

Abstract
Combination antiretroviral therapy has dramatically changed the outcome of HIV infection, turning it from a death sentence to a manageable chronic disease. However, comorbidities accompanying HIV infection, such as metabolic and cardio-vascular diseases, as well as cognitive impairment, persist despite successful virus control by combination antiretroviral therapy and pose considerable challenges to clinical management of people living with HIV. These comorbidities involve a number of pathological processes affecting a variety of different tissues and cells, making it challenging to identify a common cause(s) that would link these different diseases to HIV infection. In this article, we will present evidence that impairment of cellular cholesterol metabolism may be a common factor driving pathogenesis of HIV-associated comorbidities. Potential implications for therapeutic approaches are discussed.
Keywords: cholesterol metabolism, comorbidities, extracellular vesicles, HIV, lipid rafts, Nef
Introduction
Despite effective control of HIV infection by combination antiretroviral therapy (cART), HIV-infected people remain at high risk for developing dyslipidemia, accelerated progress of atherosclerosis, metabolic syndrome, lipodystrophy, myocardial disorder, diabetes, abnormal hematopoiesis, cognitive impairment and many other metabolic comorbidities [1–4], which pose considerable challenges to clinical management of people living with HIV (PLWH) [5]. Effective prevention or treatment of these comorbidities requires identification of the key pathogenic factors that drive development of these diseases in patients with suppressed HIV load. So far, this issue remained contentious.
Antiretroviral therapy itself has long been considered the main reason for comorbidities in HIV-infected people. Indeed, some first-generation nucleoside reverse transcriptase inhibitors, in particular thymidine analogues (stavudine, zidovudine), have pronounced metabolic side effects and were strongly implicated in the pathogenesis of HIV-related lipodystrophy [6–8]. Similarly, the early generation of protease inhibitors, including such widely used drugs as lopinavir/ritonavir combination, has been associated with a high risk of dyslipidemia, metabolic syndrome and increased fat accumulation [9–12]. Progress in antiretroviral drug development during the last 20 years produced drugs from both classes with far fewer metabolic toxicities (e.g. tenofovir, emtricitabine, atazanavir, darunavir) [13,14]. Nevertheless, although discontinuation of anti-HIV drugs with metabolic side effects improves fat distribution [15,16], the negative consequences of previous exposures (legacy effect) persist and may contribute to increased risk of metabolic comorbidities [17]. Moreover, the newest addition to antiretroviral therapy (ART) regimens – integrase strand transfer inhibitors, and in particular dalutegravir – have been associated with excessive weight gain [18–20]. With regard to HIV-associated neurocognitive disorder (HAND), some in-vitro studies suggested potential neurotoxicity of various classes of cART [21,22], although relevance of this observation to HAND pathogenesis remains controversial [23,24]. Overall, antiretroviral drugs may contribute to metabolic abnormalities, but current ART regimens are unlikely to be the primary cause of HIV comorbidities. This leaves HIV itself as a primary suspect [25]. A study of ‘elite controllers’, a small group of HIV-infected people who control HIV replication to undetectable levels without anti-HIV treatments, demonstrated significantly increased coronary atherosclerosis and monocyte activation relative to uninfected individuals [25], indicating that the cause of this comorbidity is related to HIV infection.
Therefore, to find the cause of HIV-associated comorbidities, one has to look at indirect or direct effects of HIV itself. One indirect effect of HIV infection with a known influence on pathogenesis of various comorbidities is inflammation [26]. Inflammation due to early damage to gut mucosal tissue and subsequent leakage of microflora into the blood is a characteristic feature of HIV infection [27]. Given that recovery of gut mucosal tissue after ART initiation is slow, inflammation persists in treated patients for a long time (refer to [26] for an excellent review). Similarly, in elite controllers, despite little damage to lymphoid tissues [28,29], low-level HIV replication can be detected in the gut, associated with impairment of gut barrier function [30] and inflammatory responses [31]. However, while inflammation is strongly associated with HIV comorbidities, no evidence to support its role as a primary causative factor exists. In fact, inflammation may be a marker of the disease, rather than its etiological factor.
The other potential cause of HIV comorbidities is direct effects of HIV. Given that the number of HIV-infected cells in ART-controlled infection is extremely low, to instigate systemic diseases, such as metabolic or cardio-vascular comorbidities, HIV must affect a large number of uninfected cells at various body sites. Such effects can be mediated by HIV proteins released into the blood within extracellular vesicles produced by HIV-infected cells. Since current anti-HIV drugs prevent infection of new cells, but do not block HIV transcription and translation in already infected cells, production of HIV proteins persists in ART-treated patients [32,33]. Extracellular vesicles containing HIV proteins, including Nef, are detected in a large proportion of ART-treated patients with undetectable HIV load, as well as in elite controllers [34–37]. In a recent study, Nef was detected in 83% of ART-naive individuals (median Nef level in the blood was 11.63 ng/ml and correlated with HIV load), in 47.4% of ART-treated patients with undetectable viral load (median Nef level was 8.25 ng/ml), and in 52.4% of elite controllers (median Nef level – 8.78 ng/ml) [37]. It should be noted here that due to Nef variability between the HIV isolates even within the same clade [38], immunological detection methods have inherently low sensitivity. The limit of detection of the assay used in the study by Ferdin et al.[37] was 5.46 ng/ml, so it is likely that lower levels of Nef were present in samples tested negative. In this article, we argue for the role of these Nef-containing vesicles as the pathogenic factor in HIV-associated metabolic comorbidities and propose a mechanism for this effect. We do not intend to provide a comprehensive review of existing literature on pathogenic factors in HIV comorbidities (the readers are referred to an excellent recent review [26]), but will focus on cholesterol metabolism and the role of Nef.
Nef is not the only HIV protein potentially implicated in metabolic disorders. HIV protein Vpr has been shown in mouse models to inhibit peroxisome proliferator-activated receptor gamma (PPAR-γ) [39], which is essential for adipocyte differentiation, leading to fatty acid accumulation and lipotoxicity [39]. Exposure of rat neurons to Tat led to upregulation of cholesterol biosynthesis genes and increased levels of free cholesterol and cholesteryl esters [40]. However, the role of metabolic mechanisms in the effects of other than Nef HIV proteins in HIV-associated comorbidities is not sufficiently characterized.
HIV infection, Nef, cholesterol metabolism and lipid rafts
Cholesterol is critical for HIV replication, as both HIV entry into and exit from the target cells occur through cholesterol enriched regions of the plasma membrane, lipid rafts [41–49]. Depletion of cellular cholesterol by chemical agents such as cyclodextrin [42,44,49–52] or as a consequence of genetic predisposition to high expression of ABCA1 [53,54], the cellular cholesterol transporter mediating cholesterol efflux, lead to suppression of HIV replication in vitro and control of HIV infection in vivo. Single nucleotide polymorphism in PCSK9 gene controlling expression of LDL receptor was associated with higher HIV load in HIV/hepatitis C virus coinfected women, presumably due to increased uptake of cholesterol by cells [55]. It is therefore not surprising that HIV evolved means to control cholesterol content of target cells, and Nef appears to be the main viral tool in this process.
HIV-1 protein Nef is a multifunctional protein responsible for many pathogenic effects of HIV infection. In virus producing cells, Nef inhibits an innate anti-HIV factor SERINC [56,57], thus promoting virus infectivity, and suppresses antiviral immune responses by down-modulating CD4+, major histocompatibility complex (MHC)-I, CD28 and several other immune receptors on infected cells [58]. An important, but less appreciated pathogenic effect of Nef concerns cholesterol metabolism. Nef stimulates cholesterol biosynthesis and its delivery to lipid rafts [59,60] and inhibits cholesterol efflux by suppressing activity of cholesterol transporter ABCA1 [61]. The end result of these activities is increased abundance of lipid rafts in an infected cell, benefiting production of new virions [48,62]. Importantly, lipid rafts in HIV-infected or Nef-expressing cells are not only more abundant, but are also functionally defective [63].
However, cholesterol-related effects of Nef are not limited to HIV-infected cells. Nef is released from infected cells either as a free protein coming from dying cells, or being incorporated into extracellular vesicles [35,64]. Although most published reports identify these vesicles as exosomes [36,65,66], Nef incorporation into extracellular vesicles of other origin, such as microvesicles, cannot be ruled out [34,64]. These Nef-containing extracellular vesicles can interact with uninfected cells, impairing cholesterol metabolism on a systemic level [67,68]. Our results indicate that Nef-containing extracellular vesicles downregulate ABCA1, suppress cholesterol efflux and increase abundance of lipid rafts with corresponding stimulation of inflammatory responses, similar to the effects observed for endogenously produced Nef [69].
