

Abstract
Insufficient autophagy has been reported in idiopathic pulmonary fibrosis (IPF) lungs. Specific roles of autophagy-related proteins in lung fibrosis development remain largely unknown. Here, we investigated the role of autophagy marker protein microtubule-associated protein 1 light chain 3β (LC3B) in the development of lung fibrosis. LC3B−/− mice upon aging show smaller lamellar body profiles, increased cellularity, alveolar epithelial cell type II (AECII) apoptosis, surfactant alterations, and lysosomal and endoplasmic reticulum stress. Autophagosomal soluble N-ethylmaleimide–sensitive factor attachment protein receptor syntaxin 17 is increased in the AECII of aged LC3B−/− mice and patients with IPF. Proteasomal activity, however, remained unaltered in LC3B−/− mice. In vitro knockdown of LC3B sensitized mouse lung epithelial cells to bleomycin-induced apoptosis, but its overexpression was protective. In vivo, LC3B−/− mice displayed increased susceptibility to bleomycin-induced lung injury and fibrosis. We identified cathepsin A as a novel LC3B binding partner and its overexpression in vitro drives MLE12 cells to apoptosis. Additionally, cathepsin A is increased in the AECII of aged LC3B−/− mice and in the lungs of patients with IPF. Our study reveals that LC3B mediated autophagy plays essential roles in AECII by modulating the functions of proteins like cathepsin A and protects alveolar epithelial cells from apoptosis and subsequent lung injury and fibrosis.—Kesireddy, V. S., Chillappagari, S., Ahuja, S., Knudsen, L., Henneke, I., Graumann, J., Meiners, S., Ochs, M., Ruppert, C., Korfei, M., Seeger, W., Mahavadi, P. Susceptibility of microtubule-associated protein 1 light chain 3β (MAP1LC3B/LC3B) knockout mice to lung injury and fibrosis.
Keywords: alveolar epithelial cells, lamellar bodies, autophagy, aging, lysosome
Idiopathic pulmonary fibrosis (IPF) represents one of the most aggressive forms of organ fibrosis, killing patients within a few years of diagnosis (1). IPF has a remarkable age-related onset and is characterized by a progressive decline in lung function with the histologic appearance of usual interstitial pneumonia. The pathogenesis of IPF remains to be fully elucidated but may be based on chronic epithelial injury, disturbed epithelial-mesenchymal crosstalk, activation of fibroblasts, excessive collagen deposition, and disruption of the delicate alveolar architecture, altogether resulting in progressive dyspnea, decline of lung function and, ultimately, death (2, 3). Several studies published in the past underscore the importance of the alveolar epithelium in the evolution of IPF and lung fibrosis in general; already, the very first report by Myers and Katzenstein on IPF disclosed extensive alveolar epithelial type II cell (AECII) death and suggested a pathomechanistic role for the epithelium (4). AECII are secretory cells, and lamellar bodies of the AECII are lysosome-related organelles that are responsible for the storage and secretion of lung surfactant (5, 6). Qualitative and quantitative alterations in lamellar bodies within the hypertrophic AECII are well reported in patients with IPF as well as in several animal models. Likewise, alterations in the lysosomal compartment within the AECII in both clinical as well as experimental models of lung fibrosis are well documented. Firstly, pronounced changes in surfactant properties in patients with IPF have been reported (7–11). Furthermore, giant lamellar bodies, alterations in the surfactant secretion pathway, and lysosomal stress were linked to the development of pulmonary fibrosis in Hermansky-Pudlak syndrome (HPS)-associated and in amiodarone-induced lung fibrosis (12, 13). Along the same lines, we and others have shown that autophagy, a lysosome-dependent protein quality control mechanism of a cell, is either defective as in IPF and in HPS-associated lung fibrosis or excessively active as in the amiodarone model (10, 14–16), which, in either case, indicates the pathologic relevance of autophagy in the development of lung fibrosis.
Autophagy is a tightly regulated intracellular housekeeping mechanism that degrades long-lived proteins and unnecessary/nonfunctional organelles (17). It involves the function and interaction of several autophagy-related (Atg) gene products, which result in the conversion of the cytosolic form of the microtubule-associated protein 1 light chain 3β (MAP1LC3B/LC3B) to the membrane-bound, lipidated LC3BII form that labels the double membraned structures called autophagosomes (17, 18). These autophagosomes fuse with lysosomes to degrade their contents. Recently, it was shown that mammalian orthologs of yeast Atg1 (ULK)1 and 2 double knockout mice display lung abnormalities including glycogen-laden AECII (19). In addition, autophagy was shown to be altered in the widely used bleomycin model of lung fibrosis and that mice deficient for the deconjugation protease, Atg4, display accelerated apoptosis and lung fibrosis following bleomycin challenge (20).
We previously demonstrated that LC3B is localized to the limiting membrane of lamellar bodies of AECII in 2 models of lung fibrosis: the amiodarone model and the HPS model (10, 16). In addition, in the amiodarone model, we reported that the autophagosomes and lamellar bodies share the same membrane (16) and under conditions of HPS1 knockdown, overexpression of LC3B rescued cells from defective autophagy (10). Based on these observations, we now aimed to further understand the role of LC3B in the development of lung fibrosis.
MATERIALS AND METHODS
Mice and human lung tissue
Homozygous LC3B−/− mice (B6; 129P2-Map1lc3btm1Mrab/J) and wild-type (WT) control mice (B6129PF2/J) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Both the University Animal Care Committee and the Federal Authorities for Animal Research of the Regierungspraesidium Giessen (Hessen, Germany) approved the study protocol. Mice were bred and maintained in specific pathogen-free conditions. Thirteen- and 42-wk-old mice were euthanized following the protocols previously described (12, 13). Mice that were ~13 wk old were treated with vehicle (0.9% NaCl) or 1.5 U/kg bodyweight bleomycin (Hexal AG, Holzkirchen, Germany) intratracheally. Each group consisted of about 5–10 animals: LC3B−/− or WT mice. Mice were euthanized according to the protocols previously described (10, 13). Shock-frozen lung tissue samples from 16 patients with IPF (mean age 54 ± sd: 9.14) and 6 nondiseased control subjects (donors; mean age 48.16 ± sd: 8) were used for homogenization and subsequent Western blot analysis. Additionally, formalin-fixed, paraffin-embedded lung tissue samples from 5 patients with sporadic IPF (mean age 54.8 ± sd: 11.07) and 5 nondiseased control subjects (mean age 50.8 ± sd: 9.41) were used for immunohistochemistry. All IPF diagnoses were made according to the American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus criteria (21). The Universities of Giessen and Marburg Lung Center (UGMLC) Giessen Biobank [member of the German Centre for Lung Research (DZL) Platform Biobanking] provided all human lung tissue samples. The study protocol was approved by the Ethics Committee of the Justus-Liebig University Giessen (111/08 and 58/15).
