

Abstract
Bitter taste receptor-14 (TAS2R14) is a GPCR also expressed on human airway smooth muscle cells, which signals to intracellular [Ca2+], resulting in relaxation of the airway, and is a novel target for bronchodilators. Here, we examine long-term, agonist-promoted down-regulation of TAS2R14 expression because tachyphylaxis would be an undesirable therapeutic characteristic. Five TAS2R structurally distinct full agonists were studied to ascertain biasing away from down-regulation. Agonist exposure for 18 h caused minimal desensitization by diphenhydramine (DPD) compared with ∼50% desensitization with all other agonists. Agonists evoked β-arrestin recruitment to TAS2R14, which was not seen with a phosphoacceptor-deficient mutant, TAS2R14-10A. All agonists except for DPD also caused subsequent TAS2R14 internalization and trafficking via early and late endosomes to down-regulation. TAS2R14-10A failed to undergo these events with any agonist. Molecular docking showed that DPD has specific interactions deep within a binding pocket that are not observed with the other agonists, which may lock the receptor in a conformation that does not internalize and therefore does not undergo down-regulation. Thus, TAS2R14 is subject to β-arrestin–mediated internalization and subsequent down-regulation with chronic exposure to most agonists. However, by manipulating the agonist structure, biasing toward G-protein coupling but away from long-term down-regulation can be achieved.—Woo, J. A., Castaño, M., Goss, A., Kim, D., Lewandowski, E. M., Chen, Y., Liggett, S. B. Differential long-term regulation of TAS2R14 by structurally distinct agonists.
Keywords: desensitization, internalization, down-regulation, β-arrestin, [Ca2+]i
Most GPCRs display the property of desensitization upon continuous agonist exposure (1). Homologous (also termed agonist-dependent) desensitization is defined as a waning of receptor function due to receptor occupancy by agonist (1). Within seconds to minutes of agonist exposure, a decrease in coupling of the receptor to the G-protein is observed for many GPCRs and represents the earliest component of desensitization (1). After more prolonged time periods, a net loss of the cellular complement of receptors (regardless of localization) is often encountered, a process termed down-regulation (2). Down-regulation is particularly germane to long-term agonist treatment from repetitive dosing, and it can limit therapeutic efficacy. Internalization of the receptor to the cell interior as intracellular vesicles has been described for many GPCRs (3) and occurs prior to down-regulation events and is often the mechanism that leads to receptor degradation.
The β2-adrenergic receptor (AR) has been extensively studied in regards to the mechanisms of desensitization. For this receptor, the agonist-occupied conformation promotes phosphorylation of specific serines or threonines in the third intracellular loop or carboxy-terminal tail of the receptor by GPCR kinases (GRKs). This event promotes binding of β-arrestins, which interdict between receptor and G-protein, resulting in partial uncoupling. This response is rapid (seconds), essentially becoming a part of the initial signal because it is difficult to separate the fully coupled state from the desensitized state because of this rapid mechanism (4). With more prolonged agonist exposure (minutes to hours), β2-ARs undergo internalization to the cell interior, ultimately becoming part of intracellular vesicles; this process appears to be in part due to the scaffold and adaptor effects of the bound β-arrestin (3). Receptors within the vesicles are either degraded by lysosomes or reinserted into the membrane depending on whether agonist is still present. With prolonged agonist exposure, most cells expressing β2-AR display a down-regulation of the net number of β2-AR on the order of ∼80% (5–7). Unlike uncoupling or internalization, recovery from down-regulation requires new receptor synthesis. β2-AR down-regulation can be observed as early as ∼6 h after agonist exposure and reaches steady state in most cells by 12–18 h. This paradigm for β2-AR, however, does not necessarily hold for other GPCRs. Indeed, there is evidence that most receptors evolved to meet regulatory needs specific for a given receptor or receptor subfamily. Thus, when considering a GPCR as a target for therapeutic agonists, determining whether down-regulation occurs with prolonged agonist exposure (and the mechanisms responsible for this regulation) is an important component. Recent data also indicate that differences in agonist structure can result in the stabilization of GPCRs to specific conformations that may be more, or less, susceptible to coupling to G-proteins, recruitment of β-arrestin, desensitization, or other signaling events (8–11). This phenomenon, termed the biased ligand effect (12), may be useful to tailor agonist structure for maximal therapeutic effect by mitigating long-term desensitization.
The current study focuses on long-term agonist-promoted desensitization of bitter taste receptors (TAS2Rs). These receptors are found not only on the tongue (13) but also on cell types of other organs including human airway smooth muscle (HASM) (14). Activation of HASM TAS2Rs results in marked relaxation of the muscle (15), and TAS2R agonists have been proposed as new therapeutic agents for treating airway obstruction in asthma and chronic obstructive pulmonary disease (16). On HASM, 3 of the 25 TAS2Rs (TAS2R10, 14, and 31) are expressed at levels higher than β2-ARs, and like on taste cells, they couple to an increase in intracellular [Ca2+] ([Ca2+]i), which is linked to the relaxation of smooth muscle (14). TAS2R14 appears to have one of the least-restricted requirements for agonist binding (17), so this receptor has been the focus of a number of studies relating to expression, G-protein coupling, function, and regulation. Although we have demonstrated that HASM TAS2Rs display short-term (15 min) agonist-promoted desensitization of the [Ca2+]i and the relaxation response (18), there have been no studies to date examining whether these receptors undergo down-regulation and functional desensitization with prolonged agonist exposure. Nor have there been investigations of potential biasing of certain agonists that might minimize any long-term loss of receptor expression or function during chronic exposure, which would be considered a highly favorable therapeutic feature.
