

Abstract
Tenocytes serve to synthesize and maintain collagen fibrils and other extracellular matrix proteins in tendon. Despite the high prevalence of tendon injury, the underlying biologic mechanisms of postnatal tendon growth and repair are not well understood. IGF1 plays an important role in the growth and remodeling of numerous tissues but less is known about IGF1 in tendon. We hypothesized that IGF1 signaling is required for proper tendon growth in response to mechanical loading through regulation of collagen synthesis and cell proliferation. To test this hypothesis, we conditionally deleted the IGF1 receptor (IGF1R) in scleraxis (Scx)-expressing tenocytes using a tamoxifen-inducible Cre-recombinase system and caused tendon growth in adult mice via mechanical overload of the plantaris tendon. Compared with control Scx-expressing IGF1R-positive (Scx:IGF1R+) mice, in which IGF1R is present in tenocytes, mice that lacked IGF1R in their tenocytes [Scx-expressing IGF1R-negative (Scx:IGF1RΔ) mice] demonstrated reduced cell proliferation and smaller tendons in response to mechanical loading. Additionally, we identified that both the PI3K/protein kinase B and ERK pathways are activated downstream of IGF1 and interact in a coordinated manner to regulate cell proliferation and protein synthesis. These studies indicate that IGF1 signaling is required for proper postnatal tendon growth and support the potential use of IGF1 in the treatment of tendon disorders.—Disser, N. P., Sugg, K. B., Talarek, J. R., Sarver, D. C., Rourke, B. J., Mendias, C. L. Insulin-like growth factor 1 signaling in tenocytes is required for adult tendon growth.
Keywords: tendon fibroblasts, protein synthesis, hypertrophy, extracellular matrix, IGF1
Tendon is a dense connective tissue that serves to transmit force from muscle to bone during mechanical loading. The tendon extracellular matrix (ECM) is composed mostly of type I collagen, as well as type III collagen, elastin, and proteoglycans (1). Tendon fibroblasts, or tenocytes, are the main cell type in tendon and are responsible for the synthesis, organization, and maintenance of the ECM (2). In response to high stress repetitive mechanical loading, such as that which occurs during resistance exercise, tendons adapt by undergoing hypertrophy (3). This results in an increase in tendon cross-sectional area (CSA) through an induction of cell proliferation and collagen production (4–6). Repetitive loading can also induce a chronic inflammatory condition referred to as tendinopathy (7, 8). Despite the high prevalence of tendon injury and overall importance of tendon in maintaining musculoskeletal health, the underlying biologic mechanisms that regulate postnatal tendon growth are not well understood. Further, as tendinopathy is thought to arise due to improper responses to mechanical stimuli, gaining a greater understanding of the basic biologic mechanisms of tendon growth could help in the development of new therapies for the treatment of tendon disorders.
IGF1, which can be induced by growth hormones and other mechanical signals, is an integral component of the growth and development of several different tissues (9). IGF1 is typically bound to an IGFBP carrier protein, and upon proteolytic degradation of the IGFBP, IGF1 is liberated and can interact with its receptor, IGF1R, which is a member of the receptor tyrosine kinase (RTK) family of transmembrane receptors (10). IGF1 is a potent activator of skeletal muscle cell proliferation and protein synthesis, and the deletion of IGF1 in muscle fibers results in fiber atrophy and disrupted metabolism (9, 11). In bone, overexpression of IGF1 results in increased bone mineral density, and the inactivation of IGF1R in osteoblasts impairs matrix mineralization (12). However, the role of IGF1 in tendon growth has not been fully examined. Previous studies in tendon have revealed an increase in tendon collagen synthesis following IGF1 treatment in engineered tendon tissue and healthy human tendon, as well as in horses with tendinopathies (13–15). Given the role that IGF1 plays in the growth of other tissues as well as observations of increased IGF1 expression correlated with enhanced ECM production in tendons, we sought to determine the mechanisms behind IGF1-mediated tendon growth. We hypothesized that IGF1 signaling is required for proper tendon growth in response to mechanical loading through a coordinated induction of collagen synthesis and cell proliferation. To test this hypothesis, we induced tendon growth in adult mice via mechanical overload of the plantaris tendon and deleted IGF1R in scleraxis (Scx)-expressing tenocytes. Additionally, we performed a series of in vitro experiments in which we treated tenocytes with IGF1 for various durations to examine the molecular mechanisms induced by IGF1 during postnatal tendon growth.
MATERIALS AND METHODS
Mice
All animal studies were approved by the University of Michigan and Hospital for Special Surgery Institutional Animal Care and Use Committees. We used 3 strains of mice in this study. Wild-type C57BL/6J (strain 000664) mice and IGF1Rflox mice (strain 012251) in which exon 3 of IGF1R is flanked by loxP sites (16) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). ScxCreERT2 mice in which an IRES-CreERT2 sequence was inserted between the stop codon and 3′ UTR in exon 2 of Scx (17) were kindly provided by Dr. Ronen Schweitzer (Shriners Hospitals for Children, Portland, OR, USA). Genotype of mice was determined by PCR analysis of DNA obtained from a tail tendon biopsy. After performing initial crosses between ScxCreERT2/CreERT2 and IGF1Rflox/flox mice, we generated ScxCreERT2/CreERT2:IGF1Rflox/flox mice to allow us to inactivate IGF1R in Scx-expressing cells upon treatment with tamoxifen [Scx-expressing IGF1R-negative (Scx:IGF1RΔ) mice, in which IGF1R is absent in tenocytes], whereas Scx+/+:IGF1Rflox/flox mice would maintain the expression of IGF1R in Scx-expressing cells after tamoxifen treatment [Scx-expressing IGF1R-positive (Scx:IGF1R+) mice, in which IGF1R is present in tenocytes]. The age of mice in this study was ∼4 mo. An overview of the alleles used in this study is provided in Fig. 1A. Wild-type C57BL/6J mice were used for cell culture experiments, whereas Scx:IGF1R+ and Scx:IGF1RΔ mice were used in whole animal studies.
Figure 1.

Experimental overview. A) Overview of the alleles used in this study, including the Scx wild-type (Scx+), Scx inducible Cre (ScxCreERT2), IGF1R wild-type (IGF1R+), IGF1R floxed (IGF1Rflox), and IGF1R-null (IGF1RΔ) alleles. B) Timeline of tamoxifen treatment, surgical overload, and tissue harvest. Tamoxifen was delivered on a daily basis beginning 3 d prior to surgery and continued through tissue harvest. C) Overview of surgical overload procedure, in which the Achilles tendon is removed from the animal, resulting in compensatory hypertrophy of the synergist plantaris muscle and tendon. A neotendon area of new tendon matrix forms around the original tendon. D) Relative expression of IGF1R at 7 and 14 d, with the Scx:IGF1RΔ group normalized to the Scx:IGF1R+ group at each time point. Values are means ± CV. Differences for each time point tested with a Student’s t test: a, significantly different (P < 0.05) from Scx:IGF1R+ group; n = 4 animals/group.
Synergist ablation tendon growth procedure
Mice were treated daily with an intraperitoneal injection of 1 mg of tamoxifen (T5648; MilliporeSigma, Burlington, MA, USA) dissolved in 50 µl of corn oil to activate CreERT2 recombinase in Scx-expressing cells. Tamoxifen treatment began 3 d prior to surgery and continued on a daily basis until tissue was harvested for analysis. Bilateral synergist ablation procedures were performed under isoflurane anesthesia, as previously described (6, 18). An overview of the time points and surgical procedures are shown in Fig. 1B, C. The Achilles tendon was surgically excised to prevent the gastrocnemius and soleus muscles from plantarflexing the talocrural joint, resulting in compensatory hypertrophy of the adjacent synergist plantaris muscle and tendon. A small incision was created in the skin above the posterior paw plantarflexor tendons, and the Achilles tendon was isolated and excised, leaving stumps at the myotendinous junction and calcaneus. The skin was closed with GLUture (Zoetis, Parsippany, NJ, USA), buprenorphine was administered for postoperative analgesia, and ad libitum weight bearing and cage activity were allowed in the postoperative period. Mice were closely monitored during the postoperative period for any adverse reactions. At tissue harvest, the left plantaris tendons were divided into proximal and distal halves and snap frozen at −80°C for gene expression analysis, whereas the right plantaris tendons were used for histology. After the tendons were removed, mice were euthanized by cervical dislocation. Plantaris tendons from additional nonoverloaded Scx:IGF1R+ mice were obtained as previously described for gene expression analysis.
Histology
Histology was conducted as previously described (6, 18). Plantaris tendons obtained from animals were immediately placed in 30% sucrose solution for 1 h, snap frozen in Tissue-Tek Optical Cutting Temperature (OCT) Compound (Sakura Finetek, Torrance, CA, USA), and stored at −80°C until use. Tendons were sectioned at a thickness of 10 µm in a cryostat. Sections were stained with hematoxylin and eosin to determine tendon CSA and cell density. To evaluate proliferating cells, tendon sections were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 5% goat serum, and incubated with rabbit anti-Ki67 antibodies (1:100, ab16667; Abcam,Cambridge, MA, USA) and goat anti-rabbit antibodies conjugated to AF555 (1:300, A-21429; Thermo Fisher Scientific, Waltham, MA, USA), as well as wheat germ agglutinin lectin conjugated to Alexa Fluor 488 (AF488) (1:200, W11261; Thermo Fisher Scientific) to identify the ECM, and DAPI (1:500; MilliporeSigma) to label nuclei. Slides were imaged using a BX-51 microscope and camera (Olympus, Tokyo, Japan) for the hematoxylin and eosin slides, whereas an A1 confocal laser microscope (Nikon, Tokyo, Japan) was used for the Ki67 slides. Quantification of tendon size and cell density was performed using ImageJ [National Institutes of Health (NIH), Bethesda, MD, USA].
Cell isolation
Tenocytes were isolated from the tail tendons of mice as previously described (18, 19). Mice were deeply anesthetized with isoflurane, the tail was removed, and animals were euthanized by cervical dislocation. Fascicles of tail tendons were isolated, finely minced, and placed in low-glucose DMEM (Thermo Fisher Scientific) containing 0.2% type II collagenase (Thermo Fisher Scientific) for 1 h at 37°C with constant agitation. An equal volume of growth medium (GM), which contains low-glucose DMEM with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% antibiotic antimycotic (AbAm; Thermo Fisher Scientific), was added to the digested tissue to inactivate the collagenase. Tenocytes were pelleted by centrifugation at 2500 g for 10 min, resuspended in GM and plated. All dishes or chamber slides used in the study were coated with type I collagen (Corning, Corning, NY, USA). Cells were maintained in humidified incubators at 37°C and 5% CO2. Passage 2–4 tenocytes were used in experiments.