Electron microscopy
After fixation of the lungs by instillation of 1.5% glutaraldehyde, 1.5% paraformaldehyde in 0.15 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer, lungs were subjected to a systematic uniform random sampling, and 4–6 tissue pieces per lung were sampled (22), post fixed with osmium and uranyl acetate, and embedded in epoxy resin (Epon; Miller-Stephenson, Danbury, CT, USA). Afterwards, ultrathin sections were cut (thickness 80 nm). Electron microscopic evaluation of 4 sections per organ was performed using a Morgagni 268 Electron Microscope (Thermo Fisher Scientific, Waltham, MA, USA). A systematic uniform random area sampling was performed to sample 150–200 profiles of AECII for stereological assessment of the intracellular surfactant pool defined by ultrastructural criteria as the amount of lamellar bodies. Random test points were used to count points hitting lamellar bodies or all other structures of the sampled AECII to determine the volume fraction of lamellar bodies within AECII. In addition, the point sampled intercept method was used to determine the volume-weighted mean volume of lamellar bodies, a parameter that is dependent on the number-weighted mean volume as well as the coefficient of variation of lamellar bodies (23).
Histology and immunofluorescence
Lung tissues harvested from mice were fixed in 4% formaldehyde and embedded in formalin, and serial sections of thickness 3–6 μm were prepared. Hematoxylin and eosin (H&E) and masson goldner staining were performed according to the previously described protocols (12). A ZytomedChem Plus Broad Spectrurm Kit (AP-Fast Fed, AP008RED-MS; Zytomed Systems, Berlin, Germany) was used to perform immunostaining of cleaved caspase-3 (9664; Cell Signaling Technology, Danvers, MA, USA), pro SP-C (AB3786; MilliporeSigma, Burlington, MA, USA), syntaxin 17 (PA5-40127; Thermo Fisher Scientific), and cathepisin A (ab184553; Abcam, Cambridge, United Kingdom) as per the manufacturer’s instructions and as previously described (13). Hamamatsu scanner (Nanozoomer 2.0 RS; Hamamatsu Photonics, Hamamatsu City, Japan) was used for scanning the stained sections. NDP.view 2.0 software was used for preparing and analyzing the images. For quantifying immunohistochemistry images, 5–6 regions from 5 mice per group were randomly analyzed for intensity quantification using ImageJ software (National Institutes of Health, Bethesda, MD, USA) following the instructions given in the ImageJ documentation (https://www.unige.ch/medecine/bioimaging/files/1914/1208/6000/Quantification.pdf). For staining on lung sections, prosurfactant protein C (proSP-C) antibody (AB3786; MilliporeSigma) was labeled with Alexa Fluor 488 using the Alexa Fluor 488 antibody labeling kit from Thermo Fisher Scientific (A20181), following the manufacturer’s instructions, and was used to stain AECII on mouse lung sections for costaining studies. For human sections, ATP binding cassette subfamily A member 3 (ABCA3) antibody was used to stain AECII. Microscopy was performed using a Leica M205 FA Fluorescent Stereoscope (Leica Microsystems, Buffalo Grove, IL, USA) equipped with a Leica DFC360 FX camera. Immunofluorescence images were quantified using Leica LAS X LS software on 10–15 randomly selected regions per mouse or human sections and represented as mean fluorescence intensity, using the sem. Four to 5 mice or human sections per group were used for each staining with subsequent quantification.
Western blot
Denatured lung homogenates and cell lysates were used for Western blotting as previously described (12). Antibodies used for Western blots are listed in Table 1.
TABLE 1.
List of antibodies used
| Name | Catalog no. | Company |
|---|---|---|
| Immunoblotting and immunoprecipitation | ||
| β-actin | ab8227 | Abcam |
| ATF6 | NBP1-77251 | Novus Biologicals, Centennial, CO, USA |
| Cleaved caspase-3 | 9664 | Cell Signaling Technology |
| Cathepsin D | AF1029 | R&D Systems, Minneapolis, MN, USA |
| Cathepsin A | ab184553 and ab217857 | Abcam |
| Cathepsin L () | ab58991 | Abcam |
| Gadd163 | sc-7351 HRP | Santa Cruz Biotechnology |
| GFP | ab5450 | Abcam |
| GABBARAPL1 | Genetex, Irvine, CA, USA | |
| Glyceraldehyde 3-phosphate dehydrogenase | 2118s | Cell Signaling Technology |
| LC3B | ab48394 and 2775s | Abcam and Cell Signaling Technology |
| Lamp1 | ab24170 | Abcam |
| Lamp2 | MABC40 | MilliporeSigma |
| Myc | ab9106 | Abcam |
| Mature SP-C | WRAB-76694 | Seven Hills, Cincinnati, OH, USA |
| Mature SP-B | WRAB-48604 | Seven Hills |
| PSMD11 | 14786-1-1AP | Proteintech, Rosemont, IL, USA |
| Proteasome 20s core subunit | BML-PW8155-0100 | Enzo Life Sciences, Farmingdale, NY, USA |
| ProSP-C | AB3786 | MilliporeSigma |
| Pro-SP-B | WRAB-55522 | Seven Hills |
| P62 | P0067 | MilliporeSigma |
| Syntaxin 17 () | PA5-40127 | Thermo Fisher Scientific |
| Immunohistochemistry and immunofluorescence | ||
| ABCA3 | WAMB-ABCA3-17 | Seven Hills |
| Cleaved caspase-3 | 9664 | Cell Signaling Technology |
| Cathepsin A | ab184553 and ab217857 | Abcam |
| Pro-SP-C | AB3786 | MilliporeSigma |
| Syntaxin 17 | PA5-40127 | Thermo Fisher Scientific |
| P62 | P0067 | MilliporeSigma |
Proteasome activity assay
Total lung extracts from young and old LC3B−/− and WT control mice were generated using native lysis conditions. Proteasome activity was monitored in total extracts for the chymotrypsin-like and caspase-like proteasome activities using the Promega Glo Assay (Madison, WI, USA) as previously described (24).
Cell culture
Small interfering RNA (siRNA) for LC3B was purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and transfections in MLE12 cells were performed according to the previously described protocols (16). Cells were plated 1 d before the experiment, and when they reached confluency, they were transfected with nontargeting (NT) siRNA or with siRNA against mouse LC3B (Santa Cruz Biotechnology) for 24 h followed by treatment with bleomycin for 4 h and then harvesting. Green fluorescent protein (GFP)-LC3B was generated as previously described (16), and mouse cathepsin A tagged with myc was from OriGene Technologies (Rockville, MD, USA; NM_001038492; MR207589). Cotransfection of these plasmids in MLE12 cells was performed using Lipofectamine 2000 (Thermo Fisher Scientific), and cells were harvested after 24 h. Alveolar epithelial cells from LC3B−/− or WT mice were isolated as previously described (12). Briefly, 42-wk-old LC3B−/− (5 mice) or WT mice (5 mice) were anesthetized and dissected followed by lung lavaging. Lungs were flushed with 0.9% NaCl through the left ventricle, and 1.5 ml dispase and 0.5 ml melted agarose were injected, and then the dissected lung was incubated in dispase at room temperature for 45 min. Lung tissue was then minced. Cell populations were not pooled, and cells from each mouse were analyzed separately. Following panning on antibody-coated dishes (using CD16/CD45; BD Biosciences, San Jose, CA, USA), cells were coated on noncoated dishes for 30 min at 37°C/4% CO2 incubator. Suspensions were centrifuged and pellets were frozen for further analysis. Control staining was performed using proSP-C (AB3786; MilliporeSigma) antibody as well as Nile red staining.