MATERIALS AND METHODS
Cell culture, transfections, mutagenesis
The immortalized HASM cell line D9 hTERT was utilized as previously described (19) and grown as monolayers in DMEM with 10% fetal calf serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. Human embryonic kidney (HEK)-293T cells were maintained in the same medium. Cell maintenance, transfections, and drug treatments were all at 37°C in a 95% air and 5% CO2 atmosphere. [Ca2+]i assays were performed by loading the cells in 96-well plates with Fluo-4 (Thermo Fisher Scientific, Waltham, MA, USA) and utilizing a Flex Station 3 (Molecular Devices, Sunnyvale, CA, USA). Transfections were carried out using the previously described (20) FLAG-tagged TAS2R14 (or the mutant) in pcDNA3 using Lipofectamine 2000 as previously described (19). Mutagenesis of TAS2R14 cDNA was performed in order to change the encoded serines or threonines to alanine at amino acid positions 215, 219, 223, 224, and 232 (in the third intracellular loop) and 291, 293, 308, 316, and 317 (in the cytoplasmic tail). This mutant is termed TAS2R14-10A. TAS2R14 knockdowns were performed by transfecting HASM with 100 nM On-Target SmartPool of TAS2R14 small interfering RNA from Dharmacon (Lafayette, CO, USA).
TAS2R14 desensitization
HASM cells were plated in 96-well plates at 40,000 cells per well in medium (14). At t = 0, vehicle or a TAS2R14 agonist was added to the medium. We used the highest dose for this 18-h treatment that did not cause an alteration in cell morphology or cell death. This dose, termed the treatment dose was 200 µM and represents at least 50–100-fold higher than the half maximal effective concentration (EC50) for each of the agonists (17). Agonists utilized were aristolochic acid (AA), chlorhexidine (CLX), flufenamic acid (FFA), papaverine (PAP), and diphenhydramine (DPD). Cells were incubated at 37°C, 5% CO2, and 95% air for 18 h. At t = 18 h, cells were challenged by the same agonists, and the [Ca2+]i response was immediately captured every 2.1 s for the next 100 s. This second dose, termed the challenge dose brought the final concentration to 300 µM (PAP, FFA), 500 µM (AA), or 1.0 mM (DPD, CLX). In parallel studies, the treatment dose was only with vehicle, and the challenge dose was the final concentration of the agonist as previously listed. These latter [Ca2+]i responses act as the nondesensitized control response, used to calculate the degree of homologous (i.e., agonist-specific) desensitization due to the chronic agonist exposure.
Desensitization was calculated using Eq. 1:
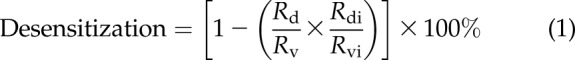 |
where Rv is the maximal [Ca2+]i observed from the acute TAS2R agonist challenge after cells were treated with vehicle alone for 18 h, and Rd is the maximal [Ca2+]i response from the drug challenge in cells that were treated 18 h with the treatment dose of the TAS2R agonist. Rdi is the acute response to ionomycin (IONO, 1.2 µM) after chronic treatment with the treatment dose of agonist, and Rvi is the response to IONO after vehicle treatment. In other wells, the cells were challenged with an agonist for another GPCR that stimulates [Ca2+]i [bradykinin (BRA, 10 µM) or endothelin1 (ET1, 200 µM)] after the 18-h treatment with vehicle or TAS2R agonist. Any changes in this [Ca2+]i response was considered heterologous desensitization. Statistical comparisons for each TAS2R agonist were made between the homologous desensitization and the heterologous desensitization results.
Immunoblots
Cells were lysed with RIPA buffer containing 10 mM Tris-HCL (pH 7.4), 30 mM NaCl, 1 mM EDTA, and 1% Nonidet P-40. Equal amounts of proteins were subjected to SDS-PAGE and transferred to nitrocellulose membrane. After blocking with 5% skim milk for 1 h, primary antibody was applied overnight at 4°C. After incubating with the primary antibody, the corresponding peroxidase-conjugated secondary antibody was detected by ECL reagents (Thermo Fisher Scientific). ECL images were captured by a Fuji LAS-4000 imager (LAS-4000; GE Healthcare Life Sciences, Pittsburgh, PA, USA) and quantified using the ImageJ software (National Institutes of Health, Bethesda, MD, USA). The primary antibodies, titers (v/v), and sources were as follows: FLAG M2 (1:1000, F1804; MilliporeSigma, Burlington, MA, USA), actin (1:3000, A1978; MilliporeSigma), and SAB502089 (TAS2R14, 1:200; MilliporeSigma). The horseradish peroxidase–linked secondary antibodies (1:1000) were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Immunochemistry and imaging
HEK-293 cells were fixed with 4% paraformaldehyde in PBS for 20 min at 25°C. Cells were washed with PBS 3 times and blocked with 3% normal goat serum and 0.2% Triton X-100 in PBS. For detection of only cell surface proteins, Triton X-100 was omitted. After blocking with 3% normal goat serum, primary antibodies were applied overnight at 4°C, and secondary antibodies were applied for 45 min at room temperature, followed by counterstaining with DAPI. The titer (v/v) and antibodies were as follows: F1804 (FLAG M2, 1:200; MilliporeSigma), ab24170 [lysosome-associated membrane glycoprotein 1 (LAMP1)], 1:100; Abcam, Cambridge, MA, USA), C45B10 (EEA1, 1:100; Cell Signaling Technology, Danvers, MA, USA), D1306 (DAPI, 1:1000; Thermo Fisher Scientific), R37117 (Alexa Fluor 594, 1:1000; Thermo Fisher Scientific), and A-11001 (Alexa Fluor 488, 1:1000; Thermo Fisher Scientific). Images were captured by FV10i confocal microscope (Olympus, Tokyo, Japan) and LSM880 confocal microscope (Carl Zeiss GmbH, Oberkochen, Germany), with the magnifications provided in the figure legends. Transfected cells were randomly chosen, with 15–20 images collected per condition, which were averaged and represent 1 experiment. The image files were stored as raw files using the Olympus or Zeiss software and then opened in ImageJ. Intensity was measured for transfected cells, and background intensity was subtracted. Background subtracted images were used to measure colocalization using Manders’ split method in ImageJ Coloc2 software (https://imagej.net/Coloc_2). Vehicle (control) intensities were subtracted from the drug-treated results and expressed in absolute values, which were then normalized to the maximal response from the indicated receptor/drug condition. Results are shown as mean ± se of 4 independent experiments. For qualitative assessment of β-arrestin recruitment, HEK-293 cells were transfected with the TAS2R14 constructs and green fluorescent protein (GFP)–β-arrestin1 or GFP–β-arrestin2. Forty-eight hours later, they were treated with the indicated agonists for 5 min at 37°C, followed by fixation with 4% paraformaldehyde in PBS. Imaging was with the Zeiss LSM880 microscope. In some studies, focal stacking confocal imaging (z stacking) was performed to confirm localization of TAS2R14 to the cell membrane by acquiring multiple consecutive images at 1.0-µm intervals.