In vitro IGF1 cell culture time course
Tenocytes were grown to 60% confluence in GM and switched to medium containing DMEM with 2% horse serum (Thermo Fisher Scientific) and 1% AbAm (Thermo Fisher Scientific), referred to as differentiation medium (DM) overnight. The next day, medium was replaced with DM containing 100 ng/ml of IGF1 (R&D Systems, Minneapolis, MN, USA) for a period of time ranging from 1, 2, 6, or 24 h. Cells that underwent the same procedure but did not receive IGF1 treatment are referred to as 0 h cells. At the end of the treatment period, RNA was isolated as described in RNA sequencing and gene expression.
In vitro signal transduction assays
Tenocytes were grown to 90% confluence in GM and serum starved in DMEM with 1% AbAm for 3 h, followed by treatment with either the MEK1/2 inhibitor PD98059 (50 μM; InvivoGen, San Diego, CA, USA) to block ERK1/2 activation or the PI3K inhibitor wortmannin (10 μM; InvivoGen) to inhibit protein kinase B (Akt) activation for 1 h. Cells were then treated with 100 ng/ml IGF1 for 5, 15, 30, or 60 min, scraped from their dishes, and homogenized in RIPA buffer (Thermo Fisher Scientific) containing 1% protease and phosphatase inhibitors (Thermo Fisher Scientific).
In vitro proliferation
Cell proliferation as measured by the uptake of bromodeoxyuridine (BrdU) was measured as previously described by Sugg et al. (18). Tenocytes at 50% confluence were incubated overnight in DM and then treated with DM containing 100 ng/ml of IGF1 (R&D Systems), PD98059 (50 μM; InvivoGen), or wortmannin (10 μM; InvivoGen). Following a 16-h overnight incubation, fresh medium was added along with 20 μM of BrdU (MilliporeSigma) for 1 h. After treatment with BrdU, cells were fixed in ice-cold methanol, permeabilized with 0.5% Triton X-100, and the BrdU epitope was exposed by denaturing DNA with 2 M HCl. Cells were then incubated with anti-BrdU antibodies (1:50, G3G4; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), and secondary antibodies were conjugated to AF555 (1:200, A-21127; Thermo Fisher Scientific) and DAPI (1:500) to identify nuclei. The number of BrdU+ nuclei as a fraction of total nuclei was quantified in 5 randomly selected fields of a single experiment from 4 independent experiments performed per group. Plates were imaged in an Evos FL microscope (Thermo Fisher Scientific) and quantified with ImageJ software.
In vitro surface sensing of translation labeling
Protein synthesis in cultured tenocytes was measured using a surface sensing of translation (SUnSET) technique, as modified from studies in C2C12 myoblast cells (20). Tenocytes were grown to 90% confluence in GM and serum starved in DMEM with 1% AbAm for 3 h, followed by treatment with either PD98059, wortmannin, or the protein synthesis inhibitor cycloheximide (R&D Systems) for 1 h. Cells were then treated for 30 min with 0.25 µM puromycin (MilliporeSigma), which is a tyrosyl-tRNA analog that is incorporated into newly translated proteins (21), followed by 100 ng/ml of IGF1 for 1 h. At the end of the treatment period, tenocytes were scraped from their dishes and homogenized in RIPA buffer (Thermo Fisher Scientific) containing 1% protease and phosphatase inhibitors (Thermo Fisher Scientific).
In vitro procollagen I labeling
To label procollagen I in proliferating or nonproliferating cells, tenocytes were cultured as described above, incubated with DM overnight, and then treated with either normal DM or DM containing 100 ng/ml of IGF1. The thymidine analog EdU was used in lieu of BrdU because the acid denaturing step required for BrdU detection degraded procollagen I epitopes. Following a 24-h incubation, fresh medium was added along with 10 µM of EdU (Thermo Fisher Scientific) for 1 h. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and EdU was detected using a Click-iT kit (Thermo Fisher Scientific). Cells were then incubated with antibodies against procollagen I (1:100, sc-30136; Santa Cruz Biotechnology, Dallas, TX, USA) and β-tubulin (1:200, ab6161; Abcam) and secondary antibodies conjugated to AF488 or AF647 (1:300, A11006 and A32733; Thermo Fisher Scientific). Slides were imaged in an LSM 880 laser scanning confocal microscope (Carl Zeiss GmbH, Oberkochen, Germany).
RNA sequencing and gene expression
RNA was isolated from tendons and cultured tenocytes as previously described (18, 22). Plantaris tendons or tenocytes were homogenized in Qiazol (Qiagen, Germantown, MD, USA), and RNA was purified with an miRNeasy Micro Kit (Qiagen) supplemented with DNase I (Qiagen). RNA concentration and quality were determined using a NanoDrop 2000 (Thermo Fisher Scientific) and a TapeStation 2200 (Agilent Technologies, Santa Clara, CA, USA).
For RNA sequencing (RNAseq), 250 ng (for whole tendons) or 500 ng (for culture cells) was delivered to the University of Michigan Sequencing Core for analysis. Sample concentrations were normalized and cDNA pools were created for each sample, and then they were subsequently tagged with a barcoded oligo adapter to allow for sample-specific resolution. Sequencing was carried out using an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) with 50 bp single end reads. Raw RNAseq data were quality checked using FastQC v.0.10.0 (Babraham Bioinformatics, Cambridge, United Kingdom). Alignment to the reference genome (mmu10; University of California, Santa Cruz, Santa Cruz, CA, USA), differential expression based on counts per million mapped reads, and postanalysis diagnostics were carried out using edgeR (23). A Benjamini-Hochberg false discovery rate procedure was applied to correct P values for multiple observations, and these false discovery rate–corrected P values are reported as q values. Sequencing data have been deposited to the NIH Gene Expression Omnibus (GSE131804; https://www.ncbi.nlm.nih.gov/geo/).
For quantitative PCR (qPCR), using iScript Reverse Transcription reagents (Bio-Rad, Hercules, CA, USA), RNA was reverse transcribed into cDNA that was amplified in a CFX96 real-time thermal cycler (Bio-Rad) using iTaq Universal SYBR Green Supermix (Bio-Rad). Target gene expression was normalized to the stable housekeeping gene cyclophilin D (Ppid) using the 2-ΔCt method. Cyclophilin D was selected as a housekeeping gene from RNAseq data and validated with qPCR. For cell culture experiments, relative copy number was calculated using the linear regression of efficiency method (24). Primer sequences are provided in Supplemental Data S1.
Biologic pathway analysis
Expression data from RNAseq measurements were imported into Ingenuity Pathway Analysis software (Qiagen) to assist in predicting cellular and molecular pathways and processes that were altered in response to manipulating IGF1 signaling in tendons.
Western blots
Western blots were performed as previously described (18, 25). Tendons and cell pellets were homogenized in ice-cold RIPA buffer (Thermo Fisher Scientific) supplemented with 1% protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). A BCA assay (Thermo Fisher Scientific) was used to determine protein concentration. Protein homogenates were diluted in Laemmli’s sample buffer, placed in boiling water for 2 min, and 20 μg of protein was separated on either 6 or 12% SDS-PAGE gels depending on the protein of interest. Proteins were transferred to nitrocellulose or PVDF membranes (Bio-Rad) using the Trans-Blot SD semidry transfer apparatus (Bio-Rad), blocked with 5% nonfat powdered milk in TTBS solution, and incubated with primary antibodies (1:1000; Cell Signaling Technology, Danvers, MA, USA) against phosphorylated (p)-IGF1RβY1135 (3918), IGF1Rβ (3025), p-IRS1S612 (2386), IRS1 (2382), p-AktT308 (13038), p-AktS473 (4060), Akt (4691), p-p70S6 kinase (p70S6K)T389 (9234), p-p70S6KT421/S424 (9204), p70S6K (2708), p-ERK1/2T202/Y204 (4370), ERK1/2 (4695); or primary antibodies from Santa Cruz Biotechnology (1:1000) against procollagen type I (sc-30136); or primary antibodies from Abcam (1:1000) against β-tubulin (ab6046), p-ELKS383 (ab34270), or ELK (ab32106); or primary antibodies from MilliporeSigma against pIRS1Y608 (1:1000) or puromycin (1:2000; 12D10). Following primary antibody incubation, membranes were rinsed and incubated with HRP-conjugated secondary antibodies (1:10,000; from either Abcam or Cell Signaling Technology). Proteins were detected using Clarity enhanced chemiluminescent reagents (Bio-Rad) and visualized using a digital chemiluminescent documentation system (Bio-Rad). Coomassie staining of membranes was performed to verify equal protein loading.
Statistics
Results are presented as means ± sd or means ± coefficient of variation (CV). Prism v.8.0 (GraphPad Software, La Jolla, CA, USA) was used to conduct statistical analyses for all data except RNAseq. A 2-way ANOVA followed by Fisher’s post hoc sorting (α = 0.05) evaluated the interaction between time after synergist ablation and IGF1R knockdown. For cell culture experiments, differences between groups were tested with an unpaired Student’s t test (α = 0.05) or a 1-way ANOVA followed by Fisher’s post hoc sorting (α = 0.05).
RESULTS
To study the role of IGF1 signaling in adult tendon hypertrophy, we treated mice with tamoxifen, induced a mechanical overload in plantaris tendons of Scx:IGF1R+ and Scx:IGF1RΔ mice, and analyzed tendons at either 7 or 14 d after creation of the growth stimulus (Fig. 1A–C). Tamoxifen treatment resulted in an over 90% reduction in IGF1R expression in Scx:IGF1RΔ mice compared with Scx:IGF1R+ mice at both 7 and 14 d (Fig. 1D). We then analyzed histologic changes in plantaris tendons. As previously observed in other models of synergist ablation-mediated mechanical overload, a neotendon tissue formed laterally around the original tendon toward the direction of the removed Achilles tendon (Fig. 2A–D). The neotendon matured and filled in with collagen over time; however, growth was blunted in the Scx:IGF1RΔ mice (Fig. 2A–D). The total tendon CSA was not different between groups at 7 d, but by 14 d the total CSA of Scx:IGF1R+ mice was twice as large as Scx:IGF1RΔ mice (Fig. 2D). This occurred due to a greater expansion of the neotendon of Scx:IGF1R+ mice over time, whereas Scx:IGF1RΔ mice displayed no change between 7 and 14 d (Fig. 2C). No differences in cell density were observed in the original tendon, neotendon, or total tendon across time or genotype (Fig. 2E–G); however, the percentage of proliferating cells was 2-fold greater in the neotendon of Scx:IGF1R+ mice compared with Scx:IGF1RΔ mice (Fig. 2H–J).