Coimmunoprecipitation
Tosylactivated dynabeads (Thermo Fisher Scientific) were used for all coimmunoprecipitation experiments, and immunoprecipitation was performed according to the manufacturer’s protocol. Briefly, dynabeads were coupled to the antibody (anti-myc, anti-cathepsin A, or rabbit IgG) by incubating them overnight at room temperature on a tube rotator. The next day, supernatant was removed by placing the tubes in a magnetic stand. After washing, antibody coupled beads were added to the sample [freshly harvested MLE12 cells using immunoprecipitated lysis buffer (Thermo Fisher Scientific)] and incubated in a tube rotator overnight at 4°C. An aliquot of 10% of the lysate, before adding it to the beads, was used as input. The next day, an unbound sample was discarded by placing the tubes in a magnetic stand. Following washing, the immunoprecipitated samples or input were then denatured at 98°C for 10 min in buffer containing SDS and were subjected to Western blotting.
Statistics
All data are expressed as means ± sd of ≥5 mice for in vivo studies. For in vitro experiments, ≥ independent experiments were conducted for bleomycin treatment and triplicate transfections were performed for siRNA/GFP-LC3B/cathepsin A-myc studies. Statistical significance was assessed using the Mann-Whitney U test. Results were significant when P < 0.05.
RESULTS
LC3B−/− mice display age-dependent alteration in lung structure
In view of our previous studies that showed a critical role of LC3B in HPS-associated lung fibrosis as well as in the amiodarone model of lung fibrosis and based on observations from other studies that reported lung-specific alterations in certain autophagy gene knockout mouse models, we now asked if LC3B knockout mice would display lung alterations. A careful histologic analysis of the lungs of 13-wk-old LC3B−/− mice (young mice) and age-matched WT control mice revealed normal lung architecture. However, 42-wk-old LC3B−/− mice (aged mice) did show areas with increased cellularity and reduced airspaces (Fig. 1A and Supplemental Fig. S1). Because immunogold labeling demonstrated a preferential binding of LC3B antibodies to the limiting membrane of lamellar bodies in our previous studies (10, 25), we closely examined the ultrastructure of AECII in LC3B−/− mice. The morphology of lamellar bodies with respect to the densely packed lipid layers as well as the limiting membrane did not differ between WT and LC3B−/− mice. At best, in some AECII of the LC3B−/− mice, there were fewer and smaller profiles of lamellar bodies compared with WT mice (Fig. 1B). Thus, we added a stereological assessment and determined the volume fraction of lamellar bodies within AECII as well as the volume-weighted mean volume of lamellar bodies. The volume fraction was significantly reduced in LC3B−/− mice compared with WT mice, meaning that per volume of AECII, the cells contained less intracellular surfactant (Fig. 1C). Also, the volume-weighted mean volume was significantly reduced in LC3B mice, indicating that lamellar bodies were smaller or characterized by a reduced size variability (Fig. 1D). Analysis of surfactant proteins revealed significant difference only in pro and mature surfactant protein B (SP-B) levels in lung homogenates between LC3B−/− and WT mice (Supplemental Fig. S2A–F).
Figure 1.

Decreased airspaces and diameter of distal air spaces in aged LC3B−/− mice. A) Representative H&E staining of complete left lungs (upper panel) and higher magnification images of the H&E staining (middle panel) and trichrome staining (lower panel) of 13- and 42-wk-old LC3B−/− and WT mice. Scale for the whole lungs, 2 mm; scale for the higher magnification pictures, 100 μm; original magnification, ×200. B) Representative transmission electron microscopic images from 42-wk-old LC3B−/− and age-matched WT mice showing an example of smaller profiles of lamellar bodies in LC3B−/− compared with WT (primary original magnification, ×8900). C, D) Stereological data revealed a reduced volume fraction of lamellar bodies in AECII [VV(LB/AECII)] (C) combined with a reduced volume-weighted mean volume of lamellar bodies [νV(LB)] (D).
Aged LC3B−/− mice display apoptosis of AECII
Because apoptosis of AECII is a hallmark in the development of lung fibrosis, we further asked if the AECII of LC3B−/− mice undergo apoptosis. Immunoblots from total lung homogenates revealed significant cleavage of caspase-3, the terminal apoptosis marker in the older but not in the younger LC3B−/− mice as compared with age-matched WT mice (Fig. 2A, B). Interestingly, histologic staining for cleaved caspase-3 as well as proSP-C, an AECII marker on serial sections revealed that AECII in the 42-wk-old LC3B−/− mice display extensive apoptosis, especially in the patchy fibrotic regions (Fig. 2C–E).
Figure 2.

AECII apoptosis in aged LC3B−/− mice. A) Western blot analysis of lung homogenates of 13-wk-old (left) or 42-wk-old (right) LC3B−/− and WT mice for cleaved caspase-3 and β-actin. B) Densitometry analysis of cleaved caspase-3/β-actin ratio. C, D) Representative immunohistochemistry images of serial lung sections of 42-wk-old LC3B−/− and WT mice for cleaved caspase-3 and proSP-C. Original magnification of images are ×200 (C) and 400, (D). Scale bars, 50 and 25 μm, respectively. Arrows in D indicate apoptotic AECII in LC3B−/− mice. E) Immunohistological staining for cleaved caspase-3 in several regions of the lungs were quantified and depicted as a bar graph. Representative blots and analysis from n = 5 mice/group and ≥3 independent experiments are shown. **P < 0.01.
LC3B−/− mice display lysosomal and endoplasmic reticulum stress and increase in syntaxin 17 within their AECII
By immunoblotting, we also analyzed the lysosomal markers, lysosome-associated membrane protein (LAMP)1 and 2, the lysosomal stress marker, cathepsin D as well as the standard autophagy substrate and receptor protein p62. Cathepsin D and LAMP1 levels did not differ between LC3B−/− and WT control mice, but p62 and LAMP2 levels were significantly increased in the older LC3B−/− mice as compared with their age-matched controls (Fig. 3A–E). Interestingly, the pro form of cathepsin D is decreased in the younger LC3B−/− mice. In the older mice, on the other hand, we observed an increase in the pro and cleaved forms of cathepsin D, albeit a nonsignificant increase (Fig. 3A, E). Furthermore, in order to analyze whether these mice develop alternate cellular stress mechanisms, we queried 2 endoplasmic reticulum (ER) stress markers, activating transcription factor 6 (ATF6), and C/EBP homologous protein (CHOP). Immunoblotting analysis revealed that the activated form (p50) of ATF6 and CHOP significantly increased in LC3B−/− mice in an age-correlated manner as compared with WT controls (Fig. 3F–H), indicating that lysosomal has a retrograde stress effect on the ER, thereby activating the ER stress proteins in aged LC3B−/− mice.
Figure 3.

