

Abstract

Identification of active sites in heterogeneous metal catalysts is critical for understanding the reaction mechanism at the molecular level and for designing more efficient catalysts. Because of their structural flexibility, subnanometric metal catalysts, including single atoms and clusters with a few atoms, can exhibit dynamic structural evolution when interacting with substrate molecules, making it difficult to determine the catalytically active sites. In this work, Pt catalysts containing selected types of Pt entities (from single atoms to clusters and nanoparticles) have been prepared, and their evolution has been followed, while they were reacting in a variety of heterogeneous catalytic reactions, including selective hydrogenation reactions, CO oxidation, dehydrogenation of propane, and photocatalytic H2 evolution reaction. By in situ X-ray absorption spectroscopy, in situ IR spectroscopy, and high-resolution electron microscopy techniques, we will show that some characterization techniques carried out in an inadequate way can introduce confusion on the interpretation of coordination environment of highly dispersed Pt species. Finally, the combination of catalytic reactivity and in situ characterization techniques shows that, depending on the catalyst–reactant interaction and metal–support interaction, singly dispersed metal atoms can rapidly evolve into metal clusters or nanoparticles, being the working active sites for those abovementioned heterogeneous reactions.
Keywords: single-atom catalysis, subnanometric clusters, size effect, platinum, hydrogenation of nitroarenes, CO oxidation, propane dehydrogenation, photocatalytic H2 evolution
1. Introduction
Heterogeneous single-atom catalysts have attracted tremendous attention in the field of catalysis in recent years, and it has been shown in some reactions that single-atom catalysts present superior catalytic performance than the conventional nanoparticulate catalysts.1−3 The recent progress made in characterization tools on physicochemical properties has introduced further knowledge for better understanding catalysis at the atomic level.4,5 By decreasing the size of metal catalysts down to subnanometric metal clusters and even atomically dispersed species, the efficiency of a metal could be, in principle, maximized, and more importantly, unique catalytic behavior may appear associated with those atomically dispersed metal species.6−9 It has been reported in the literature that, in some reactions, singly dispersed atoms are the active species, while clusters and nanoparticles are not.10,11 While in some works, metal clusters are found to be more active sites than single atoms and nanoparticles.12,13
However, to unambiguously conclude that singly dispersed metal atoms are the active species in the working catalysts is not an easy task, which can be reflected in some debates. For instance, Pt single atoms supported on Al2O3, FeOx, TiO2, and CeO2 are reported to be more active than Pt nanoparticles for CO oxidation,14−16 while in some other works, Pt atoms supported on zeolites or metal oxides are claimed to be less active than Pt nanoparticles.17 Similar issues have also been reported with Au single-atom catalysts for CO oxidation. Singly dispersed Au atoms have been presented as efficient catalysts for CO oxidation,18,19 while others have shown that Au nanoclusters and nanoparticles supported on CeO2 are much more active than singly dispersed Au atoms.20,21 Singly dispersed Pt and Pd atoms on various supports have been reported as active catalysts for the hydrogenation of nitroarenes, olefins, and ketones.22−25 However, there are some publications showing that single Pd atoms supported on FeOx show no activity in the hydrogenation of alkenes, while Pd clusters and nanoparticles on the same support are active.26
In most of the previous works, the catalytic performances of single atoms have been usually compared with nanoparticulate catalysts, without considering the presence of subnanometric metal clusters. As a transition state between single atoms and nanoparticles, metal clusters show distinct electronic and geometric structures compared to the other two entities, and consequently, a different catalytic behavior can be expected.27,28 Indeed, it has been shown that subnanometric metal clusters can catalyze reactions that neither the single-atom nor nanoparticulate counterparts can.29−31 Therefore, it is necessary to study the size effect of the metal entities on catalysis by direct comparison of the catalytic performance of single atoms, clusters, and nanoparticles for establishing a structure–reactivity relationship. This is most relevant if one takes into account that metallic species can evolve during reaction conditions. Moreover, it should be considered that if single-atom catalysts and the nanoparticulate counterpart are prepared by different methodologies, additional uncertainty can be introduced into the studies, especially when considering the complexity of heterogeneous metal catalysts.32,33 Therefore, for a fair comparative study, the model catalysts should also be better prepared by the same method and the particle size of the metal species should be controlled by the synthesis parameters. Nevertheless, their potential evolution under reaction conditions should be followed that dynamic structural transformation can occur with atomically dispersed metal species and nanoclusters under reaction conditions.34,35 This is even more critical if one takes into consideration that a small fraction of single atoms or clusters within a supported metal catalyst formed by nanoparticles can be responsible for most of the catalyst activity. On the contrary, it may also occur that for some other reactions, a small fraction of metal clusters or nanoparticles present in the catalysts containing mainly single atoms contribute most to the catalytic reactivity. Then, the size of the metal particles and metal–support interaction will become more important and even determinant for the catalytic behavior, especially when referring to single atoms.
In this work, we have prepared by the same method a series of supported Pt catalysts with particle size ranging from singly dispersed atoms to clusters to nanoparticles on different supports (TiO2, Al2O3, and CeO2). We have studied their catalytic performance for a variety of different reactions, including selective hydrogenation reactions, CO oxidation, propane dehydrogenation, and photocatalytic H2 evolution reaction. It has been found that Pt clusters and nanoparticles show higher activity than Pt single atoms in all the reactions studied. Furthermore, by in situ X-ray absorption spectroscopy (XAS),IR spectroscopy, and ex situ high-resolution scanning transmission electron microscopy (HR STEM), we have observed that the structural transformation of single Pt atoms under reaction conditions depends not only on the nature of the reactants but also on the material acting as the support. In other words, metal–reactant and metal–support interactions not only dictate the geometric and electronic properties of the metal entities, and therefore the catalytic behavior, but also are determinant for the evolution behavior of subnanometric metal species under the reaction conditions.
2. Results and Discussion
2.1. Pt Single Atoms, Clusters, and Nanoparticles for Hydrogenation Reactions
First, we have prepared a series of Pt/TiO2 catalysts by conventional wetness impregnation (see the Supporting Information for experimental details), and the size of Pt species supported on TiO2 was controlled by the Pt loading and the treatments during the preparation procedure. As shown in Figure 1a,b, Pt single atoms supported on TiO2 (named as 0.03Pt/TiO2-SA with 0.03 wt % of Pt) were directly observed by HR high-angle annular dark-field STEM (HR HAADF–STEM). After reduction by H2 at 450 °C, those singly dispersed Pt atoms agglomerated into Pt clusters of ca. 0.5–1.0 nm (see Figures 1c,d and S1). The average size of Pt particles was found to be subnanometric when the sample contained 0.1 wt % of Pt (see Figure S2). When the Pt loading was increased to 0.2 wt %, Pt nanoparticles of ∼1 nm were obtained after reduction by H2 at 450 °C (see Figures 1e,f and S3). When the Pt loading was further increased to 0.5 wt %, Pt nanoparticles of ∼1.5 nm were formed on TiO2 (see Figure S4). These STEM images suggest that Pt clusters and nanoparticles with good dispersion and narrow size distributions can be generated on TiO2 and that their particle size can be tuned by Pt loading and reduction treatment. Therefore, we can use these Pt/TiO2 catalysts as model catalysts to study the size effect on their catalytic properties for different reactions.
Figure 1.

Characterization of Pt/TiO2 catalysts with single atoms, clusters, and nanoparticles by HR HAADF–STEM imaging. (a,b) 0.03Pt/TiO2-SA sample containing singly dispersed Pt atoms. (c,d) 0.03Pt/TiO2-450H2 sample containing subnanometric Pt clusters. (e,f) 0.2Pt/TiO2-450H2 sample containing Pt nanoparticles.
The hydrogenation of 3-nitrostyrene was chosen as the first model reaction to study the catalytic performance of different types of Pt species because it can be carried out at low temperature to avoid the potential effect of reaction temperature on the size of Pt particles and also because its catalytic behavior versus the particle size of metal nanoparticles has been widely reported.36−39 As shown in Figure 2, the Pt single atoms show negligible activity. It should be mentioned that Pt single atoms supported on TiO2 remain stable after the hydrogenation of 3-nitrostyrene (see Figure S5), indicating the stability of single Pt atoms under mild reaction conditions. In the case of Pt/TiO2 catalysts containing a large fraction of metal atoms within the subnanometric clusters of ca. 0.4–0.8 nm (the 0.03%Pt/TiO2-450H2 and 0.1%Pt/TiO2-450H2 samples), the activity is significantly higher than the samples formed by Pt single atoms. Further increasing the Pt particle size to ∼1 nm will result in a higher intrinsic activity for chemoselective hydrogenation of 3-nitrostyrene to 3-aminostyrene, with a turnover frequency (TOF) of ∼2600 h–1, which is higher than any other catalyst reported so far in the literature at the same temperature and H2 pressure. It should be noted that the TOF values were calculated based on the total Pt amount in the catalysts, as in the literature.22 Moreover, further increasing the size of Pt nanoparticles to ∼1.5 nm (the 0.5%Pt/TiO2-450H2 sample) leads to a drop of the TOF, suggesting that Pt nanoparticles of ∼1 nm formed by 30–40 atoms are the most active species for the hydrogenation of 3-nitrostyrene under our conditions.
Figure 2.
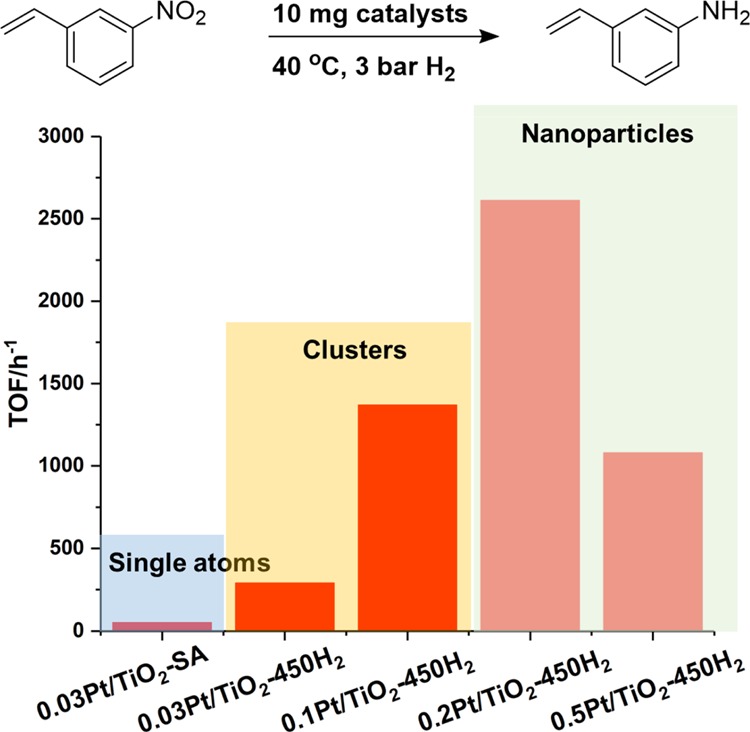
Catalytic performance of different Pt species supported on TiO2 for the hydrogenation of 3-nitrostyrene. Reaction conditions: 0.5 mmol of 3-nitrostyrene, 2 mL of toluene as the solvent, 10 mg of solid catalyst, 40 °C and 3 bar of H2. High selectivity to 3-aminostyrene has been achieved for all the Pt/TiO2 samples containing Pt clusters and nanoparticles. The TOF shown in this figure was calculated based on the total Pt amount in all the samples.
