavage employed by nucleolytic ribozymes, first laid out by Breaker et al.,3 include acquisition of in-line fitness of the 2′-OH nucleophile (α catalysis), electrostatic stabilization of the nonbridging phosphoryl oxygens (NPOs) of the dianionic phosphorane transition state (β catalysis), deprotonation and activation of the 2′-OH nucleophile (γ catalysis), and electrostatic stabilization and protonation of the 5′-O leaving group (δ catalysis). Recently, we stratified Breaker’s framework into a more precise ontology for discussion of RNA-cleaving enzymes by decomposing the β, γ, and δ strategies into primary (1°), secondary (2°), and tertiary (3°) contributions.4,5
The idea of elimination of nonproductive hydrogen bond competition as a catalytic strategy (3°γ catalysis) was recently introduced in Bingaman et al. for the glmS ribozyme5,6 and is described as follows. A requirement for nucleophile activation is that the 2′-OH nucleophile is in position to donate a hydrogen bond to the N1 position of the guanine general base (in deprotonated form). Therefore, if the nucleophile is engaged in competing (nonproductive) hydrogen bonds with the NPOs of the scissile phosphate, the occupation of productive hydrogen bonds to the general base are diminished, and activity is reduced. Hence, engineering other interactions with the NPOs that disrupt these nonproductive hydrogen bond interactions with the nucleophile correspondingly increases the occupancy of productive hydrogen bonds to the general base and leads to enhanced activity. This catalytic strategy was originally termed as gamma′′,7 and later designated as a form of 3°γ catalysis in the new ontology.4 Formally, 3°γ catalysis is the promotion of nucleophile activation (γ catalysis) through modification of the structural scaffold or hydrogen bond network (3° effect) that organizes the enzyme active site (including orienting the HO2′ nucleophile for activation by the general base). In the present case, focus is placed on modification of the hydrogen bond network (HBN) through alteration of the orientation of the HO2′ atom itself, rather than disruption of the active site structural scaffold, which gives rise to the catalytic effect, which henceforth will be referred to as 3°γ (HBN) when a precise definition is required for clarity.
In the case of the glmS ribozyme, large normal thio effects were observed in the holoribozyme at the pro-RP NPO of the scissile phosphate that were not rescuable.7 This was interpreted as arising from disruption of hydrogen bonding interactions with nucleobases and the cofactor that have a direct catalytic effect. However, a considerable (∼30-fold) inverse thio effect was observed in the aporibozyme at the same position. It was reasoned that in the aporibozyme (Figure S1), with the oxo substrate, the HO2′ forms a strong nonproductive hydrogen bond with the pro-RP NPO, which unlike in the holoribozyme, cannot receive a hydrogen bond from the cofactor. In the RP thio substrate, the sulfur atom at the pro-RP position disrupts the nonproductive HO2′–pro-RP interaction in the aporibozyme, thereby promoting the competing productive HO2′–G40:N1 interaction (where G40:N1, when in deprotonated form, is the heteroatom of the general base that abstracts the HO2′). This facilitates O2′ activation by G40:N1, and gives rise to the inverse thio effect in the aporibozyme. In subsequent studies, this “new” catalytic strategy was validated in the holoribozyme by systematically changing the hydrogen-bonding pattern at the scissile phosphate NPOs by considering different variants and a modified 1-deoxyglucosamine 6-phosphate cofactor (lacking a 2′OH observed to donate a hydrogen bond to the pro-RP NPO in the crystal structure) and measuring thio effects at the NPO positions and mutational rescue.5 In the reported study,5 it was concluded that glmS ribozyme uses an overdetermined set of hydrogen bond donors to the pro-RP position to promote nucleophile activation.