In silico molecular docking
The Swiss-Model homology-modeling server (21–25) was used to construct a homology model of TAS2R14. A model constructed from the template of C-C chemokine receptor type 5, Rubredoxin chimera (Protein Data Bank ID: 5UIW), was selected for use in molecular docking experiments. Dock 3.5.54 (26) was used to dock the structures of AA, DPD, CLX, PAP, FFA, and orphenadrine (ORP) to the TAS2R14 homology model. Residues 66, 70, 71, 89, 181, 247, and 266 were used to define an initial core ligand binding site and to generate matching spheres. A grid-based scoring function was used to score the binding poses by summing the energies calculated by Solvmap (ligand desolvation), Chemgrid (Van der Waals potential), and DelPhi (electrostatic potential) (27–30). The docked poses were visually inspected using Pymol (Schrödinger, New York, NY, USA).
Statistical analysis
Analysis was performed using Prism 7.0 software (GraphPad Software, La Jolla, CA, USA), Student’s t tests, or for multiple comparisons, 1- or 2-way ANOVA, followed by Tukey’s post hoc tests. Data are shown as the mean ± se of the indicated number of experiments or of the replicates of a single representative experiment. Values of P < 0.05 were considered significant.
RESULTS
Differential long-term desensitization of TAS2R from structurally distinct agonists
One of the earliest signals evoked by TAS2Rs in taste-cells type is an increase in [Ca2+]i (31). We have previously shown in HASM cells that this localized increase is the basis for agonist-promoted relaxation (14). Here, we established a model to explore long-term desensitization with live HASM cells in real time, in which cells are treated with vehicle or TAS2R agonist for 18 h and then challenged with an additional higher final dose of agonist with immediate readout of [Ca2+]i. In parallel wells, cellular [Ca2+]i was also stimulated by IONO to assess any nonreceptor alterations in [Ca2+]i evoked by the TAS2R agonist treatment. And finally, cells in other wells that had been exposed to vehicle or TAS2R agonist were stimulated with an agonist to another GPCR that is also coupled to stimulation of [Ca2+]i (ET1 or BRA) to ascertain any heterologous desensitization.
Figure 1 shows representative results of such experiments performed with the 5 TAS2R agonists, with the mean calculated desensitization values from multiple experiments shown in Fig. 2. For the agonist FFA, it is evident that the acute maximal [Ca2+]i response is attenuated after 18 h pre-exposure to FFA by ∼50% (green vs. blue traces, Fig. 1A). In contrast, cells treated with FFA displayed very little loss of the [Ca2+]i response to ET1 compared with control (red vs. orange traces, Fig. 1A). In addition, the response to IONO was unaffected by FFA pretreatment (bar graph, Fig. 1A). The summary graph of the calculated desensitization by FFA (Fig. 2) from multiple experiments shows 50 ± 5.0% desensitization of TAS2R responsiveness due to chronic FFA exposure, with 13 ± 3.6% loss of the ET1 response. The remaining panels of Fig. 1 and summary of Fig. 2 show results from studies with the other agonists, AA, CLX, PAP, and DPD. In the cases of CLX, AA, and PAP, there was ∼45–60% desensitization observed, with an ∼10% heterologous component. In contrast, DPD showed 25 ± 6.4% desensitization, which was not statistically different from the 12 ± 3.5% desensitization of the ET1 challenge response (P = 0.102, N = 5) (Figs. 1E and 2). Thus, from these functional studies, we conclude that the homologous desensitization response to DPD is minimal at best, particularly when compared with the other 4 TAS2R agonists. In order to exclude this distinguishing characteristic of DPD from an off-target [Ca2+]i response, we performed knockdown experiments with TAS2R14-specific small interfering RNA. Although we did not fully suppress TAS2R14 expression, we note that an ∼30% decrease in TAS2R14 expression was associated with an ∼40% decrease in the DPD-mediated [Ca2+]i response, indicating that in these experiments, DPD is acting via TAS2R14 (Supplemental Fig. S1).
Figure 1.

[Ca2+]i responsiveness of TAS2Rs is modified by long-term exposure to TAS2R agonist. HASM cells in monolayers were exposed to vehicle or the indicated TAS2R agonists FFA (A), AA (B), CLX (C), PAP (D), and DPD (E) for 18 h. Cells were then treated with a challenge dose of the TAS2R agonist (blue and green traces), another GPCR agonist that stimulates [Ca2+]i (BRA or ET1, orange and red traces), or IONO (bar graphs) and [Ca2+]i immediately determined over the next 100 s. See Materials and Methods for doses. Shown are representative data from single experiments (of 4 performed) with triplicate determinations. Desensitization of TAS2Rs was calculated from Eq. 1. See Fig. 2 for calculated desensitization results from multiple experiments. RFU, relative fluorescence unit.
Figure 2.

Differential desensitization of HASM TAS2R14 on HASM by structurally distinct agonists. Experiments were performed as described in Materials and Methods, and the raw [Ca2+]i data (Fig. 1) were utilized to calculate the extent of desensitization according to Eq. 1. The comparisons for t tests were between the desensitization when the TAS2R agonist was incubated with the cells and the challenge was with the TAS2R agonist (homologous desensitization, black bars) vs. desensitization from TAS2R agonist incubation and BRA or ET1 challenge (heterologous desensitization, white bars). All agonists except for DPD displayed significantly greater homologous as compared with heterologous desensitization. Results are from 5-8 experiments. *P < 0.005 vs. ET1 or BRA challenge conditions.