In our previous work, it has been proposed that when Pt nanoparticles were partially covered by TiO2 overlayers, they were the active sites for chemoselective hydrogenation of nitroarenes in Pt/TiO2 catalysts.40 Herein, we have studied this effect on the 0.2Pt/TiO2-450H2 sample. With the help of HR HAADF–STEM imaging and electron energy loss spectroscopy (EELS) mapping, we were able to obtain information on the structure of those small Pt particles (∼1 nm) at the atomic level. In fact, these techniques are likely the most appropriate techniques to perform such kind of analyses.41−43 As displayed in Figure 3, the hemispherical Pt nanoparticles are surrounded by TiO2 layers and the exposed surface area for each Pt nanoparticle is less than 1 × 1 nm (see more EELS mapping results in Figures S6 and S7). In other words, because of the strong metal–support interaction between Pt and TiO2, only a few Pt atoms are exposed to the reactants, and these “cluster”-type Pt sites, in the Pt nanoparticles partially covered by TiO2 overlayers, are the active sites for chemoselective hydrogenation of nitroarenes.
Figure 3.
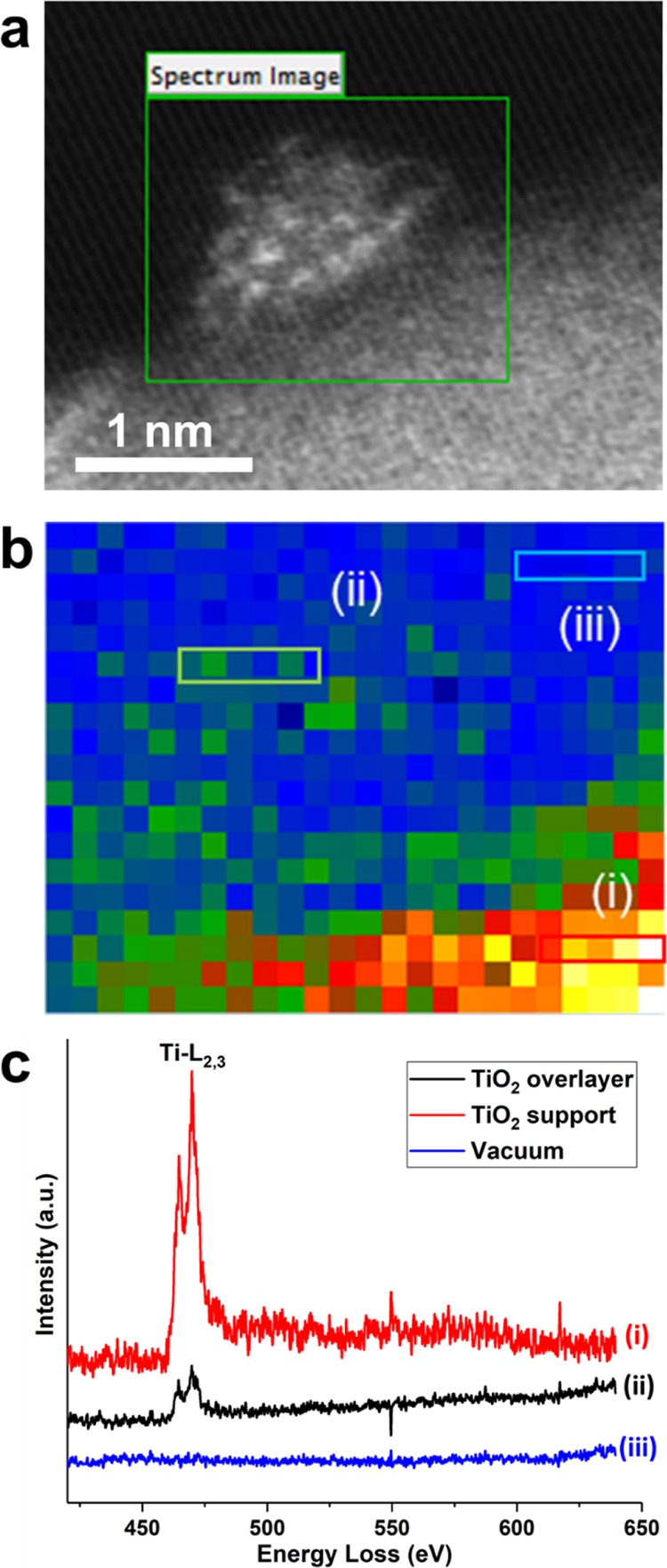
Atomic structure of the 0.2Pt/TiO2-450H2 sample by HR HAADF–STEM and EELS spectroscopy. (a) Typical HAADF–STEM image of the Pt nanoparticle supported on TiO2 with atomic resolution. (b) EELS mapping of the Ti-L edge in the selected area shown in (a). (c) Ti-L2,3 edge EELS spectra of three different areas shown in (b).
Considering that H2 activation is a key step for the hydrogenation of 3-nitrostyrene, we have measured the H2–D2 exchange to correlate the capability of the Pt single atoms, clusters, and nanoparticles for H2 activation.44 As shown in Figure S8, both the 0.03Pt/TiO2-SA sample containing single Pt atoms and 0.03Pt/TiO2-450H2 containing Pt clusters show clearly lower reactivity for H2-D2 exchange at room temperature than the 0.2Pt/TiO2-450H2 sample containing Pt nanoparticles of ∼1 nm. These results indicate that Pt nanoparticles show higher capability for the activation of H2 than single atoms and subnanometric clusters, which can explain the higher activity of the former for the hydrogenation of 3-nitrostyrene. It should be mentioned that, in a recent work, Pd3 clusters are claimed to be much more active than single Pd atoms and Pd2 dimers for the hydrogenation of alkynes.45 Though the catalytic performance of Pd particles with larger sizes is not studied in that work, the higher reactivity of metal particles with multiple atoms than single atoms for hydrogenation reactions is consistent with our findings in this work.
The drop in the activity for Pt nanoparticles of ∼1.5 nm or larger may be related with the electronic interaction between Pt and the TiO2 support. Lykhach et al. have demonstrated that the electronic charge transfer between Pt and CeO2 reaches the maximum for Pt nanoparticles of ∼50 atoms.46 In our case, the strong metal–support interaction between Pt and TiO2 could be dependent on the particle size of Pt and the coverage of TiO2 overlayers on the Pt nanoparticles may vary with the particle size of Pt, resulting in difference in the electronic and geometric structure of Pt species.47 For that reason, Pt particles in the 0.2Pt/TiO2-450H2 sample are much more active than the 0.03Pt/TiO2-450H2 sample containing Pt clusters. Because of the electronic and geometric factors, the Pt/TiO2 sample with Pt nanoparticles of ∼1 nm partially covered by TiO2 overlayers shows both high activity and chemoselectivity for the hydrogenation of nitroarenes, while Pt single atoms and small Pt clusters are sensibly less active.
To compare the catalytic properties of different Pt entities on other supports with weak metal–support interaction, we have also prepared a series of Pt/Al2O3 samples containing Pt ranging from single atoms to clusters and nanoparticles. First, a 0.2Pt/Al2O3-SA sample containing Pt single atoms was prepared by impregnation according to the literature.48 As shown in Figures 4a,b and S9, only singly dispersed Pt atoms can be observed in the HR STEM images of the Pt/Al2O3-SA sample. After reduction by H2 at 200 °C, some of the single Pt atoms agglomerate into Pt clusters, while there are still some single Pt atoms preserved in the 0.2Pt/Al2O3-200H2 sample (see Figures 4c,d and S10). When raising the reduction temperature to 300 °C (see Figure S11) and 450 °C (see Figures 4e,f and S12), the fraction of single Pt atoms in the reduced Pt/Al2O3 decreases and only very few Pt atoms can still be found in the 0.2Pt/Al2O3-450H2 sample. Therefore, the percentage of Pt single atoms can be modulated by the reduction temperature, and these Pt/Al2O3 samples can also be model catalysts for the direct comparison between single Pt atoms, clusters, and nanoparticles.
Figure 4.

Pt/Al2O3 samples with different types of Pt species. HR HAADF–STEM images of Pt/Al2O3-SA (a,b) containing only singly dispersed Pt atoms, Pt/Al2O3-200H2 (c,d) containing a mixture of both singly Pt atoms and Pt nanoclusters, and Pt/Al2O3-450H2 (e,f) containing mainly Pt nanoparticles and a few singly dispersed Pt atoms.
The chemical states and coordination environment of Pt species supported on Al2O3 obtained after different treatments were characterized by XAS. As shown in Figure 5a and Table 1, when the Pt/Al2O3 samples were directly measured by XAS without any treatments, all the three Pt/Al2O3 samples present oxidized states as revealed by X-ray absorption near-edge structure (XANES), though the 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2 samples were previously reduced by H2. The oxidized state of Pt in the two previously reduced samples should be caused by the reoxidation of Pt clusters or small Pt nanoparticles when exposed to air after the H2 reduction treatment. The oxidized nature of these samples was further confirmed by the extended X-ray absorption fine structure (EXAFS) spectra (see Figure 5b). The Pt–O contribution in the Pt/Al2O3 samples may come from O in the Al2O3 support and the oxygen directly bonded to Pt atoms.48 The presence of Pt–Al contribution in the EXAFS fit results can be ascribed to the interaction between small Pt particles and Al2O3.48,49 Interestingly, the 0.2Pt/Al2O3-200H2 sample almost shows the same EXAFS spectrum as the 0.2Pt/Al2O3-SA sample, though the size of Pt species in these two samples is quite different, as presented in the above STEM images. A large number of Pt clusters were observed in the STEM images, while the presence of these species was not reflected in the EXAFS results (see Table 1 for the fit results). The absence of Pt–Pt contribution in the EXAFS spectrum of the directly measured 0.2Pt/Al2O3-200H2 sample could be caused by the complete oxidation of Pt clusters into PtOx clusters by air. It has been observed in the literature that PtOx clusters or small PtOx nanoparticles (<5 nm) can show similar EXAFS spectra as singly dispersed Pt atoms on metal oxide supports because of the high degree of disorder of the small platinum oxide particles.50−52 Notably, when 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2 were in situ reduced by H2 before the XAS measurement, the oxidized Pt clusters or nanoparticles became partially reduced, as shown in the XANES spectra (see Figure 5c). The presence of Pt–Pt contribution at 2.7 Å can also be confirmed in the EXAFS spectra (see Figure 5d) and by the corresponding fit results. The above XAS results indicate that subnanometric Pt clusters can be easily oxidized to corresponding PtOx clusters and that the oxidized species may show similar feature as the Pt single-atom species in EXAFS spectra if the sample is measured directly without in situ prereduction treatment.