Figure 2.

Effect of IGF1R deletion on tendon growth and cell proliferation. A) Representative histology of cross-sections from Scx:IGF1R+ and Scx:IGF1RΔ mice obtained at either 7 or 14 d after mechanical overload demonstrating general morphology, cell density, and abundance of proliferating cells. The original tendon (OT) and neotendon (NT) are indicated by the hashed line. ECM, green; nuclei, blue; Ki67 (proliferating cells), red. Scale bar for all sections is 200 µm. B–D) Area measurements of tendons, with respect to the original tendon (B), neotendon (C), and total tendon (D). E–G) Cell density measurements of tendons, with respect to the original tendon (E), neotendon (F), and total tendon (G). H–J) Cell proliferation measurements, with respect to the original tendon (H), neotendon (I), and total tendon (J). Values are means ± sd, with individual data points presented. Differences tested with a 2-way ANOVA: significantly different (P < 0.05) from 7D Scx:IGF1R+ mice (a); significantly different (P < 0.05) from 7D Scx:IGF1RΔ mice (b); and significantly different (P < 0.05) from 14D Scx:IGF1R+ mice (c); n ≥ 3 mice/group. 7D, 7 d; 14D 14 d.
We then sought to identify changes in the transcriptome of plantaris tendons of Scx:IGF1R+ mice and Scx:IGF1RΔ mice in response to mechanical overload using RNAseq. At 7 d, there were 1108 transcripts that were at least 50% differentially regulated between genotypes, but there were only 159 transcripts at 14 d (Fig. 3A, B). Pathway enrichment analysis was performed to evaluate cellular functions, and signaling pathways predicted to be different between groups over time. Several of the pathways identified were involved with growth and differentiation, cytoskeletal signaling, and ECM production (Table 1). We selected several genes related to these processes to further explore and report in Fig. 3C–E, and we also performed qPCR validation of a subset of relevant genes (Table 2). All gene names have been listed in Table 3. Compared to nonoverloaded controls, several growth factors and signaling molecules including Adam12, Bmp1, Ctsd, Igf1, and Pappa were up-regulated in all overloaded groups, whereas Bmp6, Fgf2, Inhbb, and Vegfa were down-regulated at all time points (Fig. 3C). There was also an up-regulation in Bmp1, Bmp6, Igf1, Inhba, Pdgfa, Pdgfb, Tgfb1, Tgfb2, Wnt5b, and Wnt9a in 7 d Scx:IGF1RΔ mice compared with 7 d Scx:IGF1R+ mice, and by 14 d Fgf2 and Tgfb2 were significantly higher in Scx:IGF1RΔ mice (Fig. 3C). For genes involved in the liberation of IGF1 from IGFBPs, C1s2 was up-regulated at 7 d and Ctsd was up-regulated at 14 d in Scx:IGF1RΔ mice, whereas no differences in Pappa were observed between genotypes (Fig. 3C). With regards to cell proliferation, and tenocyte specification and differentiation, Acta2, Ccna2, Ccnb1, Ccne1, Cdk1, Cdk2, Cdk4, Cdk6, Cfi1, Itgav, Mki67, Pcna, Ptk2, Snai1, Trp53, and Vim were up-regulated in all overloaded groups (Fig. 3D). Cdkn1b, Itgb3, Rerg, S100a4, Scx, Snai1, and Yap1 were up-regulated in 7-d Scx:IGF1RΔ mice compared with 7-d Scx:IGF1R+ mice, whereas Acta2, Cfl1, Itgav, Mcm6, and Pcna were down-regulated (Fig. 3D). At 14 d, Rerg and Scx were up-regulated in Scx:IGF1RΔ mice (Fig. 3D). For ECM genes, Bgn, Co1a1, Co1a2, Col3a1, Col5a1, Col6a1, Col14a1, Fn1, Mmp2, Mmp3, Mmp14, Postn, Smoc2, Timp1, Tnc, Vcan, and Wisp1 were up-regulated in overloaded groups, whereas Comp was down-regulated (Fig. 3E). In Scx:IGF1RΔ mice, Bgn, Col1a1, Col1a2, Col3a1, Col5a1, Comp, Fn1, Mmp2, and Mmp14 were up-regulated at 7 d and Col5a1 at 14 d, whereas Mgp was down-regulated at 7 and 14 d compared with Scx:IGF1R+ mice (Fig. 3E).
Figure 3.

Whole tendon RNAseq. A, B) Volcano plot demonstrating log2 fold change (FC) and q values of all measured transcripts in plantaris tendons from Scx:IGF1R+ and Scx:IGF1RΔ mice at 7 (A) and 14 d (B) after surgical overload. Values >10 are shown directly on the top or side border of the graph. Genes with a >1.5-fold up-regulation in Scx:IGF1RΔ mice (log2 fold change >0.585) and q value < 0.05 (−log10 q > 1.301) are shown in red. Genes with a >1.5-fold down-regulation in Scx:IGF1RΔ mice (log2 fold change <−0.585) and q value < 0.05 (−log10 q > 1.301) are shown in blue. All other genes are shown in black. C–E) Heat maps demonstrating the log2 fold change in selected genes from RNAseq that are growth factors, cytokines, or genes involved with activating extracellular ligands (C); involved in cell proliferation and tenocyte specification and differentiation (D); or components or regulators of the ECM (E). The fold change value is displayed for each group relative to nonoverloaded Scx:IGF1R+ (control) mice. Differences between groups tested using edgeR: different (q < 0.05) in the respective overloaded group to nonoverloaded Scx:IGF1R+ mice (a); different (q < 0.05) between 7D Scx:IGF1R+ and 7D Scx:IGF1RΔ mice (b); and different (q < 0.05) between 14D Scx:IGF1R+ and 14D Scx:IGF1RΔ mice (c); n = 4 mice per group. 7D, 7 d; 14D, 14 d.
TABLE 1.
Gene enrichment analysis
| Pathway or function | 7-d Scx:IGF1R+ to Ctrl | 7-d Scx:IGF1RΔ to Ctrl | 7-d Scx:IGF1R+ to 7-d Scx:IGF1RΔ | 14-d Scx:IGF1R+ to Ctrl | 14-d Scx:IGF1RΔ to Ctrl | 14-d Scx:IGF1R+ to 14-d Scx:IGF1RΔ |
|---|---|---|---|---|---|---|
| Actin cytoskeleton signaling | 2.51E−11 | 3.98E−12 | 6.92E−06 | 1.70E−08 | 1.32E−07 | ND |
| Apoptosis | 6.17E−70 | 2.87E−59 | 5.05E−19 | 4.57E−58 | 4.09E−44 | 4.08E−08 |
| BMP signaling | 1.55E−04 | 7.59E−03 | 2.19E−02 | 7.94E−03 | 8.71E−04 | ND |
| Chromosomal replication | 3.55E−09 | 1.26E−05 | ND | 6.92E−04 | 3.63E−02 | ND |
| Cell cycle regulation | 2.82E−04 | 3.02E−03 | ND | 1.51E−03 | 3.63E−03 | ND |
| Connective tissue growth | 1.70E−34 | 8.73E−28 | 3.16E−09 | 5.45E−28 | 6.74E−22 | 2.01E−06 |
| EGF signaling | 2.19E−02 | 3.09E−02 | ND | 4.07E−02 | ND | ND |
| Ephrin A signaling | 8.51E−05 | 1.23E−03 | 2.34E−02 | 4.68E−03 | 1.70E−02 | 2.40E−02 |
| ERK/MAPK signaling | 1.12E−05 | 2.09E−04 | 9.12E−03 | 9.55E−03 | 1.82E−03 | ND |
| FAK signaling | 8.91E−06 | 6.03E−04 | 8.51E−03 | 3.63E−02 | ND | ND |
| Fatty acid β-oxidation | ND | ND | ND | 1.66E−02 | 2.09E−02 | ND |
| FGF signaling | 7.94E−04 | 2.63E−04 | 6.92E−03 | 2.40E−03 | 9.33E−03 | ND |
| Inhibition of MMPs | 7.24E−06 | 1.35E−05 | 2.51E−02 | 1.62E−08 | 3.16E−11 | 6.92E−04 |
| Integrin signaling | 1.00E−11 | 8.13E−10 | 2.51E−03 | 8.91E−06 | 1.26E−04 | ND |
| Organization of ECM | 1.82E−26 | 1.75E−27 | 3.00E−16 | 9.47E−29 | 8.64E−29 | 3.37E−11 |
| p53 signaling | 1.48E−06 | 7.76E−06 | ND | 5.89E−05 | 8.13E−05 | ND |
| p70S6K signaling | 3.72E−04 | 4.37E−03 | ND | 2.00E−02 | ND | ND |
| PAK signaling | 1.95E−04 | 4.37E−04 | 1.00E−02 | 4.57E−02 | 3.98E−02 | ND |
| PI3K signaling | 2.40E−06 | 4.90E−04 | ND | 3.98E−02 | ND | ND |
| Rac signaling | 1.70E−06 | 3.16E−04 | ND | 1.51E−02 | 4.79E−02 | ND |
| Signaling by Rho GTPases | 5.37E−09 | 1.48E−06 | ND | 2.24E−04 | 8.51E−03 | ND |
| Sonic Hedgehog signaling | 8.51E−03 | 2.34E−03 | ND | 3.09E−03 | 1.23E−02 | ND |
| TGF-β signaling | 1.20E−04 | 1.38E−02 | ND | 6.92E−03 | 3.24E−03 | ND |
The P values of select affected or differentially regulated pathways or biologic functions that were identified using Ingenuity Pathway Analysis. Ctrl, control; ND, not significantly different.
TABLE 2.