Lysosomal and ER stress in LC3B−/− mice. A) Western blot analysis of lung homogenates of 13-wk-old (left) or 42-wk-old (right) LC3B−/− and WT mice for the autophagy marker protein, p62, lysosomal markers, LAMP1 and 2, and cathepsin D and β-actin. B–E) Graphical representation of the densitometry analysis of the respective protein to β-actin ratio is shown. F) Western blot analysis of lung homogenates of 13- (left) or 42-wk-old (right) LC3B−/− and WT mice for the ER stress marker proteins, cleaved form of ATF6 and CHOP. G, H) Graphical representation of the densitometry analysis of the respective protein to β-actin ratio is shown. Representative blots and analysis from n = 5 mice/group and 3 independent experiments are shown; ns, not significant. *P < 0.05, **P < 0.01.
Of note and as indicated in Fig. 3A, B and Supplemental Fig. S3A, the autophagy substrate and receptor protein p62 accumulated in the aged mice as compared with the younger LC3B−/− mice. We interpreted this finding as an activation of compensatory mechanisms in the absence of LC3B-mediated autophagy in the younger LC3B−/− mice. We thus hypothesized that knockout of LC3B may result in a compensatory activation of the proteasome in younger LC3B−/− mice. To test this, we analyzed 2 proteasomal subunits: the 20S core subunit and the 26S proteasome non-ATPase regulatory subunit 11 (PSMD11). Interestingly, a significant increase in the 20S core subunit expression was observed in the younger mice, whereas its levels were significantly decreased in older LC3B−/− mice relative to WT controls and the levels of PSMD11 remained unaltered (Fig. 4A, B). In total lung extracts, however, we did not consistently observe differences in either chymotrypsin-like or caspase-like proteasomal activities between LC3B−/− and WT or between younger and older mice (Fig. 4C, D). This indicated that the proteasome is not activated to compensate for the loss of LC3B-mediated autophagy function in the LC3B−/− mice. In addition, we also analyzed γ-aminobutyric acid receptor-associated protein-like 1 (GABARAP1), another ATG8 family member that is involved in autophagosome maturation, to understand whether GABARAP1L would compensate for the loss of LC3B function in LC3B−/− mice. However, GABARAP1L protein levels in both young and old LC3B−/− mice did not differ from their age-matched WT mice (Supplemental Fig. S3B). This observation is in full accordance with a previous study from mouse embryonic fibroblasts of LC3B−/− mice (26).
Figure 4.

Up-regulation of syntaxin 17 in LC3B−/− mice. A) Western blot analysis of lung homogenates of 13-wk-old (left) or 42-wk-old (right) LC3B−/− and WT mice for the 20S core subunit of the 20S proteasome, the PSMD11 and β-actin. B) Graphical representation of densitometric quantification for 20S core subunit. C, D) Chymotrypsin-like (CTL) (C) and caspase-like (CL) (D) proteasome activity was measured in total lung extracts of LC3B−/− and WT control mice. E) Western blot analysis of lung homogenates of 13-wk-old (left) or 42-wk-old (right) LC3B−/− and WT mice for syntaxin 17, Atg14, and β-actin. F) Graphical representation of densitometric quantification for syntaxin 17. G) Immunofluorescence staining on paraffin sections from 42-wk-old LC3B−/− or WT control mice for the AECII marker, proSP-C (green) and syntaxin 17 (red). DAPI was used to stain the nuclei in blue. H) Syntaxin 17 fluorescence signal intensity was quantified and depicted as a bar graph. Representative blots and analysis from n = 5 mice/group and 3 independent experiments are shown; ns, not significant. *P < 0.05, **P < 0.01.
Maturation of autophagosomes and, more importantly, their fusion with lysosomes involve the autophagosomal soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) syntaxin 17. It was shown that in cells lacking the classic ATG conjugation system, syntaxin 17–positive autophagosome-like structures are formed at a reduced rate and fuse with lysosomes (27). Likewise, it has been suggested that Atg14 might also compensate for the role of LC3B to form syntaxin 17–positive autophagosomes (28). However, we did not find consistent difference in the protein levels of Atg14 in LC3B−/− mice compared with WT mice (Fig. 4E). Interestingly, syntaxin 17 protein levels did not alter in the younger LC3B−/− mice but were significantly increased in older LC3B−/− mice both in lung homogenates as well as in primary AECII (Fig. 4E, F and Supplemental Fig. S4A, B). In fact, punctate staining for syntaxin 17 was observed in WT and in younger LC3B−/− mice, but the older LC3B−/− mice displayed increased and diffused staining for syntaxin 17 within the AECII as compared with WT mice (Fig. 4G, H). Taken together, these readouts indicate that syntaxin 17–mediated autophagy may be defective in older LC3B−/− mice but not in younger mice. Next, we also evaluated syntaxin 17 in the explanted lungs of patients with IPF as well as healthy donors. Protein levels of syntaxin 17 were significantly increased in total lung extracts of patients with IPF as compared with donor lungs (Supplemental Fig. S4C, D). Likewise, immunohistochemical analysis revealed increased staining for syntaxin 17 within the AECII of patients with IPF as compared with the AECII of donor lungs (Supplemental Fig. S4E, F). This indicates that upon aging, defective autophagy, as seen in both aged LC3B−/− mice as well as in patients with IPF, results in a concomitant increase in syntaxin 17–positive structures within the AECII.
LC3B−/− mice display increased susceptibility to bleomycin-induced lung fibrosis
We further sought to study the role of LC3B in driving apoptosis of AECII and subsequent lung fibrosis both in vivo as well as in vitro. To this end, we performed siRNA-mediated knockdown of LC3B in MLE12 cells followed either by treatment with bleomycin or vehicle for 4 h or by leaving them untreated (Fig. 5A, B). Cells transfected with NT siRNA served as controls. It is well documented that bleomycin induces apoptosis of alveolar epithelial cells and subsequent lung fibrosis (29, 30). Replicating these findings, we also observed significant increase in the apoptosis marker, cleaved caspase-3 levels in MLE12 cells transfected with NT siRNA, followed by bleomycin treatment (Fig. 5A, C). Next, in cells transfected with siRNA against LC3B followed by bleomycin treatment, a further increase of the level of cleaved caspase-3 protein was observed (Fig. 5A, C). We interpreted this observation as a sensitization of MLE12 cells to bleomycin-induced apoptosis through knockdown of LC3B. On the contrary, cells overexpressing GFP-LC3B followed by bleomycin treatment displayed reduced levels of cleaved caspase-3 and thus apoptosis as signaling compared with cells overexpressing empty GFP followed by bleomycin treatment (Fig. 5D, E), indicating the protective role of GFP-LC3B upon bleomycin treatment. These data reconfirm previous studies from our group as well as from others about the protective role of LC3B in lung pathologies (16, 31, 32).
Figure 5.

