

Abstract
Growth hormone (GH) is secreted during hypoglycemia, and GH-responsive neurons are found in brain areas containing glucose-sensing neurons that regulate the counter-regulatory response (CRR). However, whether GH modulates the CRR to hypoglycemia via specific neuronal populations is currently unknown. Mice carrying ablation of GH receptor (GHR) either in leptin receptor (LepR)- or steroidogenic factor-1 (SF1)-expressing cells were studied. We also investigated the importance of signal transducer and activator of transcription 5 (STAT5) signaling in SF1 cells for the CRR. GHR ablation in LepR cells led to impaired capacity to recover from insulin-induced hypoglycemia and to a blunted CRR caused by 2-deoxy-d-glucose (2DG) administration. GHR inactivation in SF1 cells, which include ventromedial hypothalamic neurons, also attenuated the CRR. The reduced CRR was prevented by parasympathetic blockers. Additionally, infusion of 2DG produced an abnormal hyperactivity of parasympathetic preganglionic neurons, whereas the 2DG-induced activation of anterior bed nucleus of the stria terminalis neurons was reduced in mice without GHR in SF1 cells. Mice carrying ablation of Stat5a/b genes in SF1 cells showed no defects in the CRR. In summary, GHR expression in SF1 cells is required for a normal CRR, and these effects are largely independent of STAT5 pathway.—Furigo, I. C., de Souza, G. O., Teixeira, P. D. S., Guadagnini, D., Frazão, R., List, E. O., Kopchick, J. J., Prada, P. O., Donato, J., Jr. Growth hormone enhances the recovery of hypoglycemia via ventromedial hypothalamic neurons.
Keywords: autonomic nervous system, counter-regulatory response, cytokine, GH, hypothalamus
The regulation of blood glucose levels is of fundamental importance for the proper functioning of various organs. Although hyperglycemia can produce tissue damage in the long term, a sharp drop in blood glucose levels is considered a more important risk to life and can quickly lead to coma and death (1). Consequently, our physiologic systems have developed powerful responses in order to restore blood glucose levels through the activation of the hypothalamic-pituitary-adrenal axis and the sympathetic nervous system (SNS) (1). Whereas glucocorticoid secretion stimulates hepatic gluconeogenesis during hypoglycemia, SNS-mediated noradrenaline release rapidly activates glycogenolysis (2). Thus, via neuroendocrine mechanisms, the counter-regulatory response (CRR) to hypoglycemia robustly increases hepatic glucose production in order to restore blood glucose levels to a normal range (3, 4). Glucoprivic situations also lead to increases in food intake (5–7), possibly as an additional mechanism to restore body’s glucose stores.
Besides the activation of hypothalamic-pituitary-adrenal axis and SNS, other counter-regulatory hormones are secreted during hypoglycemia, including glucagon and growth hormone (GH) (3). Although the role played by glucagon increasing hepatic glucose production is well known (8), the mechanisms by which GH acts as a counter-regulatory hormone are much less studied. GH is secreted during situations of metabolic stress, such as hypoglycemia and food deprivation, and it raises blood glucose and free fatty acid levels (9–11). The metabolic effects of GH are thought to be primarily mediated by the activation of GH receptor (GHR) in the liver, adipose tissue, and skeletal muscle, all well-known direct GH-targeted tissues (12–14). However, our group recently described an extensive distribution of GH-responsive neurons in the mouse brain (15). Importantly, several hypothalamic and brainstem nuclei involved in the regulation of glucose and energy homeostasis exhibit robust phosphorylation of signal transducer and activator of transcription 5 (STAT5) after a peripheral administration of GH. Because the activation of STAT5 transcription factor is considered the major intracellular signaling pathway recruited by GHR (16), these findings indicate functional GHR expression in these neuronal populations. Among the areas that respond to the GH stimulus are the ventromedial nucleus (VMH), the arcuate nucleus (ARH), the paraventricular nucleus of the hypothalamus (PVH), and the lateral parabrachial nucleus (PBNl) (15). These brain areas either have glucose-sensing neurons or belong to the neurocircuit that triggers the CRR to hypoglycemia (17–20).
The role played by brain GH signaling to regulate metabolic responses is beginning to be revealed. GHR ablation in leptin receptor (LepR)-expressing cells leads to hepatic insulin resistance and impaired peripheral lipid metabolism (21). Another study found that GH signaling in agouti-related protein (AgRP) neurons represents a cue during food deprivation to trigger key adaptive responses to conserve limited fuel stores (22). Based on these findings, we hypothesized that the effects of GH on the CRR may require GH signaling in the brain as well. To investigate whether GH regulates the CRR via specific neuronal populations, we studied mice carrying GHR ablation in LepR-expressing cells or particularly in VMH neurons, a key relay station that regulates the CRR to hypoglycemia (17, 20). We additionally investigated the importance of STAT5 signaling pathway in VMH neurons for the CRR. Our findings revealed that GHR expression in VMH neurons is required for a normal CRR, and these effects are largely independent of STAT5 pathway.
MATERIALS AND METHODS
Animals
Tissue-specific ablation of the GHR was achieved by breeding mice carrying loxP-flanked Ghr alleles (12–14) with the LepR-internal ribosome entry site (IRES)-Cre mouse (008320; The Jackson Laboratory, Bar Harbor, ME, USA) or the steroidogenic factor-1 (SF1)-Cre mouse (012462; The Jackson Laboratory). STAT5 ablation in the VMH was achieved by breeding mice carrying loxP-flanked Stat5a/b alleles (23, 24) with the SF1-Cre mouse. We used only male mice in the in vivo experiments. In some histologic experiments, LepR-IRES-Cre and SF1-Cre mice were also crossed with the Lox-Stop-Lox (LSL) Cre-inducible tdTomato-reporter mouse (007909; The Jackson Laboratory), allowing the visualization of LepR or SF1 cells via the red fluorescent protein expression. Mice were in the C57BL/6 background, weaned at 3–4 wk of age, and their mutations confirmed by genotyping the DNA that had been previously extracted from the tail tip (RedExtract-N-Amp Tissue PCR Kit; MilliporeSigma, Burlington, MA, USA). Mice received a regular rodent chow diet (2.99 kcal/g; 9.4% calories from fat). All experiments were carried out in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA), and were previously approved by the Ethics Committee on the Use of Animals of the Institute of Biomedical Sciences at the University of São Paulo (73/2017).
Detection of GH-responsive neurons
To visualize GH-responsive cells in the brain, adult mice (n = 3/genotype) received an acute intraperitoneal injection of porcine pituitary GH (20 µg/g, from Dr. A. F. Parlow, National Hormone and Peptide Program, National Institute of Diabetes and Digestive and Kidney Diseases, University of California–Los Angeles Medical Center, Torrance, CA, USA) and were perfused 90 min later. Mice were deeply anesthetized with isoflurane and perfused transcardially with saline, followed by a 10% buffered formalin solution (∼150 ml/mouse). Brains were collected and postfixed in the same fixative for 30–60 min and cryoprotected overnight at 4°C in 0.1 M PBS containing 20% sucrose. Brains were cut in 30-µm-thick sections using a freezing microtome. Brain slices were rinsed in 0.02 M potassium PBS, pH 7.4 (KPBS), followed by pretreatment in water solution containing 1% hydrogen peroxide and 1% sodium hydroxide for 20 min. After rinsing in KPBS, sections were incubated in 0.3% glycine and 0.03% lauryl sulfate for 10 min each. Next, slices were blocked in 3% normal donkey serum for 1 h, followed by incubation in anti–phosphorylated (p)STAT5-Tyr694 primary antibody [1:1000, Resource Identification (RRID): AB_2315225; Cell Signaling Technology, Danvers, MA, USA] for 40 h. After incubation in the primary antibody, slices were submitted to fluorescence reaction or immunoperoxidase staining according to the purpose of the experiment. For the immunofluorescence reactions, sections were rinsed in KPBS and incubated for 90 min in AlexaFluor488-conjugated secondary antibody (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were mounted onto gelatin-coated slides, and the slides were covered with Fluoromount G mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA). For the immunoperoxidase staining, sections were incubated for 1 h in biotin-conjugated secondary antibody (1:1000; Jackson ImmunoResearch Laboratories) and next for 1 h with an avidin-biotin complex (1:500; Vector Laboratories, Burlingame, CA, USA). The peroxidase reaction was performed using 0.05% 3,3′-diaminobenzidine, 0.25% nickel sulfate, and 0.03% hydrogen peroxide. The slides were covered with distyrene/plasticiser/xylene (DPX) mounting medium (MilliporeSigma). Photomicrographs were acquired with a Zeiss Axiocam High-Resolution camera (HRc) coupled to a Zeiss Axioimager A1 microscope (Carl Zeiss GmbH, Oberkochen, Germany). Images were digitized using Axiovision software (Carl Zeiss GmbH). The ImageJ Cell Counter software (http://rsb.info.nih.gov/ij/) was used to manually count the number of single- or double-labeled cells in the areas of interest. The quantification was performed in 1 rostral-caudal level of the ARH (bregma: −1.46), VMH (bregma: −1.34), and PBNl (bregma: −5.20).
