

Abstract
Splenomegaly is one of the major clinical manifestations of primary myelofibrosis and is common also in other chronic Philadelphia-negative myeloproliferative neoplasms, causing symptoms and signs and affecting quality of life of patients diagnosed with these diseases. We aimed to study the impact that such alteration has on thrombotic risk and on the survival of patients with essential thrombocythemia and patients with Polycythemia Vera (PV). We studied the relationship between splenomegaly (and its grade), thrombosis and survival in 238 patients with et and 165 patients with PV followed at our center between January 1997 and May 2019.
Key words: Splenomegaly, Polycythemia Vera, essential thrombocythemia
Introduction
The 2016 revision of the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues includes new criteria for the diagnosis of Philadelphia-negative myeloproliferative neoplasms (MPNs). This revision includes Polycythemia Vera (PV), Essential Thrombocythemia (ET), and Primary Myelofibrosis (PMF), distinguished in overt and prefibrotic PMF.1
Splenomegaly is one of the major clinical manifestations of PMF. Progressive splenomegaly is significantly associated with debilitating symptoms, such as early satiety, abdominal pain, inactivity and fatigue and may cause portal hypertension and progression of cytopenias due to splenic sequestration.2 The symptoms linked to splenomegaly are associated with its grade, but it may also be asymptomatic. In one study, palpable splenomegaly was observed in 80% of the asymptomatic patients and about 10% of the patients with PMF showed severe symptomatic splenomegaly when diagnosed with PMF.3 The role of splenomegaly on quality of life and on prognosis in patients with PMF is fairly well known,4 instead the impact of splenomegaly in essential thrombocythemia and polycythemia vera is less investigated.
A mild to moderate spleen enlargement is present in about 5-20% of ET patients at diagnosis. Notwithstanding the relatively common occurrence of this feature, the prognosis of patients with spleen enlargement in ET is still unclear.5 PV is a chronic clonal myeloproliferative neoplasm characterized by increased red-cell mass; elevated white cell and platelet counts are also commonly observed in PV. PV patients have an increased risk of thrombotic and cardiovascular events and a burden of symptoms that often includes pruritus, fatigue, and night sweats.6 In a single center study with 587 patients diagnosed with PV, 155 of 506 with available data (31%) had palpable splenomegaly at diagnosis and it was associated with a higher risk of developing venous thrombosis during follow-up.7 Splenomegaly often develops at disease progression in approximately 30-40% of patients with PV.8,9
Despite the clinical relevance, increased spleen size has not been proven as a significant prognostic factor in the elaboration of major prognostic models commonly used to estimate survival, including International Prognostic Scoring System (IPSS), Dynamic IPSS (DIPSS), Dynamic IPSS plus (SIPSS-plus), MF Secondary to PV/ET-Prognostic Model (MYSEC-PM) and Mutation-enhanced IPSS70 (MIPSS- 70) in patients with MF, in risk stratification for survival in patients with PV and ET,10-14 and in traditional stratification for thrombotic risk for ET and PV,15 in International Prognostic score of thrombosis (IPSETthrombosis, and in revised international prognostic scoring system for essential thrombocythemia.16,17
In relation to the frequency and clinical relevance of splenomegaly in patients with ET or PV, we aimed to study the impact that such alteration has on thrombotic risk and on the survival of these patients.
Materials and Methods
From January 1997 to May 2019, 238 consecutive patients with diagnosis of ET and 165 patients with PV were followed at our center. Diagnosis were all made according to WHO criteria used in the respective period. The frequency of splenomegaly at diagnosis was calculated considering it positive if minimum longitudinal diameter was 15 cm at echotomography of computed tomography and was evaluated in groups of patients with diagnosis of PMF, PV and ET. Clinical features, driver-gene mutational status, age and sex were collected for all the patients with ET and PV. The frequency of thrombosis or cardiovascular events in the groups with and without splenomegaly and the number of deaths in the two groups were studied and survival was estimated using the Kaplan and Meier method. Comparison between frequencies is performed with chisquare test, comparison between medians with the Kruskal-Wallis test, while comparison between survivals using the log-rank test.
Results
In our study, 238 patients with ET and 165 with PV were followed along more than 22 years at our institution, with a median follow-up of 45.96 months (1.5-316.2) for ET patients and 58.42 months (1.2-298.39) for PV patients. The median age was respectively 65.92 years (14.56-92.09) and 62.28 (17.4-93.44). They were diagnosed according to WHO criteria used in the respective period.
Data on the driver-gene mutational status revealed that, between ET patients, 172/238 (72.26%) were JAK2V617F positive, 23/238 (9.66%) were CALR mutated and 4/238 (1.68%) were MPL mutated, while 39/238 (16.38%) were triple-negative; among PV patients, 156/165 (94.54%) were JAK2V617F positive, 5/165 (3,03%) harbored mutation on JAK2 exon 12 and 4/165 (2,42%) were negative for JAK2 mutations;
According to diagnosis subgroups, splenomegaly was present respectively in 15,54% of patients with ET and in 38.18% of PV patients. Table 1 shows gender and median age in patients with or without splenomegaly distinguished by type of MPNs. Our data show the presence of splenomegaly in 24% of males and 11.65% of females with ET, while showing splenomegaly in 45.71% of males and in 24.19 of females with PV.
The frequency of thrombosis or cardiovascular events appears to be higher in patients with splenomegaly than in patients without splenomegaly at diagnosis, both in patients with ET and in patients with PV. In fact, they occur in 39.87% of patients with ET and with splenomegaly towards 24.37% of patients without splenomegaly (P=0.04). In patients with PV thrombosis or cardiovascular events occurred in 44.44% of patients with splenomegaly and in 30.39% of patients without splenomegaly (P=0.02)
Table 1.