DPD fails to evoke receptor down-regulation or association with early or late endosomes
The minimal homologous desensitization of HASM TAS2Rs in response to DPD long-term exposure suggested that DPD might not evoke TAS2R14 loss as compared with the other TAS2R agonists studied. FLAG-tagged TAS2R14-transfected HEK-293 cells were studied by immunoblots and confocal microscopy to test this hypothesis. The extent of the change in expression was calculated by comparison to the appropriate vehicle control for each agonist. As shown in Fig. 3A, B, FFA, AA, and CLX exposure resulted in an ∼40% decrease in TAS2R14 expression in whole-cell lysates, whereas DPD exposure actually increased expression over vehicle control. Furthermore, in nonpermeabilized cells, immunofluorescent confocal microscopy showed a decrease in cell surface expression of TAS2R14 with FFA, AA, and CLX and an increase with DPD (Fig. 3C, D), consistent with the whole-cell lysate data. In additional studies, focus stacking confocal microscopy was performed, which provides Z-stack images throughout the cell. These studies in nonpermeabilized cells (Supplemental Videos S1 and S2) were performed with FLAG antibody (to identify cell surface TAS2R14) and an antibody to tubulin (an intracellular protein). The results showed expression of TAS2R14 on the cell surface in a typical 3-dimensional pattern, with the midportion of the cells revealing signals only at the cell surface in a rim-like pattern. There was no expression of tubulin (acting as a negative control). With permeabilized cells, tubulin staining showed intracellular expression as expected (a positive control, Supplemental Videos S3 and S4). Finally, these expression phenotypes (immunoblots and confocal imaging) were evident when vehicle controls (water, DMSO, or MeOH) were graphed as a group or individually (Supplemental Fig. S2).
Figure 3.

Effects of long-term agonist exposure on TAS2R14-WT expression. HEK-293T cells were transfected to express FLAG-tagged TAS2R14 and exposed to TAS2R14 agonists for 18 h in culture. A, B) Immunoblotting of whole-cell lysates reveals down-regulation of net receptor expression when cells were incubated with FFA, AA, and CLX, whereas DPD increased expression. C, D) Confocal immunofluorescent imaging of nonpermeabilized cells shows loss of cell surface TAS2R14 for all agonists except DPD. VC, vector control; Veh, vehicle. A, C) Representative experiments. B, D) Mean results from 4 independent experiments. Scale bar, 10 µm. *P < 0.05 vs. vehicle, **P < 0.005 vs. vehicle, #P < 0.0005 vs. vehicle.
To further investigate the apparent biasing of DPD away from long-term down-regulation, we tracked intracellular TAS2R14 to early endosomes (EEA1 colocalization) and late endosomes/lysosomes (LAMP1 colocalization). Typically, agonist-promoted internalization of GPCRs is associated with receptor-containing vesicles that interact with early endosomes, eventually associating with late endosomes and then degradation. We found that FFA and CLX caused a readily observable colocalization of TAS2R14 with EEA1 after 3 h of incubation of the cells with these agonists (Fig. 4A, C). In contrast, DPD displayed a relatively small extent of colocalization compared with all other drugs tested. A higher dose of DPD also failed to promote TAS2R14 association with EEA1 (Fig. 4, bottom panels). These results suggest that agonist-promoted internalization of TAS2R14 by the full agonist DPD significantly differed in extent compared with the other full agonists and that entry into the degradation pathway from early to late endosomes would be minimal, consistent with the lack of down-regulation. Results of colocalization with the late endosomal marker LAMP1 are shown in Fig. 5A, C. TAS2R14 underwent no colocalization with LAMP1 after 18 h of exposure to DPD, whereas FFA and CLX evoked extensive colocalization with this marker, again consistent with the down-regulation of TAS2R14 observed with FFA and CLX but not with DPD. A higher dose of DPD failed to promote TAS2R14-wild type (WT) association with EEA1 or LAMP1.
Figure 4.

Agonist-promoted colocalization of TAS2R14 with the early endosome marker EEA1. A, B) Transfected HEK-293T cells were exposed to vehicle or agonist for 3 h followed by confocal imaging of permeabilized cells for EEA1 (green), TAS2R14-WT (A) or TAS2R14-10A (B) (red), or DAPI (blue). Merged images (columns 2, 5) show colocalization of EEA1 and TAS2R14-WT (yellow). Original magnification: ×500 (columns 2, 5); ×2900 (columns 3, 6). Results are from a representative experiment. C) Quantification of EEA1-TAS2R14 colocalization from 4 independent experiments in which each experiment generated 15–20 images per condition. For TAS2R14-WT, FFA and CLX exposure resulted in greater colocalization than DPD. None of the agonists evoked colocalization of EEA1 with the phosphorylation-deficient TAS2R14-10A mutant receptor (columns 4–6). Scale bar, 10 µm. *P < 0.05 vs. vehicle, #P < 0.0005 vs. vehicle, +P < 0.05 vs. FFA or CLX.
Figure 5.

Agonist-promoted colocalization of TAS2R14 with the late endosome marker LAMP1. A, B) Transfected HEK-293T cells were exposed to vehicle or agonist for 15 h, followed by confocal imaging of permeabilized cells for LAMP1 (green), TAS2R14-WT (A) or TAS2R14-10A (B) (red), or DAPI (blue). Merged images (columns 2, 5) show colocalization of LAMP1 and TAS2R14 (yellow). Original magnification, ×500 (columns 2, 5) and ×2900 (columns 3, 6). Results are from a representative experiment. C) Quantification of LAMP1-TAS2R14 colocalization from 4 independent experiments in which each experiment generated 15–20 images per condition. For TAS2R14-WT, FFA and CLX exposure resulted in significant colocalization, whereas DPD did not. None of these agonists evoked colocalization of LAMP1 with the phosphorylation-deficient TAS2R14-10A mutant receptor (columns 4–6). For the 500-µM treatment with DPD, cells were exposed for 11 h. Scale bar, 10 µm. *P < 0.05 vs. vehicle.