Evaluation of glucose homeostasis
Glycemia was measured using a glucose meter (OneTouch Ultra; Johnson & Johnson, New Brunswick, NJ, USA) via blood samples collected from the tail. After 4 h without food, adult mice were subjected to a glucose tolerance test (GTT; 2 g glucose/kg body weight, i.p.). The hyperinsulinemic-euglycemic clamp was done as previously described by Zanotto et al. (25), after being unfed overnight (8–12 h). Briefly, anesthetized animals received a continuous insulin infusion at a fixed rate of 3 mU/kg/min through an inserted catheter in the jugular vein. The glucose solution (5% w/v) was infused through the same vein, but at a variable rate to keep blood glucose at euglycemia levels (close to 100 mg/dl). Blood samples were collected from the tip of the tails at intervals of 5 min throughout 120 min for blood glucose evaluation.
CRR to hypoglycemia
To evaluate the CRR to hypoglycemia, adult mice received an acute intraperitoneal injection of 1 IU/kg insulin or 0.5 g/kg 2-deoxy-d-glucose (2DG; MilliporeSigma), and their glycemia was determined in intervals during 180 min. To evaluate the participation of the autonomic nervous system in the CRR to hypoglycemia, 2DG was coinfused either with a muscarinic receptor blocker (3 mg/kg atropine; MilliporeSigma) or a combination of α and β receptor blockers (3 mg/kg phentolamine and 0.5 mg/kg propranolol, respectively; MilliporeSigma). To determine the hypoglycemia-induced hyperphagia, ad libitum–fed mice received intraperitoneal injections of either saline or 2DG (0.5 g/kg), and their food intake were recorded 2, 3, and 4 h afterward. In another experiment, mice were euthanized 90 min after intraperitoneal injections of saline or 2DG (0.5 g/kg) to determine the serum concentrations of corticosterone, glucagon, GH, and insulin, as well as the gene expression of key enzymes involved in gluconeogenesis [glucose-6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase 1 (Pck1)] in the liver. Another group of mice was perfused 2 h after intraperitoneal injections of saline or 2DG (0.5 g/kg) to determine the expression of the proto-oncogene c-Fos in the brain. The perfusion, microtomy, and immunostaining were performed as described for the pSTAT5 protocol, except that brain sections were incubated in anti–c-Fos primary antibody (1:20,000, Ab5, RRID: AB_2314043; MilliporeSigma) for 48 h. Brainstem sections were also incubated in a cocktail of anti–c-Fos and anti–choline acetyltransferase (ChAT, 1:400, RRID: AB_2079751; MilliporeSigma) antibodies to determine whether parasympathetic preganglionic neurons were activated by 2DG infusion.
Evaluation of energy balance and somatotropic axis
Body weight was weekly monitored from weaning until 13 wk of age. Then, mice were single housed, and their food intake was daily measured for ∼5 d. O2 consumption (energy expenditure) was determined using the Oxymax/Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH, USA). Body fat and lean mass were measured by time-domain nuclear magnetic resonance using the LF50 body composition mice analyzer (Bruker, Billerica, MA, USA). Body growth and somatotropic axis were assessed by the nose-anus, femur length, and serum IGF-1 concentration.
Hormone and glycogen analyses
Commercially available ELISA kits were used to determine the serum concentrations of leptin (Crystal Chem, Elk Grove Village, IL, USA), corticosterone (Arbor Assays, Ann Arbor, MI, USA), glucagon (Crystal Chem), GH (MilliporeSigma), insulin (Crystal Chem), and IGF-1 (R&D Systems, Minneapolis, MN, USA). Hepatic glycogen content was determined by a quantitative colorimetric assay (BioAssay Systems, Hayward, CA, USA).
Food restriction protocol
The food restriction applied in the experimental animals was based on the protocol used in previous studies in refs. 22 and 26. Briefly, adult mice were initially single housed, and their food intake was recorded. Then, mice were subjected to a 60% food restriction, in which each mouse received 40% of their normal intake 2 h before lights off for 5 consecutive days. The changes in body weight and glycemia were monitored at the time the food was provided.
Western blotting
Adult wild-type C57BL/6 mice received intraperitoneal injections of saline or porcine pituitary GH (20 µg/g). Mice were euthanized 15, 30, 45, 60, and 90 min after the injections (n = 4 each time). The hypothalamus was homogenized in RIPA buffer (MilliporeSigma) containing a cocktail of protease and phosphatase inhibitors (1:100; MilliporeSigma) and centrifuged (14,000 rpm, 4°C for 20 min), and the supernatants were retained. After determining total protein concentration (Pierce Bicinchoninic Acid Protein Assay; Thermo Fisher Scientific, Waltham, MA, USA), 50 µg of total protein was loaded on a 10% SDS-PAGE gel, and the separated proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% bovine serum albumin and incubated overnight at 4°C in antibodies against pSTAT5-Tyr694, pAMPK-Thr172 (RRID: AB_331250; Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; RRID: AB_10167668; Santa Cruz Biotechnology, Dallas, TX, USA). Next, we incubated the membranes for 45 min in 1:20,000 secondary antibody (IRDye 800CW; Li-Cor Biosciences, Lincoln, NE, USA). Proteins were detected and analyzed using the Li-Cor Odyssey System (Li-Cor Biosciences), and protein amounts were normalized to GAPDH.
Real-time quantitative PCR
Total RNA was extracted with Trizol (Thermo Fisher Scientific). Assessment of RNA quantity and quality was performed with an Epoch Microplate Spectrophotometer (BioTek Instruments, Winooski, VT, USA). Total RNA was incubated in DNase I RNase-Free (Roche, Basel, Switzerland). Reverse transcription was performed with 2 µg of total RNA with SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) and random hexamers (Roche). Real-time PCR was performed using the 7500TM Real-Time PCR System (Thermo Fisher Scientific) and Power Sybr Green PCR Master Mix (Thermo Fisher Scientific). Relative quantification of mRNA was calculated by 2−ΔΔCt. Data were normalized to the geometric mean of Actb, Gapdh, and Ppia and reported as fold changes compared with values obtained from the control group (set at 1.0). The following primers were used: Actb (forward: 5′-GCTCCGGCATGTGCAAAG-3′; reverse: 5′-CATCACACCCTGGTGCCTA-3′), G6pc (forward: 5′-GCTGGAGTCTTGTCAGGCAT-3′; reverse: 5′-CGGAGGCTGGCATTGTAGAT-3′), Gapdh (forward: 5′-GGGTCCCAGCTTAGGTTCAT-3′; reverse: 5′-TACGGCCAAATCCGTTCACA-3′), Pck1 (forward: 5′-CGCAAGCTGAAGAAATATGACAA-3′; reverse: 5′-TCGATCCTGGCCACATCTC-3′), and Ppia (forward: 5′-TATCTGCACTGCCAAGACTGAGT-3′; reverse: 5′-CTTCTTGCTGGTCTTGCCATTCC-3′).
Statistical analysis
The data were analyzed by the unpaired, 2-tailed Student’s t test or 2-way ANOVA, followed by Bonferroni’s multiple comparisons test. Analysis of repeated measures ANOVA was performed when applicable. Statistical analyses were performed using Prism software (GraphPad Software, La Jolla, CA, USA). We considered values of P < 0.05 to be statistically significant. All results are expressed as means ± sem.
RESULTS
Ablation of GHR in LepR-expressing neurons
Subpopulations of LepR-expressing neurons modulate the CRR to hypoglycemia (18–20, 27, 28). Previous studies have shown the presence of GH-responsive neurons in several brain areas that contain LepR-expressing cells (15, 21). To determine whether GH-responsive cells express the LepR, LepR-reporter mice received an intraperitoneal injection of GH to induce pSTAT5. We observed that 106 ± 13 (85 ± 2%) of LepR-expressing neurons coexpressed GH-induced pSTAT5 in the ARH (Fig. 1A–C). In the dorsomedial part of the VMH, we found that 41 ± 5 (45 ± 2%) of LepR neurons expressed GH-induced pSTAT5 (Fig. 1D–F). GH-responsive cells were also observed in the PBNl, a site that was previously shown to be involved in the regulation of CRR to hypoglycemia (18, 19). However, STAT5 phosphorylation was not observed in PBNl LepR-expressing cells (Fig. 1G, H). We then produced mice carrying a specific ablation of Ghr gene only in LepR-expressing cells [LepR GHR knockout (KO) mice]. As expected, a robust reduction in the number of LepR neurons expressing pSTAT5 was observed in the ARH (6 ± 1 cells; P = 0.0014 vs. control mice) and VMH (8 ± 5 cells; P = 0.013 vs. control mice), whereas pSTAT5 was still found in surrounding cells (Fig. 1).
Figure 1.

GH-responsive neurons in the hypothalamus of control and LepR GHR KO mice. LepR-expressing cells (red) and pSTAT5 (green) 90 min after an intraperitoneal injection of porcine GH (20 µg/g) in LepR-IRES-Cre::LSL-tdTomato mice (control) and LepR GHR KO::LSL-tdTomato mice. Yellow represents double-labeled cells. The inset represents a higher magnification photomicrograph of the selected area. 3V, third ventricle; AQ, cerebral aqueduct; scp, superior cerebellar peduncle; VMHdm, dorsomedial subdivision of the VMH. Scale bars, 100 µm. *P < 0.05 (unpaired, 2-tailed Student’s t test).