Gene expression data
| Gene | 7-d Scx:IGF1R+ | 7-d Scx:IGF1RΔ | 14-d Scx:IGF1R+ | 14-d Scx:IGF1RΔ |
|---|---|---|---|---|
| CCNA2 | 8.19 ± 1.90 | 4.48 ± 0.66a | 3.88 ± 1.27a | 4.38 ± 0.97a |
| CCNB1 | 3.09 ± 0.43 | 1.81 ± 0.18a | 1.19 ± 0.60a | 1.43 ± 0.36a |
| CCND1 | 3.82 ± 1.40 | 3.40 ± 0.64 | 3.00 ± 0.92 | 3.36 ± 0.53 |
| CCNE1 | 0.29 ± 0.04 | 0.23 ± 0.15 | 0.19 ± 0.02 | 0.11 ± 0.03a |
| CDKN1A | 2.07 ± 0.39 | 3.04 ± 0.85a | 2.23 ± 0.46b | 1.74 ± 0.34b |
| COL1A1 | 224 ± 72.4 | 372 ± 46.7a | 482 ± 133a | 561 ± 74.5a,b |
| COL1A2 | 889 ± 316 | 1450 ± 159a | 1890 ± 551a | 2270 ± 61.4a,b |
| COL3A1 | 1308 ± 346 | 1490 ± 324 | 2070 ± 640a | 2250 ± 472a,b |
| COL4A1 | 28.8 ± 3.93 | 35.2 ± 7.87 | 42.4 ± 11.1a | 46.5 ± 8.06a,b |
| COL5A1 | 4.66 ± 1.47 | 6.56 ± 0.84 | 7.60 ± 1.13a,c | 9.47 ± 1.42a,b |
| COL6A1 | 14.2 ± 3.53 | 32.0 ± 4.57a | 34.1 ± 11.8a | 35.3 ± 5.81a |
| EGR1 | 2.36 ± 0.97 | 1.75 ± 0.88 | 1.16 ± 0.50a | 0.93 ± 0.45a |
| EGR2 | 0.17 ± 0.04 | 0.15 ± 0.07 | 0.10 ± 0.05a | 0.05 ± 0.01a,b |
| HAS2 | 2.19 ± 0.34 | 0.79 ± 0.11a | 1.20 ± 0.42a | 0.93 ± 0.23a |
| MKI67 | 1.19 ± 0.15 | 1.62 ± 0.26 | 0.82 ± 0.36a,b | 1.03 ± 0.36a,b |
| MKX | 0.47 ± 0.12 | 0.52 ± 0.13 | 0.68 ± 0.09a | 0.78 ± 0.14a,b |
| MMP2 | 5.37 ± 1.11 | 7.95 ± 1.53a | 11.2 ± 2.17a,b | 11.4 ± 1.57a,b |
| MMP14 | 3.24 ± 0.63 | 5.12 ± 0.44a | 5.36 ± 0.89a | 4.51 ± 0.69a |
| PCNA | 22.2 ± 4.63 | 14.3 ± 3.84a | 11.9 ± 1.48a | 13.8 ± 2.61a |
| SCX | 9.97 ± 2.25 | 98.9 ± 15.6a | 19.3 ± 7.62c | 265 ± 107a,b |
| TNMD | 30.0 ± 12.9 | 36.0 ± 3.98 | 191 ± 80.0a,b | 150 ± 37.5a,b |
Expression of genes as measured by qPCR. Values are means ± sd.
Differences tested with a 2-way ANOVA (α = 0.05): adifferent (P < 0.05) from 7-d Scx:IGF1+; bdifferent (P < 0.05) from 7-d Scx:IGF1Δ; cdifferent (P < 0.05) from 14-d Scx:IGF1+.
TABLE 3.
List of gene names
| Gene abbreviation | Gene name | Gene abbreviation | Gene name |
| Acta2 | Actin α 2 |
Itgb3 | Integrin subunit β 3 |
| Adam12 | ADAM metallopeptidase domain 12 | Mcm6 | Minichromosome maintenance complex component 6 |
| Bgn | Biglycan | Mgp | Matrix Gla protein |
| Bmp1 | Bone morphogenetic protein 1 | Mki67 | Marker of proliferation Ki-67 |
| Bmp6 | Bone morphogenetic protein 6 | Mmp2 | Matrix metallopeptidase 2 |
| C1s2 | Complement C1 2 | Mmp3 | Matrix metallopeptidase 3 |
| Ccna2 | Cyclin A2 | Mmp14 | Matrix metallopeptidase 14 |
| Ccnb1 | Cyclin B1 | Pappa | IGF-dependent IGFBP-4 protease |
| Ccne1 | Cyclin E1 | Pcna | Proliferating cell nuclear antigen |
| Cdk1 | Cyclin dependent kinase 1 | Pdgfa | Platelet derived growth factor subunit A |
| Cdk2 | Cyclin dependent kinase 2 | Pdgfb | Platelet derived growth factor subunit B |
| Cdk4 | Cyclin dependent kinase 4 | Pdgfra | Platelet derived growth factor receptor α |
| Cdk6 | Cyclin dependent kinase 6 | Pdgfrb | Platelet derived growth factor receptor β |
| Cdkn1b | Cyclin dependent kinase inhibitor 1B | Postn | Periostin |
| Cfi1 | Cell division cycle 14A | Ptk2 | Protein tyrosine kinase 2 |
| Col1a1 | Collagen type I α 1 chain | Rerg | RAS like estrogen regulated growth inhibitor |
| Col1a2 | Collagen type I α 2 chain | S100a4 | S100 calcium binding protein A4 |
| Col4a1 | Collagen type IV α 1 chain | Smoc2 | Secreted modular calcium-binding protein 2 |
| Col3a1 | Collagen Type III α 1 chain | Snai1 | Snail family transcriptional repressor 1 |
| Col5a1 | Collagen type V α 1 chain | Spp1 | Osteopontin |
| Col6a1 | Collagen type VI α 1 chain | Tgfb1 | Transforming growth factor β 1 |
| Col14a1 | Collagen type XIV α 1 chain | Tgfb2 | Transforming growth factor β 2 |
| Comp | Cartilage oligomeric matrix protein | Timp1 | Tissue inhibitor of metalloproteinases 1 |
| Ctgf | Connective tissue growth factor | Tnc | Tenascin C |
| Ctsd | Cathepsin D | Trp53 | Tumor protein P53 |
| Cyr61 | Cellular communication network factor 1 | Vcan | Versican |
| Egf | Epidermal growth factor | Vegfa | Vascular endothelial growth factor A |
| Egr1 | Early growth response 1 | Vim | Vimentin |
| Egf2 | Epidermal growth factor 2 | Wisp1 | Cellular communication network factor 4 |
| Fgf2 | Fibroblast growth factor 2 | Wnt5b | Wnt family member 5B |
| Fn1 | Fibronectin 1 | Wnt9a | Wnt family member 9A |
| Has2 | Hyaluronan synthase 2 | Thbs4 | Thrombospondin 4 |
| Igf1 | Insulin-like growth factor 1 | Tnc | Tenascin C |
| Igf1r | Insulin-like growth factor receptor 1 | Tnmd | Tenomodulin |
| Inhbb | Inhibin subunit β B | Yap1 | Yes associated protein 1 |
| Itgav | Integrin subunit α V |
Based on the in vivo results that suggested a role for IGF1 in controlling cell cycle behavior and ECM synthesis involving PI3K/Akt and ERK signaling, we took a reductionist approach to evaluate IGF1 signaling in vitro using cultured primary tenocytes. We observed that IGF1 treatment resulted in IGF1RY1135 phosphorylation, leading to early downstream activation of IRS1Y608, ERK1/2T202/Y204, AktT308, AktS473, p70S6KT389, and p70S6KT421/S424 (Fig. 4A). Whereas ERK1/2T202/Y204, AktT308, and AktS473 phosphorylation decreased following 5 min of IGF1 treatment, p70S6KT389 and p70S6KT421/S424 activation was sustained from 15 through 60 min after treatment, and phosphorylation of ELK1S383 was detected at 30 and 60 min (Fig. 4A). Phosphorylation of the inhibitory IRS1S612 site was detected at 30 and 60 min following IGF1 treatment (Fig. 4A).
Figure 4.

Signaling protein activation and RNAseq in cultured tenocytes treated with IGF1. A) Representative Western blots for p-IGF1RY1135, total IGF1R, IRS1Y608, IRS1S612, total IRS1, p-ERK1/2T202/Y204, total ERK1/2, p-ELKS383, total ELK, p-AktT308, p-AktS473, total Akt, p-p70S6KT389, p-p70S6KT421/S424, and total p70S6K in cultured tenocytes that were untreated (0 min) or treated with IGF1 for 5, 15, 30, or 60 min. B–E) RNAseq analysis of untreated tenocytes (0 h) or tenocytes treated with IGF1 for 1, 2, 6, or 24 h. The fold change (FC) value displayed for each group is relative to tenocytes that were not treated with IGF1 (0 h). B) Number of genes that were significantly up-regulated (red) with a >1.5-fold up-regulation and q < 0.05, and significantly down-regulated (blue) with a >1.5-fold up-regulation and q < 0.05. C–E) Heat maps demonstrating the log2 fold change in selected genes from RNAseq that are growth factors, cytokines, or genes involved with activating extracellular ligands (C); involved in cell proliferation and tenocyte specification and differentiation (D); or components or regulators of the ECM (E). Differences between groups tested using edgeR: different (q < 0.05) from 0 h IGF1 (a); different (q < 0.05) from 1 h IGF1 (b); different (q < 0.05) from 2 h IGF1 (c); and different (q < 0.05) from 6 h IGF1 (d).
To further explore the effect of IGF1 treatment on tenocytes, we performed RNAseq using cultured tenocytes that were not treated with IGF1, or treated with IGF1 for 1, 2, 6, or 24 h. At 1 h, over 400 transcripts were at least 50% differentially regulated and q < 0.05, whereas over 1000 genes were similarly affected at 2, 6, and 24 h (Fig. 4B). We used the same panel of genes explored in whole tendons for further analysis in cultured tenocytes. Across these transcripts, Egf, Mmp2, Tnmd, Trp53, Vim, and Yap1 were not affected by IGF1 treatment (Fig. 4C–E). For the growth factors and signaling molecules that were differentially regulated in whole tendons of Scx:IGF1R+ and Scx:IGF1RΔ mice, there was an induction of Fgf2, Pdgfa, Pdgfb, Tgfb1, and Wnt9a by 2 h, whereas Igf1 was down-regulated compared with untreated cells (Fig. 4C). By 24 h, with the exception of Bmp1 and Tgfb2, nearly all other growth factors and signaling molecules that were differentially regulated in tendons were down-regulated in cultured tenocytes (Fig. 4C). Cfl1, Itgb3, Pcna, Scx, and Snai1 demonstrated an early up-regulation in response to IGF1 treatment, whereas Acta2 and Cdkn1b were initially reduced (Fig. 4D). By 24 h Mcm6, Pcna, and S100a4 were up-regulated and Acta2 was suppressed (Fig. 4D). The ECM genes Mgp, Mmp3, Spp1, Timp1, and Tnc were up-regulated by 2 h, whereas Bgn, Col1a1, Col1a2, Ctgf, Cyr61, and Smoc2 were down-regulated (Fig. 4E). At 24 h, Col3a1, Col5a1, Has2, Mgp, and Mmp14 were induced, and Bgn and Comp were reduced in IGF1-treated tenocytes.