Knockdown of LC3B sensitizes MLE12 cells to bleomycin-induced apoptosis. A) MLE12 cells were either transfected with LC3B siRNA or NT siRNA for 24 h followed by bleomycin (Bleo) or vehicle (Veh) treatment for 4 h. Representative Western blot images are shown from these cell lysates for LC3B, cleaved caspase-3, and β-actin. B, C) Graphical representation of densitometric quantification for LC3B/β-actin (B) and cleaved caspase-3/β-actin (C) ratio are shown. D) MLE12 cells were either transfected with GFP-LC3B or empty GFP vector for 24 h followed by Bleo or Veh treatment for 4 h. Representative Western blot images are shown from these cell lysates for GFP-LC3B using anti-GFP antibody or for both GFP-LC3B and endogenous LC3B using anti-LC3B antibody, cleaved caspase-3, and β-actin. E) Graphical representation of densitometric quantification for cleaved caspase-3 is shown. Representative blots and analysis from 4 independent experiments are shown; ns, not significant. *P < 0.05, **P < 0.01.
Based on these readouts and to further delineate the role of LC3B in lung fibrosis, we treated the younger LC3B−/− as well as WT mice with a single low dose (1.5 U/kg) of bleomycin and euthanized them at d 7 or 14. As indicated in Fig. 6A–D, LC3B−/− mice challenged with bleomycin at d 7 displayed increased cellularity and reduced airspaces as compared with bleomycin-challenged WT mice. Such differences were not clearly seen in mice challenged with bleomycin for 14 d (unpublished data). LC3B−/− at d 7 following bleomycin challenge displayed a nonsignificant increase in apoptosis as indicated by cleaved caspase-3 immunoblots in Supplemental Fig. S5E, F compared with WT mice challenged with bleomycin. These data supported our previous observations from Figs. 1 and 2, wherein the older LC3B−/− mice displayed increased lung alterations. In addition, we also analyzed for cellular stress (lysosomal and ER stress) markers in LC3B−/− and WT mice challenged with bleomycin. Significant differences were observed in cathepsin D levels at d 14 after bleomycin challenge of LC3B−/− vs. WT mice and ATF6 (p50) between d 7 bleomycin vs. wt mice (Supplemental Fig. S5E, G, H). In addition, p62 and LAMP2 protein levels were significantly different between bleomycin-challenged LC3B−/− mice on d 14 vs. WT mice (Supplemental Fig. S5A, D). The levels of lysosomal marker protein, LAMP1 remained unaffected (Supplemental Fig. S5A, C).
Figure 6.

LC3B−/− mice are susceptible to bleomycin-induced lung injury and fibrosis. Representative H&E (upper panel) and trichrome staining (lower panel) of complete left lungs of LC3B−/− (A) or WT mice (C) treated with bleomycin (Bleo) for 7 d and their respective higher magnification images are shown (B, D). Original magnification, ×200. Scale bars: 2.5 mm (whole lung);100 μm (high magnification).
Cathepsin A is an LC3B binding partner and is increased in the AECII of LC3B−/− mice and the lungs of patients with IPF
The results described above point toward a significant contribution of LC3B to lung fibrosis. We then sought to analyze the underlying molecular mechanisms. In this context, one intriguing observation deriving from our analysis is the increased levels of cathepsin L as well as cathepsin A levels in LC3B−/− mice lungs vs. WT control lungs (Fig. 7A–C and Supplemental Fig. S3B, C). Cathepsin L is a cysteine protease involved in functions including protein turnover and antigen presentation. Cathepsin A, on the other hand, was identified as a lysosomal protective protein because it protects β-galactosidase and neuraminidase from intralysosomal proteolysis. It is a multicatalytic enzyme with carboxypeptidase, deamidase, and esterase activities (33). Cathepsin A shares its structural homology with the yeast carboxypeptidase Y and has been shown to regulate chaperone-mediated autophagy (34). We did not observe any changes in the protein levels of LAMP2a or GABARAP (Supplemental Fig. S3B), but proSP-C–positive AECII in LC3B−/− mice showed increased cathepsin A staining as compared with the AECII of WT control mice lungs (Fig. 7D, E). In full support, primary AECII isolated from 42-wk-old LC3B−/− mice displayed significantly increased cathepsin A protein levels as compared with those of age-matched WT mice (Fig. 7F, G). We thus hypothesized that apart from the chaperone-mediated autophagy, cathepsin A might also contribute to the macroautophagy pathway. To study this, we first analyzed the protein sequence of cathepsin A. As shown in Fig. 8A, the mouse cathepsin A protein has 5 core LC3 interacting regions (LIRs), of which 3 are Y-type (YxxL), 1 is a W-type (WxxL), and 1 is F-type (FxxI) LIR. We next overexpressed myc-cathepsin A, and GFP-LC3B in MLE12 cells performed coimmunoprecipitation experiments, which revealed an interaction between the 2 overexpressed proteins (Fig. 8B). In addition, endogenous cathepsin A immunoprecipitation revealed an interaction with endogenous LC3B protein (Fig. 8C). Apart from its protective activity, cathepsin A overexpression has been shown to exert oxidative stress and cell death in cardiac tissue (35). Thus, to study the biologic significance of cathepsin A overexpression as seen in LC3B−/− mice, we overexpressed cathepsin A in MLE12 cells and analyzed cleaved caspase-3 as the terminal apoptosis marker. Cells overexpressing cathepsin A alone showed significant increase in cleaved caspase-3 as compared with cells overexpressing both LC3B as well as cathepsin A (Fig. 8D, E), indicating the importance of their interaction in regulating apoptosis. These data are fully supported by data from patients with IPF, where defective autophagy has been reported (15). As shown in Fig. 9, cathepsin A protein levels were significantly increased in the total lung extracts of patients with IPF as compared with those of donors (Fig. 9A, B). The increased cathepsin A protein levels in IPF could be ascribed to the AECII and by immunohistochemistry where serial lung sections of IPF, and donor lungs were stained for proSP-C and cathepsin A (Fig. 9C, D) as well as by immunofluorescence where the sections were costained for ABCA3, the marker of the limiting membrane of lamellar bodies in AECII and for cathepsin A (Fig. 9E, F).
Figure 7.

Increase in cathepsin A in LC3B−/− mice. A–C) Western blot analysis (A) of lung homogenates of 13-wk-old (left) or 42-wk-old (right) LC3B−/− and WT mice for cathepsin A (pro form), for cleaved form of cathepsin A and β-actin followed by their densitometric quantifications in (B, C) respectively. D) Immunofluorescence staining on paraffin sections from 42-wk-old LC3B−/− or WT control mice for the AECII marker, proSP-C (green), and cathepsin A (red). DAPI was used to stain the nuclei in blue. E) Cathepsin A fluorescence signal intensity was quantified and depicted as a bar graph. F) Representative Western blots for the indicated proteins from primary AECII isolated from 42-wk-old LC3B−/− or WT mice. G) Densitometric quantification for cathepsin A from AECII is shown. Representative blots and analysis from n = 5 mice/group and 3 independent experiments are shown. *P < 0.05, **P < 0.01.
Figure 8.