The accepted mechanism of Nef-mediated downregulation of cellular membrane proteins is Nef binding to the cytoplasmic domains and recruiting adaptor proteins to target these receptors to the endocytic machinery and degradation pathways (reviewed in [58]). Nef binds to ABCA1, however, it was found that downregulation of ABCA1 did not require a direct interaction of Nef with ABCA1 [70]. Instead, Nef-mediated transport of cholesterol to lipid rafts competed with ABCA1-dependent cholesterol efflux pathway altering functional properties of the rafts [63], displacing ABCA1 from the lipid rafts, and leading to its degradation in lysosomes and proteasomes [63,70]. In addition to this mechanism, Nef also affects de-novo production of ABCA1 by blocking the interaction between ABCA1 and calnexin, an endoplasmic reticulum chaperone necessary for proper folding and maturation of transmembrane glycosylated proteins destined for plasma membrane [71]. Nef binds to the cytoplasmic tail of calnexin causing structural changes, which affect interaction between the luminal domain of calnexin and ABCA1 [72–74]. As a result, maturation of ABCA1 and its functional activity are impaired, leading to accumulation of intracellular cholesterol and increased abundance of lipid rafts (Fig. 1). This finding presents an interesting conundrum. Given that many proteins, including HIV gp160 [75], mature through the endoplasmic reticulum, the effect of Nef on calnexin may potentially involve a large number of proteins and be detrimental both to the cell and the virus. However, there is certain selectivity in the effect of Nef: while it disrupts interaction between calnexin and ABCA1, the interaction between calnexin and gp160 was actually increased [74]. The mechanistic details of this selectivity, as well as identification of other proteins affected by the interaction of Nef with calnexin, await future studies.
Fig. 1.
Schematic representation of the effects of Nef extracellular vesicles on target cells.
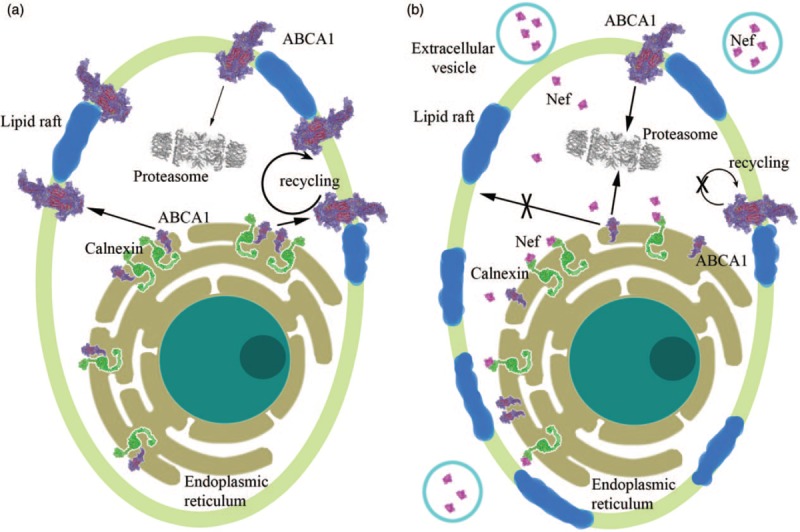
The cell plasma membrane is shown in light green, and membrane lipid rafts – in blue. Endoplasmic reticulum is shown in olive green around the cell nucleus. Nef, ABCA1, and calnexin are represented by their scaled down three-dimensional structures. (a) Cell not treated with Nef extracellular vesicles. In the endoplasmic reticulum, calnexin interacts with ABCA1 supporting ABCA1 maturation and transport to plasma membrane. ABCA1 is recycled from the cell membrane, and some is internalized to the proteasomes and degraded. (b) Cell treated with Nef extracellular vesicles. Extracellular vesicles carrying Nef molecules surround the cell and deliver Nef into the cell. Nef interacts with cytoplasmic domain of calnexin, which ostensibly causes changes in the calnexin structure, disrupting interaction of its intraendoplasmic reticulum domain with ABCA1. As a result, maturation of ABCA1 is impaired and it is targeted to proteasomes reducing cholesterol efflux. This increases cell's cholesterol content and changes abundance and properties of lipid rafts, leading to decreased recycling of the plasma membrane ABCA1 and its preferential targeting to proteasomes [63,69,73,76].
Inhibition of ABCA1 may not be the only mechanism connecting Nef with cellular cholesterol metabolism and lipid rafts. In human aortic endothelial cells, factors secreted by HIV-infected cells inhibited cholesterol efflux to high density lipoprotein (HDL) without affecting ABCA1 or other known cholesterol transporters; contribution of Nef to this effect was relatively minor [77]. In the same model Nef was responsible for increased abundance of caveolae, a subset of rafts, while disrupting caveolin-dependent cholesterol trafficking, again without affecting ABCA1 [77]. Overexpression of caveolin-1 in macrophages restored Nef-induced impairment of cholesterol efflux without restoring ABCA1 abundance [78]. Clearly, an interplay between different and often redundant pathways involved in regulation of cellular cholesterol trafficking and lipid rafts determines the eventual outcome of Nef activity.
The mechanisms described above have been demonstrated for endogenously produced Nef. How exogenous Nef affects ABCA1 is still unknown. Given that Nef, added either as free recombinant protein or with extracellular vesicles, is delivered into the cells [34,69,79], a reasonable expectation is that the mechanisms described for endogenous Nef can be functional here as well. This assumption is consistent with our unpublished results showing that the compound blocking Nef-calnexin interaction (see below) inhibits ABCA1 downregulation by Nef-containing extracellular vesicles. Future studies will be needed to fully characterize the molecular mechanisms behind the effects of exogenous Nef on cellular cholesterol metabolism.
Cholesterol metabolism, Nef and comorbidities of HIV infection
Impairment of cholesterol metabolism and overabundance of lipid rafts are common elements in the pathogenesis of almost any metabolic comorbidity associated with HIV infection and are the metabolic pathways targeted by HIV Nef, both produced intracellularly as the result of infection or secreted by the infected cells systemically. Most of Nef secreted by HIV-infected cells comes in extracellular vesicles [80,81], ensuring rapid systemic delivery of Nef to target cells and protecting it from being neutralized by anti-Nef antibodies [82]. It is therefore reasonable to suggest that disturbances of cholesterol metabolism and lipid rafts in bystander cells caused by Nef secreted by HIV-infected cells in extracellular vesicles contribute to pathogenesis of many comorbidities of HIV disease.
Inflammation
HIV infection is associated with a low-grade chronic inflammation. The reason for HIV-associated inflammation is thought to be a compromised gut mucosal epithelium leading to microbial translocation and ‘leaking’ of lipopolysaccharide (LPS) into blood [83]. Mechanistically, LPS and other microbial products induce inflammation through interaction with the inflammatory receptors. Lipid rafts host many receptors involved in inflammatory responses and play a key role in regulating their activity and, consequently, the severity of inflammatory response [84]. Many receptors are activated through re-localization to lipid rafts, including toll-like receptor 4, tumor necrosis factor receptor 1, CD11b, immune receptors B-cell receptor and T-cell receptor [85]. Augmentation of rafts results in potentiation of inflammatory responses and increased secretion of proinflammatory cytokines in response to LPS [86], while disruption of rafts is anti-inflammatory [87]. Formation of rafts stimulated by Nef may therefore potentiate inflammation in response to microbial translocation; whether or not Nef can also induce a sterile inflammation is less clear.
More generally, accumulation of cholesterol in cells involved in inflammation almost inevitably results, through the formation of rafts or other mechanisms, in a strong inflammatory response (for review see [88]). Inflammation, in turn, leads to accumulation of cholesterol in cells, forming a vicious cycle. Such vicious cycle would greatly amplify the effects of Nef on accumulation of intracellular cholesterol and inflammation, and both will feed into pathogenesis of a number of comorbidities of HIV disease, including atherosclerosis, lipodystrophy, dementia, diabetes and metabolic syndrome [83].
Dyslipidemia
HIV infection causes hypobetalipoproteinemia [low level of low density lipoprotein (LDL)], hypertiglyceridemia and hypoalphalipoproteinemia (low level of HDL) [89–91]. Hypobetalipoproteinemia is characteristic mainly for the untreated HIV infection, it is usually reversed upon commencement of modern ART regimen. Hypoalphalipoproteinemia is unaffected by ART treatment [91,92], and a leading cause of this condition is a deficiency or functional impairment of liver ABCA1, a key element in the pathway of HDL formation [93] and at the same time the very target of Nef. In addition to the reduced levels, the composition, size and functionality of HDL were affected in HIV infection [91,94,95], a finding also consistent with impairment of ABCA1. Generally, lipoprotein profile in PLWH is very similar to that in patients with Tangier disease, a monogenetic disorder where HDL fails to form due to familial ABCA1 deficiency [96], further pointing to the key role of ABCA1 deficiency in pathogenesis of HIV-associated dyslipidemia. However, no HDL turnover studies in PLWH directly supporting this notion were published and contribution of indirect mechanisms, such as inflammation and immune deficiency [94], to HIV-associated hypoalphalipoproteinemia cannot be ruled out.