GHR ablation in LepR neurons leads to impaired capacity to recover from hypoglycemia
LepR GHR KO mice exhibited a similar body weight (Fig. 2A), food intake (Fig. 2B), and energy expenditure (Vo2; Fig. 2C) compared with control mice. Similarly, as previously shown in ref. 22, a lower body adiposity was observed in LepR GHR KO mice without significant changes in lean body mass (Fig. 2D, E). To investigate the consequences of GHR ablation in LepR cells on glucose homeostasis, the LepR GHR KO mice were initially submitted to a GTT (Fig. 2F, G). We observed that LepR GHR KO mice showed normal glucose tolerance compared with control animals (Fig. 2F, G). However, LepR GHR KO mice utilized less glucose during a hyperinsulinemic-euglycemic clamp (Fig. 2H), confirming previous findings by Cady et al. (21) showing insulin resistance after GHR ablation in LepR cells. We next performed an insulin tolerance test (ITT) to determine the capacity of the animals to recover from insulin-mediated hypoglycemia. Notably, LepR GHR KO mice showed a reduced area under the curve (AUC) during the ITT compared with control animals (Fig. 2I, J). The lower AUC during the ITT together with the insulin resistance exhibited by LepR GHR KO mice suggest defects in their capacity to recover from hypoglycemia. To further test this hypothesis, mice received an intraperitoneal injection of 2DG, which causes glucopenia and consequently induces a robust CRR. Whereas control animals showed the expected increase in glycemia after 2DG infusion, LepR GHR KO mice exhibited a blunted CRR (Fig. 2K, L). As previously mentioned, the CRR induces the secretion of counter-regulatory hormones, such as corticosterone and glucagon. To determine how GH modulates the CRR, the serum levels of these counter-regulatory hormones were measured after injections of 2DG or saline. 2DG injection induced an increase in serum corticosterone levels compared with saline-injected mice (Fig. 2M). Unexpectedly, this increase was higher in LepR GHR KO mice in comparison with control animals (Fig. 2M). 2DG-induced increase in glucagon levels was similar between control and LepR GHR KO mice (Fig. 2N). Thus, impairment in the CRR observed in LepR GHR KO mice is not explained by defects in the secretion of counter-regulatory hormones.
Figure 2.

Glucose homeostasis and CRR in control and LepR GHR KO mice. A) Body weight of control and LepR GHR KO mice (n = 10–13). B) Daily food intake (n = 12–20). C) Oxygen consumption (n = 7–11). D) Body fat mass (n = 10–13; P = 0.0002). E) Lean body mass (n = 10–13). F) Blood glucose levels during a GTT (n = 12–18). G) AUC of the GTT (P = 0.2715). H) Glucose utilization during a hyperinsulinemic-euglycemic clamp (P = 0.0264; n = 8–10). I) Blood glucose levels during an ITT (n = 18–25). J) AUC of the ITT (P = 0.0487). K) Blood glucose levels during a CRR induced by 2DG infusion (n = 21–26). L) AUC of the 2DG test (P = 0.0015). M) Serum corticosterone levels {interaction between 2DG effect and GHR ablation [F(1, 13) = 3.476, P = 0.085]; n = 3–5}. N) Serum glucagon levels {interaction between 2DG effect and GHR ablation [F(1, 23) = 0.0357, P = 0.8518]; n = 5–11}. Values are means ± sem. *P < 0.05.
GHR ablation in SF1 cells causes no metabolic imbalances during normal conditions
LepR neurons are found in multiple hypothalamic and extrahypothalamic areas (29). In addition, LepR is expressed in numerous peripheral organs and tissues (30). Thus, to determine the specific neuronal population involved in GH’s effects on the CRR, we decided to induce GHR deletion in the VMH. For this purpose, GHR ablation was driven by the SF1 promoter, which in the CNS is exclusively expressed in the VMH (31–34). An acute GH stimulus induced pSTAT5 in 524 ± 88 VMH cells of control mice (Fig. 3A–C), whereas SF1 GHR KO mice exhibited only 8 ± 2 GH-responsive cells in the VMH (P = 0.0042 vs. control mice), without affecting pSTAT5 expression in surrounding nuclei (Fig. 3D–F). Before evaluating glucose homeostasis, we determined whether SF1 GHR KO mice have changes in energy balance or body growth. SF1 GHR KO mice showed similar body weight, food intake, energy expenditure, body fat mass, serum leptin levels, lean body mass, body length, femur length, and serum IGF-1 levels compared with control animals (Fig. 3G–O). Thus, GHR ablation in SF1 cells caused no apparent metabolic or growth problems.
Figure 3.

GHR ablation in SF1 cells causes no changes in energy homeostasis. A–F) Photomicrographs showing SF1 cells (red) and pSTAT5 (black) 90 min after an intraperitoneal injection of porcine GH (20 µg/g) in SF1-Cre::LSL-tdTomato mice (control) and SF1 GHR KO::LSL-tdTomato mice. The insets represent higher magnification photomicrographs of the selected areas. 3V, third ventricle; DMH, dorsomedial nucleus; fx, fornix; LHA, lateral hypothalamic area. Scale bars, 200 and 50 µm (insets). G) Body weight of control and SF1 GHR KO mice (n = 5–31). H) Daily food intake (n = 21–28). I) Oxygen consumption (n = 4). J) Body fat mass (n = 5–11). K) Serum leptin levels (n = 9–11). L) Lean body mass (n = 5–11). M) Body length (n = 8–13). N) Femur length (n = 10–12). O) Serum IGF-1 levels (n = 9–10). Values are means ± sem.
GHR ablation in SF1 cells impairs the CRR via the parasympathetic nervous system
We then evaluated possible changes in glucose homeostasis of SF1 GHR KO mice. SF1 GHR KO mice exhibited normal glucose tolerance (Fig. 4A) and insulin sensitivity during the hyperinsulinemic-euglycemic clamp (Fig. 4B). However, GHR ablation in SF1 cells impaired the recovery of hypoglycemia during the ITT (Fig. 4C, D) and attenuated the CRR after 2DG infusion (Fig. 4E, F). These results indicate that GH acts on SF1/VMH neurons to enhance the recovery of hypoglycemia. The circulating levels of counter-regulatory hormones were assessed after injection of 2DG or saline, but the 2DG-induced increase in serum corticosterone, glucagon, and GH concentrations was similarly observed in both control and SF1 GHR KO mice (Fig. 4G–I). Serum insulin levels were also similar between the experimental groups (unpublished results). Because the CRR increases the expression of gluconeogenic enzymes in the liver, including G6pc and Pck1 (18, 19), the gene expression of these enzymes was analyzed in the liver after 2DG infusion, and no differences between control and SF1 GHR KO mice were observed (Fig. 4J, K). Additionally, 2DG infusion reduced hepatic glycogen content in control mice, whereas saline-treated SF1 GHR KO mice already exhibited a lower glycogen content compared with control mice, and 2DG was unable to further decrease hepatic glycogen content in SF1 GHR KO mice (Fig. 4L).
Figure 4.

GHR ablation in SF1 cells impairs the CRR to hypoglycemia. A) Blood glucose levels during a GTT (n = 17–24). B) Glucose utilization during the hyperinsulinemic-euglycemic clamp (P = 0.7356; n = 7–8). C) Blood glucose levels during an ITT (n = 21–28). D) AUC of the ITT (P = 0.0193). E) Blood glucose levels during a CRR induced by 2DG infusion (n = 17–27). F) AUC of the 2DG test (P = 0.0024). G) Serum corticosterone levels {interaction between 2DG effect and GHR ablation [F(1, 21) = 0.0183, P = 0.8936]; n = 6–7}. H) Serum glucagon levels {interaction between 2DG effect and GHR ablation [F(1, 23) = 0.6025, P = 0.4455]; n = 5–9}. I) Serum GH levels {interaction between 2DG effect and GHR ablation [F(1, 12) = 0.0928, P = 0.7658]; n = 4}. J) Hepatic G6pc mRNA levels (interaction between 2DG effect and GHR ablation [F(1, 18) = 0.6377, P = 0.4349]; n = 4–7). K) Hepatic Pck1 mRNA levels {interaction between 2DG effect and GHR ablation [F(1, 19) = 1.151, P = 0.2968]; n = 5–7}. L) Hepatic glycogen content {GHR ablation effect [F(1, 20) = 12.22, P = 0.0023]; interacion between 2DG effect and GHR ablation [F(1, 20) = 2.078, P = 0.1649]; n = 6}. Values are means ± sem. *P < 0.05.
The autonomic nervous system also plays a key role regulating hepatic metabolism and glucose production (2, 35, 36). The importance of the autonomic nervous system in the CRR was investigated in mice that received coinfusion of 2DG with pharmacological inhibitors of the SNS (phentolamine and propranolol) or parasympathetic nervous system (PNS) (atropine). Initially, the inhibition of the SNS or PNS during the CRR was investigated in normal mice (Fig. 5A, B). Coinfusion of α and β blockers reduced the CRR produced by 2DG (Fig. 5A, B), which is in accordance with the key role played by the SNS increasing hepatic glucose production during the CRR (2, 35, 36). On the other hand, the parasympathetic blocker atropine amplified the CRR (Fig. 5A, B), confirming the inhibitory action of the vagal tone in the hepatic glucose production (37, 38). To determine the role of the autonomic nervous system in the blunted CRR exhibited by animals carrying ablation of GHR in the VMH, SNS, or PNS, blockers were coinfused with 2DG in control and SF1 GHR KO mice. Remarkably, although the glycemia of SF1 GHR KO mice still remained lower than control animals after the coinfusion of α and β blockers (Fig. 5C, D), atropine coinfusion prevented the impaired response exhibited by SF1 GHR KO mice (Fig. 5E, F). Thus, the impaired CRR caused by the ablation of GHR in SF1/VMH cells is likely related to changes in the activity of the PNS rather than caused by defects in the secretion of counter-regulatory hormones or the SNS.
Figure 5.