Figure 5.

Characterizations of Pt/Al2O3 samples by XAS. (a) XANES spectra and (b) EXAFS spectra (not phase-corrected) of the as-prepared Pt/Al2O3-SA, Pt/Al2O3-200H2, and Pt/Al2O3-450H2 samples. The spectra presented in (a,b) were directly recorded without any treatments in the synchrotron. (c) XANES spectra and (d) EXAFS spectra (not phase-corrected) of the Pt/Al2O3-200H2 and Pt/Al2O3-450H2 samples obtained after in situ reduction treatment in the synchrotron. The fit curves of the EXAFS spectra are presented as dashed red curves in (b,d).
Table 1. Fit Results the EXAFS Spectra of Pt/Al2O3 Samples Prepared under Different Conditionsa.
| Pt–O |
Pt–Pt |
Pt–Al |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| sample | CNPt–O | R (Å) | σ2 (Å2) | CNPt–Pt | R (Å) | σ2 (Å2) | CNPt–Al | R (Å) | σ2 (Å2) |
| Pt foil | 12 | 2.77 | |||||||
| PtO2 | 6 | 1.99 | |||||||
| 0.2Pt/Al2O3-SAb | 6.2 ± 0.9 | 2.00 ± 0.01 | 0.003 ± 0.002 | 3 ± 2 | 2.92 ± 0.05 | 0.01 | |||
| 0.2Pt/Al2O3-200H2b | 5.7 ± 0.9 | 1.99 ± 0.01 | 0.002 ± 0.002 | 2 ± 1 | 2.90 ± 0.07 | 0.01 | |||
| 0.2Pt/Al2O3-450H2b | 3.0 ± 0.6 | 2.01 ± 0.01 | 0.003 ± 0.003 | 3 ± 1 | 2.76 ± 0.03 | 0.01 | |||
| 0.2Pt/Al2O3-200H2-in situc | 2.0 ± 0.5 | 1.99 ± 0.01 | 0.003 ± 0.003 | 5 ± 2 | 2.75 ± 0.02 | 0.009 ± 0.004 | |||
| 0.2Pt/Al2O3-450H2-in situc | 2 ± 1 | 2.01 ± 0.04 | 0.01 ± 0.02 | 7 ± 2 | 2.72 ± 0.01 | 0.009 ± 0.003 | |||
ΔE0 = 9, Rfactor = 0.7. To decrease the number of parameters, the Debye–Waller (σ2) was kept free but the same for all paths with exception of the second shell contribution in the Pt/Al2O3 samples measured directly. In this case, it was fixed to 0.01.
The sample was directly measured by XAS without any treatment.
The sample was reduced by H2 at a given temperature before the XAS measurement.
The importance of utilization of in situ XAS to characterize the chemical states and coordination environment of supported Pt catalysts has also been reflected in the case of the Pt/TiO2 sample containing Pt nanoparticles of ∼1 nm. As shown in Figure S13, when 0.2Pt/TiO2-450H2 was measured directly by XAS without any pretreatment, the XANES spectrum corresponding to partially oxidized Pt species was observed. The presence of the Pt–O contribution at ∼2.0 Å in the EXAFS spectrum (Figure S14) of the directly measured 0.2Pt/TiO2-450H2 sample indicated that Pt seemed to be atomically dispersed, which was inconsistent with the STEM images shown in Figure 1. As discussed above, the reoxidation of Pt particles by air can cause the transformation of small metallic Pt nanoparticles into disordered PtOx particles, which may be inadequately interpreted as singly dispersed Pt atoms. Notably, after in situ reduction treatment by H2 at 50 °C, significant reduction of PtOx into metallic Pt was observed in the XANES spectrum, and the Pt–Pt bonding at ∼2.7 Å was also detected in the EXAFS spectrum. More interestingly, when the prereduction temperature was increased to 450 °C, a higher contribution of Pt–O bonding was observed in the EXAFS compared to the spectrum obtained after prereduction treatment at lower temperatures (see fit results in Table S1). The observation of the increased Pt–O contribution can be associated with the formation of TiO2 overlayers on the surface of Pt nanoparticles because of the strong metal–support interaction.53 On the basis of the EXAFS characterization results on Pt/Al2O3 and Pt/TiO2 samples, we can conclude that because of their high reactivity with O2, the characterization techniques using in situ treatment facilities are necessary to obtain reliable information on the chemical states and coordination environment of supported Pt clusters or small nanoparticles.
Catalytic Behavior of Pt Single Atoms Supported on Al2O3 for Hydrogenation Reactions
First, we have tested the catalytic performance of Pt/Al2O3 catalysts for the hydrogenation of 3-nitrostyrene. As shown in Figure 6a, similar to the situation of Pt/TiO2 catalysts, the 0.2Pt/Al2O3-SA sample shows negligible activity for the hydrogenation of 3-nitrostyrene. Similar to the case of 0.03Pt/TiO2-SA, Pt atoms supported on Al2O3 remain stable after the hydrogenation reaction under such mild conditions (see Figure S15). In the case of reduced Pt/Al2O3 samples, considerable conversion of 3-nitrostyrene can be observed and the reactivity increases with the reduction temperature, which corresponds to the higher percentage of Pt nanoparticles in the 0.2Pt/Al2O3-450H2 catalyst. Although the Pt/Al2O3 catalysts are not chemoselective for 3-aminostyrene (∼60% selectivity to 3-aminostyrene on all the three Pt/Al2O3 catalysts, see Figure S16), the ranking of the activity for the hydrogenation of 3-nitrostyrene is similar to Pt/TiO2 catalysts, indicating that Pt clusters and nanoparticles show higher reactivity than Pt single atoms.
Figure 6.

Catalytic performance of Pt/Al2O3 catalysts for hydrogenation reactions. (a) Hydrogenation of 3-nitrostyrene with various Pt/Al2O3 catalysts. Reaction conditions: 0.5 mmol of 3-nitrostyrene, 2 mL of toluene as the solvent, 20 mg of solid catalyst, 50 °C and 3 bar of H2. The product distributions of the hydrogenation of 3-nitrostyrene with Pt/Al2O3 catalysts are shown in Figure S16. (b) Hydrogenation of styrene with various Pt/Al2O3 catalysts. Reaction conditions: 1.0 mmol of styrene, 2 mL of toluene as the solvent, 10 mg of solid catalyst, 50 °C and 3 bar of H2. The TOF in this figure was calculated based on the total Pt amount in all the Pt/Al2O3 samples.
Because it has been reported in the literature that singly dispersed Pt atoms can be used for the hydrogenation of C=C bonds, we have also tested the reactivity of Pt/Al2O3 catalysts for the hydrogenation of styrene. As can be seen in Figure 6b, singly dispersed Pt atoms also show negligible activity toward the hydrogenation of C=C bonds. The Pt/Al2O3 catalysts with agglomerated Pt species show much higher activity and the activity increases with the percentage of Pt nanoparticles in the catalyst, indicating that Pt nanoparticles are more active than single Pt atoms and clusters for the hydrogenation of C=C bonds.
One may argue that the single-atom catalysts shown above are not reduced, although the single-atom Pt catalysts are also usually positively charged in most of the reported works.23,54,55 We have tried to reduce the Pt single atoms supported on Al2O3 by NaBH4 and aqueous N2H4 (see the Experimental Section for details). After such mild reduction treatment, the activity for the hydrogenation of styrene and 3-nitrostyrene remained in both cases unchanged as the pristine Pt/Al2O3-SA sample.
From a mechanistic point of view, the hydrogenation of −NO2 and C=C may follow different reaction mechanisms on metal catalysts.40,56,57 It has been reported that the hydrogenation of C=C on nanoparticulate Pt catalysts is a structure-insensitive reaction, while the hydrogenation of −NO2 is a structure-sensitive reaction. Nevertheless, our experimental results show that Pt nanoparticles are more active than Pt clusters and single Pt atoms for the two hydrogenation reactions. In other words, the above results indicate that singly dispersed Pt atoms supported on TiO2 and Al2O3 show much lower activity for the hydrogenation of −NO2 and C=C groups.
Taking into account that residual Cl may be present in the Pt/TiO2 and Pt/Al2O3 catalysts when using H2PtCl6 as the Pt precursor, we have tried to measure the Pt–Cl bonding interaction in Pt/Al2O3 samples by XANES.58 As shown in Figure S17, the feature corresponding to the Pt–Cl bond is not present in our samples.59 Furthermore, in order to exclude the influence of Cl, we have prepared Pt/TiO2 catalysts using platinum(II) acetylacetonate as the precursor by impregnating it on TiO2. When the sample is activated in air (350 °C in air) for the decomposition of the acetylacetonate ligand bonded to Pt, we can obtain a Pt/TiO2 sample containing atomically dispersed Pt species (see Figures S18–S21). Further reduction treatment at 450 °C by H2 will lead to the agglomeration of the Pt atoms into Pt clusters or nanoparticles, showing a similar evolution behavior as observed when using H2PtCl6 as the precursor. When increasing the Pt loading from 0.04 to 0.5 wt %, the size of Pt particles also increases from 0.5 to ∼1 nm. It should be noted that the size of Pt species is smaller in the Pt/TiO2 samples prepared with Pt(acac)2 than those prepared with H2PtCl6.
We have tested the catalytic performance of these Cl-free Pt/TiO2 samples for the hydrogenation of 3-nitrostyrene. As shown in Figure S22, the initial TOF increases with the Pt loading, which means the TOF also increases with the size of Pt particles supported on TiO2. This trend is consistent with the results obtained with Pt/TiO2 samples prepared with H2PtCl6, among which the Pt particles of ∼1 nm show the highest activity. Notably, the absolute TOF values obtained with Pt/TiO2 catalysts prepared with Pt(acac)2 are lower than the Pt/TiO2 samples prepared with H2PtCl6, indicating that the Pt precursor indeed has influence on the catalytic performance of supported Pt catalysts.