Low HDL levels are commonly associated with high triglyceride levels due to lack of sufficient acceptor for triglycerides transferred from very low density lipoprotein (VLDL) to HDL through action of cholesteryl ester transfer protein. Deficiency of ABCA1 has been implicated in overproduction of VLDL by liver [97]. HIV-associated hypertriglyceridemia may, however, have additional contributing mechanisms due to insulin resistance prevalent in HIV infection [96,98], which, in turn, may be related to the impairment of cholesterol metabolism (see below). Remarkably, hypetriglyceridemia and hypoalphalipoproteinemia were mitigated in patients infected with Nef-deficient strain of HIV [99]. Mice injected with recombinant Nef displayed hypoalphalipoproteinemia, hypetriglyceridemia and reduced level of ABCA1 in liver homogenates [68], reproducing the observations in HIV-infected people and simian immunodeficiency virus-infected monkeys [67]. Mice injected with exosomes containing Nef also displayed hypoalphalipoproteinemia as well as reduced levels of ABCA1 in liver and peritoneal macrophages [69], providing in-vivo evidence for the effect of Nef on ABCA1.
Atherosclerosis
Hypoalphalipoproteinemia and hypertriglyceridemia caused by HIV, often combined with hyperbetacholesterolemia caused by some ART regimens, constitute a classical proatherogenic lipoprotein profile associated with elevated risk of atherosclerosis in HIV infection [100]. However, cardiovascular risk prediction based entirely on changes in lipoprotein profile underestimates the actual cardiovascular disease risk in HIV infection [101], pointing to the contribution of additional factors, both local and systemic. One of the systemic factors is elevated inflammation (see above), an important element in atherosclerosis [102]. A key local element in pathogenesis of atherosclerosis is accumulation of cholesteryl esters in the cells of vessel wall, macrophages and smooth muscle cells, with formation of foam cells. ABCA1 deficiency and impairment of cholesterol efflux are key causes of the formation of foam cells and development of atherosclerosis [103] and we have demonstrated that ABCA1 deficiency triggered by both intracellular and extracellular Nef causes formation of foam cells in vitro and in vivo and development of atherosclerosis in vivo[61,68,104]. Lipid rafts also play a direct role in accumulation of cholesterol in macrophages and formation of foam cells. Rafts are a location of TREM-1, an important contributor to the foam cell formation and atherogenesis [105]. Further, the current understanding of the mechanisms of ABCA1-mediated cholesterol efflux is that ABCA1 moves cholesterol from rafts to the ‘activated lipid domains’ where it becomes accessible to extracellular cholesterol acceptors [106]. Excessive rafts contribute to the impairment of cholesterol efflux by affecting this capacity of ABCA1. Lipid rafts also harbor CD36, a putative oxLDL receptor responsible for the uncontrolled cellular uptake of modified LDL [107]. Disruption of rafts with methyl-β-cyclodextrin and with apolipoprotein A-I (apoA-I) binding protein (AIBP) is antiatherogenic [108,109]. Thus, HIV Nef may contribute to the development of atherosclerosis in HIV infection through four interrelated mechanisms: causing dyslipidemia, potentiating systemic and local inflammation, and inducing accumulation of cholesterol and elevating the abundance of lipid rafts in macrophages and smooth muscle cells.
Diabetes
Type 2 diabetes is highly prevalent in HIV infection [110]. Defects in cholesterol homeostasis and impairment of lipid rafts are intimately linked to two key elements of pathogenesis of type 2 diabetes: impairment of insulin secretion from pancreatic β cells and insulin resistance. β Cells are very sensitive to excessive cholesterol, and impairment of cholesterol metabolism causes a sharp decline in their ability to regulate insulin secretion in response to changes in blood glucose levels. Specifically, studies in mice have shown that increases in cholesterol levels in pancreatic β cells conditionally lacking Abca1 led to markedly impaired insulin secretion [111]. Many effects of impaired cholesterol metabolism are thought to be mediated by overabundance of lipid rafts. In β cells, lipid rafts are critically involved in regulation of glucose sensing and insulin secretion through regulating the activity of glucose transporters [112], shifting neuronal nitric oxide synthase into a dimeric form [113], and regulating activity of SNAP receptor complexes [114]. Increased abundance of lipid rafts is associated with reduced glucose sensing and reduced insulin secretion [115]. Furthermore, accumulation of excessive cholesterol in insulin-containing β-cell secretory granules impairs their secretion [116]. Cholesterol enriched secretory granules and impaired glucose-stimulated insulin secretion were observed in cells lacking ABCA1 [117]. Lipid rafts play a critical role in proper compartmentalization of insulin signaling in adipocytes, important players in maintaining insulin sensitivity [118]; extracellular Nef inhibited glucose transporter type 4 (GLUT4) trafficking and glucose uptake in these cells [119]. In skeletal muscle cells, another tissue significantly contributing to insulin sensitivity, lipid rafts are involved in regulation of insulin-stimulated glucose uptake, influencing translocation of GLUT4 from perinuclear stores to plasma membrane [120]. Another contributor to pathogenesis of type 2 diabetes is dyslipidemia: both hypoalphalipoproteinemia and hypetriglyceridemia characteristic for the HIV infection (see above) are also important factors regulating insulin secretion and insulin sensitivity [121]. Taken together, these observations suggest that Nef-induced changes to cholesterol metabolism and lipid rafts are an important pathogenic factor in HIV-associated diabetes.
Hematopoiesis
Hematological abnormality is another important comorbidity of HIV infection [122]. It is characterized by reduced growth and differentiation of multiple hematopoietic lineages suggesting impaired functionality of early hematopoietic progenitors. Available data suggest an important role of HIV protein Nef in pathogenesis of hematopoietic abnormalities of HIV infection [123,124]. Prost et al.[123] described a mechanism where Nef acts as a PPAR-γ agonist reducing expression of signal transducer and activator of transcription 5. However, the molecular details of this mechanism remain unknown. Several lines of evidence support an important role of cholesterol metabolism in hematopoiesis and point to a possibility that impairment of cholesterol metabolism by circulating Nef may contribute to the abnormal hematopoiesis. Mice deficient in the two key transporters maintaining cellular cholesterol efflux, ABCA1 and ABCG1, displayed a myeloproliferative disorder manifested in profound leukocytosis and an expansion of the population of hematopoietic stem progenitor cells (HSPC) in bone marrow [125]. Significantly, this phenotype favored hematopoietic lineage decisions toward granulocytes rather than macrophages in the bone marrow, leading to impaired support for osteoblasts and decreased Cxcl12/SDF-1 production by mesenchymal progenitors [126]. Conversely, stimulation of cholesterol efflux by elevating expression of ABCA1 or levels of ABCA1 ligands, apoA-I or apolipoprotein E, had an opposite effect reducing HSPC proliferation and monocytosis [127]. Stimulation of cholesterol efflux and disruption of lipid rafts by apoA-I-binding protein (AIBP)-regulated HSPC emergence from hematogenic endothelium [128]. AIBP, ABCA1 and cholesterol efflux are important regulators of lipid rafts that control functionality of hematopoietic stem cells as many receptors involved in regulation of hematopoiesis are localized in rafts [129]. Quiescent HSPC contain very few rafts, and formation of rafts is a prerequisite for HSPC re-entry into the cell cycle; inhibition of rafts induces hibernation of HSPC [130]. On the other hand, disruption of lipid rafts with phospholipase C-β2 promotes egress of cells from bone marrow niches, implying that rafts are required for retention of HSPC in bone marrow [131]. Taken together, these findings are consistent with a suggestion that impairment of reverse cholesterol transport, accumulation of cellular cholesterol and increased abundance of rafts, possibly due to action of Nef, produce a phenotype that mimics at least some elements of HIV-associated hematological disorder, such as anemia and thrombocytopenia. Furthermore, some other elements of hematological abnormality, such as leukocytosis, may have their origin in impaired hematopoiesis, contributing to, rather than originating from, HIV-associated inflammation.
Cognitive impairment
HAND is a frequent comorbidity of HIV infection. Effective treatment of HIV infection has reduced the rate of progression and severity of HAND symptoms, but the overall incidence of HAND (about 50% of HIV-infected subjects) remains unchanged [24,132,133]. HAND has all clinical hallmarks of a neurodegenerative disorder with progressive chronic loss of neurons, a spectrum of declining cognitive functions, together with behavioral changes and motor impairment. Neuroinflammation, demyelination, apoptosis and accelerated development of Alzheimer's disease were implicated as possible pathogenic mechanisms of HAND [134]. All these mechanisms have abnormality of cholesterol metabolism as a key element of their action.