Changes in the activity of the PNS explain the blunted CRR exhibited by SF1 GHR KO mice. A) Blood glucose levels in normal mice after injection of 2DG (n = 59), 2DG + α(phentolamine)/β (propranolol) blockers (n = 43), and 2DG + atropine (n = 44). *2DG + α/β blockers is different than the other groups (P < 0.05); #2DG + atropine is different than the 2DG group (P < 0.001). B) AUC after injection of 2DG, 2DG + α(phentolamine)/β (propranolol) blockers, and 2DG + atropine. C) Blood glucose levels in control and SF1 GHR KO mice after coinfusion of 2DG and α/β blockers (n = 20–25). D) AUC after coinfusion of 2DG and α/β blockers (P = 0.0103). E) Blood glucose levels in control and SF1 GHR KO mice after coinfusion of 2DG and atropine (n = 9–13). F) AUC after coinfusion of 2DG and atropine (P = 0.7544). Values are means ± sem. *P < 0.05 (1-way ANOVA and Bonferroni’s multiple comparisons test).
GHR ablation in SF1 cells affects the activity of the neurocircuit that regulates the PNS
The dorsal motor nucleus of the vagus (DMX) is composed of cholinergic neurons whose axons form the efferent vagus nerve and innervate parasympathetic ganglia that regulate liver function. Because our previous findings indicated the participation of the PNS in the impaired CRR exhibited by SF1 GHR KO mice, we measured c-Fos expression, a marker of neuronal activity, in the DMX of saline- or 2DG-treated mice (Fig. 6A–C). Saline-treated mice showed very low c-Fos expression in the DMX (Fig. 6A). Conversely, 2DG-treated SF1 GHR KO mice exhibited increased c-Fos expression in the DMX compared with control animals (Fig. 6A–C). We colocalized c-Fos with ChAT and confirmed that 83 ± 5% of 2DG-induced c-Fos expression in SF1 GHR KO mice was restricted to parasympathetic preganglionic neurons of the DMX, representing 50 ± 3% of the neuronal population (Fig. 6D–F). Thus, GHR ablation in the VMH causes an abnormal hyperactivity of DMX preganglionic neurons during the CRR.
Figure 6.

GHR ablation in SF1 cells affects the activity of the neurocircuit that regulates the PNS. A) Number of c-Fos–positive cells in the DMX 2 h after intraperitoneal injections of saline or 2DG {interaction between 2DG effect and GHR ablation [F(1, 11) = 6.039, P = 0.0318]; n = 3–5}. B, C) Photomicrographs showing 2DG-induced c-Fos expression in the DMX of control and SF1 GHR KO mice. D–F) Epifluorescence photomicrographs showing the colocalization between ChAT (red) and c-Fos (green) in the DMX of SF1 GHR KO mice. G, H) Epifluorescence photomicrographs showing the distribution of SF1/tdTomato axons in the vagus complex (G) or the BSTa (H) using the in SF1-Cre::LSL-tdTomato reporter mouse. I) Number of c-Fos–positive cells in the BSTa 2 h after intraperitoneal injections of saline or 2DG {interaction between 2DG effect and GHR ablation [F(1, 12) = 5.079, P = 0.0437]; n = 3–5}. J–L) Photomicrographs showing 2DG-induced c-Fos expression in the BSTa of saline-treated SF1 GHR KO (J), 2DG-treated control (K), and 2DG-treated SF1 GHR KO mice (L). Aco, anterior commissure; AP, area postrema; cc, central canal of the spinal cord; NTS, nucleus of the solitary tract; XII, hypoglossal nucleus. Values are means ± sem. Scale bars: 200 µm (B, C); 100 µm (D–F); 400 µm (G); 200 µm (H, J–L). *P < 0.05 (Bonferroni’s multiple comparisons test).
To determine whether SF1/VMH neurons send direct projections to regulate DMX cells, SF1-Cre mice were crossed with LSL-tdTomato reporter mouse. Thus, SF1 axons could be visualized through tdTomato expression (Fig. 6G, H). In the vagus complex, SF1/VMH axons were mostly found in the lateral part of the nucleus of the solitary tract and surrounding the central canal of the spinal cord, whereas very few SF1/VMH axons innervate the DMX (Fig. 6G). Thus, it is likely that VMH SF1 neurons regulate the activity of DMX preganglionic neurons via an indirect, polysynaptic circuit. In this sense, previous studies have identified the anterior bed nucleus of the stria terminalis (BSTa) as a key downstream target of VMH neurons to regulate the CRR to hypoglycemia (20, 27). Indeed, BSTa is heavily innervated by VMH SF1 neurons (Fig. 6H). VMH sends excitatory inputs to BSTa neurons, inducing their activation during a CRR (20, 27). Therefore, we determined the c-Fos expression in the BSTa of saline- or 2DG-treated mice and confirmed the results of previous studies showing activation of BSTa during a CRR induced by 2DG infusion (20, 27). Notably, SF1 GHR KO mice exhibited an attenuated BSTa activation compared with 2DG-treated control animals (Fig. 6I–L). c-Fos expression was also analyzed in other brain sites involved in stress or CRR, but control and SF1 GHR KO mice showed similar numbers of c-Fos–positive cells in the PVH, central nucleus of the amygdala, and PBNl after the injection of saline or 2DG (unpublished results).
GH signaling in SF1 cells does not regulate metabolism during food deprivation
Our group recently found that GHR ablation in AgRP neurons increases the weight loss and impairs the capacity of mice to maintain blood glucose levels during food deprivation (22). To evaluate whether GH signaling in SF1 cells is also necessary to maintain blood glucose levels during food deprivation, control and SF1 GHR KO mice were subjected to 5 d of 60% food restriction. Differently than that observed in mice carrying ablation of GHR in AgRP neurons (22), SF1 GHR KO mice exhibited a similar weight loss rate (Fig. 7A) and glycemia (Fig. 7B) during food restriction. We also assessed the 2DG-induced hyperphagia because GHR ablation in AgRP neurons reduces this response (22). 2DG injection increased food intake similarly in control and SF1 GHR KO mice (Fig. 7C).
Figure 7.
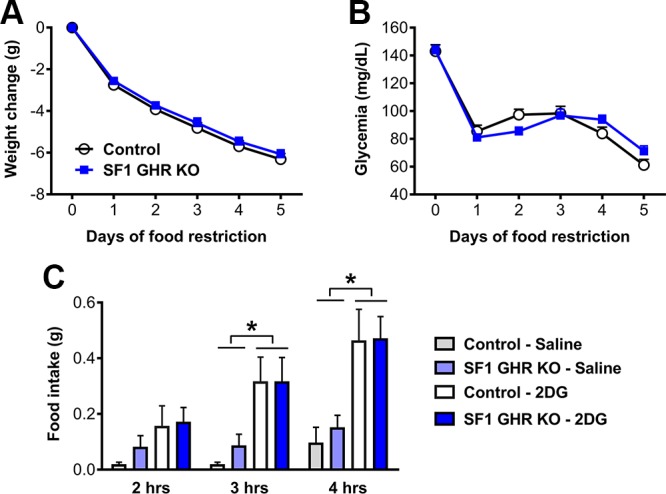
Changes in body weight and glycemia during food restriction, and glucoprivic-induced hyperphagia in control and SF1 GHR KO mice. A, B) Daily changes in body weight {interaction between food restriction effect and GHR ablation [F(5, 235) = 0.3783, P = 0.8633]; n = 21–28} and glycemia {interaction between food restriction effect and GHR ablation [F(5, 210) = 2.515, P = 0.0309]; n = 19–25} in mice that received 40% of their normal intake 2 h before lights off for 5 consecutive days. C) Food intake 2 h {main effect of 2DG [F(1, 12) = 4.651, P = 0.052], main effect of GHR ablation [F(1, 12) = 0.5398, P = 0.4766], and interaction [F(1, 12) = 0.2028, P = 0.6605]; n = 4}, 3 h {main effect of 2DG [F(1, 12) = 14.83, P = 0.0023], main effect of GHR ablation [F(1, 12) = 0.2429, P = 0.631], and interaction [F(1, 12) = 0.2429, P = 0.631]}, and 4 h {main effect of 2DG [F(1, 12) = 18.22, P = 0.0011], main effect of GHR ablation [F(1, 12) = 0.1506, P = 0.7047], and interaction [F(1, 12) = 0.087, P = 0.7731]} after intraperitoneal injection of either saline or 2DG in control and SF1 GHR KO mice. Values are means ± sem. *P < 0.05 (Bonferroni’s multiple comparisons test).
GH induces the phosphorylation of STAT5 but not AMPK in the hypothalamus
GHR is member of type I cytokine receptor family and recruits several intracellular signaling pathways (39). To determine possible molecular mechanisms recruited by GH to modulate the CRR, we assessed the capacity of an acute intraperitoneal GH injection to activate specific signaling molecules in the hypothalamus. In particular, we were interested to determine whether GH signaling can phosphorylate the AMPK because previous studies have shown that activation of AMPK in the VMH is critical for the CRR to hypoglycemia and secretion of counter-regulatory hormones (40–43). As expected, GH stimulus induced a robust and sustained phosphorylation of STAT5 in the hypothalamus 30, 45, 60, and 90 min after the injection (P = 0.0004; Fig. 8A, B). However, no statistically significant change was observed in AMPK phosphorylation after GH injection compared with saline-treated mice (P = 0.419; Fig. 8A, C). Thus, GH induces the phosphorylation of STAT5 but not AMPK in the hypothalamus.
Figure 8.