One may argue that the reduction treatment used in the preparation procedure may influence the surface properties of the oxide support. In order to exclude the reduction treatment on the support, we have prepared a PtNP/TiO2 catalyst by loading colloid Pt nanoparticles (0.2 wt % of Pt) on the surface of TiO2. In such a way, Pt nanoparticles can be generated on TiO2 without reduction treatment. Furthermore, a calcination treatment at 450 °C in air was carried out with the PtNP/TiO2 sample to remain consistent with the preparation of the 0.03Pt/TiO2-SA sample (see Figures S23 and S24).
We have tested the PtNP/TiO2 samples for the hydrogenation of 3-nitrostyrene under the same conditions as used in the article. As shown in Figures S25 and S26, both PtNP/TiO2-air and PtNP/TiO2-450H2 are active, though PtNP/TiO2-450H2 gives a slightly higher initial TOF. Notably, though the PtNP/TiO2-air sample has been calcined in air, as for the Pt/TiO2-SA sample, its activity is much higher than the Pt/TiO2-SA sample containing singly dispersed Pt atoms. These results confirm the vital role of agglomerated Pt species for the hydrogenation reaction. Importantly, the selectivity to 3-aminostyrene is much higher with the reduced PtNP/TiO2-450H2 sample, which should be related to the formation of TiO2–x overlayers on Pt nanoparticles because of strong metal–support interaction.40
2.2. CO Oxidation Reaction with Pt Single Atoms, Clusters, and Nanoparticles
CO oxidation is another important model reaction to study the structure–reactivity correlations of supported metal catalysts. It has been reported in the literature that Pt single atoms supported on Al2O3 are active for CO oxidation. In this work, we have tested the catalytic activity of Pt single atoms supported on Al2O3 and followed the evolution of Pt single atoms during the CO oxidation reaction. The presence of singly dispersed Pt atoms is confirmed by low-temperature IR bands at 2120–2150 cm–1 (see Figure S27), which correspond to CO adsorbed on cationic Pt atoms.16,17 As shown in Figures 7b and S28, it is found that Pt single atoms agglomerate into Pt nanoparticles under reaction conditions. Moreover, the size of the in situ formed Pt nanoparticles also increases with the reaction temperature from 150 to 325 °C, indicating that Pt species suffer dynamic structural transformation under CO oxidation conditions (see Figures 7a and S29–S32).
Figure 7.

Different types of Pt species supported on Al2O3 for CO oxidation. (a) Catalytic result of the 0.2Pt/Al2O3-SA sample for CO oxidation and the evolution of Pt single atoms to Pt nanoparticles under CO oxidation reaction conditions. The average sizes of Pt nanoparticles at different reaction temperatures are shown in this figure. (b) Typical HAADF–STEM image of the 0.2Pt/Al2O3-SA sample after the CO oxidation reaction, showing the presence of Pt nanoparticles in the used catalyst. (c) Comparison of the catalytic performance of Pt/Al2O3 catalysts with different sizes, ranging from single atoms to clusters to nanoparticles. (d) TOF values of different Pt/Al2O3 catalysts in the low-temperature range (150–225 °C) for the CO oxidation reaction. The TOF in this figure was calculated based on the total Pt amount in all the Pt/Al2O3 samples.
The structural transformation of Pt single atoms into Pt nanoparticles has also been performed, followed by in situ CO IR spectroscopy. As displayed in Figure 8, the formation of Pt nanoparticles after the CO + O2 reaction at 225 °C can be identified by the CO adsorption bands at 2075–2050 cm–1. The CO IR band at 2075 and 2050 cm–1 corresponds to CO adsorbed on terrace and corner Pt sites, respectively. Interestingly, after the CO oxidation reaction at higher temperature (325 °C), the contribution of CO adsorbed on corner Pt sites increases, implying that a surface reconstruction or disintegration of Pt nanoparticles into smaller particles occurs at high reaction temperature, which is consistent with our previous study on Pt particles confined in zeolite.34 The structural transformation behavior of Pt nanoparticles also indicates that the CO oxidation reaction is a structure-sensitive reaction and the active sites could be varied at different working temperatures.
Figure 8.
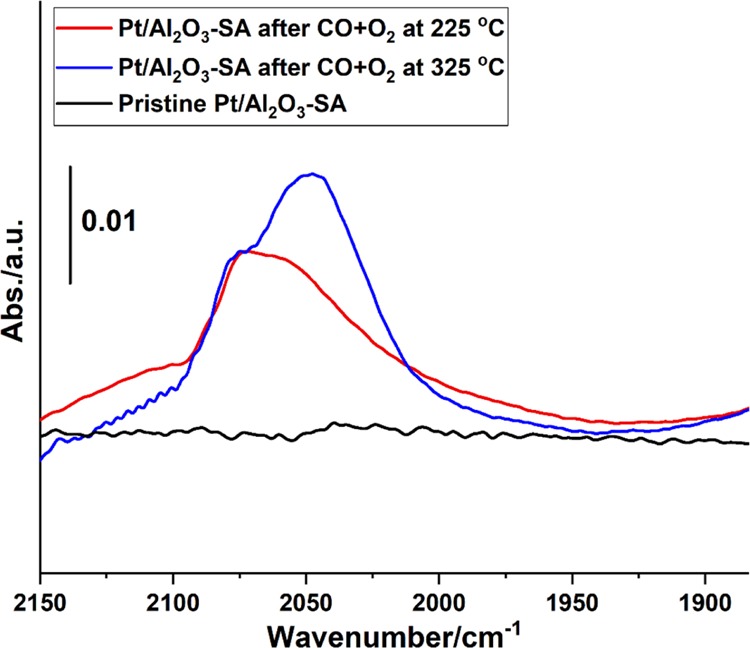
Evolution of Pt single atoms under CO oxidation reaction conditions, followed by in situ CO IR spectroscopy. The pristine 0.2Pt/Al2O3-SA sample containing singly dispersed Pt atoms shows negligible absorption of CO at room temperature because of the weak adsorption of CO on cationic Pt atoms. After the CO oxidation reaction at 225 and 320 °C, typical CO adsorption bands on Pt nanoparticles can be observed, indicating the agglomeration of Pt single atoms into nanoparticles under reaction conditions, which is consistent with the images obtained by electron microscopy.
The sintering of single Pt atoms into Pt clusters and nanoparticles is confirmed by in situ XAS. As shown in Figure 9a, the white line intensity of Pt-edge XANES decreased gradually in the CO + O2 atmosphere when increasing the reaction temperature from 25 to 225 °C, indicating the reduction of Pt single atoms under reaction conditions. Furthermore, according to the |FT| of Pt-edge EXAFS spectra (see Figure 9b and Table 2), a new contribution at 2.7 Å can be associated with Pt–Pt bonding, suggesting the transformation of singly dispersed Pt species into agglomerated Pt particles, which has also been observed in a very recent publication.60 In order to compare the reactivity of Pt single atoms, clusters, and nanoparticles, we have tested the Pt/Al2O3 catalysts for CO oxidation on different types of Pt species generated by controlled reduction treatment. As shown in Figure 7c,d, both Pt clusters and Pt nanoparticles show significantly higher TOFs (normalized to the amount of Pt atoms in the catalyst) than Pt single atoms. 0.2Pt/Al2O3-300H2 and 0.2Pt/Al2O3-450H2 samples show similar activity, while 0.2Pt/Al2O3-200H2 shows intermediate activity between these two samples. It should be mentioned that the TOF values shown in Figure 7d are calculated based on all the Pt species in the catalyst. If we calculate the TOF based on surface-exposed atoms, then Pt clusters and nanoparticles may show even higher TOF, while the TOF for Pt single atoms will remain unchanged. On the basis of the catalytic results shown in Figure 7 and the size distribution of Pt species in various Pt/Al2O3 samples, it can be concluded that the 0.2Pt/Al2O3-SA sample with a higher fraction of single Pt atoms shows lower activity for CO oxidation than Pt nanoparticles and clusters supported on Al2O3.
Figure 9.

Evolution of Pt single atoms supported on Al2O3 under CO + O2 reaction conditions, followed by in situ XAS. (a) XANES spectra and (b) EXAFS spectra (not phase-corrected) of the pristine 0.2Pt/Al2O3-SA sample and the corresponding sample after the CO + O2 reaction at 225 and 325 °C, respectively. The fit curves of the EXAFS spectra are presented as dashed red curves in (b).
Table 2. Fit Results of the Pristine Pt/Al2O3-SA Sample and the Sample after in Situ XAS Experiments under CO + O2 Reaction Conditionsa.
| Pt–O |
Pt–Pt |
Pt–Al |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| sample | CNPt–O | R (Å) | σ2 (Å2) | CNt | Rt (Å) | σt2 (Å2) | CNt | R (Å) | σt2 (Å2) |
| 0.2Pt/Al2O3-SAb | 6.2 ± 0.9 | 2.00 ± 0.01 | 0.003 ± 0.002 | 3 ± 2 | 2.92 ± 0.05 | 0.01 | |||
| 0.2Pt/Al2O3-SA-225CO + O2c | 2.4 ± 0.8 | 1.99 ± 0.02 | 0.008 ± 0.006 | 8 ± 3 | 2.77 ± 0.01 | 0.01 ± 0.003 | |||
| 0.2Pt/Al2O3-SA-325CO + O2c | 1.5 ± 0.7 | 1.96 ± 0.02 | 0.005 ± 0.006 | 9 ± 3 | 2.76 ± 0.01 | 0.01 ± 0.003 | |||
ΔE0 = 9, Rfactor = 0.7. To decrease the number of parameters, the Debye–Waller (σ2) was kept free but the same for all paths with exception of the second shell contribution in the Pt/Al2O3 samples measured directly. In this case, it was fixed to 0.01.
The sample was directly measured by XAS without any treatment.
The sample was treated under CO + O2 conditions at a given temperature before the XAS measurement.