Abnormal cellular cholesterol metabolism has been documented for classical neurodegenerative diseases: Alzheimer's disease, Parkinson disease, prion diseases and Niemann-Pick C disease [135–138]. A key element of neurodegeneration is neuronal dysfunction leading to neuronal death, and there is overwhelming evidence that protein misfolding and subsequent oligomerization/aggregation is a primary cause of neuronal dysfunction and death in many neurodegenerative diseases, such as Alzheimer's disease, Parkinson disease and prion diseases [139]. Many, if not most, neurodegenerative diseases have a ‘prion-like’ feature in their pathogenesis, where a misfolded protein causes cascading misfolding of other copies of itself [140,141] or of a different protein [142]. Two conditions are required for the misfolding process to spread throughout the brain, causing progressive neuronal dysfunction and death. The first is the initial presence of a misfolded copy of an amyloid-like protein, which is a result of a mutation, an infection, a trauma, or is a spontaneous event. The second is a high local concentration of normal protein to allow nucleation and propagation of misfolding cascade to occur [141]. Accumulation of various amyloidogenic proteins is a common feature of many neurodegenerative diseases, and most amyloid proteins involved in neurodegeneration are raft proteins [136,143–145]. Clustering of amyloidogenic proteins in rafts makes them susceptible to modification (e.g. phosphorylation), misfolding and aggregation when a misfolded copy of the protein becomes available. It is plausible that the abundance of rafts, and consequently the availability of sites where proteins are present at high local concentration, is a key ‘permissive’ element, or a risk factor, in the pathogenesis of neurodegenerative diseases, including HAND. The role of lipid rafts in inflammation has been described above, and these considerations apply to neuroinflammation characteristic to HAND. In addition, increased abundance and altered composition of lipid rafts may induce apoptosis of neurons and glia by triggering apoptotic signaling [146–148]. Finally, defects in myelination of axons is a common finding in postmortem analysis of brain samples from HAND patients [149]. Myelin is produced by oligodendrocytes from cholesterol, which in the brain is synthesized almost exclusively by glia [150,151]. Brain cholesterol is extensively recycled from broken down myelin [152]. This recycling, which depends on ABCA1 [153], is essential for both repair and production of new myelin sheaths [154–156]. Therefore, Nef-induced impairment of ABCA1 is expected to affect myelination.
These possibilities are not mutually exclusive, and Nef via its effects on cholesterol metabolism and lipid rafts may contribute to the development of HAND through all of these mechanisms.
Nef in the brain may originate from blood or may be secreted directly into CSF by HIV-infected cells in the brain [65,80,157]. HIV stays in the brain in microglial cells and astrocytes, as well as in perivascular macrophages migrating through blood–brain barrier [158]. Remarkably, HIV infection in the brain persists in treated patients without viremia [24]. The proposed lipid rafts-centered hypothesis unifies many of the known mechanisms of HAND, such as neuroinflammation, neuroapoptosis and connection with the Alzheimer's disease, demyelination, as well as common features of neurodegeneration in general, such as presence of misfolded proteins and impairment of lipid metabolism.
Potential therapeutic strategies
The prominent role played by cholesterol metabolism and lipid rafts in pathogenesis of HIV-associated comorbidities opens a possibility for new therapeutic approaches. Current approaches for treatment or prevention of HIV-associated atherosclerosis do not differ much from approaches used for general population (e.g. using statins). A more specific, and probably more effective, treatment would be to target the cause of HIV-associated cholesterol metabolism impairment, that is either the Nef-induced downregulation of ABCA1, or its downstream effect, modification of lipid rafts.
The first goal can be accomplished by stimulating the ABCA1 expression to counteract the effect of Nef, or by inhibiting the Nef activity. Among the most potent stimulators of ABCA1 expression are agonists of liver X receptor (LXR) [159]. LXR agonists are being developed for the treatment of atherosclerosis, as synthetic LXR agonists have been shown to inhibit the progression [160,161] and even promote the regression [162] of atherosclerosis in mouse models. They also were shown to attenuate inflammation and improve prognosis of neurodegenerative diseases in animal models [163]. However, introduction of LXR agonists into clinical practice was impeded by a significant limitation: LXR activation leads to increased fatty acid synthesis, accumulation of triglycerides and the development of fatty liver [164,165]. A new generation of LXR agonists that do not induce lipogenic effects but preserve ABCA1-inducing activity has been described [166–170], but so far, these drugs have not yet moved beyond Phase I clinical trials.
A search for inhibitors of Nef-mediated downregulation of ABCA1 has just started. Given that work on Nef inhibitors has produced a number of promising compounds [171–174], it appears that a large choice of potential candidates should be available. However, the N-terminal region of Nef responsible for ABCA1 downregulation [74] is distinct from regions of the protein responsible for downregulation of MHC-I, CD4+ and SERINC5, which are targeted in most screens [171–174]. Modeling of Nef-calnexin interaction (Fig. 2) provided an opportunity for virtual screening of potential inhibitors and resulted in identification of the first compounds that specifically inhibit Nef interaction with calnexin and release the Nef-mediated blockage of calnexin-assisted ABCA1 maturation [73,74]. Development of these compounds for clinical use is ahead.
Fig. 2.
Model of Nef-calnexin interaction.
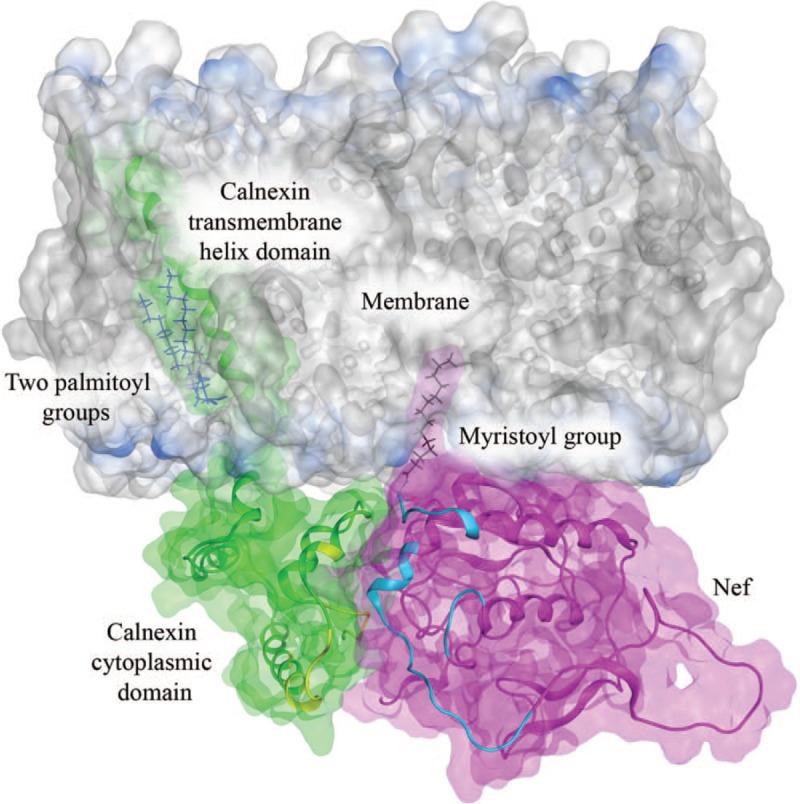
Interacting structures of Nef (green) and the calnexin cytoplasmic domain (magenta) are shown relative to the endoplasmic reticulum membrane. The interaction sites (shown in yellow for calnexin and cyan for Nef) were targeted in the virtual screening aimed at identifying potential small molecule inhibitors of this interaction [72,73]. The model has been adopted from our earlier results [73] and enhanced using molecular dynamics simulation (unpublished data).
Another potential therapeutic approach to alleviate Nef-mediated comorbidities is to reduce and normalize lipid rafts affected by Nef-induced changes to cholesterol metabolism. This appears to be a difficult proposition given essential functions that lipid rafts play in cell physiology, which requires therapeutic agents to discriminate between affected and unaffected cells and provide a measured effect that does not reduce rafts below physiological levels. Currently available lipid raft-targeting agents, such as cyclodextrins, do not fit these criteria and thus cannot be used for long-term treatment necessary to control comorbidities associated with chronic HIV infection. However, a recently identified innate factor, AIBP [175–177], ideally fits this requirement. This protein enhances apoA-I-mediated cholesterol efflux specifically from cells challenged by proinflammatory agents while sparing nonactivated cells [109,175,178–180]. Furthermore, AIBP appears to selectively target lipid rafts on activated cells, normalizing their abundance and function activated by inflammatory stimuli [178]. Our unpublished results demonstrated that AIBP reverses Nef-mediated effects on lipid rafts in monocyte-derived macrophages and normalizes their responses to inflammatory stimuli. Further work is warranted to evaluate the therapeutic potential of AIBP-derived agents for treatment of HIV-associated comorbidities.