GH induces the phosphorylation of STAT5 but not AMPK in the hypothalamus. A–C) Western blotting of hypothalamic samples collected from wild-type C57BL/6 mice that received either saline (n = 5) or GH injection (i.p.; n = 4 each time) 15, 30, 45, 60, and 90 min prior to tissue collection. The phosphorylation of STAT5-Tyr694 (B), AMPK-Thr172 (C), and GAPDH (for normalization) was determined. Values are means ± sem. *P < 0.05 vs. saline injection (Bonferroni’s multiple comparisons test).
STAT5 signaling in SF1 cells is not required for the effects of GH on the CRR
STAT5 transcription factors are known to be critical mediators of GH’s effects in different tissues (16, 39, 44). To determine which intracellular signaling pathway is recruited by GH to modulate the CRR via SF1 cells, we produced mice carrying ablation of Stat5a/b genes specifically in SF1 cells (SF1 STAT5 KO mice). Conditional inactivation of Stat5a/b genes was achieved as previously described by our group in refs. 23, 24, and 45. Whereas control mice exhibited a large number of GH-responsive cells in the VMH, SF1 STAT5 KO mice showed very few pSTAT5-positive cells in the VMH after an acute GH injection (Fig. 9A, B). Then, possible changes in energy balance were evaluated. SF1 STAT5 KO mice showed a similar body weight, food intake, energy expenditure, body fat mass, and lean body mass compared with control mice (Fig. 9C–G). SF1 STAT5 KO mice also showed normal glucose tolerance (Fig. 10A) and insulin sensitivity during the hyperinsulinemic-euglycemic clamp (Fig. 10B). Differently than that observed in SF1 GHR KO mice, SF1 STAT5 KO mice exhibited an equivalent drop in blood glucose levels after insulin administration (Fig. 10C, D) and a similar CRR induced by 2DG (Fig. 10E, F) compared with control animals. On the other hand, as seen in SF1 GHR KO mice, coinfusion of α and β blockers with 2DG reduced the AUC in SF1 STAT5 KO mice (Fig. 10G, H), whereas blood glucose levels of control and SF1 STAT5 KO mice were similar after the coinfusion of atropine and 2DG (Fig. 10I, J). SF1 STAT5 KO mice also showed similar weight loss and glycemia during food restriction (Fig. 10K, L) and an equivalent 2DG-induced hyperphagia compared with control mice (Fig. 10M).
Figure 9.

STAT5 ablation in SF1 cells causes no changes in energy homeostasis. A, B) Photomicrographs showing GH-responsive cells (pSTAT5 staining) 90 min after an intraperitoneal injection of porcine GH in control and SF1 STAT5 KO mice. 3V, third ventricle; LHA, lateral hypothalamic area. Scale bar, 200 µm. C) Body weight of control and SF1 STAT5 KO mice (n = 5–9). D) Daily food intake (n = 19–23). E) Oxygen consumption (n = 8). F) Body fat mass (n = 21–23). G) Lean body mass (n = 21–23). Values are means ± sem.
Figure 10.

STAT5 ablation in SF1 cells attenuates the CRR induced by 2DG infusion. A) Blood glucose levels during a GTT (n = 12). B) Glucose utilization during a hyperinsulinemic-euglycemic clamp (P = 0.4274; n = 3–4). C) Blood glucose levels during an ITT (n = 18–22). D) AUC of the ITT (P = 0.6876). E) Blood glucose levels during a CRR induced by 2DG infusion (n = 19–21). F) AUC of the 2DG test (P = 0.2256). G) Blood glucose levels after coinfusion of 2DG and α(phentolamine)/β (propranolol) blockers (n = 7–11). H) AUC after coinfusion of 2DG and α/β blockers (P = 0.0240). I) Blood glucose levels after coinfusion of 2DG and atropine (n = 5–11). J) AUC after coinfusion of 2DG and atropine (P = 0.2057). K, L) Daily changes in body weight {interaction between food restriction effect and STAT5 ablation [F(5, 85) = 0.8211, P = 0.5381]; n = 8–11} and glycemia {interaction between food restriction effect and STAT5 ablation [F(5, 85) = 1.309, P = 0.2678]; n = 8–11} in mice that received 40% of their normal intake 2 h before lights off for 5 consecutive days. M) Food intake after intraperitoneal injection of either saline or 2DG in control and SF1 STAT5 KO mice. Values are means ± sem. *P < 0.05.
DISCUSSION
Hypoglycemia is a powerful stimulus to induce GH secretion (9), and previous studies have shown that GH deficiency leads to impaired CRR and spontaneous hypoglycemia (26, 46, 47). However, the mechanisms of action recruited by GH to help in the recovery of hypoglycemia remain largely unknown. Because classic GH-target tissues are major regulators of glucose homeostasis, there is a general assumption that the counter-regulatory effects of GH could be mediated by peripheral tissues. In the present study, we investigated whether GH modulates the CRR via VMH neurons.
The mechanisms that regulate GH secretion during situations of metabolic stress are complex. A previous study by Stanley et al. (48) demonstrated the glucose-sensing capacity of GH-releasing hormone (GHRH) neurons. In this sense, GHRH neurons are activated during hypoglycemia, explaining the increased GH release during this condition (9, 48). Food restriction or food deprivation also changes the pattern of GH secretion (26, 49, 50). Whereas food restriction suppresses GH pulsatility in mice (49, 50), baseline GH levels increase during prolonged food restriction (22, 26). The decreased GH pulsatility during food restriction is mediated by neuropeptide Y because KO mice for neuropeptide Y or Y1 receptor maintain the pulsatile secretion of GH (49). Conversely, ghrelin secretion is partially responsible for increasing baseline GH levels during prolonged starvation (26, 51) but not in short-term food restriction (50). Interestingly, some differences between humans and mice are observed in terms of GH physiology during food restriction. Differently than mice, humans increase the frequency of GH pulses during prolonged food restriction (52). Additionally, although food restriction–induced increase in GH levels drives the release of free fatty acids in humans (53), the increase in free fatty acids during food restriction in mice is likely not mediated by GH (50). Finally, changes from a pulsatile secretion pattern to a continuous release may affect the biologic effects of GH (54, 55). Pulsatile infusions of GH are more effective in stimulating growth in hypophysectomized male rats than continuous GH infusion (55). Pulsatile administration of placental GH in female C57BL/6J mice also causes a higher growth and hyperinsulinemia compared with continuous infusion (54). Nonetheless, either continuous infusion or daily injections of GH produce similar effects in GH-deficient patients (56). However, it is not known how GH secretion pattern affects the activity of GH-responsive neurons.
Recent studies investigated the metabolic phenotype of mice deficient in GHR in LepR cells (21, 22). These studies found that LepR GHR KO mice have normal food intake and body weight, but they present hepatic insulin resistance (21). Here, we confirmed the insulin resistance exhibited by LepR GHR KO mice (21) and further showed that these animals exhibit impaired capacity to recover from insulin-induced hypoglycemia and have a blunted CRR. LepR-expressing cells are found in multiple brain sites and peripheral tissues (29, 30). Of note, hepatic cells express functional LepR, and therefore, genetic deletions driven by Lepr promoter likely target the liver (57). Because liver-specific GHR ablation increases blood glucose and insulin levels in male mice, causing insulin resistance (13, 58), a partial disruption of GH signaling in the liver may explain the hepatic insulin resistance of LepR GHR KO mice. However, no change in Ghr mRNA expression was found in the liver of LepR GHR KO mice (21). In addition, neurons that regulate GH secretion by negative feedback also express the LepR (59, 60). This includes somatostatin and GHRH neurons of the ARH (59, 60). Therefore, LepR GHR KO mice may present changes in GH secretion by the lack of negative feedback on LepR-expressing neurons. However, the impact of this deletion in the somatotropic axis seems to be modest because LepR GHR KO mice have normal body weight and lean mass, although we previously showed that LepR GHR KO mice have a slight increase in body length (22). Therefore, other mechanisms are able to compensate for the loss of GH action in LepR-expressing cells to avoid hypersecretion of GH in LepR GHR KO mice. Altogether, the use of LepR-specific ablation of GHR may affect different tissues and neural populations, so the evaluation of additional mouse models with less confounding factors is of interest to establish the role of central GH action in the regulation of the CRR.
LepR is expressed by cholecystokinin cells of the PBNl, and these neurons were recently implicated in the regulation of the CRR to hypoglycemia (18, 19). However, despite the presence of GH-responsive cells in the PBNl, these neurons are not positive for LepR. Thus, other LepR cells mediate the defects in the CRR exhibited by LepR GHR KO mice. In this sense, the VMH is a key relay station that regulates the CRR (17, 20) and contains a large amount of GH-responsive neurons (15). Former studies have used the SF1-Cre mouse to induce genetic deletions targeting VMH neurons because SF1 expression in the CNS is exclusively found in the VMH (31–34). SF1 is also expressed in pituitary gonadotrophs, adrenal cortex, and gonads (61). However, SF1 GHR KO mice have no reproductive problems (Donato J, personal communications, May 24, 2019), as well as normal basal and 2DG-induced corticosterone secretion. Thus, GHR ablation in SF1 cells causes no apparent endocrine problems. Additionally, SF1 GHR KO mice exhibit normal energy balance and somatic growth, suggesting that GH signaling in SF1 cells plays no metabolic role at normal conditions. However, GHR ablation in SF1 cells also led to impaired capacity to recover from insulin-induced hypoglycemia and a blunted CRR. Because the coexpression between LepR and SF1 is only found in the VMH, our findings indicate that GH acts on VMH neurons to enhance the CRR to hypoglycemia.