We have also observed that the average size of Pt nanoparticles in Pt/Al2O3-SA after the CO oxidation reaction is larger than those in the 0.2Pt/Al2O3-300H2 and 0.2Pt/Al2O3-450H2 samples after being used for the CO + O2 reaction (see Figures S33–S36). As discussed above, Pt single atoms are not stable in the CO + O2 atmosphere and they agglomerate to Pt nanoparticles probably through an Ostwald ripening mechanism, in which single Pt atoms migrate to the Pt clusters or nanoparticles to form larger particles. In the case of 0.2Pt/Al2O3-300H2 and 0.2Pt/Al2O3-450H2 samples, Pt clusters or small nanoparticles are stabilized by the Al2O3 support and the Ostwald ripening process under CO oxidation reaction conditions is not as significant as in the 0.2Pt/Al2O3-SA sample. This structural transformation behavior can be related to the stability of different Pt species under CO oxidation reaction conditions and to the fact that Ostwald ripening tends to occur between particles of different sizes.61−63
Catalytic Behavior of Pt Single Atoms Supported on TiO2 and CeO2 for CO Oxidation
In the case of 0.03Pt/TiO2-SA, a similar agglomeration behavior from single atoms to nanoparticles is observed during the CO oxidation test (see Figures 10a,b and S37). Then, we have compared the activity of Pt clusters and nanoparticles with that of single Pt atoms. As shown in Figure 10c, Pt clusters show the highest TOF value, followed by Pt nanoparticles. Similar to the situation with 0.2Pt/Al2O3 samples, Pt single atoms supported on TiO2 exhibit the lowest TOF. It should be noted that the variation tendency of CO oxidation activity versus Pt particle size is different on Al2O3 and TiO2, implying that the reactivity of different types of metal entities may depend on the support. In the case of Pt/CeO2 containing single Pt atoms (0.2Pt/CeO2-SA, see Figure S38), the activity is quite low, which is in line with the reported work.16 After the CO oxidation reaction with 0.2Pt/CeO2-SA, the agglomeration of Pt atoms into clusters or nanoparticles has not been observed (see Figures 10d,e and S39), implying that Pt atoms are stabilized by CeO2 under reaction conditions. The behavior of Pt atoms supported on CeO2 is dramatically different from that of Pt atoms supported on Al2O3 and TiO2, suggesting that the evolution behavior of Pt single atoms also depends on the interaction between Pt and the support. In any case, while single Pt atoms are stable on CeO2, they are not active for the CO oxidation reaction. Afterward, as done before, the 0.2Pt/CeO2-SA sample was reduced by H2 at 500 °C to generate Pt nanoparticles of 1–2 nm on CeO2 (see Figure S40). As shown in Figure 10f, the 0.2Pt/CeO2-500H2 sample exhibits much higher activity than 0.2Pt/CeO2-SA, which is consistent with a very recent publication.64 Those Pt nanoparticles of 1–2 nm in the 0.2Pt/CeO2-500H2 sample remained stable after the CO + O2 reaction (see Figure S41). The above results suggest that for low-temperature CO oxidation reaction, Pt clusters and nanoparticles are more active than Pt single atoms on Al2O3, TiO2, and CeO2. Furthermore, the reactivity of Pt clusters and nanoparticles and the evolution behavior are strongly relevant to the support, implying the importance of considering the role of the support when discussing the catalytic behavior of heterogeneous single-atom catalysts.65,66
Figure 10.

Size effect of Pt species supported on TiO2 and CeO2 for CO oxidation. (a) HR STEM image of fresh 0.03Pt/TiO2-SA sample, showing the presence of single Pt atoms dispersed on TiO2. (b) STEM image of the 0.03Pt/TiO2-SA catalyst after the CO oxidation reaction, showing the transformation of singly dispersed Pt atoms into Pt nanoparticles under reaction conditions. (c) Comparison of the activity of Pt single atoms, clusters, and nanoparticles for low-temperature CO oxidation. (d) HR STEM image of fresh 0.2Pt/CeO2-SA sample. Because of the low contrast between Pt single atoms and CeO2, it is difficult to visualize Pt single atoms. Nevertheless, neither Pt nanoparticles nor clusters are observed in this sample, implying that Pt species are atomically dispersed on CeO2. (e) STEM image of the 0.2Pt/CeO2-SA catalyst after the CO oxidation reaction. Pt nanoparticles are not found in the used catalyst. (f) Comparison of the activity of Pt single atoms (0.2Pt/CeO2-SA) and nanoparticles (0.2Pt/CeO2-500H2) for CO oxidation.
The higher capability for O2 activation on the 0.2Pt/CeO2-500H2 sample is confirmed by in situ IR experiments. As presented in Figure S42, peroxide species can only be detected on the 0.2Pt/CeO2-500H2 sample containing Pt nanoparticles but not on the 0.2Pt/CeO2-SA sample. The activation of O2 on the metal/ceria catalyst has been proposed to occur at the interface between metal nanoparticles and ceria, with the participation of oxygen vacancies, which is verified by the presence of Ce3+ in the X-ray photoelectron spectroscopy spectrum of the reduced 0.2Pt/CeO2-500H2 sample (see Figure S43).67,68 The IR results imply that O2 activation is facilitated at the interface between Pt nanoparticles and CeO2 rather than between Pt single atoms and CeO2, which can explain why the 0.2Pt/CeO2-500H2 sample is more active for CO oxidation. It has been shown by both experimental and theoretical studies that Cu nanoparticles are more active than Cu single atoms or clusters for O2 activation.69,70 Although the metal element (Cu) studied in the above reports is different, taking into account the catalytic results presented here, one could make the hypothesis that if multiple metal atoms are required for O2 activation, it may also be applied to Pt. Nevertheless, the adsorption of CO behavior on Pt species could also be related to the particle size.71
In summary, the results presented above in this work indicate that single Pt atoms may evolve into Pt clusters or nanoparticles under CO oxidation reaction conditions if the interaction between Pt atoms and the support is not strong enough. Moreover, regardless of the reducibility of the support, the agglomerated Pt species (clusters or nanoparticles) are more active than the Pt single atoms on Al2O3, TiO2, and CeO2. It should be noted that the active sites and the kinetically relevant steps on support Pt catalysts may vary with the support and the reaction conditions.72−74 Considering the complexity of the reaction mechanism for the CO oxidation reaction and its dependence on the size and the support material, the reasons that cause the different catalytic behavior of single Pt atoms and the corresponding agglomerates are not in the scope of this study, which requires more systematic investigations in the future.
2.3. Propane Dehydrogenation with Pt Single Atoms, Clusters, and Nanoparticles
It has been reported that propane dehydrogenation to propylene is a structure-sensitive reaction, in which the activity on Pt nanoparticles increases when decreasing the particle size in the range of 1–9 nm.75 Recently, Xiong et al. have reported the application of single-atom Pt/CeO2 and Pt–Sn/CeO2 catalysts for the propane dehydrogenation reaction, and it has been found that singly dispersed Pt atoms can undergo agglomeration into Pt or PtSn clusters under the reaction conditions.76 It is hence not clear how Pt single atoms perform when compared to Pt clusters and nanoparticles for the propane dehydrogenation reaction.
Herein, the reactivity of different types of Pt species supported on Al2O3 has also been tested. As shown in Figure 11, the 0.2Pt/Al2O3-SA sample is initially active for the catalytic dehydrogenation of propane to propylene, although the activity is low. However, a drop of activity is observed in the starting stage, and then the propane conversion increases again. These changes on reactivity imply that Pt species may undergo structural transformation under reaction conditions. Indeed, we have measured the used 0.2Pt/Al2O3-SA sample after the propane dehydrogenation reaction, and the formation of a large fraction of Pt nanoparticles is confirmed by electron microscopy (see Figure S44). Furthermore, the catalytic performance of 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2 for the propane dehydrogenation reaction has also been measured, and as can be seen in Figure 11, the Pt/Al2O3 catalysts that contain Pt clusters and nanoparticles in the starting catalysts are initially more active than Pt single atoms. Interestingly, a decrease in activity and a subsequent activity recovery have also been observed with both 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2. According to the STEM images of the used catalysts, the particle size of Pt species increases slightly compared to that of the corresponding pristine 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2 samples (see Figures S45 and S46), suggesting a slight sintering of Pt species in these two catalysts during the propane dehydrogenation reaction. Therefore, it can be speculated that Pt nanoparticles with larger size than those in the 0.2Pt/Al2O3-450H2 sample may be more active. Following that hypothesis, 0.2Pt/Al2O3-SA was reduced by H2 at 600 °C to form a 0.2Pt/Al2O3-600H2 sample, in which a larger amount of Pt clusters and nanoparticles are formed compared to the 0.2Pt/Al2O3-200H2 and 0.2Pt/Al2O3-450H2 samples (see Figures S47 and S48). The catalytic results shown in Figure 11 clearly indicate that a higher percentage of Pt clusters and nanoparticles of ∼1 nm can give higher activity for the propane dehydrogenation reaction and the propane conversion keeps decreasing with time on stream. On the basis of the above results, it can be concluded that Pt single atoms supported on Al2O3 are active but not stable for the propane dehydrogenation reaction, probably because of the sintering of single atoms to clusters and nanoparticles in the reductive atmosphere, while Pt clusters and nanoparticles are more active than Pt single atoms.
Figure 11.

Catalytic performance of different Pt/Al2O3 catalysts for the propane dehydrogenation reaction at 600 °C. (a) 0.2Pt/Al2O3-SA, (b) 0.2Pt/Al2O3-200H2, (c) 0.2Pt/Al2O3-450H2, and (d) 0.2Pt/Al2O3-600H2. The TOF values shown in (e) are calculated based on all Pt atoms in the catalyst. Reaction conditions: 140 mg of solid catalyst, mixture of propane and N2 as the feed gas (5 mL/min of propane and 16 mL/min of N2) at 600 °C.
2.4. Photocatalytic H2 Evolution with Pt Single Atoms, Clusters, and Nanoparticles
In the above examples, conventional heterogeneous catalysis involving activation of small molecules was employed to study the catalytic behavior of different types of Pt species. Pt is also widely used for photocatalysis, in which Pt is usually thought to serve as the cocatalyst. The photogenerated electrons in the semiconductors will transfer to Pt sites, which catalyze the half reaction between protons and electrons to form H2.77 It has been reported in the literature that the size of Pt cocatalysts has significant influence on the photocatalytic reactivity because of their size-dependent electronic structures.78,79 Recently, it has been reported that single-site-based metal catalysts can also be used for photocatalysis.80,81 In the following example, we have studied three Pt/TiO2 catalysts with different sizes of Pt species for photocatalytic H2 evolution from ethanol–H2O mixture under UV light.
As shown in Figure 12, all three Pt/TiO2 catalysts are active for the photocatalytic H2 evolution reaction and the 0.03Pt/TiO2-450H2 sample containing Pt clusters shows the highest activity, while the other two samples show similar activity. The three used Pt/TiO2 catalysts after the photocatalytic reaction have also been measured by STEM. As presented in Figure 13, a large fraction of Pt nanoparticles and some Pt clusters can be found in the 0.03Pt/TiO2-SA sample, implying the sintering of singly dispersed Pt atoms under the photocatalytic reaction conditions. In the case of the other two samples, slight sintering is also observed, implying the instability of Pt species during the photocatalytic process under UV light. On the basis of the above results, it can be concluded that Pt clusters and nanoparticles show higher activity when working as cocatalysts for the photocatalytic H2 evolution reaction. Furthermore, singly dispersed Pt atoms on TiO2 are not stable under the photocatalytic conditions, and their sintering behavior may be associated with the surface reaction (oxidation of ethanol by photogenerated holes and the reduction of H+ to H2 by photogenerated electrons) under UV light irradiation.