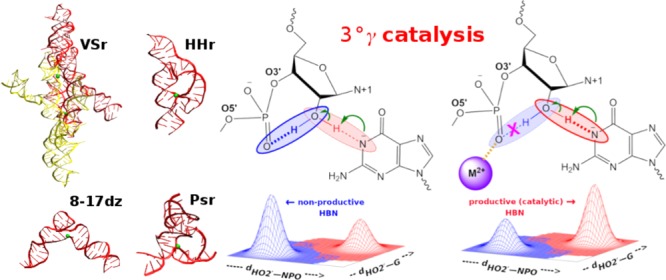
An alternate mechanism to facilitate nucleophile activation through promotion of productive hydrogen bonding (3°γ catalysis) that can come into play, which has not yet been systematically investigated, is one in which a divalent metal ion interacts with one of the NPOs of the scissile phosphate (Figure 1). In the absence of the metal ion, the O2′ can form a favorable hydrogen bond to either of the NPO positions (nonproductive hydrogen bonds). However, metal ions potentially can play multiple roles (structural organization, tuning of nucleobase pKa, or direct involvement in the chemical reaction) and as such may contribute to more than one catalytic strategy. It is extremely challenging to design experimental measurements that can unambiguously deconstruct these individual catalytic contributions; however, computational methods can impose constraints that experiments cannot, enabling in some cases their isolation and providing a way to investigate them independently. As described above, we distinguish this precise mode of catalysis as 3°γ (HBN) as it arises from the alteration of the hydrogen bond network in the active site that supports γ catalysis.
Figure 1.

Tertiary γ (HBN) catalysis induced by metal ion binding. Schematic illustrating 3°γ (HBN) catalysis induced by a divalent metal ion. In the absence of any metal ions (left) the HO2′ can form a nonproductive interaction with a NPO of the scissile phosphate that competes with productive hydrogen bonding with the Watson–Crick edge of the general base guanine in deprotonated (activated) form. In the presence of a metal ion interacting with that NPO (right), the nonproductive interaction is weakened and consequently productive interactions are promoted in which the HO2′ hydrogen bonds to the N1 position of the deprotonated general base guanine as required for nucleophile activation. Here “HBN” is used to distinguish a specific form of 3°γ catalysis that more generally involves the promotion of nucleophile activation through modification of the structural scaffold or hydrogen bond network that organizes the enzyme active site.
We investigate the elimination of nonproductive hydrogen bonding to promote nucleophile activation, that is, 3°γ (HBN) catalysis, induced by metal ion binding in the hammerhead (HHr), pistol (Psr), and Varkud satellite (VSr) ribozymes and the recently crystallized 8-17 DNAzyme (8-17dz) (Figure 2). These systems all catalyze site-specific RNA strand cleavage by 2′-O-transphosphorylation and share remarkable similarities in their catalytic sites and mechanisms,8−10 including a conserved guanine implicated as a general base and a functionally critical divalent metal ion bound at the scissile phosphate. We quantify the 3°γ (HBN) effect for each system using a novel computational approach that enables free energy mapping of the active and inactive hydrogen bonding probability distributions and their coupling with divalent metal ion binding.
Figure 2.

Structures and active sites of VSr, HHr, Psr, and 8-17dz. Shown are the crystal structures and representative active sites in their active protonation state (e.g., general base guanine deprotonated at the N1 position) and conformational state derived from MD simulations in solution. (A, B) VSr dimer crystal structure12 (PDB ID 5V3I) and MD active state; (C, D) HHr crystal structure13 (PDB ID 2OEU) and MD active state;8 (E, F) Psr crystal structure14 (PDB ID 5K7C) and MD active state;9 (G, H) 8-17dz crystal structure15 (PDB ID 5XM8) and MD active state.10
VSr, HHr, Psr, and 8-17dz share a common active site “L-platform” architecture and a functionally critical metal ion recruiting pocket (Figure 2) as part of the surrounding scaffold. The “L-platform” motif was first identified by Piccirilli, Lilley and co-workers in VSr, hairpin ribozyme (HPr), and HHr11 and later expanded to include key “L-scaffold” residues and illustrated to be a framework for design utilized also in the twister (Twr) ribozyme, Psr, and 8-17dz. The L-platform/L-scaffold framework can be designed to contain a metal binding pocket (the “L-pocket”) that recruits a functionally critical divalent metal ion. HHr, Psr, and 8-17dz belong to the “G+M+” catalytic paradigm; they use guanine as the general base and a metal ion (Mg2+ in case of HHr and Psr, Pb2+ in case of 8-17dz) directly in the catalytic reaction. VSr belongs to the “G+A” catalytic paradigm that exclusively uses nucleobases in the chemical steps of catalysis, but unlike other “G+A” ribozymes (e.g., Twr and HPr), in VSr, a metal ion (Mg2+) plays a critical organizational role in the active site to anchor and tune the pKa of the general base, without taking direct part in the catalytic reaction.