β-Arrestin recruitment of TAS2R14 and a TAS2R14 lacking phosphorylation sites
GPCR internalization evoked by agonist is typically dependent on binding of β-arrestin to the GRK-phosphorylated receptor, which occurs in the third intracellular loop or cytoplasmic tail of the protein. TAS2R14 has a total of 10 serines or threonines in the third intracellular loop and cytoplasmic tail. These potential phosphoacceptor sites were mutated to alanine and HEK-293 cells transfected with WT or mutant (termed TAS2R14-10A) constructs and GFP–β-arrestin1 or GFP–β-arrestin2. In the absence of agonist, GFP–β-arrestin1, 2 displays a homogenous expression pattern throughout the intracellular space (4). As previously described for other GPCRs, upon agonist exposure, the distribution of GFP–β-arrestin1, 2 takes on a distinct punctate pattern with a loss of the homogenous pattern and greater localization to the cell surface. These features have been shown to be consistent with β-arrestin recruitment to cell surface receptors (4). For CLX, AA, FFA, and DPD, these changes in GFP–β-arrestin1 distribution were found with cells expressing TAS2R14-WT after a 5-min exposure to these agonists (Fig. 6A). Similar results were obtained when GFP–β-arrestin2 was transfected (Fig. 6B). This suggests that despite minimal internalization from DPD exposure, β-arrestin1 and β-arrestin2 are in fact recruited to TAS2R14 by this agonist, but the receptor does not proceed further down the intracellular degradation pathway. We further tested this proposed series of events with the mutated receptor. As shown in Fig. 6A, B (bottom panels), none of the agonists caused β-arrestin1 or β-arrestin2 recruitment to TAS2R14-10A. We also examined higher concentrations of DPD for purposes of comparison with other studies. DPD at 500 µM and 1.0 mM also evoked β-arrestin1 and 2 of TAS2R14-WT but not TAS2R14-10A (Fig. 6C, D). The EEA1 colocalization studies were also performed with TAS2R-10A to further validate the proposed agonist-promoted pathway toward down-regulation (Fig. 4B). As with TAS2R-WT, DPD had little effect on colocalization of TAS2R-10A and EEA1. However, treatment with CLX and FFA also failed to display colocalization of TAS2R14-10A, consistent with the lack of agonist-promoted β-arrestin1 or β-arrestin2 recruitment observed with this mutant with any agonist. Because all of the agonists failed to promote early endosome association of TAS2R-10A, we expected that there would be no association with late endosomes, as indicated by colocalization with LAMP1. This indeed was the case for all agonists studied (Fig. 5B, C). In addition, there was no significant down-regulation of TAS2R14-10A density evoked by FFA, AA, CLX, or DPD observed in whole-cell lysates or at the cell surface (Fig. 7).
Figure 6.

β-Arrestin recruitment by TAS2R14 agonists. HEK-293T cells were transfected to express GFP–β-arrestin1 or GFP–β-arrestin2 and either TAS2R14-WT or TAS2R14-10A and exposed to the indicated agonists for 5 min. A) All agonists caused a redistribution of GFP–β-arrestin1 from a homogenous intracellular pattern to a punctate cell surface pattern with TAS2R14-WT, whereas none of the agonists evoked these changes with TAS2R14-10A. B) This same pattern was observed with GFP–β-arrestin2. C, D). Additional doses of 500 µM and 1.0 mM were used with DPD, revealing redistribution of both β-arrestins with TAS2R14-WT but not TAS2R14-10A. As a positive control, the β2-AR was transfected with either of the β-arrestins, and cells were treated with 1.0 µM isoproterenol (Iso). Veh, vehicle. Results are representative of 4 independent experiments. Scale bar, 5 µm.
Figure 7.

Effects of long-term agonist exposure on TAS2R14-10A expression. HEK-293T cells were transfected to express FLAG-tagged TAS2R14 and exposed to TAS2R14 agonists for 18 h in culture. A, B) Immunoblotting of whole-cell lysates reveals no down-regulation of TAS2R14-10A receptor expression when cells were treated with any agonist. C, D) Confocal immunofluorescent imaging of nonpermeabilized cells shows no change in the cell surface TAS2R14-10A expression for any of the agonists. Vec, vector control; Veh, vehicle. A, C) Representative experiments. B, D) Results from 4 independent experiments. Scale bar, 10 µm
Molecular docking reveals unique interactions of DPD deep within the binding pocket
To ascertain the basis for the biased action of DPD, in which coupling is preserved while down-regulation is minimal due to a lack of receptor internalization, we developed a model for in silico docking of the 5 compounds to TAS2R14. The model was developed through homology modeling and is based upon the crystal structure of C-C chemokine receptor type 5 (Protein Data Bank ID: 5UIW). This template was selected, as the resulting model had the highest Global Model Quality Estimation score (0.51), which reflects the overall expected accuracy of a model built from a selected template. Docking into the model’s binding pocket was attempted with all 5 compounds, but AA was unable to be docked successfully. Of the 4 other compounds, DPD was found to bind deepest into the pocket, and it situated into a subpocket formed by Trp66, Phe71, Trp89, and Gln266 (Fig. 8). The other 3 docked compounds were not able to engage this subpocket, particularly because of poor interactions with Trp66 and Phe71. Consequently, they were unable to penetrate as deeply into the overall binding pocket and displayed poorer shape complementarity with the protein, potentially leading to increased conformational variability of both the ligand and the receptor. The unique binding pose of DPD prompted us to dock a related compound, ORP, to TAS2R14. Comparison of the 2 structures is shown in Supplemental Fig. S3A. When ORP was docked to the same model, it also revealed binding deep within the pocket, with interactions at the same 4 amino acids as DPD (Supplemental Fig. S3B). We thus expected a similar phenotype (lack of down-regulation) with ORP, as was seen with DPD, because both compounds display similar poses. Exposure of cells to ORP indeed failed to evoke down-regulation of TAS2R14 (Supplemental Fig. S3C), consistent with a cellular phenotype similar to that of DPD.
Figure 8.