As we observed differences in cell proliferation in vivo and that IGF1 affected the expression of cell cycle control genes in vitro, we next determined if IGF1 directly impacts the proliferation of cultured tenocytes, and the role of the PI3K/Akt and ERK pathways in this process. We sought to validate the findings for Mki67 expression in RNAseq data, and using qPCR we observed a slight >1.5-fold transient induction at 2 h after IGF1 treatment, and a near 8-fold induction in Mki67 at 24 h (Fig. 5A). In support of these findings, we observed a 2.5-fold increase in the number of BrdU+ tenocytes in response to IGF1 treatment (Fig. 5B). Inhibiting the PI3K/Akt pathway with wortmannin did not impact IGF1-mediated tenocyte proliferation or Mki67 expression, whereas blocking ERK1/2T202/Y204 with PD98059 reduced cell proliferation and Mki67 expression (Fig. 5B, C).
Figure 5.

Proliferation in tenocytes treated with IGF1. A) Mki67 expression in untreated tenocytes (0 h) or in tenocytes treated with IGF1 for 1, 2, 6, or 24 h, measured with qPCR. Differences between groups tested using a 1-way ANOVA: different (P < 0.05) from 0 h (a); different (P < 0.05) from 1 h (b); different (P < 0.05) from 2 h (c); and different (P < 0.05) from 6 h (d). B) The abundance of proliferating tenocytes (expressed as a percentage of total tenocytes) and Mki67 expression (C) in untreated cells or in cells treated with IGF1, IGF1 and wortmannin, or IGF1 and PD98059 for 2 h. Inset (B) is a representative image demonstrating BrdU+ nuclei (red) and total nuclei (blue). Scale bar, 30 µm. Differences tested with a 1-way ANOVA: significantly different (P < 0.05) from control (a); significantly different (P < 0.05) from IGF1 (b); and significantly different (P < 0.05) from IGF1 and wortmannin (c). Values are means ± CV; n ≥ 4 replicates/group.
Based on the differences in tendon size between Scx:IGF1R+ and Scx:IGF1RΔ mice, the differential expression of Col1a1 and Col1a2 in whole tendons, and the cell culture RNAseq data, we sought to determine whether IGF1 directly induces type I collagen expression in tenocytes. Using qPCR, there were no differences in Col1a1 and Col1a2 expression following treatment with IGF1 (Fig. 6A, B). We also did not observe dose- or time-dependent effects of IGF1 on type I collagen protein abundance in tenocytes, although we did note that cells that are in the S phase, as indicated by the presence of EdU+ nuclei, reduce type I collagen production (Fig. 6C–E).
Figure 6.

Type I collagen production in tenocytes treated with IGF1. Col1a1 (A) and Col1a2 (B) expression in untreated tenocytes (0 h) or in tenocytes treated with IGF1 for 1, 2, 6, or 24 h, measured with qPCR. Values are means ± CV. Differences between groups tested using a 1-way ANOVA found no significant differences. C) Representative immunocytochemistry of cultured tenocytes treated with 0 or 100 ng/ml of IGF1 for 24 h. β-tubulin, green; EdU, red; Procol Iα1, magenta. Scale bar, 50 µm in all images. D) Representative Western blot for procollagen type Iα1 (Procol Iα1) in untreated tenocytes (0 h) or tenocytes treated with 100 ng/ml of IGF1 for 6 or 24 h. E) Representative Western blot for Procol Iα1 in tenocytes treated with 0, 10, or 100 ng/ml of IGF1 for 24 h.
Finally, we sought to determine the effect of IGF1 signaling on general protein synthesis. Using the Sunset method in which the tyrosyl-tRNA analog puromycin is incorporated into newly synthesized proteins, we treated tenocytes with IGF1 and observed a 2-fold increase in puromycin incorporation. The broad-spectrum translation inhibitor cycloheximide as well as wortmannin blocked IGF1-mediated protein synthesis in tenocytes, but surprisingly ERK1/2 inhibition resulted in a nearly 6-fold increase in protein synthesis (Fig. 7A, B). To investigate this phenomenon in more detail, we probed for the presence of permissive and inhibitory phosphorylation sites on components of the PI3K/Akt and ERK1/2 signaling pathways. As anticipated PD98059 blocked ERK1/2T202/Y204 phosphorylation as well as phosphorylation of the downstream transcription factor ELK1S383, and wortmannin inhibited AktT308 phosphorylation (Fig. 7C). However, inhibition of ERK1/2 surprisingly increased AktT308 phosphorylation independent of IRS1Y608 phosphorylation, suggesting ERK1/2 is acting to inhibit protein synthesis downstream of IRS1 (Fig. 7C). Further, wortmannin decreased ERK1/2T202/Y204 and ELK1S383 phosphorylation and abolished phosphorylation of the inhibitory IRS1S612 site, leading to increased phosphorylation of the permissive IRS1Y608 site (Fig. 7C). Based on these findings, we propose a model in which IGF1 regulates tenocyte proliferation and protein synthesis through coordinated and intersecting actions of the PI3K/Akt and ERK1/2 pathways (Fig. 8).
Figure 7.

Protein synthesis in tenocytes treated with IGF1. Representative Western blot for proteins that have incorporated puromycin (A) and quantification of band densitometry for tenocytes (B) that were untreated or were treated with IGF1, IGF1 and cycloheximide, IGF1 and wortmannin, or IGF1 and PD98059. C) Representative Western blots for p-ERK1/2T202/Y204, total ERK1/2, p-ELKS383, total ELK, IRS1Y608, IRS1S612, total IRS1, p-AktT308, and total Akt in untreated cells or in cells treated with IGF1, IGF1 and wortmannin, or IGF1 and PD98059. Differences tested with a 1-way ANOVA: significantly different (P < 0.05) from −IGF1 controls. Coomassie stained membranes are shown as total protein loading controls. Values are means ± CV; n ≥ 4 replicates/group.
Figure 8.
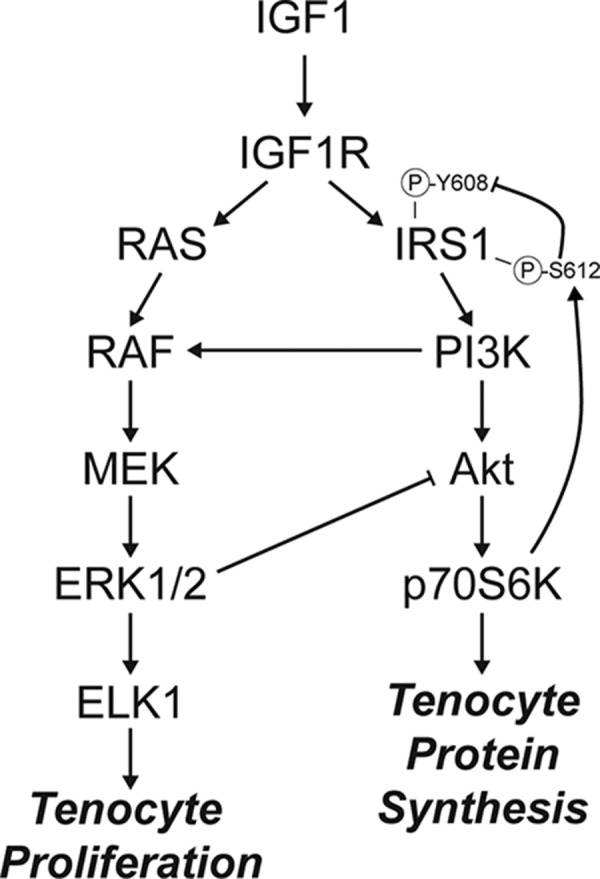
IGF1 signaling in tenocytes. Diagram of the proposed regulation of tenocyte proliferation and protein synthesis by IGF1.
DISCUSSION
IGF1 is known to play a critical role in the growth and adaptation of various musculoskeletal tissues (including skeletal muscle, bone, and cartilage), but less is known about tendon. To study tendon growth, we used the synergist ablation technique, in which the Achilles tendon is surgically removed resulting in a supraphysiological growth stimulus to the synergist plantaris tendon and muscle (6, 22, 26–29). A neotendon matrix consisting of immature collagen and other ECM proteins forms around the plantaris tendon, and over a 1-mo period this ECM matures and resembles the original tendon matrix (22). The neotendon is enriched in a population of Scx-expressing tenocytes that appear to arise from CD146+ progenitor cells in the perivascular region of tendons (6, 22, 30). The increase in tenocytes in this model appears to come about due to recruitment of CD146+ progenitor cells into the tenogenic lineage and expansion of recruited tenocytes prior to terminal differentiation (6, 22). By applying the synergist ablation model in a line of mice in which IGF1R was deleted in tenocytes, we demonstrated that IGF1 signaling is required for the proper growth of tendons in adult animals. Compared with Scx:IGF1R+ mice, the plantaris tendons of Scx:IGF1RΔ mice had reduced CSAs and numbers of proliferating cells, and displayed alterations in growth factors, signaling molecules, cell cycle control genes, and ECM components. Using in vitro studies, we observed that IGF1 promotes a delayed increase in cell proliferation and directs protein synthesis, but surprisingly did not directly regulate type I collagen expression. IGF1 also induced the expression of Egr1, Egr2, and Scx, which are transcription factors that direct the expression of genes that are important for tendon development and growth (31, 32), as well as Snai1, which is involved in fibroblast-mediated tissue growth (33). Furthermore, we identified that both the PI3K/Akt and ERK pathways are activated downstream of IGF1 and work in a coordinated manner to regulate cell proliferation and protein synthesis. These results have important implications for our understanding of the basic biology of tendon growth and may inform the treatment of tendinopathies.