We designed a novel computational approach to gain predictive insight into the extent of 3°γ (HBN) catalytic effects in the HHr, VSr, Psr, and 8-17dz induced by divalent metal ion binding at the scissile phosphate (L-pocket). Our approach uses molecular dynamics (MD) simulation with enhanced sampling techniques16 and recently developed models for divalent metal ion binding to nucleic acids17 to construct free energy maps of two-dimensional probability distribution functions (2D PDFs) (Figure 3) using coordinates that report on productive (catalytic) and nonproductive hydrogen bond networks in the active site (Figure 1). Productive hydrogen bonds are tracked as the distance of the HO2′ (hydrogen attached to the O2′ nucleophile) from the Watson–Crick (WC) edge of the catalytic guanine general base (dHO2′–G), and nonproductive hydrogen bonds are tracked as the distance of the HO2′ from the NPOs of the scissile phosphate (dHO2′–NPO). These maps were created for each ribozyme system both in the presence and in the absence of a divalent metal ion bound in the L-pocket (Figure 3). Metal ion binding modes to the NPO in the case of VSr, HHr, and 8-17dz involved direct (inner-sphere) coordination, and in the case of Psr involved indirect (outer-sphere) coordination. These binding modes are consistent with the current body of experimental thio/rescue effect and metal ion titration experiments18−20 and predictions from molecular dynamics simulations.8−10,21 Full details of the simulations are provided in Supporting Information.
Figure 3.
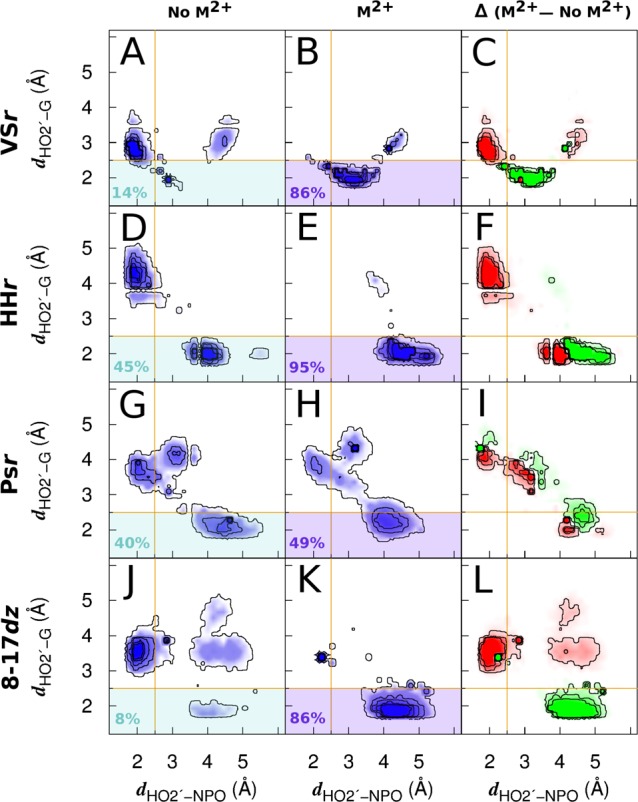
Evidence of 3°γ (HBN) effect in VSr, HHr, Psr, and 8-17dz. Two dimensional probability distribution functions (2D PDFs) constructed from the unbiased and reweighted umbrella sampling simulations (see Supporting Information for full details) are shown for VSr [A–C], HHr [D–F], Psr [G–I], and 8-17dz [J–L]. The 2D PDFs are constructed along the coordinates dHO2′–G and dHO2′–NPO, where the dHO2′–G coordinate corresponds to the distance of the HO2′ from the WC edge of the catalytic guanine (minimal distance to either N1 or O6 positions) and the dHO2′–NPO coordinate corresponds to the distance of the HO2′ from the NPO of the scissile phosphate to which the metal ion is bound (pro-RP NPO in the case of HHr, 8-17dz, and Psr and pro-SP NPO in the case of VSr). Panels A, D, G, and J correspond to 2D PDFs obtained in the absence of the divalent metal ion, panels B, E, H, and K correspond to 2D PDFs obtained in the presence of the divalent metal ion, and panels C, F, I, and L correspond to difference plots of the respective 2D PDFs obtained in the presence and absence of the metal ion (red and green indicate negative and positive values of the difference maps, respectively). Threshold value of the dHO2′–G and dHO2′–NPO coordinates (2.5 Å) beyond which the respective interactions are not considered to exist is indicated by gold colored lines, and percentages of productive hydrogen bonding are indicated at the bottom left of each PDF.