Age and sex in patients with myeloproliferative disease with or without splenomegaly.
| Age | Sex | |
|---|---|---|
| (median) | (m/f) | |
| Essential thrombocythemia | 65,92 | 75/163 |
| Splenomegaly | 67,94 | 18/19 |
| No splenomegaly | 65,85 | 57/144 |
| Polycythemia vera | 62,28 | 103/62 |
| Splenomegaly | 60,55 | 48/15 |
| No splenomegaly | 62,48 | 55/47 |
In patients with splenomegaly at diagnosis we found a higher number of deaths compared to patients who did not present splenomegaly both in ET and PV patients, and respectively 32.43% versus 8.42% (P=0.0001) and 22.22% versus 7.84% (P<0.004). Finally, survival in patients presenting with splenomegaly at diagnosis appears significantly worse, as it can be seen in Figure 1.
Discussion and Conclusions
Splenomegaly is one of the most common manifestations among those present at diagnosis of MPNs. In our experience, splenomegaly is can be revealed in about 15.54% of patients with ET an 38.18% of PV patients at diagnosis. Furthermore, our data show an important correlation with the thrombotic risk, with the frequency of deaths and with survival.
Ruxolitinib, an oral JAK1/JAK2 inhibitor, is approved for the treatment of patients with IPSS intermediate or high-risk PMF and patients with PV who have had an inadequate response to or are intolerant to hydroxyurea. Ruxolitinib has been shown to be effective in reducing splenomegaly both in patients with PMF and in patients with PV,18-20 significantly improving the life quality of the patients. Current therapies available for ET do not significantly affect splenomegaly.
Despite the clinical relevance, increased spleen size has not been proven as a significant prognostic factor in major prognostic models. In accordance with our data and our experience, we believe that the prognostic value of splenomegaly is underestimated in ET and PV and that should be evaluated the possibility to include it as an item of a prognostic scoring system. Further welldesigned clinical studies are needed to evaluate the significance of splenomegaly in ET and PV patients and its impact on overall survival and on thrombotic risk.
Figure 1.
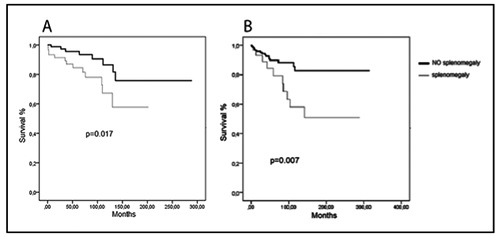
Survival in patients with splenomegaly versus patients without splenomegaly at diagnosis. In PV patients (A) and ET patients (B).
Funding Statement
Funding: None.
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405. [DOI] [PubMed] [Google Scholar]
- 2.Mesa RA, Nagorney DS, Schwager S, et al. Palliative goals, patient selection, and perioperative platelet management: Outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006;107:361-70. [DOI] [PubMed] [Google Scholar]
- 3.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113:2895-901. [DOI] [PubMed] [Google Scholar]
- 4.Bose P, Verstovsek S. Prognosis of primary myelofibrosis in the genomic era. Clin Lymphoma Myeloma Leuk 2016;16:S105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andriani A, Latagliata R, Anaclerico B, et al. Spleen enlargement is a risk factor for thrombosis in essential thrombocythemia: Evaluation on 1,297 patients. Am J Hematol 2016;91:318-21. [DOI] [PubMed] [Google Scholar]
- 6.Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J 2018;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J 2017;7:662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radia D, Geyer HL. Thrombocytemia patients. Hematology Am Soc Hematol Educ Program 2015;2015:340. [DOI] [PubMed] [Google Scholar]
- 9.Bryan JC, Verstovsek S. Overcoming treatment challenges in Myelofibrosis and polycythemia vera: the role of ruxolitinib. Cancer Chemother Pharmacol 2016;77:1125-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (international working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010;115:1703-8. [DOI] [PubMed] [Google Scholar]
- 11.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: A refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011;29:392-7. [DOI] [PubMed] [Google Scholar]
- 12.Passamonti F, Giorgino T, Mora B, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017;31:2726-31. [DOI] [PubMed] [Google Scholar]
- 13.Guglielmelli P, Lasho TL, Rotunno G, et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelo-fibrosis. J Clin Oncol 2018;36:310-8. [DOI] [PubMed] [Google Scholar]
- 14.Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013; 7:1874-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbui T, Barosi G, Birgegard G, et al. Philadelphia negative classical mieloproliferative neoplasm: critical concepts amd management recommendation from European LeukemiaNet. J Clin Oncol 2011;29:761-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an international Prognostic score of thrombosis in World Healt Organization-essential thrombocythemia (IPSET thrombosis). Blood 2012;120:5128-33. [DOI] [PubMed] [Google Scholar]
- 17.Haider M, Gangat N1, Lasho T, et al. Validation of the revised International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSETthrombosis) in 585 Mayo Clinic patients. Am J Hematol 2016;91:390-4. [DOI] [PubMed] [Google Scholar]
- 18.Harrison C, Kiladjian JJ, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012;366:787-98. [DOI] [PubMed] [Google Scholar]
- 19.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy fomyelofibrosis. Blood 2013;122:4047-53. [DOI] [PubMed] [Google Scholar]
- 20.Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow- up from the RESPONSE trial. Haematologica 2016;101:821-9. [DOI] [PMC free article] [PubMed] [Google Scholar]