Conclusion
Findings described in this review point to an important role that impairment of cholesterol metabolism and downstream changes to lipid rafts play in pathogenesis of HIV-associated comorbidities. While this pathogenic mechanism likely contributes to most metabolic diseases, there is a clear specificity in HIV infection related to the prominent role of Nef. Therefore, potential treatment strategies may combine HIV-specific, Nef-targeting agents, with approaches stimulating cholesterol efflux and reducing lipid rafts in a nonspecific fashion.
Acknowledgements
D.S. and N.M. wrote the part of the review devoted to comorbidities, A.A.M. and A.A. described the studies of Nef-calnexin interaction and therapeutics, and M.B. conceived the review and finalized the article. We are grateful to Dr Gary Simon for advice on antiretroviral drugs. This work was supported by NIH grants R01NS102163, R01HL131473 and P30AI117970, grant from AHA 17GRNT33630163, and grant from Russian Foundation for Basic Research 17-54-30021.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 2011; 17:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samaras K. The burden of diabetes and hyperlipidemia in treated HIV infection and approaches for cardiometabolic care. Curr HIV/AIDS Rep 2012; 9:206–217. [DOI] [PubMed] [Google Scholar]
- 3.Muyanja D, Muzoora C, Muyingo A, Muyindike W, Siedner MJ. High prevalence of metabolic syndrome and cardiovascular disease risk among people with HIV on stable ART in Southwestern Uganda. AIDS Patient Care STDS 2016; 30:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willig AL, Overton ET. Metabolic complications and glucose metabolism in HIV infection: a review of the evidence. Curr HIV/AIDS Rep 2016; 13:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myerson M, Malvestutto C, Aberg JA. Management of lipid disorders in patients living with HIV. J Clin Pharmacol 2015; 55:957–974. [DOI] [PubMed] [Google Scholar]
- 6.Brown TT, Li X, Cole SR, Kingsley LA, Palella FJ, Riddler SA, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS 2005; 19:1375–1383. [DOI] [PubMed] [Google Scholar]
- 7.Tien PC, Schneider MF, Cole SR, Levine AM, Cohen M, DeHovitz J, et al. Antiretroviral therapy exposure and incidence of diabetes mellitus in the Women's Interagency HIV Study. AIDS 2007; 21:1739–1745. [DOI] [PubMed] [Google Scholar]
- 8.Lagathu C, Bereziat V, Gorwood J, Fellahi S, Bastard JP, Vigouroux C, et al. Metabolic complications affecting adipose tissue, lipid and glucose metabolism associated with HIV antiretroviral treatment. Expert Opin Drug Saf 2019; 18:829–840. [DOI] [PubMed] [Google Scholar]
- 9.Echecopar-Sabogal J, D’Angelo-Piaggio L, Chaname-Baca DM, Ugarte-Gil C. Association between the use of protease inhibitors in highly active antiretroviral therapy and incidence of diabetes mellitus and/or metabolic syndrome in HIV-infected patients: a systematic review and meta-analysis. Int J STD AIDS 2018; 29:443–452. [DOI] [PubMed] [Google Scholar]
- 10.Young L, Wohl DA, Hyslop WB, Lee YZ, Napravnik S, Wilkin A. Effects of raltegravir combined with tenofovir/emtricitabine on body shape, bone density, and lipids in African-Americans initiating HIV therapy. HIV Clin Trials 2015; 16:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Worm SW, Sabin C, Weber R, Reiss P, El-Sadr W, Dabis F, et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the data collection on adverse events of anti-HIV drugs (D:A:D) study. J Infect Dis 2010; 201:318–330. [DOI] [PubMed] [Google Scholar]
- 12.Maggi P, Bellacosa C, Carito V, Perilli F, Lillo A, Volpe A, et al. Cardiovascular risk factors in patients on long-term treatment with nevirapine- or efavirenz-based regimens. J Antimicrob Chemother 2011; 66:896–900. [DOI] [PubMed] [Google Scholar]
- 13.Overton ET, Arathoon E, Baraldi E, Tomaka F. Effect of darunavir on lipid profile in HIV-infected patients. HIV Clin Trials 2012; 13:256–270. [DOI] [PubMed] [Google Scholar]
- 14.Margolis AM, Heverling H, Pham PA, Stolbach A. A review of the toxicity of HIV medications. J Med Toxicol 2014; 10:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin A, Smith DE, Carr A, Ringland C, Amin J, Emery S, et al. Reversibility of lipoatrophy in HIV-infected patients 2 years after switching from a thymidine analogue to abacavir: the MITOX Extension Study. AIDS 2004; 18:1029–1036. [DOI] [PubMed] [Google Scholar]
- 16.McComsey GA, Ward DJ, Hessenthaler SM, Sension MG, Shalit P, Lonergan JT, et al. Improvement in lipoatrophy associated with highly active antiretroviral therapy in human immunodeficiency virus-infected patients switched from stavudine to abacavir or zidovudine: the results of the TARHEEL study. Clin Infect Dis 2004; 38:263–270. [DOI] [PubMed] [Google Scholar]
- 17.Ketlogetswe KS, Post WS, Li X, Palella FJ, Jr, Jacobson LP, Margolick JB, et al. Lower adiponectin is associated with subclinical cardiovascular disease among HIV-infected men. AIDS 2014; 28:901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norwood J, Turner M, Bofill C, Rebeiro P, Shepherd B, Bebawy S, et al. Brief report: weight gain in persons with HIV switched from efavirenz-based to integrase strand transfer inhibitor-based regimens. J Acquir Immune Defic Syndr 2017; 76:527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menard A, Meddeb L, Tissot-Dupont H, Ravaux I, Dhiver C, Mokhtari S, et al. Dolutegravir and weight gain: an unexpected bothering side effect?. AIDS 2017; 31:1499–1500. [DOI] [PubMed] [Google Scholar]
- 20.Bourgi K, Rebeiro PF, Turner M, Castilho JL, Hulgan T, Raffanti SP, et al. Greater weight gain in treatment naive persons starting dolutegravir-based antiretroviral therapy. Clin Infect Dis 2019; [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson K, Liner J, Meeker RB. Antiretroviral neurotoxicity. J Neurovirol 2012; 18:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Underwood J, Robertson KR, Winston A. Could antiretroviral neurotoxicity play a role in the pathogenesis of cognitive impairment in treated HIV disease?. AIDS 2015; 29:253–261. [DOI] [PubMed] [Google Scholar]
- 23.Etherton MR, Lyons JL, Ard KL. HIV-associated neurocognitive disorders and antiretroviral therapy: current concepts and controversies. Curr Infect Dis Rep 2015; 17:485. [DOI] [PubMed] [Google Scholar]
- 24.Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder – pathogenesis and prospects for treatment. Nat Rev Neurol 2016; 12:234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereyra F, Lo J, Triant VA, Wei J, Buzon MJ, Fitch KV, et al. Increased coronary atherosclerosis and immune activation in HIV-1 elite controllers. AIDS 2012; 26:2409–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, et al. Immune activation, inflammation, and non-AIDS co-morbidities in HIV-infected patients under long-term ART. Viruses 2019; 11:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol 2008; 1:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buranapraditkun S, Pissani F, Teigler JE, Schultz BT, Alter G, Marovich M, et al. Preservation of peripheral T follicular helper cell function in HIV controllers. J Virol 2017; 91: e00497-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaardbo JC, Ronit A, Hartling HJ, Gjerdrum LM, Springborg K, Ralfkiaer E, et al. Immunoregulatory T cells may be involved in preserving CD4 T cell counts in HIV-infected long-term nonprogressors and controllers. J Acquir Immune Defic Syndr 2014; 65:10–18. [DOI] [PubMed] [Google Scholar]
- 30.Hatano H, Somsouk M, Sinclair E, Harvill K, Gilman L, Cohen M, et al. Comparison of HIV DNA and RNA in gut-associated lymphoid tissue of HIV-infected controllers and noncontrollers. AIDS 2013; 27:2255–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olson GS, Moore SW, Richter JM, Garber JJ, Bowman BA, Rawlings CA, et al. Increased frequency of systemic pro-inflammatory Vdelta1(+) gammadelta T cells in HIV elite controllers correlates with gut viral load. Sci Rep 2018; 8:16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiegand M, Moller AA, Schreiber W, Krieg JC, Holsboer F. Alterations of nocturnal sleep in patients with HIV infection. Acta Neurol Scand 1991; 83:141–142. [DOI] [PubMed] [Google Scholar]
- 33.Baxter AE, O’Doherty U, Kaufmann DE. Beyond the replication-competent HIV reservoir: transcription and translation-competent reservoirs. Retrovirology 2018; 15:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNamara RP, Costantini LM, Myers TA, Schouest B, Maness NJ, Griffith JD, et al. Nef secretion into extracellular vesicles or exosomes is conserved across human and simian immunodeficiency viruses. mBio 2018; 9: e02344-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olivetta E, Arenaccio C, Manfredi F, Anticoli S, Federico M. The contribution of extracellular Nef to HIV-induced pathogenesis. Curr Drug Targets 2016; 17:46–53. [DOI] [PubMed] [Google Scholar]