The CRR relies not only on the secretion of counter-regulatory hormones but also on the activation of the SNS. In fact, we showed that coinfusion of 2DG with sympathetic blockers strongly suppresses the glycemic curve during the CRR. However, SF1 GHR KO mice still maintained a lower glycemic curve during the coinfusion of 2DG and α/β blockers. On the other hand, parasympathetic blockers completely eliminated the differences between control and SF1 GHR KO mice in the glycemic curve during the CRR, suggesting that GHR ablation in VMH neurons possibly affected hepatic glucose metabolism via the PNS. This hypothesis is corroborated by an abnormal hyperactivity observed in DMX preganglionic neurons of SF1 GHR KO mice during the CRR. Thus, although we generally assume that only the SNS plays a role during the CRR, it is also important for a coordinated change of the PNS in order to achieve a precise regulation of blood glucose levels. Interestingly, previous studies have shown that VMH neurons regulate the activity of both the SNS (62–64) and PNS (65–68). In this sense, VMH lesions increase the activity of the PNS, whereas the sympathetic tone is reduced (67). In accordance with our results, a previous study showed that VMH neurons can induce glucoregulatory effects through cholinergic fibers of the vagus nerves (69).
The CRR produces a robust increase in hepatic glucose production. This effect can be achieved by an activation of gluconeogenesis (3, 4). SF1 GHR KO mice showed the expected 2DG-induced increase in the hepatic expression of G6pc and Pck1 mRNA. The CRR also increases hepatic glucose production via glycogenolysis. Of note, VMH stimulation is capable of influencing glycogenolysis and glycogen content (70, 71). The activation of glycogenolysis explains the lower glycogen content in 2DG-treated control mice compared with saline-injected animals. Thus, the incapacity to reduce glycogen content after 2DG infusion together with an apparent normal activation of gluconeogenesis suggest that defects in glycogenolysis are the probable underlying cause of the blunted CRR exhibited by SF1 GHR KO mice.
Classic neuroanatomical studies described projections from VMH neurons to the nucleus of the solitary tract (72). However, as observed in the present study, these terminal fields are sparse and do not target the DMX directly. Thus, it is likely that VMH neurons influence the activity of parasympathetic preganglionic neurons via indirect circuits. In this sense, VMH neurons send very dense projections to the BSTa (72), as shown here. Additionally, recent studies have found that BSTa is a critical area activated by VMH neurons during hypoglycemia to induce the CRR (20, 27). Because a reduced c-Fos expression in the BSTa of SF1 GHR KO mice was found after 2DG injection, our results indicate that GH acts as a cue in VMH neurons during hypoglycemia to induce the activation of the VMH→BSTa neurocircuit in order to enhance the recovery of hypoglycemia. Whereas BSTa neurons send a small number of axons to the vagus complex, dense BSTa projections are found in forebrain areas that regulate the autonomic nervous system, including the PVH and central nucleus of the amygdala (73). However, no changes in 2DG-induced c-Fos expression were found in these areas comparing control and SF1 GHR KO mice. Thus, it remains unknown how the VMH→BSTa neurocircuit modulates the activity of the PNS during the CRR.
GHR is a membrane receptor and relies on the activation of several intracellular signaling pathways to induce its cellular effects (39). In the present study, we sought to identify the intracellular mechanisms recruited by GHR signaling in order to regulate the CRR to hypoglycemia. Activation of AMPK in the VMH plays a key role to induce the CRR to hypoglycemia (40–43). However, we found no evidence that an acute GH stimulus is able to induce the phosphorylation of AMPK-Thr172 in the hypothalamus. STAT5 transcriptional factors are well-known downstream targets of GH (16). However, SF1 STAT5 KO mice showed normal CRR to 2DG, indicating that the action of GH regulating the recovery of hypoglycemia is largely independent of STAT5.
We found that neither GHR nor STAT5 ablation in SF1 cells affected the acute hyperphagic behavior induced by 2DG (5–7, 74, 75). Conversely, inactivation of GHR in LepR or AgRP cells reduces 2DG-induced hyperphagia (22). GH circulating levels increase during food restriction, and GH secretion is important to prevent hypoglycemia during this situation (26). GHR ablation in AgRP cells impairs the maintenance of blood glucose levels during food restriction (22). However, disruption of GHR in SF1 cells did not affect the maintenance of glycemia during food restriction. These results highlight the importance of GH signaling in SF1 cells for the regulation of the CRR but not for other aspects of energy and glucose homeostasis.
In summary, here we showed that GH signaling in SF1 cells modulates the CRR to improve the recovery of hypoglycemia. Thus, our study provides additional evidence that GH signaling in the brain, independently of IGF-1 circulating levels, regulates important physiologic responses. The findings of the present and our former study (22) allow us to hypothesize that, in addition to its classic effect on growth, GH also plays a key role during situations of metabolic stress by acting on specific neuronal populations to produce appropriate physiologic adjustments in order restore the homeostasis. In this sense, while GH acts on AgRP neurons to signal energy deficiency during chronic food deprivation, triggering key neuroendocrine responses to conserve limited fuel stores (22), SF1/VMH neurons sense the increased GH circulating levels during hypoglycemia to induce neural responses that will restore blood glucose levels. Therefore, our study identified a novel mechanism of action recruited by GH to enhance the recovery of hypoglycemia. These findings also contribute to the understanding of the causes of the recurrent hypoglycemia exhibited by GH-deficient individuals.
ACKNOWLEDGMENTS
The authors thank Ana Maria P. Campos (Department of Physiology and Biophysics, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil) for expert technical assistance. This work was supported by the São Paulo Research Foundation (FAPESP; Grants 17/02983-2 to J.D.; 16/09679-4 to I.C.F.; 17/18498-6 and 18/20087-7 to P.O.P.; and 17/21840-8 to R.F.), Pfizer, Inc. [2017 Global Advancing Science Through Pfizer–Investigator Research Exchange (ASPIRE) Young Investigator Research Award to J.D.], and U.S. National Institutes of Health, National Institute on Aging, Grant R01AG059779 (to J.J.K. and E.O.L.). The authors declare no conflicts of interest.
Glossary
- 2DG
2-deoxy-d-glucose
- AgRP
agouti-related protein
- ARH
arcuate nucleus
- AUC
area under the curve
- BSTa
anterior bed nucleus of the stria terminalis
- ChAT
choline acetyltransferase
- CRR
counter-regulatory response
- DMX
dorsal motor nucleus of the vagus
- G6pc
glucose-6-phosphatase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GH
growth hormone
- GHR
GH receptor
- GHRH
GH-releasing hormone
- GTT
glucose tolerance test
- IRES
internal ribosome entry site
- ITT
insulin tolerance test
- KO
knockout
- KPBS
0.02 M potassium PBS, pH 7.4
- LepR
leptin receptor
- LSL
Lox-Stop-Lox
- PBNl
lateral parabrachial nucleus
- Pck1
phosphoenolpyruvate carboxykinase 1
- PNS
parasympathetic nervous system
- PVH
paraventricular nucleus of the hypothalamus
- RRID
resource identification
- SF1
steroidogenic factor-1
- SNS
sympathetic nervous system
- STAT5
signal transducer and activator of transcription 5
- VMH
ventromedial nucleus
AUTHOR CONTRIBUTIONS
I. C. Furigo and J. Donato conceived the project, designed the experiments, interpreted the results, and wrote the manuscript; D. Guadagnini and P. O. Prada performed the insulin clamp studies; I. C. Furigo, G. O. de Souza, P. D. S. Teixeira, and R. Frazão performed the remaining experiments; E. O. List and J. J. Kopchick provided essential equipment, reagents, and expertise; and all authors revised and approved the final version of the manuscript.