For each of the systems, in the absence of a divalent metal ion bound to the NPO, significant nonproductive hydrogen bonding is observed as illustrated in the left “No M2+” panels A, D, G, and J in Figure 3 by substantial probability density in the unshaded region corresponding to the hydrogen bonding of the nucleophile to the NPOs (dHO2′–NPO < 2.5 Å). For VSr and 8-17dz, occupancy of productive hydrogen bonding in the absence of the metal is quite small (14% and 8%, respectively), whereas for HHr and Psr, it is more substantial (45% and 40%, respectively). This can be explained by the prediction from molecular simulations that the divalent metal in VSr and 8-17dz10 plays a more critical role in organizing the active site for γ catalysis than that in HHr8,21 and Psr.9 In the presence of a divalent metal ion bound to the NPO, the probability distributions shift by depleting the probability density for nonproductive hydrogen bonding with the NPOs (shown in red) to productive hydrogen bonding with the Watson–Crick edge of the general base guanine (shown in green). This shift is most pronounced for VSr, HHr, and 8-17dz, where the metal ion is inner-sphere bound to the NPO, and least pronounced for Psr, where the metal ion is outer-sphere bound to the NPO.
The 3°γ (HBN) induced by metal ion binding in HHr, VSr, 8-17dz, and Psr is quantified in Figure 4 and summarized in Table 1. For each system, the one-dimensional probability distribution functions (1D PDFs) along the productive hydrogen bonding coordinates dHO2′–G in the presence of the metal ion are compared to analogous 1D PDFs in absence of the metal ion. We define the 3°γ (HBN) factor (shown in red) as the ratio of the areas under the 1D PDFs that reside below a threshold value (2.5 Å) of the HO2′–guanine distance, obtained in the presence and absence of the metal ion.
Figure 4.

Quantitative determination of 3°γ (HBN) effect induced by metal ion binding in VSr, HHr, Psr, and 8-17dz. One-dimensional probability distribution functions (1D PDFs) constructed from the unbiased and reweighted umbrella sampling simulations (see Supporting Information for full details) are shown for VSr (A), HHr (B), Psr (C), and 8-17dz (D). The 1D PDFs are constructed along the coordinates dHO2′–G that correspond to the distance of the HO2′ from the WC edge of the catalytic N1-deprotonated guanine (minimal distance to either N1 or O6 positions). 1D PDFs obtained in the presence of the metal ion are shown in purple and 1D PDFs obtained in absence of metal ion are shown in cyan. The 3°γ (HBN) factor is defined as the ratio of the areas under the 1D PDFs obtained in the presence and absence of the metal ion that reside below a threshold value (2.5 Å marked by gold line) of the HO2′–guanine distance (shaded region).