Binding pose of DPD in the TAS2R14 homology model binding pocket. DPD binds in a subpocket formed by Trp66, Phe71, Trp89, and Gln266, which the other docked compounds were only able to access partially.
DISCUSSION
GPCR desensitization is a mechanism whereby cells regulate individual receptor activity and integrate the multiple signals being received by the cell from other receptors and events. In normal physiologic circumstances, this regulation provides for homeostasis in response to events over seconds to days. In pathologic conditions, GPCR desensitization can have a positive adaptive effect or can be deleterious and contribute to the disease (32–34). There are some GPCRs that appear to be resistant to short- or long-term desensitization, which may be due to their critical function. For example, the β3-AR of brown adipose tissue does not desensitize to short-term agonists, potentially because of its importance in mediating sympathetic nonshivering thermogenesis in the neonate (35). Similarly, the α2c-AR, which regulates presynaptic release of norepinephrine at cardiac presynaptic terminals, does not desensitize (9, 36). This may protect the heart from catecholamine overload, which can have acute and chronic cardiotoxicity. Thus, to fully appreciate the physiologic role of a given GPCR in a specific cell type and its potential to be a therapeutic target, agonist-promoted desensitization and the mechanisms responsible for this regulation should be explored. TAS2Rs were initially thought to have a highly restricted expression to taste cells of the tongue. However, it has now become clear that 1 or more of the 25 human TAS2R subtypes are expressed on multiple cell types of many organs (37–39). Although the function of many of these extraoral TAS2Rs has not been elucidated, the role of these receptors on smooth muscle cells of the airway has been intensely investigated. One motivation for these studies is the poor control of moderate and severe asthmatics (40–42) and the lack of any direct bronchodilators for treatment of airway obstruction except for β2-AR agonists (43).
There are a number of attractive features of TAS2Rs for being potential drug targets for asthma and chronic obstructive lung disease. There are currently thousands of bitter substances (from plants or existing drugs) that may be agonists at these receptors (17). TAS2Rs promote an equal or greater extent of airway relaxation than β2-AR agonists (15) and exert their action via a non-cAMP mechanism, distinguishing them from β2-AR agonists. Furthermore, because delivery by inhalation is possible, systemic effects of a TAS2R agonist given for the treatment of obstructive lung disease could be minimized. Although acute treatment of airway obstruction is an important goal, the use of bronchodilators to maintain airway caliber on an ongoing basis (often termed “controller” therapy) improves clinical outcome (44). Thus, the effects of chronic TAS2R agonist exposure on receptor function and regulation is an important issue for potential new drugs acting at these targets. We have previously shown that HASM TAS2Rs undergo ∼30% desensitization after 15 min of exposure to some agonists (18). In the current work, we have explored long-term agonist exposure, examining receptor coupling to [Ca2+]i, β-arrestin recruitment, internalization, and trafficking, with a more prolonged and clinically relevant exposure time. We utilized a number of structurally distinct agonists, which act at TAS2R14, because of the potential for biased ligand activity. This concept is shown in Eq. 2, where R is the unbound receptor, A1 and A2 are different agonists, and R*1 and R*2 are distinct conformations stabilized by the given agonist (Eq. 2).
Implicit in the biased ligand concept is the fact that a given GPCR can evoke more than 1 event. The existence or amplitude of these events could vary depending on the structure of the agonist because agonist structure could stabilize the receptor in 1 or more specific conformations. As an example, one agonist could activate receptor coupling to the G-protein as well as promote internalization, whereas another could be biased away from internalization but maintain G-protein coupling.
Initial studies were in HASM and examined desensitization to the 5 agonists after 18 h of drug exposure, measuring [Ca2+]i as the product of TAS2R coupling to G-αi (43). All but 1 agonist (DPD) evoked 45–60% desensitization. DPD desensitization was ∼25%, with an ∼12% heterologous component. This provided the first indication that DPD might largely circumvent long-term agonist-promoted down-regulation. And, indeed, in immunoblots of cell lysates and whole-cell immunofluorescence confocal imaging, we observed a net loss of receptors as well as cell surface receptors for the tested agonists except for DPD. There was no decrease of TAS2R14 with long-term DPD treatment, and in fact, there was typically an increase in the net expression and the cell surface expression with this agonist. For most GPCRs, the internalized receptor in vesicles is routed to early endosomes, which can be identified with antibodies to EEA1. Imaging studies aimed at colocalization of TAS2R14 and EEA1 in transfected HEK cells revealed extensive colocalization for all tested agonists except for DPD. Interestingly, DPD did evoke recruitment of β-arrestin1 and β-arrestin2, which typically precedes internalization. Taken together, it appears that DPD does establish a receptor conformation that leads to GRK phosphorylation and subsequent β-arrestin recruitment, but further assembly into intracellular vesicles and entry into the degradation pathway does not occur. Consistent with this notion is the lack of colocalization of TAS2R14 with the late endosomal marker LAMP1 under long-term exposure to DPD, whereas the other agonists were found to promote colocalization with the late endosomes and display a net decrease of cellular TAS2R14.
The mutated TAS2R14 lacking GRK phosphoacceptor sites was utilized in several ways. This receptor failed to recruit β-arrestin by any agonist, indicating that TAS2R14 uses this common GPCR pathway for agonist-promoted internalization. This was further validated by the lack of cell surface receptor loss, or colocalization with early endosomes, of TAS2R14-10A when exposed to the agonists CLX, AA, and FFA. The link between this common pathway and ultimate loss of receptor was established by noting the lack of colocalization with the late endosomal marker and no detectable down-regulation of TAS2R14-10A by any of the agonists.