REFERENCES
- 1.Verberne A. J., Sabetghadam A., Korim W. S. (2014) Neural pathways that control the glucose counterregulatory response. Front. Neurosci. 8, 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizuno K., Ueno Y. (2017) Autonomic nervous system and the liver. Hepatol. Res. 47, 160–165 [DOI] [PubMed] [Google Scholar]
- 3.Rizza R. A., Cryer P. E., Gerich J. E. (1979) Role of glucagon, catecholamines, and growth hormone in human glucose counterregulation. Effects of somatostatin and combined alpha- and beta-adrenergic blockade on plasma glucose recovery and glucose flux rates after insulin-induced hypoglycemia. J. Clin. Invest. 64, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seoane-Collazo P., Fernø J., Gonzalez F., Diéguez C., Leis R., Nogueiras R., López M. (2015) Hypothalamic-autonomic control of energy homeostasis. Endocrine 50, 276–291 [DOI] [PubMed] [Google Scholar]
- 5.Thompson C. I., Zagon I. S., McLaughlin P. J. (1979) Hypophagia follows the initial hyperphagia produced by 2-deoxy-D-glucose in rats. Physiol. Behav. 23, 187–190 [DOI] [PubMed] [Google Scholar]
- 6.Ikeda H., Nishikawa K., Matsuo T. (1980) Feeding responses of Zucker fatty rat to 2-deoxy-D-glucose, norepinephrine, and insulin. Am. J. Physiol. 239, E379–E384 [DOI] [PubMed] [Google Scholar]
- 7.Clegg D. J., Edwards G. L., Martin R. J. (2003) Central insulin potentiates eating elicited by 2-deoxy-D-glucose. Physiol. Behav. 78, 331–336 [DOI] [PubMed] [Google Scholar]
- 8.Liljenquist J. E., Mueller G. L., Cherrington A. D., Keller U., Chiasson J.-L., Perry J. M., Lacy W. W., Rabinowitz D. (1977) Evidence for an important role of glucagon in the regulation of hepatic glucose production in normal man. J. Clin. Invest. 59, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth J., Glick S. M., Yalow R. S., Bersonsa (1963) Hypoglycemia: a potent stimulus to secretion of growth hormone. Science 140, 987–988 [DOI] [PubMed] [Google Scholar]
- 10.Greenwood F. C., Landon J. (1966) Growth hormone secretion in response to stress in man. Nature 210, 540–541 [DOI] [PubMed] [Google Scholar]
- 11.Winkler B., Steele R., Altszuler N., Debodo R. C. (1964) Effect of growth hormone on free fatty acid metabolism. Am. J. Physiol. 206, 174–178 [DOI] [PubMed] [Google Scholar]
- 12.List E. O., Berryman D. E., Funk K., Gosney E. S., Jara A., Kelder B., Wang X., Kutz L., Troike K., Lozier N., Mikula V., Lubbers E. R., Zhang H., Vesel C., Junnila R. K., Frank S. J., Masternak M. M., Bartke A., Kopchick J. J. (2013) The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol. Endocrinol. 27, 524–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.List E. O., Berryman D. E., Funk K., Jara A., Kelder B., Wang F., Stout M. B., Zhi X., Sun L., White T. A., LeBrasseur N. K., Pirtskhalava T., Tchkonia T., Jensen E. A., Zhang W., Masternak M. M., Kirkland J. L., Miller R. A., Bartke A., Kopchick J. J. (2014) Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology 155, 1793–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.List E. O., Berryman D. E., Ikeno Y., Hubbard G. B., Funk K., Comisford R., Young J. A., Stout M. B., Tchkonia T., Masternak M. M., Bartke A., Kirkland J. L., Miller R. A., Kopchick J. J. (2015) Removal of growth hormone receptor (GHR) in muscle of male mice replicates some of the health benefits seen in global GHR-/- mice. Aging (Albany N.Y.) 7, 500–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furigo I. C., Metzger M., Teixeira P. D., Soares C. R., Donato J., Jr (2017) Distribution of growth hormone-responsive cells in the mouse brain. Brain Struct. Funct. 222, 341–363 [DOI] [PubMed] [Google Scholar]
- 16.Teglund S., McKay C., Schuetz E., van Deursen J. M., Stravopodis D., Wang D., Brown M., Bodner S., Grosveld G., Ihle J. N. (1998) Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93, 841–850 [DOI] [PubMed] [Google Scholar]
- 17.Routh V. H. (2010) Glucose sensing neurons in the ventromedial hypothalamus. Sensors (Basel) 10, 9002–9025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flak J. N., Patterson C. M., Garfield A. S., D’Agostino G., Goforth P. B., Sutton A. K., Malec P. A., Wong J. T., Germani M., Jones J. C., Rajala M., Satin L., Rhodes C. J., Olson D. P., Kennedy R. T., Heisler L. K., Myers M. G., Jr (2014) Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balance. Nat. Neurosci. 17, 1744–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garfield A. S., Shah B. P., Madara J. C., Burke L. K., Patterson C. M., Flak J., Neve R. L., Evans M. L., Lowell B. B., Myers M. G., Jr., Heisler L. K. (2014) A parabrachial-hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell Metab. 20, 1030–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meek T. H., Nelson J. T., Matsen M. E., Dorfman M. D., Guyenet S. J., Damian V., Allison M. B., Scarlett J. M., Nguyen H. T., Thaler J. P., Olson D. P., Myers M. G., Jr., Schwartz M. W., Morton G. J. (2016) Functional identification of a neurocircuit regulating blood glucose. Proc. Natl. Acad. Sci. USA 113, E2073–E2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cady G., Landeryou T., Garratt M., Kopchick J. J., Qi N., Garcia-Galiano D., Elias C. F., Myers M. G., Jr., Miller R. A., Sandoval D. A., Sadagurski M. (2017) Hypothalamic growth hormone receptor (GHR) controls hepatic glucose production in nutrient-sensing leptin receptor (LepRb) expressing neurons. Mol. Metab. 6, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furigo I. C., Teixeira P. D. S., de Souza G. O., Couto G. C. L., Romero G. G., Perelló M., Frazão R., Elias L. L., Metzger M., List E. O., Kopchick J. J., Donato J., Jr (2019) Growth hormone regulates neuroendocrine responses to weight loss via AgRP neurons. Nat. Commun. 10, 662; erratum: 980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furigo I. C., Melo H. M., Lyra E Silva N. M., Ramos-Lobo A. M., Teixeira P. D. S., Buonfiglio D. C., Wasinski F., Lima E. R., Higuti E., Peroni C. N., Bartolini P., Soares C. R. J., Metzger M., de Felice F. G., Donato J., Jr (2018) Brain STAT5 signaling modulates learning and memory formation. Brain Struct. Funct. 223, 2229–2241 [DOI] [PubMed] [Google Scholar]
- 24.Buonfiglio D. C., Ramos-Lobo A. M., Silveira M. A., Furigo I. C., Hennighausen L., Frazão R., Donato J., Jr (2015) Neuronal STAT5 signaling is required for maintaining lactation but not for postpartum maternal behaviors in mice. Horm. Behav. 71, 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zanotto T. M., Quaresma P. G. F., Guadagnini D., Weissmann L., Santos A. C., Vecina J. F., Calisto K., Santos A., Prada P. O., Saad M. J. A. (2016) Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab. 6, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao T. J., Liang G., Li R. L., Xie X., Sleeman M. W., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Goldstein J. L., Brown M. S. (2010) Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA 107, 7467–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faber C. L., Matsen M. E., Velasco K. R., Damian V., Phan B. A., Adam D., Therattil A., Schwartz M. W., Morton G. J. (2018) Distinct neuronal projections from the hypothalamic ventromedial nucleus mediate glycemic and behavioral effects. Diabetes 67, 2518–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’souza A. M., Neumann U. H., Glavas M. M., Kieffer T. J. (2017) The glucoregulatory actions of leptin. Mol. Metab. 6, 1052–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos-Lobo A. M., Donato J., Jr (2017) The role of leptin in health and disease. Temperature (Austin) 4, 258–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin J., Barb C. R., Matteri R. L., Kraeling R. R., Chen X., Meinersmann R. J., Rampacek G. B. (2000) Long form leptin receptor mRNA expression in the brain, pituitary, and other tissues in the pig. Domest. Anim. Endocrinol. 19, 53–61 [DOI] [PubMed] [Google Scholar]
- 31.Dhillon H., Zigman J. M., Ye C., Lee C. E., McGovern R. A., Tang V., Kenny C. D., Christiansen L. M., White R. D., Edelstein E. A., Coppari R., Balthasar N., Cowley M. A., Chua S., Jr., Elmquist J. K., Lowell B. B. (2006) Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203 [DOI] [PubMed] [Google Scholar]
- 32.Lindberg D., Chen P., Li C. (2013) Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J. Comp. Neurol. 521, 3167–3190 [DOI] [PubMed] [Google Scholar]
- 33.Pedroso J. A. B., de Mendonca P. O. R., Fortes M. A. S., Tomaz I., Pecorali V. L., Auricino T. B., Costa I. C., Lima L. B., Furigo I. C., Bueno D. N., Ramos-Lobo A. M., Lotfi C. F. P., Donato J., Jr (2017) SOCS3 expression in SF1 cells regulates adrenal differentiation and exercise performance. J. Endocrinol. 235, 207–222 [DOI] [PubMed] [Google Scholar]
- 34.Ramos-Lobo A. M., Teixeira P. D. S., Furigo I. C., Donato J., Jr (2017) SOCS3 ablation in SF1 cells causes modest metabolic effects during pregnancy and lactation. Neuroscience 365, 114–124 [DOI] [PubMed] [Google Scholar]
- 35.Yi C. X., la Fleur S. E., Fliers E., Kalsbeek A. (2010) The role of the autonomic nervous liver innervation in the control of energy metabolism. Biochim. Biophys. Acta 1802, 416–431 [DOI] [PubMed] [Google Scholar]
- 36.Kalsbeek A., Bruinstroop E., Yi C. X., Klieverik L. P., La Fleur S. E., Fliers E. (2010) Hypothalamic control of energy metabolism via the autonomic nervous system. Ann. N. Y. Acad. Sci. 1212, 114–129 [DOI] [PubMed] [Google Scholar]