Table 1. Summary of 3°γ (HBN) Effect in VSr, HHr, Psr, and 8-17dza.
| productive
hydrogen bonding (%) |
|||
|---|---|---|---|
| system | M2+ | no M2+ | 3°γ (HBN) |
| VSr | 86 | 14 | 6.1 |
| HHr | 95 | 45 | 2.1 |
| Psr | 49 | 40 | 1.2 |
| Psr* | 78 | 40 | 2.0 |
| 8-17dz | 86 | 8 | 10.8 |
| productive
hydrogen bonding (%) |
|||
|---|---|---|---|
| oxo | RP thio | inv. thio | |
| VSr DM | 23 | 88 | 3.8 |
| expt | 6 | ||
Percentages of productive hydrogen bonding and corresponding 3°γ (HBN) factors for VSr, HHr, Psr, and 8-17dz (Figure 4). For Psr, the divalent metal ion is outer-sphere bound to the NPO as suggested by experiment20 and theory;9 however, for discussion we also consider an inner-sphere binding mode indicated as “Psr*” (Figure S3). Also shown are results in Figure 3 for the inverse thio effect for oxo and RP thio substrates interpreted as the 3°γ (HBN) for the VSr G638I/A756(3cP) double mutant (“VSr DM”, Figure S2) and compared with experiment (expt).
The 3°γ (HBN) factor is highest in 8-17dz (10.8), followed by VSr (6.1), indicative of the pronounced 3°γ (HBN) effect in these enzymes. In HHr, the 3°γ (HBN) factor is 2.1, despite that, like VSr and 8-17dz, the metal ion in the simulations makes an inner sphere contact with the NPO. This can be explained by the result that, in the absence of a divalent metal ion bound at the NPO, the HHr retains a significant degree of productive hydrogen bonding (45%) that increases the denominator of the 3°γ (HBN) factor reducing its magnitude. In fact, in the presence of the bound divalent ion, HHr has the highest productive H-bond occupancy (95%), relative to VSr and 8-17dz (86%). Simulation results suggest that, unlike in VSr and 8-17dz, the divalent metal ion in HHr is not as important for positioning the general base for nucleophile activation. This result is consistent with experiments that show the HHr retains a high degree of activity at high monovalent salt concentrations even in the absence of divalent metal ions.22,23 The situation for Psr is similar to that of HHr. Recent experimental20 and computational9 work has shown that Psr and HHr are functionally related. For Psr, productive H-bond occupancies in the presence and absence of the divalent metal ion (49% and 40%, respectively) lead to a 3°γ (HBN) factor of 1.2. This slight normal 3°γ (HBN) catalytic effect in Psr is, at least in part, related to the fact that the metal ion makes an outer-sphere contact with the NPO. While outer-sphere binding of a Mg2+ ion in Psr can still make hydrogen bond contact through a metal-coordinated water molecule with the pro-RP NPO, this interaction alone would not constitute “overdetermined” hydrogen bonding at this position, as suggested is required for a significant 3°γ (HBN) effect.5 To support this hypothesis, we carried out analogous simulations on Psr with an active site model in which the metal ion makes an inner sphere contact with the NPO (Figure S3). When the metal ion is inner-sphere coordinated to the NPO, the 3°γ (HBN) factor in Psr increases to 2.0, which is very similar to that observed in HHr.