Figure 12.
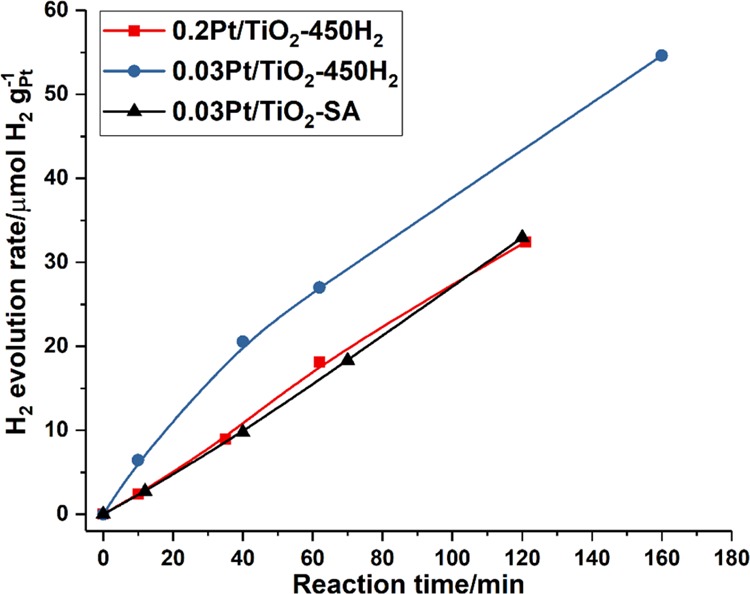
Catalytic performance of Pt single atoms, clusters, and nanoparticles supported on TiO2 for photocatalytic H2 evolution under UV light. The same amount of solid Pt/TiO2 catalysts was used as the catalyst, and the H2 evolution rate was normalized to the Pt mass in different catalysts.
Figure 13.

HAADF–STEM images of various Pt/TiO2 catalysts after the photocatalytic H2 evolution reaction. (a,b) 0.03Pt/TiO2-SA, (c,d) 0.03Pt/TiO2-450H2, and (e,f) 0.2Pt/TiO2-450H2. Pt clusters and nanoparticles are formed in the 0.03Pt/TiO2-SA sample after the photocatalytic H2 evolution reaction. In the other two cases, the size of Pt clusters and nanoparticles also slightly increases, indicating the instability of Pt species under the photocatalytic reaction conditions.
3. Conclusions
In summary, the results presented in this work demonstrate that when studying the catalytic properties of highly dispersed heterogeneous metal catalysts containing subnanometric metal species (single atoms or clusters) or small nanoparticles of ∼1 nm, it is critical to characterize the coordination environment of the metal entities by the combination of adequate ex situ and in situ characterization techniques. Furthermore, in terms of single-atom catalysts, the evolution of the atomically dispersed metal species under reaction conditions should also be carefully followed by in situ electron microscopy or spectroscopy techniques, in order to verify the nature of the active sites under reaction conditions. Our results suggest that in a variety of reactions, single atoms can sinter into clusters or nanoparticles during the reaction, and the evolution behavior depends on the metal–support and metal–reactant interactions. Nevertheless, we have found that a small portion of metal clusters or nanoparticles present together with the single-atom species can contribute to the reactivity in a dominant manner, indicating the importance of performing an appropriate comparison between different types of metal entities (from single atoms to clusters and nanoparticles).
Acknowledgments
This work has been supported by the European Union through the European Research Council (grant ERC-AdG-2014-671093, SynCatMatch) and the Spanish government through the “Severo Ochoa Program” (SEV-2016-0683). L.L. thanks ITQ for providing a contract. The authors also thank Microscopy Service of UPV for the TEM and STEM measurements. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the U.S. DOE under contract no. DE-AC02-06CH11357 and the Canadian Light Source and its funding partners. The HR STEM and STEM–EELS studies were conducted at the Laboratorio de Microscopias Avanzadas, Universidad de Zaragoza, Spain. R.A. acknowledges support from Spanish MINECO grant MAT2016-79776-P (AEI/FEDER, UE), from the Government of Aragon and the European Social Fund (grant number E13_17R, FEDER, UE), and from the European Union H2020 program “ESTEEM3” (grant number 823717). A.V.P. thanks the Spanish Government (Agencia Estatal de Investigación) and the European Union (European Regional Development Fund) for a grant for young researchers (CTQ2015-74138-JIN, AEI/FEDER/UE).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b04214.
Experimental details on the sample preparation, characterization, catalytic tests, and additional characterization and catalytic results (PDF)
Author Present Address
¶ Departament d’Enginyeria Química, Universitat Rovira i Virgili, Avinguda dels Països Catalans, 26, 43007 Tarragona, Spain.
The authors declare no competing financial interest.
Supplementary Material
References
- Liu J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. 10.1021/acscatal.6b01534. [DOI] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Chen Y.; Ji S.; Chen C.; Peng Q.; Wang D.; Li Y. Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications. Joule 2018, 2, 1242–1264. 10.1016/j.joule.2018.06.019. [DOI] [Google Scholar]
- Flytzani-Stephanopoulos M.; Gates B. C. Atomically dispersed supported metal catalysts. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 545–574. 10.1146/annurev-chembioeng-062011-080939. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang X.; Zhang J.; Huang Y.; Liu B. In Situ/Operando Techniques for Characterization of Single-Atom Catalysts. ACS Catal. 2019, 9, 2521–2531. 10.1021/acscatal.8b04937. [DOI] [Google Scholar]
- Gates B. C.; Flytzani-Stephanopoulos M.; Dixon D. A.; Katz A. Atomically Dispersed Supported Metal Catalysts: Perspectives and Suggestions for Future Research. Catal. Sci. Technol. 2017, 7, 4259–4275. 10.1039/c7cy00881c. [DOI] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe S.; Koyasu K.; Tsukuda T. Nonscalable Oxidation Catalysis of Gold Clusters. Acc. Chem. Res. 2014, 47, 816–824. 10.1021/ar400209a. [DOI] [PubMed] [Google Scholar]
- Du Y.; Sheng H.; Astruc D.; Zhu M. Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 2019, 10.1021/acs.chemrev.8b00726. [DOI] [PubMed] [Google Scholar]
- Malta G.; Kondrat S. A.; Freakley S. J.; Davies C. J.; Lu L.; Dawson S.; Thetford A.; Gibson E. K.; Morgan D. J.; Jones W.; et al. Identification of Single-Site Gold Catalysis in Acetylene Hydrochlorination. Science 2017, 355, 1399–1403. 10.1126/science.aal3439. [DOI] [PubMed] [Google Scholar]
- Chung H. T.; Cullen D. A.; Higgins D.; Sneed B. T.; Holby E. F.; More K. L.; Zelenay P. Direct Atomic-Level Insight into the Active Sites of a High-Performance PGM-Free ORR Catalyst. Science 2017, 357, 479–484. 10.1126/science.aan2255. [DOI] [PubMed] [Google Scholar]
- Lin J.; Chen Y.; Zhou Y.; Li L.; Qiao B.; Wang A.; Liu J.; Wang X.; Zhang T. More Active Ir Subnanometer Clusters than Single-Atoms for Catalytic Oxidation of CO at Low Temperature. AIChE J. 2017, 63, 4003–4012. 10.1002/aic.15756. [DOI] [Google Scholar]
- Herzing A. A.; Kiely C. J.; Carley A. F.; Landon P.; Hutchings G. J. Identification of Active Gold Nanoclusters on Iron Oxide Supports for CO Oxidation. Science 2008, 321, 1331–1335. 10.1126/science.1159639. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Wang A.; Yang X.; Allard L. F.; Jiang Z.; Cui Y.; Liu J.; Li J.; Zhang T. Single-Atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. 10.1038/nchem.1095. [DOI] [PubMed] [Google Scholar]
- DeRita L.; Dai S.; Lopez-Zepeda K.; Pham N.; Graham G. W.; Pan X.; Christopher P. Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150–14165. 10.1021/jacs.7b07093. [DOI] [PubMed] [Google Scholar]
- Jones J.; Xiong H.; DeLaRiva A. T.; Peterson E. J.; Pham H.; Challa S. R.; Qi G.; Oh S.; Wiebenga M. H.; Pereira Hernandez X. I.; Wang Y.; Datye A. K. Thermally Stable Single-Atom Platinum-on-Ceria Catalysts via Atom Trapping. Science 2016, 353, 150–154. 10.1126/science.aaf8800. [DOI] [PubMed] [Google Scholar]
- Ding K.; Gulec A.; Johnson A. M.; Schweitzer N. M.; Stucky G. D.; Marks L. D.; Stair P. C. Identification of Active Sites in CO Oxidation and Water-Gas Shift over Supported Pt Catalysts. Science 2015, 350, 189–192. 10.1126/science.aac6368. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Liu J.; Wang Y.-G.; Lin Q.; Liu X.; Wang A.; Li J.; Zhang T.; Liu J. Highly Efficient Catalysis of Preferential Oxidation of CO in H2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249–6254. 10.1021/acscatal.5b01114. [DOI] [Google Scholar]
- Qiao B.; Liang J.-X.; Wang A.; Xu C.-Q.; Li J.; Zhang T.; Liu J. J. Ultrastable Single-Atom Gold Catalysts with Strong Covalent Metal-Support Interaction (CMSI). Nano Res. 2015, 8, 2913–2924. 10.1007/s12274-015-0796-9. [DOI] [Google Scholar]
- Wang J.; Tan H.; Yu S.; Zhou K. Morphological Effects of Gold Clusters on the Reactivity of Ceria Surface Oxygen. ACS Catal. 2015, 5, 2873–2881. 10.1021/cs502055r. [DOI] [Google Scholar]