Cathepsin A is an LC3B interacting protein. A) Alignment of 5 putative LIRs of mouse cathepsin A, highlighting the N- and C-terminal amino acids of the core motif in yellow. B) Cathepsin A is coimmunoprecipitated with LC3B in MLE12 cells coexpressing cathepsin A (myc tagged) and LC3B (GFP tagged). C) Representative Western blots for cathepsin A and LC3B following immunoprecipitation of endogenous cathepsin A from MLE12 cells. D) Overexpression of cathepsin A, LC3B, empty myc, or GFP plasmids followed by Western blots using anti-myc, GFP, cleaved caspase-3, and β-actin. E) Densitometric quantification for cleaved caspase-3 is shown. Representative blots and analysis from 3 independent experiments are shown. *P < 0.05.
Figure 9.

Increase in cathepsin A in AECII of IPF lungs. A, B) Western blot analysis of lung homogenates of patients with IPF and age-matched donors for cathepsin A and glyceraldehyde 3-phosphate dehydrogenase as loading control (A) followed by densitometric quantification for cathesin A (B). C, D) Serial paraffin sections from IPF and donor lungs were stained for cathepsin A and the AECII marker proSP-C and pictomicrographs were performed for the same areas from both types of staining. High magnification images (D) show several AECII cells also stained positively for cathepsin A, as indicated by the arrows. E) Immunofluorescence staining for the AECII lamellar body limiting membrane marker, ABCA3 (green) and cathepsin A (red). DAPI was used to stain nuclei in blue. F) Cathepsin A fluorescence signal intensity was quantified and depicted as a bar graph. Scale bars, 100 μm. Representative pictures are shown from ~10–15 patients with IPF and donors. **P < 0.01.
DISCUSSION
In the current study, we report that the LC3B−/− mice at 42 wk display reduced airspaces and diameter of distal airspaces and increased cellularity. These older mice also display AECII apoptosis and increased lysosomal and ER stress without any concomitant increase in the proteasomal activity. However, syntaxin 17, the lysosomal SNARE that is involved in the autophagosome-lysosome fusion is accumulated in the AECII of aged LC3B−/− mice (Fig. 10) as well as in the AECII of human patients with IPF. Furthermore, in vitro knockdown of LC3B followed by bleomycin treatment in MLE12 cells resulted in increased apoptosis. Likewise, in vivo, LC3B−/− mice upon bleomycin challenge showed increased susceptibility to lung injury as reflected by reduced airspaces, increased cellularity, as well as increased apoptosis. In addition to this, we identified cathepsin A as a novel interacting partner of LC3B and its protein levels to be increased in the AECII of LC3B−/− mice as well as in patients with IPF. Furthermore, in vitro overexpression of cathepsin A resulted in increased apoptosis, which was reduced in the presence of overexpressed LC3B. To our knowledge, this is the first study to reveal lung defects in LC3B−/− mice and their susceptibility to lung injury upon bleomycin treatment as well as the significance of the cathepsin A overexpression in fibrotic lungs.
Figure 10.
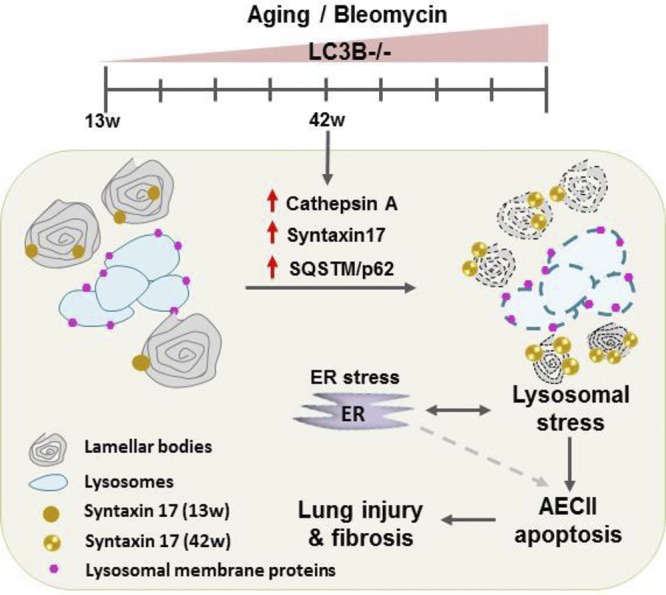
Schematic representation of pathomechanistic events in LC3B−/− mice. Aged LC3B−/− mice (42 w) lungs show increased cellularity and AECII apoptosis with smaller lamellar body profiles. AECII show increase in syntaxin 17, lysosomal, and ER stress with a concomitant increase in the novel LC3B interacting partner, cathepsin A that contributes to AECII apoptosis. Furthermore, LC3B−/− mice display increased susceptibility to bleomycin-induced lung injury and fibrosis. Similar observations from the lungs of patients with IPF emphasized the pathomechanistic role of autophagy in lung fibrosis.
Our current observations are in line with our previous study where we showed that overexpression of GFP-LC3B protects lung epithelial cells from HPS1 knockdown–induced defective autophagy (16). Although protective effects of LC3B in several settings have been reported (16, 31, 32), this is the first study to show that its loss can result in lung injury. Our data also complement a recent study by Cheong and colleagues (19) who reported that serine/threonine-protein kinases, ULK1/2 double knockout mice, and ATG5 knockout mice display lung defects at a very early age. ULK1 and 2 are mammalian orthologs of yeast ATG1, which is the first protein in the autophagy pathway (36, 37). ULK1/2 double knockout results in the deactivation of all the downstream autophagy gene products, thus resulting in a cumulative knockout situation of the autophagy pathway that may have resulted in the neonatal mortality of ULK1/2−/− mice. Here, we show that the loss of LC3B, the distal protein in the autophagy pathway is sufficient to result in lung injury, albeit in older mice. That said, we cannot exclude nonautophagy functions of LC3B to contribute to the phenotype observed in LC3B−/− mice.
It has been reported that LC3B−/− mice develop normally without any compensatory increase in LC3α or proteins associated with fibronectin, the synthesis of which is used to study LC3-regulated mRNA translational control. The same study emphasized the activation of novel compensatory mechanisms in the absence of LC3B (26). In line with this notion, one striking observation of our current study is that the autophagy substrate protein p62 is increased slightly in younger, but significantly in older, LC3B−/− mice as compared with age-matched WT control mice. A straightforward explanation of that observation is that compensatory autophagy pathways might be activated in the younger mice in the absence of LC3B. Indeed, it has been suggested that other proteins that are involved in the homeostatic processes might compensate for the loss of LC3B (31, 38). However, we did not observe significant increase in GABARAP proteins that could compensate for the loss of LC3B-mediated autophagy in LC3B−/− mice.
It is well established that the ubiquitin proteasome system and autophagy are the 2 fundamental protein quality control mechanisms of the cell (39). In fact, under certain settings of defective proteasomal degradation, autophagic degradation has been shown to be activated in order to alleviate proteotoxic stress (40–42). Although there is no direct evidence showing the inverse, recent reports reinforce the notion that coordinated and complementary action of these pathways is extremely important to rescue cells from cellular stress (39). Hence, we hypothesized that the increased accumulation of p62 in older as compared with younger LC3B−/− mice may be a result of the compensatory function of the proteasome in the younger mice. Although we observed an increase in the 20S core subunit protein levels in the younger LC3B−/− mice, we did not observe an increase in the proteasome activity. This led us to study the regulation of other autophagy-related proteins potentially compensating for the loss of LC3B in the younger LC3B−/− mice. It has been shown that the lysosomal SNARE protein syntaxin 17 is recruited to the completed autophagosomes and aids in the fusion of autophagosomes and lysosomes also in the absence of the classic ATG conjugation mechanisms. Agreeing with this notion, we also identified that syntaxin 17 was accumulated in the AECII of older LC3B−/− mice but not in the younger mice, indicating that syntaxin 17–mediated autophagy may compensate part of LC3B functions in the younger mice, but upon aging, its functions are discounted. Likewise, in the AECII of patients with IPF where defective autophagy has been reported, we now identified elevated syntaxin 17 protein levels thereby supporting the data from LC3B−/− mice.