The active site divalent metal ions considered here play multiple structural and catalytic roles, making direct verification of the predicted 3°γ (HBN) effect experimentally challenging. Nonetheless, in an effort to place the calculated 3°γ (HBN) factor in context of experimentally measured reaction rates, we consider the possibility of a 3°γ effect that arises due to a thio substitution at the pro-RP position of the scissile phosphate in a double mutant (DM) variant of VSr in which the purported general base, G638, and general acid, A756, have been mutated to inosine (G638I) and 3-deazapurine (A756(3cP)), respectively. The G638I mutation eliminates the exocyclic amine of the guanine, while A756(3cP) eliminates the exocyclic amine of the adenine as well as replacing the nitrogen atom at the N3 position with a carbon atom (see Figure S2). Recently, we measured a 6-fold inverse thio effect at the pro-RP position of the DM and reasoned that while in the wild-type VSr the pro-RP NPO receives two H-bonds from the G638 and A756 exocyclic amines, in the DM, these H-bonds are absent, which allows the HO2′ to form a favorable interaction with the pro-RP NPO. In a manner similar to the glmS aporibozyme, a thio substitution at the pro-RP position in the DM disrupts the HO2′–pro-RP interaction giving rise to the inverse thio effect. The results of our calculation on the VSr DM are summarized in Figure 5. The calculated 3°γ (HBN) factor for VSr is 3.8, which is reasonably aligned with the experimentally observed 6-fold effect, but slightly underestimated, although this distinction between 3.8 and 6 corresponds to <0.3 kcal/mol in free energy and is likely beyond the precision level of the experiment and estimated error of the simulations (∼1/2kBT at 298 K).
Figure 5.

Quantitative determination of 3°γ (HBN) effect in VSr G638I/A756(3cP) double mutant. 2D PDFs [A–C] obtained for the RP thio and oxo substrates of the double mutant indicate that in the oxo substrate productive H-bonding occupancy is only 23% (A), as the HO2′ can form a favorable interaction with the pro-RP NPO. In the RP thio substrate, the HO2′–pro-RP interaction is disrupted resulting in the increase of productive H-bonding occupancy to 88% (B). The 1D PDFs of productive H-bonding obtained in case of the RP thio and oxo substrates indicate a 3°γ (HBN) factor of 3.8.
The predicted 3°γ effects induced by metal ion binding are also somewhat smaller in magnitude than the 30-fold effect interpreted for the glmS ribozyme based on the inverse thio effect of the aporibozyme.6 It is notable that this inverse thio effect is diminished by increasing Mg2+ ion concentration (from 1 to 50 mM), due to an increase in activity of the oxo substrate, leading to only a 6-fold enhancement at 50 mM. The interpretation of this result was that divalent metal ion binding in the aporibozyme with oxo substrate was achieving the same 3°γ (HBN) effect as thio substitution at the pro-RP position.6 This 6-fold catalytic effect of increasing the Mg2+ ion concentration from 1 to 50 mM in the aporibozyme with the oxo substrate is substantially less than the 30-fold effect inferred from thio substitution experiments and is in the range of values predicted here for 3°γ (HBN) catalysis induced by metal ion binding. The factors that would lead to more precise quantitative agreement will require further investigation, but the qualitative agreement shown here is suggestive that the calculated 3°γ (HBN) factors can be considered as a lower bound to the observed effect for the systems considered.
The results herein provide the first quantitative evidence for 3°γ (HBN) catalytic effect induced by metal ion binding across a series of related ribo- and deoxyribozymes and evidence for a new role for metal ions in RNA catalysis. We observe that direct divalent metal ion binding to the NPOs can lead to a 3°γ (HBN) catalytic effect of up to an order of magnitude speed-up, particularly if, in the absence of the bound metal, H-bonding to these positions by other nucleobase functional groups is limited. Indirect divalent metal ion interactions with the NPOs, as predicted in Psr, lead to only a very slight 3°γ (HBN) catalytic effect. Finally, we explain the majority of the recently measured 6-fold thio rescue effect in the VSr double mutant as arising from a 3°γ (HBN) effect.