- 37.Pocai A., Obici S., Schwartz G. J., Rossetti L. (2005) A brain-liver circuit regulates glucose homeostasis. Cell Metab. 1, 53–61 [DOI] [PubMed] [Google Scholar]
- 38.Matsuhisa M., Yamasaki Y., Shiba Y., Nakahara I., Kuroda A., Tomita T., Iida M., Ikeda M., Kajimoto Y., Kubota M., Hori M. (2000) Important role of the hepatic vagus nerve in glucose uptake and production by the liver. Metabolism 49, 11–16 [DOI] [PubMed] [Google Scholar]
- 39.Dehkhoda F., Lee C. M. M., Medina J., Brooks A. J. (2018) The growth hormone receptor: mechanism of receptor activation, cell signaling, and physiological aspects. Front. Endocrinol. (Lausanne) 9, 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCrimmon R. J., Fan X., Ding Y., Zhu W., Jacob R. J., Sherwin R. S. (2004) Potential role for AMP-activated protein kinase in hypoglycemia sensing in the ventromedial hypothalamus. Diabetes 53, 1953–1958 [DOI] [PubMed] [Google Scholar]
- 41.Han S. M., Namkoong C., Jang P. G., Park I. S., Hong S. W., Katakami H., Chun S., Kim S. W., Park J. Y., Lee K. U., Kim M. S. (2005) Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 48, 2170–2178 [DOI] [PubMed] [Google Scholar]
- 42.McCrimmon R. J., Fan X., Cheng H., McNay E., Chan O., Shaw M., Ding Y., Zhu W., Sherwin R. S. (2006) Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes 55, 1755–1760 [DOI] [PubMed] [Google Scholar]
- 43.McCrimmon R. J., Shaw M., Fan X., Cheng H., Ding Y., Vella M. C., Zhou L., McNay E. C., Sherwin R. S. (2008) Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes 57, 444–450 [DOI] [PubMed] [Google Scholar]
- 44.Furigo I. C., Ramos-Lobo A. M., Frazão R., Donato J., Jr (2016) Brain STAT5 signaling and behavioral control. Mol. Cell. Endocrinol. 438, 70–76 [DOI] [PubMed] [Google Scholar]
- 45.Silveira M. A., Furigo I. C., Zampieri T. T., Bohlen T. M., de Paula D. G., Franci C. R., Donato J., Jr., Frazao R. (2017) STAT5 signaling in kisspeptin cells regulates the timing of puberty. Mol. Cell. Endocrinol. 448, 55–65 [DOI] [PubMed] [Google Scholar]
- 46.Hussain K., Hindmarsh P., Aynsley-Green A. (2003) Spontaneous hypoglycemia in childhood is accompanied by paradoxically low serum growth hormone and appropriate cortisol counterregulatory hormonal responses. J. Clin. Endocrinol. Metab. 88, 3715–3723 [DOI] [PubMed] [Google Scholar]
- 47.Tennese A. A., Wevrick R. (2011) Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology 152, 967–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stanley S., Domingos A. I., Kelly L., Garfield A., Damanpour S., Heisler L., Friedman J. (2013) Profiling of glucose-sensing neurons reveals that GHRH neurons are activated by hypoglycemia. Cell Metab. 18, 596–607 [DOI] [PubMed] [Google Scholar]
- 49.Huang L., Tan H. Y., Fogarty M. J., Andrews Z. B., Veldhuis J. D., Herzog H., Steyn F. J., Chen C. (2014) Actions of NPY, and its Y1 and Y2 receptors on pulsatile growth hormone secretion during the fed and fasted state. J. Neurosci. 34, 16309–16319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steyn F. J., Leong J. W., Huang L., Tan H. Y., Xie T. Y., Nelson C., Waters M. J., Veldhuis J. D., Epelbaum J., Chen C. (2012) GH does not modulate the early fasting-induced release of free fatty acids in mice. Endocrinology 153, 273–282 [DOI] [PubMed] [Google Scholar]
- 51.Li R. L., Sherbet D. P., Elsbernd B. L., Goldstein J. L., Brown M. S., Zhao T. J. (2012) Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. J. Biol. Chem. 287, 17942–17950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan J. L., Heist K., DePaoli A. M., Veldhuis J. D., Mantzoros C. S. (2003) The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Invest. 111, 1409–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakharova A. A., Horowitz J. F., Surya S., Goldenberg N., Harber M. P., Symons K., Barkan A. (2008) Role of growth hormone in regulating lipolysis, proteolysis, and hepatic glucose production during fasting. J. Clin. Endocrinol. Metab. 93, 2755–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao S., Vickers M. H., Evans A., Stanley J. L., Baker P. N., Perry J. K. (2016) Comparison of pulsatile vs. continuous administration of human placental growth hormone in female C57BL/6J mice. Endocrine 54, 169–181 [DOI] [PubMed] [Google Scholar]
- 55.Bick T., Hochberg Z., Amit T., Isaksson O. G., Jansson J. O. (1992) Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology 131, 423–429 [DOI] [PubMed] [Google Scholar]
- 56.Laursen T., Jørgensen J. O., Jakobsen G., Hansen B. L., Christiansen J. S. (1995) Continuous infusion versus daily injections of growth hormone (GH) for 4 weeks in GH-deficient patients. J. Clin. Endocrinol. Metab. 80, 2410–2418 [DOI] [PubMed] [Google Scholar]
- 57.Mittenbühler M. J., Sprenger H. G., Gruber S., Wunderlich C. M., Kern L., Brüning J. C., Wunderlich F. T. (2018) Hepatic leptin receptor expression can partially compensate for IL-6Rα deficiency in DEN-induced hepatocellular carcinoma. Mol. Metab. 17, 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan Y., Menon R. K., Cohen P., Hwang D., Clemens T., DiGirolamo D. J., Kopchick J. J., Le Roith D., Trucco M., Sperling M. A. (2009) Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J. Biol. Chem. 284, 19937–19944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell J. N., Macosko E. Z., Fenselau H., Pers T. H., Lyubetskaya A., Tenen D., Goldman M., Verstegen A. M., Resch J. M., McCarroll S. A., Rosen E. D., Lowell B. B., Tsai L. T. (2017) A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rupp A. C., Allison M. B., Jones J. C., Patterson C. M., Faber C. L., Bozadjieva N., Heisler L. K., Seeley R. J., Olson D. P., Myers M. G., Jr (2018) Specific subpopulations of hypothalamic leptin receptor-expressing neurons mediate the effects of early developmental leptin receptor deletion on energy balance. Mol. Metab. 14, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim K. W., Donato J., Jr., Berglund E. D., Choi Y. H., Kohno D., Elias C. F., Depinho R. A., Elmquist J. K. (2012) FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Invest. 122, 2578–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakaguchi T., Bray G. A. (1990) Ventromedial hypothalamic lesions attenuate responses of sympathetic nerves to carotid arterial infusions of glucose and insulin. Int. J. Obes. 14, 127–133 [PubMed] [Google Scholar]
- 63.Balkan B., Strubbe J. H., Bruggink J. E., Steffens A. B. (1991) Altered sympathetic control of nutrient mobilization during physical exercise after lesions in the VMH. Am. J. Physiol. 260, R368–R372 [DOI] [PubMed] [Google Scholar]
- 64.Minokoshi Y., Haque M. S., Shimazu T. (1999) Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 48, 287–291 [DOI] [PubMed] [Google Scholar]
- 65.Rohner-Jeanrenaud F., Ionescu E., Jeanrenaud B. (1983) The origins and role of efferent vagal nuclei in hyperinsulinemia in hypothalamic and genetically obese rodents. J. Auton. Nerv. Syst. 9, 173–184 [DOI] [PubMed] [Google Scholar]
- 66.Valensi P., Doaré L., Perret G., Germack R., Pariès J., Mesangeau D. (2003) Cardiovascular vagosympathetic activity in rats with ventromedial hypothalamic obesity. Obes. Res. 11, 54–64 [DOI] [PubMed] [Google Scholar]
- 67.Paes A. M., Carniatto S. R., Francisco F. A., Brito N. A., Mathias P. C. (2006) Acetylcholinesterase activity changes on visceral organs of VMH lesion-induced obese rats. Int. J. Neurosci. 116, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 68.Suzuki Y., Shimizu H., Ishizuka N., Kubota N., Kubota T., Senoo A., Kageyama H., Osaka T., Hirako S., Kim H. J., Matsumoto A., Shioda S., Mori M., Kadowaki T., Inoue S. (2014) Vagal hyperactivity due to ventromedial hypothalamic lesions increases adiponectin production and release. Diabetes 63, 1637–1648 [DOI] [PubMed] [Google Scholar]
- 69.Szabo A. J., Iguchi A., Burleson P. D., Szabo O. (1983) Vagotomy or atropine blocks hypoglycemic effect of insulin injected into ventromedial hypothalamic nucleus. Am. J. Physiol. 244, E467–E471 [DOI] [PubMed] [Google Scholar]
- 70.Coutinho E. A., Okamoto S., Ishikawa A. W., Yokota S., Wada N., Hirabayashi T., Saito K., Sato T., Takagi K., Wang C. C., Kobayashi K., Ogawa Y., Shioda S., Yoshimura Y., Minokoshi Y. (2017) Activation of SF1 neurons in the ventromedial hypothalamus by DREADD technology increases insulin sensitivity in peripheral tissues. Diabetes 66, 2372–2386 [DOI] [PubMed] [Google Scholar]
- 71.Shimazu T., Fukuda A., Ban T. (1966) Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature 210, 1178–1179 [DOI] [PubMed] [Google Scholar]
- 72.Canteras N. S., Simerly R. B., Swanson L. W. (1994) Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J. Comp. Neurol. 348, 41–79 [DOI] [PubMed] [Google Scholar]
- 73.Dong H. W., Swanson L. W. (2006) Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J. Comp. Neurol. 494, 142–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luquet S., Phillips C. T., Palmiter R. D. (2007) NPY/AgRP neurons are not essential for feeding responses to glucoprivation. Peptides 28, 214–225 [DOI] [PubMed] [Google Scholar]
- 75.Ozawa Y., Arima H., Watanabe M., Shimizu H., Ito Y., Banno R., Sugimura Y., Ozaki N., Nagasaki H., Oiso Y. (2011) Repeated glucoprivation delayed hyperphagic responses while activating neuropeptide Y neurons in rats. Peptides 32, 763–769 [DOI] [PubMed] [Google Scholar]