- Guo L.-W.; Du P.-P.; Fu X.-P.; Ma C.; Zeng J.; Si R.; Huang Y. Y.; Jia C. J.; Zhang Y. W.; Yan C. H. Contributions of Distinct Gold Species to Catalytic Reactivity for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 13481. 10.1038/ncomms13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Liu X.; Wang A.; Zhang L.; Qiao B.; Yang X.; Huang Y.; Miao S.; Liu J.; Zhang T. FeOx-Supported Platinum Single-Atom and Pseudo-Single-Atom Catalysts for Chemoselective Hydrogenation of Functionalized Nitroarenes. Nat. Commun. 2014, 5, 5634. 10.1038/ncomms6634. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Asakura H.; Zhang J.; Zhang J.; De S.; Yan N. Stabilizing a Platinum1 Single-Atom Catalyst on Supported Phosphomolybdic Acid without Compromising Hydrogenation Activity. Angew. Chem., Int. Ed. 2016, 55, 8319–8323. 10.1002/anie.201602801. [DOI] [PubMed] [Google Scholar]
- Vilé G.; Albani D.; Nachtegaal M.; Chen Z.; Dontsova D.; Antonietti M.; López N.; Pérez-Ramírez J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chem., Int. Ed. 2015, 54, 11265–11269. 10.1002/anie.201505073. [DOI] [PubMed] [Google Scholar]
- Liu P.; Zhao Y.; Qin R.; Mo S.; Chen G.; Gu L.; Chevrier D. M.; Zhang P.; Guo Q.; Zang D.; Wu B.; Fu G.; Zheng N. Photochemical Route for Synthesizing Atomically Dispersed Palladium Catalysts. Science 2016, 352, 797–800. 10.1126/science.aaf5251. [DOI] [PubMed] [Google Scholar]
- Rossell M. D.; Caparrós F. J.; Angurell I.; Muller G.; Llorca J.; Seco M.; Rossell O. Magnetite-Supported Palladium Single-Atoms Do Not Catalyse the Hydrogenation of Alkenes but Small Clusters Do. Catal. Sci. Technol. 2016, 6, 4081–4085. 10.1039/c6cy00596a. [DOI] [Google Scholar]
- Boronat M.; Leyva-Pérez A.; Corma A. Theoretical and Experimental Insights into the Origin of the Catalytic Activity of Subnanometric Gold Clusters: Attempts to Predict Reactivity with Clusters and Nanoparticles of Gold. Acc. Chem. Res. 2014, 47, 834–844. 10.1021/ar400068w. [DOI] [PubMed] [Google Scholar]
- Vajda S.; White M. G. Catalysis Applications of Size-Selected Cluster Deposition. ACS Catal. 2015, 5, 7152–7176. 10.1021/acscatal.5b01816. [DOI] [Google Scholar]
- Gates B. C. Supported Metal Clusters: Synthesis, Structure, and Catalysis. Chem. Rev. 1995, 95, 511–522. 10.1021/cr00035a003. [DOI] [Google Scholar]
- Corma A.; Concepción P.; Boronat M.; Sabater M. J.; Navas J.; Yacaman M. J.; Larios E.; Posadas A.; López-Quintela M. A.; Buceta D.; Mendoza E.; Guilera G.; Mayoral A. Exceptional Oxidation Activity with Size-Controlled Supported Gold Clusters of Low Atomicity. Nat. Chem. 2013, 5, 775–781. 10.1038/nchem.1721. [DOI] [PubMed] [Google Scholar]
- Oliver-Meseguer J.; Liu L.; Garcia-Garcia S.; Canos-Gimenez C.; Dominguez I.; Gavara R.; Domenech-Carbo A.; Concepcion P.; Leyva-Perez A.; Corma A. Stabilized naked sub-nanometric Cu clusters within a polymeric film catalyze C-N, C-C, C-O, C-S, and C-P bond-forming reactions. J. Am. Chem. Soc. 2015, 137, 3894–3900. 10.1021/jacs.5b00222. [DOI] [PubMed] [Google Scholar]
- Hoffman A. S.; Debefve L. M.; Zhang S.; Perez-Aguilar J. E.; Conley E. T.; Justl K. R.; Arslan I.; Dixon D. A.; Gates B. C. Beating Heterogeneity of Single-Site Catalysts: MgO-Supported Iridium Complexes. ACS Catal. 2018, 8, 3489–3498. 10.1021/acscatal.8b00143. [DOI] [Google Scholar]
- He Q.; Freakley S. J.; Edwards J. K.; Carley A. F.; Borisevich A. Y.; Mineo Y.; Haruta M.; Hutchings G. J.; Kiely C. J. Population and Hierarchy of Active Species in Gold Iron Oxide Catalysts for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 12905. 10.1038/ncomms12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Zakharov D. N.; Arenal R.; Concepcion P.; Stach E. A.; Corma A. Evolution and Stabilization of Subnanometric Metal Species in Confined Space by in situ TEM. Nat. Commun. 2018, 9, 574. 10.1038/s41467-018-03012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moliner M.; Gabay J. E.; Kliewer C. E.; Carr R. T.; Guzman J.; Casty G. L.; Serna P.; Corma A. Reversible Transformation of Pt Nanoparticles into Single Atoms inside High-Silica Chabazite Zeolite. J. Am. Chem. Soc. 2016, 138, 15743–15750. 10.1021/jacs.6b10169. [DOI] [PubMed] [Google Scholar]
- Hartfelder U.; Kartusch C.; Makosch M.; Rovezzi M.; Sá J.; van Bokhoven J. A. Particle Size and Support Effects in Hydrogenation over Supported Gold Catalysts. Catal. Sci. Technol. 2013, 3, 454–461. 10.1039/c2cy20485a. [DOI] [Google Scholar]
- Zhao F.; Ikushima Y.; Arai M. Hydrogenation of Nitrobenzene with Supported Platinum Catalysts in Supercritical Carbon Dioxide: Effects of Pressure, Solvent, and Metal Particle Size. J. Catal. 2004, 224, 479–483. 10.1016/j.jcat.2004.01.003. [DOI] [Google Scholar]
- Coq B.; Tijani A.; Figuéras F. Particle Size Effect on the Kinetics of p-Chloronitrobenzene Hydrogenation over Platinum/Alumina Catalysts. J. Mol. Catal. 1991, 68, 331–345. 10.1016/0304-5102(91)80091-g. [DOI] [Google Scholar]
- Lyu J.; Wang J.; Lu C.; Ma L.; Zhang Q.; He X.; Li X. Size-Dependent Halogenated Nitrobenzene Hydrogenation Selectivity of Pd Nanoparticles. J. Phys. Chem. C 2014, 118, 2594–2601. 10.1021/jp411442f. [DOI] [Google Scholar]
- Corma A.; Serna P.; Concepción P.; Calvino J. J. Transforming Nonselective into Chemoselective Metal Catalysts for the Hydrogenation of Substituted Nitroaromatics. J. Am. Chem. Soc. 2008, 130, 8748–8753. 10.1021/ja800959g. [DOI] [PubMed] [Google Scholar]
- Arenal R.; March K.; Ewels C. P.; Rocquefelte X.; Kociak M.; Loiseau A.; Stéphan O. Atomic Configuration of Nitrogen-Doped Single-Walled Carbon Nanotubes. Nano Lett. 2014, 14, 5509–5516. 10.1021/nl501645g. [DOI] [PubMed] [Google Scholar]
- Liu L.; Díaz U.; Agostini G.; Concepción P.; Corma A. Generation of Subnanometric Platinum with High Stability during Transformation of a 2D Zeolite into 3D. Nat. Mater. 2017, 16, 132–138. 10.1038/nmat4757. [DOI] [PubMed] [Google Scholar]
- Vargas J. A.; Petkov V.; Nouh E. S. A.; Ramamoorthy R. K.; Lacroix L.-M.; Poteau R.; Viau G.; Lecante P.; Arenal R. Ultrathin Gold Nanowires with the Polytetrahedral Structure of Bulk Manganese. ACS Nano 2018, 12, 9521–9531. 10.1021/acsnano.8b05036. [DOI] [PubMed] [Google Scholar]
- Serna P.; Concepción P.; Corma A. Design of highly active and chemoselective bimetallic gold-platinum hydrogenation catalysts through kinetic and isotopic studies. J. Catal. 2009, 265, 19–25. 10.1016/j.jcat.2009.04.004. [DOI] [Google Scholar]
- Vorobyeva E.; Fako E.; Chen Z.; Collins S. M.; Johnstone D.; Midgley P. A.; Hauert R.; Safonova O. V.; Vilé G.; López N.; Mitchell S.; Pérez-Ramírez J. Atom-by-Atom Resolution of Structure-Function Relations over Low-Nuclearity Metal Catalysts. Angew. Chem., Int. Ed. 2019, 58, 8724–8729. 10.1002/anie.201902136. [DOI] [PubMed] [Google Scholar]
- Lykhach Y.; Kozlov S. M.; Skála T.; Tovt A.; Stetsovych V.; Tsud N.; Dvořák F.; Johánek V.; Neitzel A.; Mysliveček J.; Fabris S.; Matolín V.; Neyman K. M.; Libuda J. Counting electrons on supported nanoparticles. Nat. Mater. 2016, 15, 284. 10.1038/nmat4500. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Wagner T. Interaction of nanostructured metal overlayers with oxide surfaces. Surf. Sci. Rep. 2007, 62, 431–498. 10.1016/j.surfrep.2007.07.001. [DOI] [Google Scholar]
- Moses-DeBusk M.; Yoon M.; Allard L. F.; Mullins D. R.; Wu Z.; Yang X.; Veith G.; Stocks G. M.; Narula C. K. CO oxidation on supported single Pt atoms: experimental and ab initio density functional studies of CO interaction with Pt atom on theta-Al2O3(010) surface. J. Am. Chem. Soc. 2013, 135, 12634–12645. 10.1021/ja401847c. [DOI] [PubMed] [Google Scholar]
- Sanchez S. I.; Menard L. D.; Bram A.; Kang J. H.; Small M. W.; Nuzzo R. G.; Frenkel A. I. The Emergence of Nonbulk Properties in Supported Metal Clusters: Negative Thermal Expansion and Atomic Disorder in Pt Nanoclusters Supported on gmma-Al2O3. J. Am. Chem. Soc. 2009, 131, 7040–7054. 10.1021/ja809182v. [DOI] [PubMed] [Google Scholar]
- Derevyannikova E. A.; Kardash T. Y.; Stadnichenko A. I.; Stonkus O. A.; Slavinskaya E. M.; Svetlichnyi V. A.; Boronin A. I. Structural Insight into Strong Pt–CeO2 Interaction: From Single Pt Atoms to PtOx Clusters. J. Phys. Chem. C 2018, 123, 1320–1334. 10.1021/acs.jpcc.8b11009. [DOI] [Google Scholar]
- Jiang Z.; Sun Z.; Yang Y.; Chen S.; Shangguan W.; Wu X. The Role of Metal Oxide Interactions: Revisiting Pt Growth on the TiO2 Surface in the Process of Impregnation Method. Nanoscale 2017, 9, 14272–14279. 10.1039/c7nr02913f. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Li Y.; Zheng L.; Yan Y.; Zhang Y.; Chen G.; Sun S.; Zhang J. Highly Active, Stable Oxidized Platinum Clusters as Electrocatalysts for the Hydrogen Evolution Reaction. Energy Environ. Sci. 2017, 10, 2450–2458. 10.1039/c7ee02537h. [DOI] [Google Scholar]