The 3°γ (HBN) catalysis appears to be a likely a strategy used by ribozymes and deoxyribozymes to maximize catalytic efficiency, and the approach described here can serve as a useful tool in estimating their contributions and aid in our predictive understanding of their mechanisms. While the focus of the current work is to make quantitative predictions about the degree to which divalent metal ions may contribute to enhancement of activity via 3°γ (HBN) catalysis, this is also a strategy that has been observed for the glmS ribozyme that does not have a catalytic metal ion requirement. Rather, the glmS ribozyme uses a strategy of overdetermined hydrogen bonding to the pro-RP NPO to eliminate competition of nonproductive hydrogen bonding of the nucleophile that prevents its activation by the general base. A similar strategy could be employed by protein enzymes such as RNase A, where, for example, the pro-RP NPO of the substrate can receive hydrogen bonds from His12, His119, and Phe120 at different stages along the reaction coordinate.24
Acknowledgments
The authors are grateful for financial support provided by the National Institutes of Health (GM62248 to D.M.Y. and GM131568 to J.A.P.). Computational resources were provided by the National Institutes of Health under Grant No. S10OD012346, the Office of Advanced Research Computing (OARC) at Rutgers, the State University of New Jersey, Rutgers Discovery Information Institute (RDI2), the State University of New Jersey, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. OCI-1053575 (Project No. TG-MCB110101). This research is also part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (Awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana–Champaign and its National Center for Supercomputing Applications. B.W. acknowledges support from the Predoctoral Tranining Program in Chemistry and Biology (T32-GM008720).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b02035.
Additional details for computational methods, theory underlying free energy simulations approach, 3°γ (HBN) effect observed in glmS ribozyme, and structures of modified nucleotides employed in VSr double mutant (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lilley D. M. Structure, Folding and Catalysis of the Small Nucleolytic Ribozymes. Curr. Opin. Struct. Biol. 1999, 9, 330–338. 10.1016/S0959-440X(99)80044-X. [DOI] [PubMed] [Google Scholar]
- Lilley D. M. J. Mechanisms of RNA Catalysis. Philos. Trans. R. Soc., B 2011, 366, 2910–2917. 10.1098/rstb.2011.0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson G. M.; Nakamura S.; Roth A.; Breaker R. R. Ribozyme Speed Limits. RNA 2003, 9, 907–918. 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua P. C.; Harris M. E.; Piccirilli J. A.; Gaines C.; Ganguly A.; Kostenbader K.; Ekesan Ş.; York D. M. An Ontology for Facilitating Discussion of Catalytic Strategies of RNA-Cleaving Enzymes. ACS Chem. Biol. 2019, 14, 1068–1076. 10.1021/acschembio.9b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingaman J. L.; Gonzalez I. Y.; Wang B.; Bevilacqua P. C. Activation of the glmS Ribozyme Nucleophile via Overdetermined Hydrogen Bonding. Biochemistry 2017, 56, 4313–4317. 10.1021/acs.biochem.7b00662. [DOI] [PubMed] [Google Scholar]
- Bingaman J. L.; Zhang S.; Stevens D. R.; Yennawar N. H.; Hammes-Schiffer S.; Bevilacqua P. C. The GlcN6P Cofactor Plays Multiple Catalytic Roles in the glmS Ribozyme. Nat. Chem. Biol. 2017, 13, 439. 10.1038/nchembio.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seith D. D.; Bingaman J. L.; Veenis A. J.; Button A. C.; Bevilacqua P. C. Elucidation of Catalytic Strategies of Small Nucleolytic Ribozymes from Comparative Analysis of Active Sites. ACS Catal. 2018, 8, 314–327. 10.1021/acscatal.7b02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.-S.; Lopez C. S.; Giambaşu G. M.; Martick M.; Scott W. G.; York D. M. Role of Mg2+ in Hammerhead Ribozyme Catalysis from Molecular Simulation. J. Am. Chem. Soc. 2008, 130, 3053–3064. 