The actions of DPD, which are biased away from agonist-promoted internalization and down-regulation, were further explored using in silico molecular docking. There is no crystal structure for TAS2Rs, so a homology model was constructed by using the Swiss-Model homology-modeling server. Although AA could not be docked, all the other agonists fit into a binding pocket that included residues Trp66, Phe71, Trp89, Leu181, Phe247, Gln266, and others. DPD was found to bind the deepest into a pocket formed by Phe71, Trp66, Trp89, and Gln266 (Fig. 8), with particularly extensive favorable interactions with Trp66 and Phe71, which was not found with the other compounds. The TAS2R14 agonist ORP, which is structurally related to DPD, also docked in a similar manner to DPD. ORP also failed to evoke down-regulation. It thus appears that the biasing is dependent on 1 or more of these deep-pocket interactions. Despite the exploratory nature of these modeling results and the limitations of docking to GPCR crystal structures and homology models (45), they point to potential unique interactions of DPD and ORP with the receptor. These compounds may lock the receptor–β-arrestin complex in some way to mitigate internalization. The extended effect, then, would be the lack of down-regulation of TAS2R14 by these agonists during long-term agonist exposure with minimal functional desensitization compared with the other agonists studied.
CONCLUSIONS
TAS2R14 is subject to long-term agonist-promoted down-regulation of receptor protein leading to functional desensitization. The mechanism is similar to a number of other GPCRs, in which the agonist-stabilized conformation of the receptor promotes binding of β-arrestins to the phosphorylated receptor. Subsequently, the receptor internalizes to the cell interior and undergoes degradation following an early to late endosomal pathway. The clinical correlate to this long-term regulation, tachyphylaxis, would thus be expected for TAS2R14 agonists that evoke this aforementioned set of events. We considered, though, that manipulation of the molecular structure of the ligand might result in a biased agonist, which stabilizes the receptor conformation to a state that promotes receptor coupling (and thus physiologic function) but not long-term down-regulation. This scenario was indeed found with the agonist DPD, and molecular docking reveals unique contact points for this agonist within the ligand binding pocket. These results indicate that DPD or similar TAS2R14-biased agonists may be particularly useful therapeutic agents for treating airway obstruction in asthma and chronic obstructive lung disease in that they evoke signaling yet may be associated with minimal tachyphylaxis due to structural features that limit the down-regulation effect.
ACKNOWLEDGMENTS
The authors thank Melissa Steward (University of South Florida Morsani College of Medicine) for manuscript preparations. This work was funded by U.S. National Institutes of Health, National Heart, Lung, and Blood Institute Grants HL114471 and HL045967. The authors declare no conflicts of interest.
Glossary
- [Ca2+]i
intracellular [Ca2+]
- AA
aristolochic acid
- AR
adrenergic receptor
- BRA
bradykinin
- CLX
chlorhexidine
- DPD
diphenhydramine
- ET1
endothelin1
- FFA
flufenamic acid
- GFP
green fluorescent protein
- GRK
GPCR kinase
- HASM
human airway smooth muscle
- HEK
human embryonic kidney
- IONO
ionomycin
- LAMP1
lysosome-associated membrane glycoprotein 1
- ORP
orphenadrine
- PAP
papaverine
- TAS2R
bitter taste receptor
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
J. A. Woo, D. Kim, E. M. Lewandowski, Y. Chen, and S. B. Liggett conceived the project, directed or performed the experiments, analyzed and interpreted the data, and wrote the paper; and M. Castaño and A. Goss performed and analyzed the experiments.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Gainetdinov R. R., Premont R. T., Bohn L. M., Lefkowitz R. J., Caron M. G. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144 [DOI] [PubMed] [Google Scholar]
- 2.Rajagopal S., Shenoy S. K. (2018) GPCR desensitization: acute and prolonged phases. Cell. Signal. 41, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irannejad R., Tsvetanova N. G., Lobingier B. T., von Zastrow M. (2015) Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol. 35, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W. C., Mihlbachler K. A., Brunnett A. C., Liggett S. B. (2009) Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway β2-adrenergic receptor physiologic signaling. Proc. Natl. Acad. Sci. USA 106, 15007–15012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turki J., Green S. A., Newman K. B., Meyers M. A., Liggett S. B. (1995) Human lung cell β 2-adrenergic receptors desensitize in response to in vivo administered β-agonist. Am. J. Physiol. 269, L709–L714 [DOI] [PubMed] [Google Scholar]
- 6.Wang W. C., Juan A. H., Panebra A., Liggett S. B. (2011) MicroRNA let-7 establishes expression of beta2-adrenergic receptors and dynamically down-regulates agonist-promoted down-regulation. Proc. Natl. Acad. Sci. USA 108, 6246–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green S. A., Turki J., Bejarano P., Hall I. P., Liggett S. B. (1995) Influence of β 2-adrenergic receptor genotypes on signal transduction in human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 13, 25–33 [DOI] [PubMed] [Google Scholar]
- 8.Eason M. G., Jacinto M. T., Liggett S. B. (1994) Contribution of ligand structure to activation of alpha 2-adrenergic receptor subtype coupling to Gs. Mol. Pharmacol. 45, 696–702 [PubMed] [Google Scholar]
- 9.Jewell-Motz E. A., Small K. M., Theiss C. T., Liggett S. B. (2000) Alpha 2A/alpha 2C-adrenergic receptor third loop chimera show that agonist interaction with receptor subtype backbone establishes G protein-coupled receptor kinase phosphorylation. J. Biol. Chem. 275, 28989–28993 [DOI] [PubMed] [Google Scholar]
- 10.Swift S. M., Schwarb M. R., Mihlbachler K. A., Liggett S. B. (2007) Pleiotropic β-agonist-promoted receptor conformations and signals independent of intrinsic activity. Am. J. Respir. Cell Mol. Biol. 36, 236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D., Cho S., Castaño M. A., Panettieri R. A., Woo J. A., Liggett S. B. (2019) Biased TAS2R bronchodilators inhibit airway smooth muscle growth by downregulating phosphorylated extracellular signal-regulated kinase 1/2. Am. J. Respir. Cell Mol. Biol. 60, 532–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wisler J. W., Xiao K., Thomsen A. R., Lefkowitz R. J. (2014) Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandrashekar J., Mueller K. L., Hoon M. A., Adler E., Feng L., Guo W., Zuker C. S., Ryba N. J. (2000) T2Rs function as bitter taste receptors. Cell 100, 703–711 [DOI] [PubMed] [Google Scholar]