Numerous growth factors have been studied in the context of tenocyte proliferation in vitro. In the current study, we observed a minor increase in markers of cell proliferation within 2 h of IGF1 treatment but did not see notable increases until 24 h. In mechanically stimulated tendons, cell proliferation was reduced at 7 d in Scx:IGF1RΔ mice. The CSA of tendons was similar between mice at 7 d, but there was no appreciable growth in the tendons of Scx:IGF1RΔ mice between 7 and 14 d, whereas the tendons of Scx:IGF1R+ mice continued to grow thereafter. This suggests that IGF1 is regulating tenocyte proliferation in an indirect manner, perhaps through controlling the expression of other growth factors and signaling molecules that directly regulate cell cycle machinery. TGF-β1 signals through the TGF-βRII and TGF-βRI receptors to activate the Smad2/3 and MAPK signaling pathways in tenocytes (1), and treating tenocytes with recombinant TGF-β1 increases tenocyte proliferation (34, 35). PDGFa and PDGFb, which are activated by mechanical loading and signal through the PDGFRα and PDGFRβ receptors that are members of the RTK family, also promote tenocyte proliferation and are required for proper tendon growth (18, 36). Related to PDGFa and PDGFb, FGF2 induces fibroblast growth and proliferation also through RTK signaling, and in human renal fibroblasts, treatment with TGF-β1 increased cell proliferation through induction of FGF2 expression (37). In our data, the expression of TGFβ1, PDGFa, PDGFb, and FGF2 was up-regulated by 2 h following IGF1 treatment in cultured tenocytes, which corresponded to modest increases in Fos, Jun, Mki67, Pcna, cyclin E (Ccne1), Cdk2, and Cdk6, along with a decrease in p27 (Cdkn1b). By 24 h, Bmp6 which inhibits cell proliferation (38, 39) was down-regulated, along with an up-regulation in Fos, Jun, Mki67, and Pcna. Several cyclins (Ccna2, Ccnb1, and Ccne1) and cyclin-dependent kinases (Cdk1, Cdk2, Cdk4, and Cdk6) were also up-regulated, and a down-regulation in p27 was observed. At the whole tissue level, Bmp6 was elevated and Pcna was decreased at d 7 in Scx:IGF1RΔ mice compared with Scx:IGF1R+ mice, and several other growth factors and signaling molecules with direct roles in regulating cell proliferation were also differentially regulated. Therefore, it is likely that IGF1 chiefly acts in an indirect manner to regulate tenocyte proliferation through the regulation of other growth factors that act locally. The delayed effect of IGF1 on inducing tenocyte proliferation may explain why the neotendon areas were not different between Scx:IGF1R+ and Scx:IGF1RΔ mice at d 7, but at d 14 the neotendon of the Scx:IGF1R+ mice was 2-fold larger than Scx:IGF1RΔ mice. There may also be an effect of IGF1 on recruitment of progenitor cells into the tenogenic lineage; however, we suspect that once progenitor cells express Scx they are likely committed to tenogenesis. Additionally, IGF1 is known to have an antiapoptotic role in numerous cell types (40), which might explain why altered cell proliferation at d 7 does not translate to differential cell density at d 14.
In addition to reducing cell proliferation, the loss of IGF1R in tenocytes resulted in smaller tendons of mechanically loaded mice. Because type I collagen is the major constituent of the tendon ECM, and previous studies have demonstrated that direct injection of IGF1 into tendons increases collagen synthesis (41), and patients with acromegaly that have elevated levels of IGF1 also demonstrate an up-regulation of Col1a1 in their tendons (42), we sought to determine whether IGF1 could directly regulate collagen synthesis in tenocytes. Based on these studies, we anticipated that IGF1 would directly induce type I collagen expression and that Scx:IGF1RΔ mice would have reduced expression in their tendons in response to mechanical overload. However, Col1a1 and Col1a2, as well as Col3a1 and Col5a1, were up-regulated in Scx:IGF1RΔ mice compared with Scx:IGF1R+ mice, and treatment of tenocytes with IGF1 did not induce Col1a1 or Col1a2 expression and resulted in no change in type I procollagen protein abundance. These results indicate that IGF1 does not directly induce type I collagen gene expression, nor does it increase type I collagen translation. In support of our observations of a delayed growth defect in tendons of Scx:IGF1RΔ mice, 3-dimensional tissue engineered tendon constructs treated with IGF1 demonstrated increased size and hydroxyproline content, but this increase was not observed until at least 2 wk after beginning treatment with IGF1 (15). Numerous other genes that encode minor but important proteins that constitute or modify the ECM such as Ctgf, Fn, Mmp2, Mmp3, Mmp14, Postn, Smoc2, Spp1, Thbs4, Timp1, Tnc, Tnmd, Vcan, and Wisp1 were induced by mechanical loading of tendons, and Ctgf, Cyr61, Has2, Postn, Spp1, Thbs4, Timp1, Tnc, and Vcan appeared to be directly or indirectly regulated by IGF1 in cultured tenocytes. Although type I collagen is the major protein in tendon ECM, mature collagen rich matrices require the presence of proteoglycans and matricellular proteins for proper construction and assembly (43), and this may explain differences in CSA observed at d 14.
To further explore the difference in size between tendons of Scx:IGF1R+ and Scx:IGF1RΔ mice, we determined whether IGF1 regulated general protein synthesis. IGF1 is known to activate the PI3K/Akt pathway, which increases protein synthesis in skeletal muscle (1), and local IGF1 can increase the protein fractional synthesis rate in tendons of elderly men and patients with Ehlers-Danlos syndrome (13, 44). Using the SUnSET technique (20, 21), we observed that treating tenocytes with IGF1 doubled protein synthesis rates, and blocking Akt phosphorylation returned protein synthesis levels to baseline. Based on these findings and our observations of IGF1 and type I collagen synthesis, we propose that the larger tendons in the Scx:IGF1R+ mice occurred in part due to an increase in overall protein synthesis compared with Scx:IGF1RΔ mice, rather than a direct increase in collagen production. There are also other signaling molecules regulated by IGF1 or mechano-sensing pathways in whole tissue that influence collagen production. Further, the regulation of Col1a1 and Col1a2 translation is a complex process. Although translation requires that active ribosomes bind to Col1a1 and Col1a2 mRNAs, translation initiation also requires the interaction of regulatory cofactors with the 5′ stem loop and 3′ UTR of the transcripts and the binding of these transcripts to nonmuscle myosin and association with vimentin filaments (45). Combined, our results indicate that IGF1 signaling contributes to protein synthesis in tenocytes through an Akt-dependent mechanism, but IGF1 does not appear sufficient to induce type I collagen translation and likely works in a coordinated fashion with other signaling pathways to regulate this process.
ERK is a well-known signaling pathway that is activated in response to mechanical loading, often through the integrin αV/β3/FAK pathway (46). In whole tendons, integrin αV (Itgav) and β3 (Itgab3) were induced in response to synergist ablation, and RNAseq pathway enrichment analysis predicted activation of integrin, FAK, and ERK pathways, with a differential response between Scx:IGF1R+ and Scx:IGF1RΔ mice. IGF1 is known to work in coordination with integrin αV/β3/FAK signal transduction, with ERK as a common and important downstream effector kinase for both pathways (47). Although mechanical loading is known to increase both ERK and PI3K/Akt activation in tendons (18, 48–50), the role of IGF1 in modulating ERK and PI3K/Akt in tendons was not known. In this study, we observed that IGF1 activates both ERK and PI3K pathways in tenocytes, and that ERK activation is required for the IGF1-mediated increase in tenocyte proliferation. ELK1 is a transcription factor downstream of ERK that directs the expression of genes involved in cell proliferation, such as Fos and Jun that subsequently regulate expression of cyclins and cyclin-dependent kinases that control cell cycle progression (51–54). ELK1S383 was phosphorylated in response to IGF1 treatment, and as expected this process was blocked by inhibiting ERKT202/Y204 phosphorylation. We also observed that ELK1S383 phosphorylation was inhibited by blocking PI3K. In other cell types, the PI3K pathway can amplify ERKT202/Y204 phosphorylation through activating the MAP3K RAF (55), and based on our results, there is likely a similar mechanism occurring in tenocytes.
In addition to promoting cell proliferation, IGF1 also increased protein synthesis by ∼2-fold in tenocytes, and this process was dependent upon PI3K/Akt activation. However, inhibiting ERKT202/Y204 phosphorylation resulted in a nearly 6-fold induction in protein synthesis, which was also a phenomenon that was not anticipated because ERK activation is often associated with mild increases in protein synthesis. This led us to look at potential interactions between the PI3K/Akt and ERK pathways. IGF1R phosphorylates IRS1Y608, which is the principal site of interaction between IRS1 and the SH2 domain of PI3K. This causes the localization of PI3K to the plasma membrane where it can catalyze the conversion of PIP2 into PIP3 (56). Akt then binds membrane-bound PIP3 through an N-terminal pleckstrin homology domain, and it is activated by PDK1. The recruitment of Akt to the membrane and the phosphorylation of AktT308 through PDK1 allows Akt to be released into cytosol to activate mTOR, p70S6K, and other downstream effectors (56). IRS1 can also be phosphorylated at S612, which blocks phosphorylation of its own Y608 residue, and therefore IRS1S612 phosphorylation inhibits the ability of IRS1 to activate PI3K (57). In some cell types, p-ERKT202/Y204 can phosphorylate IRS1S612 (57, 58); however, we did not observe this response in tenocytes. Instead, p-ERKT202/Y204 appears to modulate protein synthesis downstream of IRS1 by inhibiting phosphorylation of AktT308 either directly or by targeting a process downstream of IRS1Y608. This inhibitory role of ERK on the PI3K pathway explains the pronounced increase in protein synthesis when ERK signaling is inhibited. Although p-ERKT202/Y204 did not play a role in IRS1S612 or IRS1Y608 phosphorylation, inhibition of Akt activation abolished IRS1S612 phosphorylation and increased IRS1Y608 phosphorylation, suggesting that Akt or a downstream effector molecule acts to negatively regulate the PI3K pathway at IRS1. Previous studies have indicated that p70S6KT389 can phosphorylate several inhibitory serine residues on IRS1 (59), and we propose that p70S6KT389 is acting in a similar way to phosphorylate IRS1S612 and provide feedback inhibition on IGF1-mediated PI3K/Akt activation in tenocytes. Combined, these results indicate that the PI3K/Akt and ERK pathways interact downstream of IGF1 to control tenocyte proliferation and protein synthesis.