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenbader K.; York D. M. Molecular Simulations of the Pistol Ribozyme: Unifying the Interpretation of Experimental Data and Establishing Functional Links with the Hammerhead Ribozyme. RNA 2019, rna.071944.119. 10.1261/rna.071944.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekesan Ş; York D. M. Dynamical Ensemble of the Active State and Transition State Mimic for the RNA-cleaving 8-17 DNAzyme in Solution. Nucleic Acids Res. 2019, 10.1093/nar/gkz773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suslov N. B.; DasGupta S.; Huang H.; Fuller J. R.; Lilley D. M. J.; Rice P. A.; Piccirilli J. A. Crystal Structure of the Varkud Satellite Ribozyme. Nat. Chem. Biol. 2015, 11, 840–846. 10.1038/nchembio.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta S.; Suslov N. B.; Piccirilli J. A. Structural Basis for Substrate Helix Remodeling and Cleavage Loop Activation in the Varkud Satellite Ribozyme. J. Am. Chem. Soc. 2017, 139, 9591–9597. 10.1021/jacs.7b03655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martick M.; Lee T.-S.; York D. M.; Scott W. G. Solvent Structure and Hammerhead Ribozyme Catalysis. Chem. Biol. 2008, 15, 332–342. 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A.; Vusurovic N.; Gebetsberger J.; Gao P.; Juen M.; Kreutz C.; Micura R.; Patel D. Pistol Ribozyme Adopts a Pseudoknot Fold Facilitating Site-specific In-line Cleavage. Nat. Chem. Biol. 2016, 12, 702–708. 10.1038/nchembio.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Yu X.; Chen Y.; Zhang J.; Wu B.; Zheng L.; Haruehanroengra P.; Wang R.; Li S.; Lin J.; Li J.; Sheng J.; Huang Z.; Ma J.; Gan J. Crystal Structure of an RNA-Cleaving DNAzyme. Nat. Commun. 2017, 8, 2006. 10.1038/s41467-017-02203-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteva M. T.; Dissanayake T.; Chen H.; Radak B. K.; Kuechler E. R.; Giambaşu G. M.; Lee T.-S.; York D. M.. Multiscale Methods for Computational RNA Enzymology. In Methods in Enzymology; Chen S.-J., Burke-Aguero D. H., Eds.; Academic Press, 2014; pp 335–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteva M. T.; Giambasu G. M.; York D. M. Force Field for Mg2+, Mn2+, Zn2+, and Cd2+ Ions that have Balanced Interactions with Nucleic Acids. J. Phys. Chem. B 2015, 119, 15460–15470. 10.1021/acs.jpcb.5b10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward W. L.; Plakos K.; DeRose V. J. Nucleic Acid Catalysis: Metals, Nucleobases, and other Cofactors. Chem. Rev. 2014, 114, 4318–4342. 10.1021/cr400476k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.-J. J.; Liu J. Rational Evolution of Cd2+-specific DNAzymes with Phosphorothioate Modified Cleavage Junction and Cd2+ Sensing. Nucleic Acids Res. 2015, 43, 6125–6133. 10.1093/nar/gkv519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. J.; Liu Y.; Li N. S.; Dai Q.; Piccirilli J. A.; Lilley D. M. Comparison of the Structures and Mechanisms of the Pistol and Hammerhead Ribozymes. J. Am. Chem. Soc. 2019, 141, 7865–7875. 10.1021/jacs.9b02141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Giese T. J.; Golden B. L.; York D. M. Divalent Metal Ion Activation of a Guanine General Base in the Hammerhead Ribozyme: Insights from Molecular Simulations. Biochemistry 2017, 56, 2985–2994. 10.1021/acs.biochem.6b01192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rear J. L.; Wang S.; Feig A. L.; Beigelman L.; Uhlenbeck O. C.; Herschlag D. Comparison of the Hammerhead Cleavage Reactions Stimulated by Monovalent and Divalent Cations. RNA 2001, 7, 537–545. 10.1017/S1355838201002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis E. A.; Bartel D. P. The Hammerhead Cleavage Reaction in Monovalent Cations. RNA 2001, 7, 546–552. 10.1017/S1355838201002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formoso E.; Matxain J. M.; Lopez X.; York D. M. Molecular Dynamics Simulation of Bovine Pancreatic Ribonuclease A-CpA and Transition state-like Complexes. J. Phys. Chem. B 2010, 114, 7371–7382. 10.1021/jp909004y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.