- 36.Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, et al. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010; 11:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferdin J, Goricar K, Dolzan V, Plemenitas A, Martin JN, Peterlin BM, et al. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS One 2018; 13:e0191613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rolland M, Frahm N, Nickle DC, Jojic N, Deng W, Allen TM, et al. Increased breadth and depth of cytotoxic T lymphocytes responses against HIV-1-B Nef by inclusion of epitope variant sequences. PLoS One 2011; 6:e17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shrivastav S, Kino T, Cunningham T, Ichijo T, Schubert U, Heinklein P, et al. Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor {gamma} and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy. Mol Endocrinol 2008; 22:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohseni Ahooyi T, Shekarabi M, Torkzaban B, Langford TD, Burdo TH, Gordon J, et al. Dysregulation of neuronal cholesterol homeostasis upon exposure to HIV-1 Tat and cocaine revealed by RNA-sequencing. Sci Rep 2018; 8:16300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol 2000; 74:3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses 2001; 17:1009–1019. [DOI] [PubMed] [Google Scholar]
- 43.Campbell SM, Crowe SM, Mak J. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J Clin Virol 2001; 22:217–227. [DOI] [PubMed] [Google Scholar]
- 44.Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol 2002; 76:4709–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holm K, Weclewicz K, Hewson R, Suomalainen M. Human immunodeficiency virus type 1 assembly and lipid rafts: Pr55(gag) associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100. J Virol 2003; 77:4805–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao Z, Graham DR, Hildreth JE. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res Hum Retroviruses 2003; 19:675–687. [DOI] [PubMed] [Google Scholar]
- 47.Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J Virol 2005; 79:12088–12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilflingseder D, Stoiber H. Float on: lipid rafts in the lifecycle of HIV. Front Biosci 2007; 12:2124–2135. [DOI] [PubMed] [Google Scholar]
- 49.Carter GC, Bernstone L, Sangani D, Bee JW, Harder T, James W. HIV entry in macrophages is dependent on intact lipid rafts. Virology 2009; 386:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiner DB, Williams WV, Weisz PB, Greene MI. Synthetic cyclodextrin derivatives inhibit HIV infection in vitro. Pathobiology 1992; 60:206–212. [DOI] [PubMed] [Google Scholar]
- 51.Manes S, del Real G, Lacalle RA, Lucas P, Gomez-Mouton C, Sanchez-Palomino S, et al. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep 2000; 1:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viard M, Parolini I, Sargiacomo M, Fecchi K, Ramoni C, Ablan S, et al. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J Virol 2002; 76:11584–11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rappocciolo G, Jais M, Piazza P, Reinhart TA, Berendam SJ, Garcia-Exposito L, et al. Alterations in cholesterol metabolism restrict HIV-1 trans infection in nonprogressors. mBio 2014; 5:e01031–e010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prasad VR, Bukrinsky MI. New clues to understanding HIV nonprogressors: low cholesterol blocks HIV trans infection. mBio 2014; 5:e01396–e013914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuniholm MH, Liang H, Anastos K, Gustafson D, Kassaye S, Nowicki M, et al. Association of a 3′ untranslated region polymorphism in proprotein convertase subtilisin/kexin type 9 with HIV viral load and CD4+ levels in HIV/hepatitis C virus coinfected women. AIDS 2017; 31:2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Usami Y, Wu Y, Gottlinger HG. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015; 526:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015; 526:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pereira EA, daSilva LL. HIV-1 Nef: taking control of protein trafficking. Traffic 2016; 17:976–996. [DOI] [PubMed] [Google Scholar]
- 59.van’t Wout AB, Swain JV, Schindler M, Rao U, Pathmajeyan MS, Mullins JI, et al. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J Virol 2005; 79:10053–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng YH, Plemenitas A, Fielding CJ, Peterlin BM. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A 2003; 100:8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng YH, Plemenitas A, Linnemann T, Fackler OT, Peterlin BM. Nef increases infectivity of HIV via lipid rafts. Curr Biol 2001; 11:875–879. [DOI] [PubMed] [Google Scholar]
- 63.Cui HL, Grant A, Mukhamedova N, Pushkarsky T, Jennelle L, Dubrovsky L, et al. HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J Lipid Res 2012; 53:696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellwanger JH, Veit TD, Chies JAB. Exosomes in HIV infection: a review and critical look. Infect Genet Evol 2017; 53:146–154. [DOI] [PubMed] [Google Scholar]
- 65.Khan MB, Lang MJ, Huang MB, Raymond A, Bond VC, Shiramizu B, et al. Nef exosomes isolated from the plasma of individuals with HIV-associated dementia (HAD) can induce Abeta1-42 secretion in SH-SY5Y neural cells. J Neurovirol 2016; 22:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puzar Dominkus P, Ferdin J, Plemenitas A, Peterlin BM, Lenassi M. Nef is secreted in exosomes from Nef.GFP-expressing and HIV-1-infected human astrocytes. J Neurovirol 2017; 23:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asztalos BF, Mujawar Z, Morrow MP, Grant A, Pushkarsky T, Wanke C, et al. Circulating Nef induces dyslipidemia in simian immunodeficiency virus-infected macaques by suppressing cholesterol efflux. J Infect Dis 2010; 202:614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui HL, Ditiatkovski M, Kesani R, Bobryshev YV, Liu Y, Geyer M, et al. HIV protein Nef causes dyslipidemia and formation of foam cells in mouse models of atherosclerosis. FASEB J 2014; 28:2828–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mukhamedova N, Hoang A, Dragoljevic D, Dubrovsky L, Pushkarsky T, Low H, et al. Exosomes containing HIV protein Nef reorganize lipid rafts potentiating inflammatory response in bystander cells. PLoS Pathog 2019; 15:e1007907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mujawar Z, Tamehiro N, Grant A, Sviridov D, Bukrinsky M, Fitzgerald ML. Mutation of the ATP cassette binding transporter A1 (ABCA1) C-terminus disrupts HIV-1 Nef binding but does not block the Nef enhancement of ABCA1 protein degradation. Biochemistry 2010; 49:8338–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCaffrey K, Braakman I. Protein quality control at the endoplasmic reticulum. Essays Biochem 2016; 60:227–235. [DOI] [PubMed] [Google Scholar]
- 72.Adzhubei AA, Anashkina AA, Tkachev YV, Kravatsky YV, Pushkarsky T, Kulkarni A, et al. Modelling interaction between HIV-1 Nef and calnexin. AIDS 2018; 32:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hunegnaw R, Vassylyeva M, Dubrovsky L, Pushkarsky T, Sviridov D, Anashkina AA, et al. Interaction between HIV-1 Nef and calnexin: from modeling to small molecule inhibitors reversing HIV-induced lipid accumulation. Arterioscler Thromb Vasc Biol 2016; 36:1758–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jennelle L, Hunegnaw R, Dubrovsky L, Pushkarsky T, Fitzgerald ML, Sviridov D, et al. HIV-1 protein Nef inhibits activity of ATP-binding cassette transporter A1 by targeting endoplasmic reticulum chaperone calnexin. J Biol Chem 2014; 289:28870–28884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Land A, Braakman I. Folding of the human immunodeficiency virus type 1 envelope glycoprotein in the endoplasmic reticulum. Biochimie 2001; 83:783–790. [DOI] [PubMed] [Google Scholar]
- 76.Mukhamedova N, Hoang A, Cui HL, Carmichael I, Fu Y, Bukrinsky M, et al. Small GTPase ARF6 regulates endocytic pathway leading to degradation of ATP-binding cassette transporter A1. Arterioscler Thromb Vasc Biol 2016; 36:2292–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin S, Nadeau PE, Mergia A. HIV inhibits endothelial reverse cholesterol transport through impacting subcellular caveolin-1 trafficking. Retrovirology 2015; 12:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin S, Nadeau PE, Wang X, Mergia A. Caveolin-1 reduces HIV-1 infectivity by restoration of HIV Nef mediated impairment of cholesterol efflux by apoA–I. Retrovirology 2012; 9:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quaranta MG, Mattioli B, Spadaro F, Straface E, Giordani L, Ramoni C, et al. HIV-1 Nef triggers Vav-mediated signaling pathway leading to functional and morphological differentiation of dendritic cells. FASEB J 2003; 17:2025–2036. [DOI] [PubMed] [Google Scholar]