There are several limitations to this study. We only included male mice, and additional experiments evaluating the role of sex will likely provide further insight into the role of IGF1 in tendon adaptation. The plantaris overload synergist ablation technique used in this study is a supraphysiological growth stimulus that exceeds the normal growth signals tendons experience with exercise training. We only evaluated through 2 wk after mechanical overload, and it is likely that IGF1 continues to have an influence on neotendon matrix growth and maturation beyond this point. Tendon mechanical properties were not assessed in this study, which limits interpretations about functional changes in mechanically overloaded tendons. We did not measure cell proliferation in nonoverloaded mice in this study, but we and others have previously shown very low proliferation rates of tenocytes in tendons of adult mice and rats at this age (6, 22, 60). We focused our analysis on the PI3K/Akt and ERK pathways based on the bioinformatics results, but it is likely IGF1 also regulates other downstream pathways in tenocytes. IGF1R was not completely abolished in tendons because cells other than tenocytes may also express this receptor. Despite these limitations, we feel that this study provided novel insight into the role of IGF1 signaling in regulating tendon growth.
The ability of tendon to grow and respond to mechanical and biochemical cues requires the coordination of multiple biologic processes. Studies of 3-dimensional tissue engineered tendon constructs in culture and exercise training in human subjects have shown that tendons grow best in response to intermittent mechanical loading with adequate rest periods built in between loading regimes (3–5, 61, 62). Failure of tendons to respond to these cues can lead to the development of tendinopathies or tendon ruptures (8). The results in the current study demonstrate an important role for IGF1 signaling in the growth of tendons in adult animals and provide mechanistic support for the potential use of IGF1 in the treatment of tendinopathies. This is further supported by results from a small trial that demonstrated IGF1 increased collagen content and cell proliferation in horses with tendinopathy (14), and local injection of IGF1 into the tendons of elderly men increased local collagen production (13). However, it is unlikely that the use of growth factors alone would be sufficient to treat tendon disorders. High-load eccentric resistance training can reduce pain and improve ECM structural organization in patients with tendinopathy, although this training does not lead to full symptomatic resolution for many patients (63, 64). Recent studies have demonstrated that tendons synthesize collagen during rest phases at night and assemble collagen into mature fibrils during the active day phases (65, 66). Combining the use of recombinant growth factors with properly timed mechanical loading interventions and appropriate rest periods could lead to the improved treatment of tendon disorders. Although IGF1 appears to be a critical pathway in modulating tendon growth, further studies that integrate molecular genetics, cell biology, and mechanotransduction will provide additional insight into the basic mechanisms of tendon growth and likely lead to the development of novel biomarkers of early tendinopathic changes and improved treatments of painful and debilitating tendon disorders.
ACKNOWLEDGMENTS
The authors acknowledge technical contributions from Dr. David Oliver (Hospital for Special Surgery) and Dr. James Markworth (University of Michigan). The ScxCreERT2 mice were kindly provided by Dr. Ronen Schweitzer (Shriners Hospital for Children, Portland, OR, USA). This work was supported by U.S. National Institutes of Health (NIH) Grants R01-AR063649 and F32-AR067086. The authors declare no conflicts of interest.
Glossary
- AbAm
antibiotic antimycotic
- Akt
protein kinase B
- BrdU
bromodeoxyuridine
- CSA
cross-sectional area
- CV
coefficient of variation
- DM
differentiation medium
- ECM
extracellular matrix
- EdU
5-ethynyl-2'-deoxyuridine
- ELK
ETS like-1 protein
- FAK
focal adhesion kinase
- GM
growth medium
- IRS1
insulin receptor substrate 1
- p70S6K
p70S6 kinase
- qPCR
quantitative PCR
- RNAseq
RNA sequencing
- RTK
receptor tyrosine kinase
- Scx
scleraxis
- Scx:IGF1R+
Scx-expressing IGF1R-positive
- Scx:IGF1RΔ
Scx-expressing IGF1R-negative
- SUnSET
surface sensing of translation
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
N. P. Disser, K. B. Sugg, and C. L. Mendias designed research; N. P. Disser, K. B. Sugg, J. R. Talarek, D. C. Sarver, B. J. Rourke, and C. L. Mendias performed research; N. P. Disser, K. B. Sugg, and C. L. Mendias analyzed data; and N. P. Disser, K. B. Sugg, and C. L. Mendias wrote the manuscript.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Gumucio J. P., Sugg K. B., Mendias C. L. (2015) TGF-β superfamily signaling in muscle and tendon adaptation to resistance exercise. Exerc. Sport Sci. Rev. 43, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J. H.-C. (2006) Mechanobiology of tendon. J. Biomech. 39, 1563–1582 [DOI] [PubMed] [Google Scholar]
- 3.Couppé C., Kongsgaard M., Aagaard P., Hansen P., Bojsen-Moller J., Kjaer M., Magnusson S. P. (2008) Habitual loading results in tendon hypertrophy and increased stiffness of the human patellar tendon. J. Appl. Physiol. 105, 805–810 [DOI] [PubMed] [Google Scholar]
- 4.Svensson R. B., Heinemeier K. M., Couppé C., Kjaer M., Magnusson S. P. (2016) Effect of aging and exercise on the tendon. J. Appl. Physiol. 121, 1237–1246 [DOI] [PubMed] [Google Scholar]
- 5.Geremia J. M., Baroni B. M., Bobbert M. F., Bini R. R., Lanferdini F. J., Vaz M. A. (2018) Effects of high loading by eccentric triceps surae training on Achilles tendon properties in humans. Eur. J. Appl. Physiol. 118, 1725–1736 [DOI] [PubMed] [Google Scholar]
- 6.Gumucio J. P., Phan A. C., Ruehlmann D. G., Noah A. C., Mendias C. L. (2014) Synergist ablation induces rapid tendon growth through the synthesis of a neotendon matrix. J. Appl. Physiol. 117, 1287–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millar N. L., Murrell G. A. C., McInnes I. B. (2017) Inflammatory mechanisms in tendinopathy - towards translation. Nat. Rev. Rheumatol. 13, 110–122 [DOI] [PubMed] [Google Scholar]
- 8.Mead M. P., Gumucio J. P., Awan T. M., Mendias C. L., Sugg K. B. (2018) Pathogenesis and management of tendinopathies in sports medicine. Transl. Sports Med. 1, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinemeier K. M., Mackey A. L., Doessing S., Hansen M., Bayer M. L., Nielsen R. H., Herchenhan A., Malmgaard-Clausen N. M., Kjaer M. (2012) GH/IGF-I axis and matrix adaptation of the musculotendinous tissue to exercise in humans. Scand. J. Med. Sci. Sports 22, e1–e7 [DOI] [PubMed] [Google Scholar]
- 10.Lelbach A., Muzes G., Feher J. (2005) The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol. Hung. 92, 97–107 [DOI] [PubMed] [Google Scholar]
- 11.Vassilakos G., Lei H., Yang Y., Puglise J., Matheny M., Durzynska J., Ozery M., Bennett K., Spradlin R., Bonanno H., Park S., Ahima R. S., Barton E. R. (2019) Deletion of muscle IGF-I transiently impairs growth and progressively disrupts glucose homeostasis in male mice. FASEB J. 33, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yakar S., Courtland H.-W., Clemmons D. (2010) IGF-1 and bone: new discoveries from mouse models. J. Bone Miner. Res. 25, 2543–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R. H., Holm L., Malmgaard-Clausen N. M., Reitelseder S., Heinemeier K. M., Kjaer M. (2014) Increase in tendon protein synthesis in response to insulin-like growth factor-I is preserved in elderly men. J. Appl. Physiol. 116, 42–46 [DOI] [PubMed] [Google Scholar]
- 14.Dahlgren L. A., van der Meulen M. C., Bertram J. E., Starrak G. S., Nixon A. J. (2002) Insulin-like growth factor-I improves cellular and molecular aspects of healing in a collagenase-induced model of flexor tendinitis. J. Orthop. Res. 20, 910–919 [DOI] [PubMed] [Google Scholar]
- 15.Herchenhan A., Bayer M. L., Eliasson P., Magnusson S. P., Kjaer M. (2015) Insulin-like growth factor I enhances collagen synthesis in engineered human tendon tissue. Growth Horm. IGF Res. 25, 13–19 [DOI] [PubMed] [Google Scholar]
- 16.Dietrich P., Dragatsis I., Xuan S., Zeitlin S., Efstratiadis A. (2000) Conditional mutagenesis in mice with heat shock promoter-driven cre transgenes. Mamm. Genome 11, 196–205 [DOI] [PubMed] [Google Scholar]
- 17.Howell K., Chien C., Bell R., Laudier D., Tufa S. F., Keene D. R., Andarawis-Puri N., Huang A. H. (2017) Novel model of tendon regeneration reveals distinct cell mechanisms underlying regenerative and fibrotic tendon healing. Sci. Rep. 7, 45238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugg K. B., Markworth J. F., Disser N. P., Rizzi A. M., Talarek J. R., Sarver D. C., Brooks S. V., Mendias C. L. (2018) Postnatal tendon growth and remodeling require platelet-derived growth factor receptor signaling. Am. J. Physiol. Cell Physiol. 314, C389–C403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudgens J. L., Sugg K. B., Grekin J. A., Gumucio J. P., Bedi A., Mendias C. L. (2016) Platelet-rich plasma activates proinflammatory signaling pathways and induces oxidative stress in tendon fibroblasts. Am. J. Sports Med. 44, 1931–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman C. A., Mabrey D. M., Frey J. W., Miu M. H., Schmidt E. K., Pierre P., Hornberger T. A. (2011) Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 25, 1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman C. A., Hornberger T. A. (2013) Measuring protein synthesis with SUnSET: a valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 41, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz A. J., Sarver D. C., Sugg K. B., Dzierzawski J. T., Gumucio J. P., Mendias C. L. (2015) p38 MAPK signaling in postnatal tendon growth and remodeling. PLoS One 10, e0120044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson M. D., McCarthy D. J., Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rutledge R. G., Stewart D. (2010) Assessing the performance capabilities of LRE-based assays for absolute quantitative real-time PCR. PLoS One 5, e9731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gumucio J. P., Qasawa A. H., Ferrara P. J., Malik A. N., Funai K., McDonagh B., Mendias C. L. (2019) Reduced mitochondrial lipid oxidation leads to fat accumulation in myosteatosis. FASEB J. 33, 7863–7881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olesen J. L., Heinemeier K. M., Haddad F., Langberg H., Flyvbjerg A., Kjaer M., Baldwin K. M. (2006) Expression of insulin-like growth factor I, insulin-like growth factor binding proteins, and collagen mRNA in mechanically loaded plantaris tendon. J. Appl. Physiol. 101, 183–188 [DOI] [PubMed] [Google Scholar]