Next, we observed that the younger LC3B−/− mice are susceptible to bleomycin-induced lung injury. The most pronounced effects were observed 7 d after a single low-dose bleomycin application in which LC3B−/− mice display increased cellularity and patchy fibrotic consolidations as compared with the WT mice treated with bleomycin. A note of caution here is that this time point coincides with inflammatory response to bleomycin. LC3B−/− and WT mice treated with bleomycin showed no perceivable differences at d 14 posttreatment. Our observation regarding the susceptibility of LC3B−/− mice supports the findings of a previous study in which deficiency of ATG4B resulted in increased sensitivity of mice to bleomycin-induced lung fibrosis (20). ATG4B is a cysteine protease that cleaves LC3B at its C terminus, a cleavage that is essential to conjugate it to phosphatidylethanolamine and insert it on the membranes (43) and absence of ATG4B might result in the inhibition of LC3B cleavage and thereby autophagy.
Another interesting observation from this study is the increase of cathepsin A protein levels in the aged LC3B−/− mice and in the AECII of the lungs of patients with IPF, as well as the interaction between these 2 proteins. To our knowledge, the interaction between these 2 proteins has not yet been reported. Our data indicate that the increase in cathepsin A in the absence of LC3B may contribute to the apoptosis of AECII. Cathepsin A has been suggested to elicit oxidative stress and cell death in cardiomyocytes. Although not shown directly in our study, it is plausible to speculate that the apoptosis of mouse lung epithelial cells upon cathepsin A overexpression in vitro or the overexpression of cathepsin A in the AECII of LC3B−/− mice may retrogradely elicit oxidative stress and subsequent apoptosis. Our data do suggest, however that modulation of cathepsin A protein levels by LC3B is important in defining the physiologic roles of cathepsin A in AECII. Our in vitro as well as in vivo observations are fully supported by the data from the lungs of patients with IPF, altogether suggesting that the increased cathepsin A protein levels may contribute to the increased cellular stress and apoptosis of AECII in lung fibrosis.
Our current study ascertains the pathomechanistic role of autophagy in general and LC3B in particular in the development of pulmonary fibrosis. This study also raises the possibility that in IPF, LC3B-mediated regulation of cellular mechanisms may play an integral role in eliciting cellular stress in the AECII, which, in addition to the activation of concomitant stress pathways, subsequently results in pulmonary fibrosis.
ACKNOWLEDGMENTS
The authors thank the German Research Foundation for supporting this study through an independent research grant to P.M. The authors also thank Silke Haendel, Stefanie Hezel [Department of Internal Medicine, Justus-Liebig University (JLU) Giessen], and Susanne Faßbender (Institute of Functional and Applied Anatomy, Hannover Medical School) for their excellent technical support. The authors also thank Prof. Dr. Saverio Bellusci (Department of Internal Medicine, JLU Giessen) and laboratory members for help with fluorescence miscroscopy. The authors declare no conflicts of interest.
Glossary
- ABCA3
ATP binding cassette subfamily A member 3
- AECII
alveolar epithelial cell type II
- ATF6
activating transcription factor 6
- Atg
autophagy-related
- CHOP
C/EBP homologous protein
- ER
endoplasmic reticulum
- GABARAP1
γ-aminobutyric acid receptor-associated protein-like 1
- GFP
green fluorescent protein
- H&E
hematoxylin and eosin
- HPS
Hermansky-Pudlak syndrome
- IPF
idiopathic pulmonary fibrosis
- LAMP
lysosome-associated membrane protein
- LIR
LC3 interacting region
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3β
- NT
nontargeting
- proSP-C
prosurfactant protein C
- PSMD11
26S proteasome non-ATPase regulatory subunit 11
- siRNA
small interfering RNA
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- SP-B
surfactant protein B
- ULK
mammalian orthologs of yeast Atg1
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
V. S. Kesireddy, S. Chillappagari, S. Ahuja, I. Henneke, J. Graumann, C. Ruppert, and M. Korfei performed experiments; V. S. Kesireddy and P. Mahavadi developed the project, analyzed and interpreted the data, and wrote the article; S. Chillappagari and J. Graumann critically read the manuscript; L. Knudsen and M. Ochs performed electron microscopy and interpreted the data; S. Meiners performed proteasome analysis; W. Seeger and P. Mahavadi supervised the study; P. Mahavadi designed the study; and all authors approved the final version of the manuscript.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.King T. E., Jr., Pardo A., Selman M. (2011) Idiopathic pulmonary fibrosis. Lancet 378, 1949–1961 [DOI] [PubMed] [Google Scholar]
- 2.Selman M., Pardo A. (2006) Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc. Am. Thorac. Soc. 3, 364–372 [DOI] [PubMed] [Google Scholar]
- 3.American Thoracic Society ; European Respiratory Society (2002) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. [This joint statement of the American Thoracic Society (ATS) and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001.] Am. J. Respir. Crit. Care Med. 165, 277–304; erratum: 166, 426 [DOI] [PubMed] [Google Scholar]
- 4.Myers J. L., Katzenstein A. L. (1988) Epithelial necrosis and alveolar collapse in the pathogenesis of usual interstitial pneumonia. Chest 94, 1309–1311 [DOI] [PubMed] [Google Scholar]
- 5.Fehrenbach H. (2001) Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weaver T. E., Na C. L., Stahlman M. (2002) Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol. 13, 263–270 [DOI] [PubMed] [Google Scholar]
- 7.Lutz D., Gazdhar A., Lopez-Rodriguez E., Ruppert C., Mahavadi P., Günther A., Klepetko W., Bates J. H., Smith B., Geiser T., Ochs M., Knudsen L. (2015) Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 52, 232–243 [DOI] [PubMed] [Google Scholar]
- 8.Thomas A. Q., Lane K., Phillips J., III, Prince M., Markin C., Speer M., Schwartz D. A., Gaddipati R., Marney A., Johnson J., Roberts R., Haines J., Stahlman M., Loyd J. E. (2002) Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165, 1322–1328 [DOI] [PubMed] [Google Scholar]
- 9.Schmitz G., Müller G. (1991) Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 32, 1539–1570 [PubMed] [Google Scholar]
- 10.Mahavadi P., Knudsen L., Venkatesan S., Henneke I., Hegermann J., Wrede C., Ochs M., Ahuja S., Chillappagari S., Ruppert C., Seeger W., Korfei M., Guenther A. (2015) Regulation of macroautophagy in amiodarone-induced pulmonary fibrosis. J. Pathol. Clin. Res. 1, 252–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller B. E., Hook G. E. (1990) Hypertrophy and hyperplasia of alveolar type II cells in response to silica and other pulmonary toxicants. Environ. Health Perspect. 85, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahavadi P., Korfei M., Henneke I., Liebisch G., Schmitz G., Gochuico B. R., Markart P., Bellusci S., Seeger W., Ruppert C., Guenther A. (2010) Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am. J. Respir. Crit. Care Med. 182, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahavadi P., Henneke I., Ruppert C., Knudsen L., Venkatesan S., Liebisch G., Chambers R. C., Ochs M., Schmitz G., Vancheri C., Seeger W., Korfei M., Guenther A. (2014) Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone-induced lung fibrosis. Toxicol. Sci. 142, 285–297 [DOI] [PubMed] [Google Scholar]