- Ahmadi M.; Timoshenko J.; Behafarid F.; Roldan Cuenya B. Tuning the Structure of Pt Nanoparticles through Support Interactions: An in Situ Polarized X-ray Absorption Study Coupled with Atomistic Simulations. J. Phys. Chem. C 2019, 123, 10666–10676. 10.1021/acs.jpcc.9b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X.; Junge K.; Dai X.; Kreyenschulte C.; Pohl M.-M.; Wohlrab S.; Shi F.; Brückner A.; Beller M. Synthesis of Single Atom Based Heterogeneous Platinum Catalysts: High Selectivity and Activity for Hydrosilylation Reactions. ACS Cent. Sci. 2017, 3, 580–585. 10.1021/acscentsci.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Zhu Y.; Asakura H.; Zhang B.; Zhang J.; Zhou M.; Han Y.; Tanaka T.; Wang A.; Zhang T.; Yan N. Thermally Stable Single Atom Pt/m-Al2O3 for Selective Hydrogenation and CO Oxidation. Nat. Commun. 2017, 8, 16100. 10.1038/ncomms16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Rioux R. M.; Hoefelmeyer J. D.; Komor R.; Niesz K.; Grass M.; Yang P.; Somorjai G. A. Hydrothermal Growth of Mesoporous SBA-15 Silica in the Presence of PVP-Stabilized Pt Nanoparticles: Synthesis, Characterization, and Catalytic Properties. J. Am. Chem. Soc. 2006, 128, 3027–3037. 10.1021/ja057383r. [DOI] [PubMed] [Google Scholar]
- Claus P.; Brückner A.; Mohr C.; Hofmeister H. Supported Gold Nanoparticles from Quantum Dot to Mesoscopic Size Scale: Effect of Electronic and Structural Properties on Catalytic Hydrogenation of Conjugated Functional Groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. 10.1021/ja0012974. [DOI] [Google Scholar]
- Gracia F. J.; Miller J. T.; Kropf A. J.; Wolf E. E. Kinetics, FTIR, and Controlled Atmosphere EXAFS Study of the Effect of Chlorine on Pt-Supported Catalysts during Oxidation Reactions. J. Catal. 2002, 209, 341–354. 10.1006/jcat.2002.3601. [DOI] [Google Scholar]
- Ankudinov A. L.; Rehr J. J.; Bare S. R. Hybridization Peaks in Pt–Cl XANES. Chem. Phys. Lett. 2000, 316, 495–500. 10.1016/s0009-2614(99)01358-5. [DOI] [Google Scholar]
- Dessal C.; Len T.; Morfin F.; Rousset J.-L.; Aouine M.; Afanasiev P.; Piccolo L. Dynamics of Single Pt Atoms on Alumina During CO Oxidation Monitored by Operando X-ray and Infrared Spectroscopies. ACS Catal. 2019, 9, 5752–5759. 10.1021/acscatal.9b00903. [DOI] [Google Scholar]
- Bliem R.; van der Hoeven J. E. S.; Hulva J.; Pavelec J.; Gamba O.; de Jongh P. E.; Schmid M.; Blaha P.; Diebold U.; Parkinson G. S. Dual Role of CO in the Stability of Subnano Pt Clusters at the Fe3O4(001) Surface. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 8921–8926. 10.1073/pnas.1605649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson G. S.; Novotny Z.; Argentero G.; Schmid M.; Pavelec J.; Kosak R.; Blaha P.; Diebold U. Carbon Monoxide-Induced Adatom Sintering in a Pd-Fe3O4 Model Catalyst. Nat. Mater. 2013, 12, 724. 10.1038/nmat3667. [DOI] [PubMed] [Google Scholar]
- Campbell C. T. The Energetics of Supported Metal Nanoparticles: Relationships to Sintering Rates and Catalytic Activity. Acc. Chem. Res. 2013, 46, 1712–1719. 10.1021/ar3003514. [DOI] [PubMed] [Google Scholar]
- Pereira-Hernández X. I.; DeLaRiva A.; Muravev V.; Kunwar D.; Xiong H.; Sudduth B.; Engelhard M.; Kovarik L.; Hensen E. J. M.; Wang Y.; et al. Tuning Pt-CeO2 Interactions by High-Temperature Vapor-Phase Synthesis for Improved Reducibility of Lattice Oxygen. Nat. Commun. 2019, 10, 1358. 10.1038/s41467-019-09308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi M.; Mistry H.; Roldan Cuenya B. Tailoring the Catalytic Properties of Metal Nanoparticles via Support Interactions. J. Phys. Chem. Lett. 2016, 7, 3519–3533. 10.1021/acs.jpclett.6b01198. [DOI] [PubMed] [Google Scholar]
- Lou Y.; Liu J. CO Oxidation on Metal Oxide Supported Single Pt atoms: The Role of the Support. Ind. Eng. Chem. Res. 2017, 56, 6916–6925. 10.1021/acs.iecr.7b01477. [DOI] [Google Scholar]
- Liu H.-H.; Wang Y.; Jia A.-P.; Wang S.-Y.; Luo M.-F.; Lu J.-Q. Oxygen Vacancy Promoted CO Oxidation over Pt/CeO2 Catalysts: A Reaction at Pt–CeO2 Interface. Appl. Surf. Sci. 2014, 314, 725–734. 10.1016/j.apsusc.2014.06.196. [DOI] [Google Scholar]
- Artiglia L.; Orlando F.; Roy K.; Kopelent R.; Safonova O.; Nachtegaal M.; Huthwelker T.; van Bokhoven J. A. Introducing Time Resolution to Detect Ce3+ Catalytically Active Sites at the Pt/CeO2 Interface through Ambient Pressure X-ray Photoelectron Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 102–108. 10.1021/acs.jpclett.6b02314. [DOI] [PubMed] [Google Scholar]
- Liu L.; Matsushita T.; Concepción P.; Leyva-Pérez A.; Corma A. Facile Synthesis of Surface-Clean Monodispersed CuOx Nanoparticles and Their Catalytic Properties for Oxidative Coupling of Alkynes. ACS Catal. 2016, 6, 2211. 10.1021/acscatal.5b02935. [DOI] [Google Scholar]
- Fernández E.; Boronat M.; Corma A. Trends in the Reactivity of Molecular O2 with Copper Clusters: Influence of Size and Shape. J. Phys. Chem. C 2015, 119, 19832–19846. 10.1021/acs.jpcc.5b05023. [DOI] [Google Scholar]
- Laletina S. S.; Mamatkulov M.; Shor E. A.; Kaichev V. V.; Genest A.; Yudanov I. V.; Rösch N. Size-Dependence of the Adsorption Energy of CO on Pt Nanoparticles: Tracing Two Intersecting Trends by DFT Calculations. J. Phys. Chem. C 2017, 121, 17371–17377. 10.1021/acs.jpcc.7b05580. [DOI] [Google Scholar]
- Allian A. D.; Takanabe K.; Fujdala K. L.; Hao X.; Truex T. J.; Cai J.; Buda C.; Neurock M.; Iglesia E. Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. 10.1021/ja110073u. [DOI] [PubMed] [Google Scholar]
- Cargnello M.; Doan-Nguyen V. V. T.; Gordon T. R.; Diaz R. E.; Stach E. A.; Gorte R. J.; Fornasiero P.; Murray C. B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–773. 10.1126/science.1240148. [DOI] [PubMed] [Google Scholar]
- Liu A.; Liu X.; Liu L.; Pu Y.; Guo K.; Tan W.; Gao S.; Luo Y.; Yu S.; Si R.; et al. Getting Insights into the Temperature-Specific Active Sites on Platinum Nanoparticles for CO Oxidation: A Combined in Situ Spectroscopic and ab Initio Density Functional Theory Study. ACS Catal. 2019, 9, 7759–7768. 10.1021/acscatal.9b02552. [DOI] [Google Scholar]
- Zhu J.; Yang M.-L.; Yu Y.; Zhu Y.-A.; Sui Z.-J.; Zhou X.-G.; Holmen A.; Chen D. Size-Dependent Reaction Mechanism and Kinetics for Propane Dehydrogenation over Pt Catalysts. ACS Catal. 2015, 5, 6310–6319. 10.1021/acscatal.5b01423. [DOI] [Google Scholar]
- Xiong H.; Lin S.; Goetze J.; Pletcher P.; Guo H.; Kovarik L.; Artyushkova K.; Weckhuysen B. M.; Datye A. K. Thermally Stable and Regenerable Platinum-Tin Clusters for Propane Dehydrogenation Prepared by Atom Trapping on Ceria. Angew. Chem., Int. Ed. 2017, 56, 8986–8991. 10.1002/anie.201701115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Wang D.; Han H.; Li C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. 10.1021/ar300227e. [DOI] [PubMed] [Google Scholar]
- Berr M. J.; Schweinberger F. F.; Döblinger M.; Sanwald K. E.; Wolff C.; Breimeier J.; Crampton A. S.; Ridge C. J.; Tschurl M.; Heiz U.; et al. Size-Selected Subnanometer Cluster Catalysts on Semiconductor Nanocrystal Films for Atomic Scale Insight into Photocatalysis. Nano Lett. 2012, 12, 5903–5906. 10.1021/nl3033069. [DOI] [PubMed] [Google Scholar]
- Schweinberger F. F.; Berr M. J.; Döblinger M.; Wolff C.; Sanwald K. E.; Crampton A. S.; Ridge C. J.; Jäckel F.; Feldmann J.; Tschurl M.; et al. Cluster Size Effects in the Photocatalytic Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 13262–13265. 10.1021/ja406070q. [DOI] [PubMed] [Google Scholar]
- Xing J.; Chen J. F.; Li Y. H.; Yuan W. T.; Zhou Y.; Zheng L. R.; Wang H. F.; Hu P.; Wang Y.; Zhao H. J.; et al. Stable Isolated Metal Atoms as Active Sites for Photocatalytic Hydrogen Evolution. Chem.—Eur. J. 2014, 20, 2138–2144. 10.1002/chem.201303366. [DOI] [PubMed] [Google Scholar]
- Li X.; Bi W.; Zhang L.; Tao S.; Chu W.; Zhang Q.; Luo Y.; Wu C.; Xie Y. Single-Atom Pt as Co-Catalyst for Enhanced Photocatalytic H2 Evolution. Adv. Mater. 2016, 28, 2427–2431. 10.1002/adma.201505281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.