- 27.Hamilton D. L., Philp A., MacKenzie M. G., Patton A., Towler M. C., Gallagher I. J., Bodine S. C., Baar K. (2014) Molecular brakes regulating mTORC1 activation in skeletal muscle following synergist ablation. Am. J. Physiol. Endocrinol. Metab. 307, E365–E373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarthy J. J., Mula J., Miyazaki M., Erfani R., Garrison K., Farooqui A. B., Srikuea R., Lawson B. A., Grimes B., Keller C., Van Zant G., Campbell K. S., Esser K. A., Dupont-Versteegden E. E., Peterson C. A. (2011) Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 138, 3657–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugg K. B., Korn M. A., Sarver D. C., Markworth J. F., Mendias C. L. (2017) Inhibition of platelet-derived growth factor signaling prevents muscle fiber growth during skeletal muscle hypertrophy. FEBS Lett. 591, 801–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C. H., Lee F. Y., Tarafder S., Kao K., Jun Y., Yang G., Mao J. J. (2015) Harnessing endogenous stem/progenitor cells for tendon regeneration. J. Clin. Invest. 125, 2690–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murchison N. D., Price B. A., Conner D. A., Keene D. R., Olson E. N., Tabin C. J., Schweitzer R. (2007) Regulation of tendon differentiation by scleraxis distinguishes force-transmitting tendons from muscle-anchoring tendons. Development 134, 2697–2708 [DOI] [PubMed] [Google Scholar]
- 32.Léjard V., Blais F., Guerquin M.-J., Bonnet A., Bonnin M.-A., Havis E., Malbouyres M., Bidaud C. B., Maro G., Gilardi-Hebenstreit P., Rossert J., Ruggiero F., Duprez D. (2011) EGR1 and EGR2 involvement in vertebrate tendon differentiation. J. Biol. Chem. 286, 5855–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalluri R., Weinberg R. A. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendias C. L., Gumucio J. P., Lynch E. B. (2012) Mechanical loading and TGF-β change the expression of multiple miRNAs in tendon fibroblasts. J. Appl. Physiol. 113, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spindler K. P., Imro A. K., Mayes C. E., Davidson J. M. (1996) Patellar tendon and anterior cruciate ligament have different mitogenic responses to platelet-derived growth factor and transforming growth factor beta. J. Orthop. Res. 14, 542–546 [DOI] [PubMed] [Google Scholar]
- 36.Thomopoulos S., Das R., Silva M. J., Sakiyama-Elbert S., Harwood F. L., Zampiakis E., Kim H. M., Amiel D., Gelberman R. H. (2009) Enhanced flexor tendon healing through controlled delivery of PDGF-BB. J. Orthop. Res. 27, 1209–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strutz F., Zeisberg M., Renziehausen A., Raschke B., Becker V., van Kooten C., Müller G. (2001) TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 59, 579–592 [DOI] [PubMed] [Google Scholar]
- 38.Arndt S., Karrer S., Hellerbrand C., Bosserhoff A. K. (2019) Bone morphogenetic protein-6 inhibits fibrogenesis in scleroderma offering treatment options for fibrotic skin disease. [E-pub ahead of print] J. Invest. Dermatol. [DOI] [PubMed] [Google Scholar]
- 39.Kersten C., Sivertsen E. A., Hystad M. E., Forfang L., Smeland E. B., Myklebust J. H. (2005) BMP-6 inhibits growth of mature human B cells; induction of Smad phosphorylation and upregulation of Id1. BMC Immunol. 6, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gehmert S., Sadat S., Song Y.-H., Yan Y., Alt E. (2008) The anti-apoptotic effect of IGF-1 on tissue resident stem cells is mediated via PI3-kinase dependent secreted frizzled related protein 2 (Sfrp2) release. Biochem. Biophys. Res. Commun. 371, 752–755 [DOI] [PubMed] [Google Scholar]
- 41.Hansen M., Boesen A., Holm L., Flyvbjerg A., Langberg H., Kjaer M. (2013) Local administration of insulin-like growth factor-I (IGF-I) stimulates tendon collagen synthesis in humans. Scand. J. Med. Sci. Sports 23, 614–619 [DOI] [PubMed] [Google Scholar]
- 42.Doessing S., Holm L., Heinemeier K. M., Feldt-Rasmussen U., Schjerling P., Qvortrup K., Larsen J. O., Nielsen R. H., Flyvbjerg A., Kjaer M. (2010) GH and IGF1 levels are positively associated with musculotendinous collagen expression: experiments in acromegalic and GH deficiency patients. Eur. J. Endocrinol. 163, 853–862 [DOI] [PubMed] [Google Scholar]
- 43.Mouw J. K., Ou G., Weaver V. M. (2014) Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 15, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen R. H., Holm L., Jensen J. K., Heinemeier K. M., Remvig L., Kjaer M. (2014) Tendon protein synthesis rate in classic Ehlers-Danlos patients can be stimulated with insulin-like growth factor-I. J. Appl. Physiol. 117, 694–698 [DOI] [PubMed] [Google Scholar]
- 45.Stefanovic B. (2013) RNA protein interactions governing expression of the most abundant protein in human body, type I collagen. Wiley Interdiscip. Rev. RNA 4, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tahimic C. G. T., Wang Y., Bikle D. D. (2013) Anabolic effects of IGF-1 signaling on the skeleton. Front. Endocrinol. (Lausanne) 4, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tahimic C. G. T., Long R. K., Kubota T., Sun M. Y., Elalieh H., Fong C., Menendez A. T., Wang Y., Vilardaga J.-P., Bikle D. D. (2016) Regulation of ligand and shear stress-induced insulin-like growth factor 1 (IGF1) signaling by the integrin pathway. J. Biol. Chem. 291, 8140–8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott A., Cook J. L., Hart D. A., Walker D. C., Duronio V., Khan K. M. (2007) Tenocyte responses to mechanical loading in vivo: a role for local insulin-like growth factor 1 signaling in early tendinosis in rats. Arthritis Rheum. 56, 871–881 [DOI] [PubMed] [Google Scholar]
- 49.Scott A., Khan K. M., Duronio V. (2005) IGF-I activates PKB and prevents anoxic apoptosis in Achilles tendon cells. J. Orthop. Res. 23, 1219–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Havis E., Bonnin M.-A., Esteves de Lima J., Charvet B., Milet C., Duprez D. (2016) TGFβ and FGF promote tendon progenitor fate and act downstream of muscle contraction to regulate tendon differentiation during chick limb development. Development 143, 3839–3851 [DOI] [PubMed] [Google Scholar]
- 51.Bahrami S., Drabløs F. (2016) Gene regulation in the immediate-early response process. Adv. Biol. Regul. 62, 37–49 [DOI] [PubMed] [Google Scholar]
- 52.Cook S. J., Aziz N., McMahon M. (1999) The repertoire of fos and jun proteins expressed during the G1 phase of the cell cycle is determined by the duration of mitogen-activated protein kinase activation. Mol. Cell. Biol. 19, 330–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boros J., Donaldson I. J., O’Donnell A., Odrowaz Z. A., Zeef L., Lupien M., Meyer C. A., Liu X. S., Brown M., Sharrocks A. D. (2009) Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 19, 1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao J., Hodge C., Meyer D., Ho P. S., Rosenspire K., Schwartz J. (1997) Growth hormone regulates ternary complex factors and serum response factor associated with the c-fos serum response element. J. Biol. Chem. 272, 25951–25958 [DOI] [PubMed] [Google Scholar]
- 55.Ebi H., Costa C., Faber A. C., Nishtala M., Kotani H., Juric D., Della Pelle P., Song Y., Yano S., Mino-Kenudson M., Benes C. H., Engelman J. A. (2013) PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Natl. Acad. Sci. USA 110, 21124–21129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gumucio J. P., Mendias C. L. (2013) Atrogin-1, MuRF-1, and sarcopenia. Endocrine 43, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Fea K., Roth R. A. (1997) Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 272, 31400–31406 [DOI] [PubMed] [Google Scholar]
- 58.Andreozzi F., Laratta E., Sciacqua A., Perticone F., Sesti G. (2004) Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ. Res. 94, 1211–1218 [DOI] [PubMed] [Google Scholar]
- 59.Copps K. D., White M. F. (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan Q., Lui P. P. Y., Lee Y. W. (2013) In vivo identity of tendon stem cells and the roles of stem cells in tendon healing. Stem Cells Dev. 22, 3128–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paxton J. Z., Hagerty P., Andrick J. J., Baar K. (2012) Optimizing an intermittent stretch paradigm using ERK1/2 phosphorylation results in increased collagen synthesis in engineered ligaments. Tissue Eng. Part A 18, 277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huisman E., Lu A., McCormack R. G., Scott A. (2014) Enhanced collagen type I synthesis by human tenocytes subjected to periodic in vitro mechanical stimulation. BMC Musculoskelet. Disord. 15, 386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kongsgaard M., Kovanen V., Aagaard P., Doessing S., Hansen P., Laursen A. H., Kaldau N. C., Kjaer M., Magnusson S. P. (2009) Corticosteroid injections, eccentric decline squat training and heavy slow resistance training in patellar tendinopathy. Scand. J. Med. Sci. Sports 19, 790–802 [DOI] [PubMed] [Google Scholar]
- 64.Kongsgaard M., Qvortrup K., Larsen J., Aagaard P., Doessing S., Hansen P., Kjaer M., Magnusson S. P. (2010) Fibril morphology and tendon mechanical properties in patellar tendinopathy: effects of heavy slow resistance training. Am. J. Sports Med. 38, 749–756 [DOI] [PubMed] [Google Scholar]
- 65.Pickard A., Chang J., Alachkar N., Calverley B., Garva R., Arvan P., Meng Q.-J., Kadler K. E. (2019) Preservation of circadian rhythms by the protein folding chaperone, BiP. FASEB J. 33, 7479–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yeung C. C., Kadler K. E. (2019) Importance of the circadian clock in tendon development. Curr. Top. Dev. Biol. 133, 309–342 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.