- 14.Patel A. S., Lin L., Geyer A., Haspel J. A., An C. H., Cao J., Rosas I. O., Morse D. (2012) Autophagy in idiopathic pulmonary fibrosis. PLoS One 7, e41394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bueno M., Lai Y. C., Romero Y., Brands J., St Croix C. M., Kamga C., Corey C., Herazo-Maya J. D., Sembrat J., Lee J. S., Duncan S. R., Rojas M., Shiva S., Chu C. T., Mora A. L. (2015) PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest. 125, 521–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahuja S., Knudsen L., Chillappagari S., Henneke I., Ruppert C., Korfei M., Gochuico B. R., Bellusci S., Seeger W., Ochs M., Guenther A., Mahavadi P. (2016) MAP1LC3B overexpression protects against Hermansky-Pudlak syndrome type-1-induced defective autophagy in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L519–L531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 18.Codogno P., Meijer A. J. (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12 (Suppl 2), 1509–1518 [DOI] [PubMed] [Google Scholar]
- 19.Cheong H., Wu J., Gonzales L. K., Guttentag S. H., Thompson C. B., Lindsten T. (2014) Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy 10, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabrera S., Maciel M., Herrera I., Nava T., Vergara F., Gaxiola M., López-Otín C., Selman M., Pardo A. (2015) Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 11, 670–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghu G., Collard H. R., Egan J. J., Martinez F. J., Behr J., Brown K. K., Colby T. V., Cordier J. F., Flaherty K. R., Lasky J. A., Lynch D. A., Ryu J. H., Swigris J. J., Wells A. U., Ancochea J., Bouros D., Carvalho C., Costabel U., Ebina M., Hansell D. M., Johkoh T., Kim D. S., King T. E., Jr., Kondoh Y., Myers J., Müller N. L., Nicholson A. G., Richeldi L., Selman M., Dudden R. F., Griss B. S., Protzko S. L., Schünemann H. J.; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tschanz S., Schneider J. P., Knudsen L. (2014) Design-based stereology: planning, volumetry and sampling are crucial steps for a successful study. Ann. Anat. 196, 3–11 [DOI] [PubMed] [Google Scholar]
- 23.Gundersen H. J., Bagger P., Bendtsen T. F., Evans S. M., Korbo L., Marcussen N., Møller A., Nielsen K., Nyengaard J. R., Pakkenberg B., et al. (1988) The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS 96, 857–881 [DOI] [PubMed] [Google Scholar]
- 24.Welk V., Coux O., Kleene V., Abeza C., Trümbach D., Eickelberg O., Meiners S. (2016) Inhibition of proteasome activity induces formation of alternative proteasome complexes. J. Biol. Chem. 291, 13147–13159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahuja S., Knudsen L., Chillappagari S., Henneke I., Ruppert C., Korfei M., Gochuico B. R., Bellusci S., Seeger W., Ochs M., Guenther A., Mahavadi P. (2016) MAP1LC3B overexpression protects against Hermansky-Pudlak syndrome type-1-induced defective autophagy in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L519–L531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cann G. M., Guignabert C., Ying L., Deshpande N., Bekker J. M., Wang L., Zhou B., Rabinovitch M. (2008) Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev. Dyn. 237, 187–195 [DOI] [PubMed] [Google Scholar]
- 27.Tsuboyama K., Koyama-Honda I., Sakamaki Y., Koike M., Morishita H., Mizushima N. (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041 [DOI] [PubMed] [Google Scholar]
- 28.Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y., Amano A., Yoshimori T. (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 [DOI] [PubMed] [Google Scholar]
- 29.Wang R., Ibarra-Sunga O., Verlinski L., Pick R., Uhal B. D. (2000) Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L143–L151 [DOI] [PubMed] [Google Scholar]
- 30.Lee V. Y., Schroedl C., Brunelle J. K., Buccellato L. J., Akinci O. I., Kaneto H., Snyder C., Eisenbart J., Budinger G. R., Chandel N. S. (2005) Bleomycin induces alveolar epithelial cell death through JNK-dependent activation of the mitochondrial death pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L521–L528 [DOI] [PubMed] [Google Scholar]
- 31.Lee S. J., Smith A., Guo L., Alastalo T. P., Li M., Sawada H., Liu X., Chen Z. H., Ifedigbo E., Jin Y., Feghali-Bostwick C., Ryter S. W., Kim H. P., Rabinovitch M., Choi A. M. (2011) Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 183, 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lahm T., Petrache I. (2012) LC3 as a potential therapeutic target in hypoxia-induced pulmonary hypertension. Autophagy 8, 1146–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiraiwa M. (1999) Cathepsin A/protective protein: an unusual lysosomal multifunctional protein. Cell. Mol. Life Sci. 56, 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuervo A. M., Mann L., Bonten E. J., d’Azzo A., Dice J. F. (2003) Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 22, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrera A., Kern U., Linz D., Gomez-Auli A., Hohl M., Gassenhuber J., Sadowski T., Schilling O. (2016) Proteomic profiling of cardiomyocyte-specific cathepsin a overexpression links cathepsin a to the oxidative stress response. J. Proteome Res. 15, 3188–3195 [DOI] [PubMed] [Google Scholar]
- 36.Alers S., Löffler A. S., Wesselborg S., Stork B. (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alers S., Löffler A. S., Wesselborg S., Stork B. (2012) The incredible ULKs. Cell Commun. Signal. 10, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martens S. (2016) No ATG8s, no problem? How LC3/GABARAP proteins contribute to autophagy. J. Cell Biol. 215, 761–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nedelsky N. B., Todd P. K., Taylor J. P. (2008) Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu K., Dunner K., Jr., McConkey D. J. (2010) Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29, 451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang B., Cai J., Sun L., Li Y., Qu J., Snider B. J., Wu S. (2014) Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS One 9, e103364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jänen S. B., Chaachouay H., Richter-Landsberg C. (2010) Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 58, 1766–1774 [DOI] [PubMed] [Google Scholar]
- 43.Skytte Rasmussen M., Mouilleron S., Kumar Shrestha B., Wirth M., Lee R., Bowitz Larsen K., Abudu Princely Y., O’Reilly N., Sjøttem E., Tooze S. A., Lamark T., Johansen T. (2017) ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 13, 834–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.