- 80.Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC, et al. HIV type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res Hum Retroviruses 2011; 27:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JH, Schierer S, Blume K, Dindorf J, Wittki S, Xiang W, et al. HIV-Nef and ADAM17-containing plasma extracellular vesicles induce and correlate with immune pathogenesis in chronic HIV infection. EBioMedicine 2016; 6:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013; 200:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013; 39:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol 2011; 187:1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sorci-Thomas MG, Thomas MJ. Microdomains, inflammation, and atherosclerosis. Circ Res 2016; 118:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lai L, Azzam KM, Lin WC, Rai P, Lowe JM, Gabor KA, et al. MicroRNA-33 regulates the innate immune response via ATP binding cassette transporter-mediated remodeling of membrane microdomains. J Biol Chem 2016; 291:19651–19660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murphy AJ, Woollard KJ, Suhartoyo A, Stirzaker RA, Shaw J, Sviridov D, et al. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A–I in in vitro and in vivo models of inflammation. Arterioscler Thromb Vasc Biol 2011; 31:1333–1341. [DOI] [PubMed] [Google Scholar]
- 88.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015; 15:104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.El Sadr WM, Mullin CM, Carr A, Gibert C, Rappoport C, Visnegarwala F, et al. Effects of HIV disease on lipid, glucose and insulin levels: results from a large antiretroviral-naive cohort. HIV Med 2005; 6:114–121. [DOI] [PubMed] [Google Scholar]
- 90.Oh J, Hegele RA. HIV-associated dyslipidaemia: pathogenesis and treatment. Lancet Infect Dis 2007; 7:787–796. [DOI] [PubMed] [Google Scholar]
- 91.Low H, Hoang A, Pushkarsky T, Dubrovsky L, Dewar E, Di Yacovo MS, et al. HIV disease, metabolic dysfunction and atherosclerosis: a three year prospective study. PLoS One 2019; 14:e0215620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rose H, Woolley I, Hoy J, Dart A, Bryant B, Mijch A, et al. HIV infection and high-density lipoprotein: the effect of the disease vs the effect of treatment. Metabolism 2006; 55:90–95. [DOI] [PubMed] [Google Scholar]
- 93.Attie AD, Kastelein JP, Hayden MR. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res 2001; 42:1717–1726. [PubMed] [Google Scholar]
- 94.Tort O, Escriba T, Egana-Gorrono L, de Lazzari E, Cofan M, Fernandez E, et al. Cholesterol efflux responds to viral load and CD4 counts in HIV+ patients and is dampened in HIV exposed. J Lipid Res 2018; 59:2108–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Siegel MO, Borkowska AG, Dubrovsky L, Roth M, Welti R, Roberts AD, et al. HIV infection induces structural and functional changes in high density lipoproteins. Atherosclerosis 2015; 243:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Serfaty-Lacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, Janus ED, et al. Homozygous Tangier disease and cardiovascular disease. Atherosclerosis 1994; 107:85–98. [DOI] [PubMed] [Google Scholar]
- 97.Chung S, Timmins JM, Duong M, Degirolamo C, Rong S, Sawyer JK, et al. Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J Biol Chem 2010; 285:12197–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vu CN, Ruiz-Esponda R, Yang E, Chang E, Gillard B, Pownall HJ, et al. Altered relationship of plasma triglycerides to HDL cholesterol in patients with HIV/HAART-associated dyslipidemia: further evidence for a unique form of metabolic syndrome in HIV patients. Metabolism 2013; 62:1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Low H, Cheng L, Di Yacovo MS, Churchill MJ, Meikle P, Bukrinsky M, et al. Lipid metabolism in patients infected with Nef-deficient HIV-1 strain. Atherosclerosis 2016; 244:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shah ASV, Stelzle D, Lee KK, Beck EJ, Alam S, Clifford S, et al. Global burden of atherosclerotic cardiovascular disease in people living with HIV. Circulation 2018; 138:1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Triant VA, Perez J, Regan S, Massaro JM, Meigs JB, Grinspoon SK, et al. Cardiovascular risk prediction functions underestimate risk in HIV infection. Circulation 2018; 137:2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002; 105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 103.Singaraja RR, Brunham LR, Visscher H, Kastelein JJ, Hayden MR. Efflux and atherosclerosis: the clinical and biochemical impact of variations in the ABCA1 gene. Arterioscler Thromb Vasc Biol 2003; 23:1322–1332. [DOI] [PubMed] [Google Scholar]
- 104.Pushkarsky T, Shilov E, Kruglova N, Naumann R, Brichacek B, Jennelle L, et al. Short communication: accumulation of neutral lipids in liver and aorta of Nef-transgenic mice. AIDS Res Hum Retroviruses 2017; 33:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zysset D, Weber B, Rihs S, Brasseit J, Freigang S, Riether C, et al. TREM-1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat Commun 2016; 7:13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem 2014; 289:24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rios FJ, Ferracini M, Pecenin M, Koga MM, Wang Y, Ketelhuth DF, et al. Uptake of oxLDL and IL-10 production by macrophages requires PAFR and CD36 recruitment into the same lipid rafts. PLoS One 2013; 8:e76893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med 2016; 8:333ra350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schneider DA, Choi SH, Agatisa-Boyle C, Zhu L, Kim J, Pattison J, et al. AIBP protects against metabolic abnormalities and atherosclerosis. J Lipid Res 2018; 59:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Naidu S, Ponnampalvanar S, Kamaruzzaman SB, Kamarulzaman A. Prevalence of metabolic syndrome among people living with HIV in developing countries: a systematic review. AIDS Patient Care STDS 2017; 31:1–13. [DOI] [PubMed] [Google Scholar]
- 111.Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 2007; 13:340–347. [DOI] [PubMed] [Google Scholar]
- 112.Ohtsubo K, Takamatsu S, Gao C, Korekane H, Kurosawa TM, Taniguchi N. N-Glycosylation modulates the membrane sub-domain distribution and activity of glucose transporter 2 in pancreatic beta cells. Biochem Biophys Res Commun 2013; 434:346–351. [DOI] [PubMed] [Google Scholar]
- 113.Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta-cell dysfunction. Diabetes 2007; 56:2328–2338. [DOI] [PubMed] [Google Scholar]
- 114.Xia F, Gao X, Kwan E, Lam PP, Chan L, Sy K, et al. Disruption of pancreatic beta-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem 2004; 279:24685–24691. [DOI] [PubMed] [Google Scholar]
- 115.Dirkx R, Jr, Solimena M. Cholesterol-enriched membrane rafts and insulin secretion. J Diabetes Investig 2012; 3:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bogan JS, Xu Y, Hao M. Cholesterol accumulation increases insulin granule size and impairs membrane trafficking. Traffic 2012; 13:1466–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kruit JK, Wijesekara N, Fox JE, Dai XQ, Brunham LR, Searle GJ, et al. Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes 2011; 60:3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, et al. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci U S A 2007; 104:13678–13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cheney L, Hou JC, Morrison S, Pessin J, Steigbigel RT. Nef inhibits glucose uptake in adipocytes and contributes to insulin resistance in human immunodeficiency virus type I infection. J Infect Dis 2011; 203:1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fecchi K, Volonte D, Hezel MP, Schmeck K, Galbiati F. Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J 2006; 20:705–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.von Eckardstein A, Sibler RA. Possible contributions of lipoproteins and cholesterol to the pathogenesis of diabetes mellitus type 2. Curr Opin Lipidol 2011; 22:26–32. [DOI] [PubMed] [Google Scholar]
- 122.Vishnu P, Aboulafia DM. Haematological manifestations of human immune deficiency virus infection. Br J Haematol 2015; 171:695–709. [DOI] [PubMed] [Google Scholar]
- 123.Prost S, Le DM, Auge S, Le GR, Derdouch S, Auregan G, et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest 2008; 118:1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Calenda V, Graber P, Delamarter JF, Chermann JC. Involvement of HIV nef protein in abnormal hematopoiesis in AIDS: in vitro study on bone marrow progenitor cells. Eur J Haematol 1994; 52:103–107. [DOI] [PubMed] [Google Scholar]
- 125.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010; 328:1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, et al. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 2012; 11:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Investig 2011; 121:4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gu Q, Yang X, Lv J, Zhang J, Xia B, Kim JD, et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 2019; 363:1085–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ratajczak MZ, Adamiak M. Membrane lipid rafts, master regulators of hematopoietic stem cell retention in bone marrow and their trafficking. Leukemia 2015; 29:1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yamazaki S, Iwama A, Takayanagi S, Morita Y, Eto K, Ema H, et al. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J 2006; 25:3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Adamiak M, Poniewierska-Baran A, Borkowska S, Schneider G, Abdelbaset-Ismail A, Suszynska M, et al. Evidence that a lipolytic enzyme – hematopoietic-specific phospholipase C-beta2 – promotes mobilization of hematopoietic stem cells by decreasing their lipid raft-mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia 2016; 30:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cysique LA, Brew BJ. Prevalence of nonconfounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J Neurovirol 2011; 17:176–183. [DOI] [PubMed] [Google Scholar]