- 14.Deshpande D. A., Wang W. C. H., McIlmoyle E. L., Robinett K. S., Schillinger R. M., An S. S., Sham J. S. K., Liggett S. B. (2010) Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat. Med. 16, 1299–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshpande D. A., Robinett K. S., Wang W. C., Sham J. S., An S. S., Liggett S. B. (2011) Bronchodilator activity of bitter tastants in human tissue. Nat. Med. 17, 776–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liggett S. B. (2013) Bitter taste receptors on airway smooth muscle as targets for novel bronchodilators. Expert Opin. Ther. Targets 17, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyerhof W., Batram C., Kuhn C., Brockhoff A., Chudoba E., Bufe B., Appendino G., Behrens M. (2010) The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 35, 157–170 [DOI] [PubMed] [Google Scholar]
- 18.Robinett K. S., Deshpande D. A., Malone M. M., Liggett S. B. (2011) Agonist-promoted homologous desensitization of human airway smooth muscle bitter taste receptors. Am. J. Respir. Cell Mol. Biol. 45, 1069–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim D., Woo J. A., Geffken E., An S. S., Liggett S. B. (2017) Coupling of airway smooth muscle bitter taste receptors to intracellular signaling and relaxation is via Gαi1,2,3. Am. J. Respir. Cell Mol. Biol. 56, 762–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D., Pauer S. H., Yong H. M., An S. S., Liggett S. B. (2016) β2-adrenergic receptors chaperone trapped bitter taste receptor 14 to the cell surface as a heterodimer and exert unidirectional desensitization of taste receptor function. J. Biol. Chem. 291, 17616–17628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bienert S., Waterhouse A., de Beer T. A. P., Tauriello G., Studer G., Bordoli L., Schwede T. (2017) The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. 45, D313–D319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guex N., Peitsch M. C., Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30 (Suppl 1), S162–S173 [DOI] [PubMed] [Google Scholar]
- 24.Benkert P., Biasini M., Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertoni M., Kiefer F., Biasini M., Bordoli L., Schwede T. (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 7, 10480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorber D. M., Shoichet B. K. (2005) Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 5, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorber D. M., Shoichet B. K. (1998) Flexible ligand docking using conformational ensembles. Protein Sci. 7, 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang N., Shoichet B. K., Irwin J. J. (2006) Benchmarking sets for molecular docking. J. Med. Chem. 49, 6789–6801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng E. C., Gschwend D. A., Blaney J. M., Kuntz I. D. (1993) Orientational sampling and rigid-body minimization in molecular docking. Proteins 17, 266–278 [DOI] [PubMed] [Google Scholar]
- 30.Shoichet B. K., Leach A. R., Kuntz I. D. (1999) Ligand solvation in molecular docking. Proteins 34, 4–16 [DOI] [PubMed] [Google Scholar]
- 31.Adler E., Hoon M. A., Mueller K. L., Chandrashekar J., Ryba N. J., Zuker C. S. (2000) A novel family of mammalian taste receptors. Cell 100, 693–702 [DOI] [PubMed] [Google Scholar]
- 32.Wettschureck N., Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 33.Rockman H. A., Koch W. J., Lefkowitz R. J. (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 [DOI] [PubMed] [Google Scholar]
- 34.Salazar N. C., Chen J., Rockman H. A. (2007) Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 1768, 1006–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bengtsson T., Redegren K., Strosberg A. D., Nedergaard J., Cannon B. (1996) Down-regulation of beta3 adrenoreceptor gene expression in brown fat cells is transient and recovery is dependent upon a short-lived protein factor. J. Biol. Chem. 271, 33366–33375 [DOI] [PubMed] [Google Scholar]
- 36.Eason M. G., Liggett S. B. (1992) Subtype-selective desensitization of α 2-adrenergic receptors. Different mechanisms control short and long term agonist-promoted desensitization of α 2C10, α 2C4, and α 2C2. J. Biol. Chem. 267, 25473–25479 [PubMed] [Google Scholar]
- 37.Clark A. A., Liggett S. B., Munger S. D. (2012) Extraoral bitter taste receptors as mediators of off-target drug effects. FASEB J. 26, 4827–4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.An S. S., Liggett S. B. (2018) Taste and smell GPCRs in the lung: evidence for a previously unrecognized widespread chemosensory system. Cell. Signal. 41, 82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liggett S. B. (2014) Bitter taste receptors in the wrong place: novel airway smooth muscle targets for treating asthma. Trans. Am. Clin. Climatol. Assoc. 125, 64–74; discussion 74–75 [PMC free article] [PubMed] [Google Scholar]
- 40.Malmstrom K., Rodriguez-Gomez G., Guerra J., Villaran C., Piñeiro A., Wei L. X., Seidenberg B. C., Reiss T. F.; Montelukast/Beclomethasone Study Group (1999) Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma. A randomized, controlled trial. Ann. Intern. Med. 130, 487–495 [DOI] [PubMed] [Google Scholar]
- 41.Drazen J. M., Silverman E. K., Lee T. H. (2000) Heterogeneity of therapeutic responses in asthma. Br. Med. Bull. 56, 1054–1070 [DOI] [PubMed] [Google Scholar]
- 42.Sharma S., Litonjua A. A., Tantisira K. G., Fuhlbrigge A. L., Szefler S. J., Strunk R. C., Zeiger R. S., Murphy A. J., Weiss S. T.; Childhood Asthma Management Program Research Group (2008) Clinical predictors and outcomes of consistent bronchodilator response in the childhood asthma management program. J. Allergy Clin. Immunol. 122, 921–928.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matera M. G., Page C. P., Cazzola M. (2011) Novel bronchodilators for the treatment of chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 32, 495–506 [DOI] [PubMed] [Google Scholar]
- 44.Shrewsbury S., Pyke S., Britton M. (2000) Meta-analysis of increased dose of inhaled steroid or addition of salmeterol in symptomatic asthma (MIASMA). BMJ 320, 1368–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim V. J. Y., Du W., Chen Y. Z., Fan H. (2018) A benchmarking study on virtual ligand screening against homology models of human GPCRs. Proteins 86, 978–989 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.