- 133.Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G. Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol 2009; 4:163–174. [DOI] [PubMed] [Google Scholar]
- 134.Canet G, Dias C, Gabelle A, Simonin Y, Gosselet F, Marchi N, et al. HIV neuroinfection and Alzheimer's disease: similarities and potential links?. Front Cell Neurosci 2018; 12:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pani A, Mandas A, Dessi S. Cholesterol, Alzheimer's disease, prion disorders: a menage a trois?. Curr Drug Targets 2010; 11:1018–1031. [DOI] [PubMed] [Google Scholar]
- 136.Kim KS, Kim JS, Park JY, Suh YH, Jou I, Joe EH, et al. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum Mol Genet 2013; 22:4805–4817. [DOI] [PubMed] [Google Scholar]
- 137.Cui HL, Guo B, Scicluna B, Coleman BM, Lawson VA, Ellett L, et al. Prion infection impairs cholesterol metabolism in neuronal cells. J Biol Chem 2014; 289:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vance JE, Karten B. Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res 2014; 55:1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature 2006; 443:796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, et al. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci U S A 2014; 111:10329–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Holmes BB, Diamond MI. Prion-like properties of tau protein: the importance of extracellular tau as a therapeutic target. J Biol Chem 2014; 289:19855–19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luo J, Wärmländer SKTS, Gräslund A, Abrahams JP. Cross-interactions between the alzheimer disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J Biol Chem 2016; 291:16485–16493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of alzheimer's β-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry 2014; 53:4489–4502. [DOI] [PubMed] [Google Scholar]
- 144.Valencia A, Reeves PB, Sapp E, Li X, Alexander J, Kegel KB, et al. Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington's disease. J Neurosci Res 2010; 88:179–190. [DOI] [PubMed] [Google Scholar]
- 145.Malchiodi-Albedi F, Paradisi S, Matteucci A, Frank C, Diociaiuti M. Amyloid oligomer neurotoxicity, calcium dysregulation, and lipid rafts. Int J Alzheimers Dis 2011; 2011:906964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mollinedo F, Gajate C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Future Oncol 2010; 6:811–821. [DOI] [PubMed] [Google Scholar]
- 147.Gargalovic P, Dory L. Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J Lipid Res 2003; 44:1622–1632. [DOI] [PubMed] [Google Scholar]
- 148.Michel V, Bakovic M. Lipid rafts in health and disease. Biol Cell 2007; 99:129–140. [DOI] [PubMed] [Google Scholar]
- 149.Liu H, Xu E, Liu J, Xiong H. Oligodendrocyte injury and pathogenesis of HIV-1-associated neurocognitive disorders. Brain Sci 2016; 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci 2005; 8:468–475. [DOI] [PubMed] [Google Scholar]
- 151.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001; 12:105–112. [DOI] [PubMed] [Google Scholar]
- 152.Ando S, Tanaka Y, Toyoda Y, Kon K. Turnover of myelin lipids in aging brain. Neurochem Res 2003; 28:5–13. [DOI] [PubMed] [Google Scholar]
- 153.Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015; 6:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bartzokis G, Lu PH, Geschwind DH, Edwards N, Mintz J, Cummings JL. Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Arch Gen Psychiatry 2006; 63:63–72. [DOI] [PubMed] [Google Scholar]
- 155.Bartzokis G, Lu PH, Geschwind DH, Tingus K, Huang D, Mendez MF, et al. Apolipoprotein E affects both myelin breakdown and cognition: implications for age-related trajectories of decline into dementia. Biol Psychiatry 2007; 62:1380–1387. [DOI] [PubMed] [Google Scholar]
- 156.Bjorkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 2004; 24:806–815. [DOI] [PubMed] [Google Scholar]
- 157.Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis 2017; 8:e2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alexaki A, Wigdahl B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog 2008; 4:e1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li X, Yeh V, Molteni V. Liver X receptor modulators: a review of recently patented compounds (2007–2009). Expert Opin Ther Pat 2010; 20:535–562. [DOI] [PubMed] [Google Scholar]
- 160.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A 2002; 99:7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D, et al. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett 2003; 536:6–11. [DOI] [PubMed] [Google Scholar]
- 162.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, et al. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol 2005; 25:135–142. [DOI] [PubMed] [Google Scholar]
- 163.Xu P, Li D, Tang X, Bao X, Huang J, Tang Y, et al. LXR agonists: new potential therapeutic drug for neurodegenerative diseases. Mol Neurobiol 2013; 48:715–728. [DOI] [PubMed] [Google Scholar]
- 164.Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem 2002; 277:34182–34190. [DOI] [PubMed] [Google Scholar]
- 165.Chisholm JW, Hong J, Mills SA, Lawn RM. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 2003; 44:2039–2048. [DOI] [PubMed] [Google Scholar]
- 166.Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, Miyazaki M, et al. Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res 2009; 50:312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li N, Wang X, Zhang J, Liu C, Li Y, Feng T, et al. Identification of a novel partial agonist of liver X receptor alpha (LXRalpha) via screening. Biochem Pharmacol 2014; 92:438–447. [DOI] [PubMed] [Google Scholar]
- 168.van der Hoorn J, Linden D, Lindahl U, Bekkers M, Voskuilen M, Nilsson R, et al. Low dose of the liver X receptor agonist, AZ876, reduces atherosclerosis in APOE∗3Leiden mice without affecting liver or plasma triglyceride levels. Br J Pharmacol 2011; 162:1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kirchgessner TG, Martin R, Sleph P, Grimm D, Liu X, Lupisella J, et al. Pharmacological characterization of a novel liver X receptor agonist with partial LXRalpha activity and a favorable window in nonhuman primates. J Pharmacol Exp Ther 2015; 352:305–314. [DOI] [PubMed] [Google Scholar]
- 170.Li N, Wang X, Xu Y, Lin Y, Zhu N, Liu P, et al. Identification of a novel liver X receptor agonist that regulates the expression of key cholesterol homeostasis genes with distinct pharmacological characteristics. Mol Pharmacol 2017; 91:264–276. [DOI] [PubMed] [Google Scholar]
- 171.Dekaban GA, Dikeakos JD. HIV-I Nef inhibitors: a novel class of HIV-specific immune adjuvants in support of a cure. AIDS Res Ther 2017; 14:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Moonsamy S, Bhakat S, Ramesh M, Soliman ME. Identification of binding mode and prospective structural features of novel nef protein inhibitors as potential anti-HIV drugs. Cell Biochem Biophys 2017; 75:49–64. [DOI] [PubMed] [Google Scholar]
- 173.Emert-Sedlak LA, Loughran HM, Shi H, Kulp JL, 3rd, Shu ST, Zhao J, et al. Synthesis and evaluation of orally active small molecule HIV-1 Nef antagonists. Bioorg Med Chem Lett 2016; 26:1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Smithgall TE, Thomas G. Small molecule inhibitors of the HIV-1 virulence factor. Nef Drug Discov Today Technol 2013; 10:e523–e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fang L, Choi SH, Baek JS, Liu C, Almazan F, Ulrich F, et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013; 498:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fang L, Liu C, Miller YI. Zebrafish models of dyslipidemia: relevance to atherosclerosis and angiogenesis. Transl Res 2014; 163:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhu L, Fang L. AIBP: a novel molecule at the interface of cholesterol transport, angiogenesis, and atherosclerosis. Methodist DeBakey Cardiovasc J 2015; 11:160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Woller SA, Choi SH, An EJ, Low H, Schneider DA, Ramachandran R, et al. Inhibition of neuroinflammation by AIBP: spinal effects upon facilitated pain states. Cell Rep 2018; 23:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Choi SH, Wallace AM, Schneider DA, Burg E, Kim J, Alekseeva E, et al. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight 2018; 3: e120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang M, Li L, Xie W, Wu JF, Yao F, Tan YL, et al. Apolipoprotein A-1 binding protein promotes macrophage cholesterol efflux by facilitating apolipoprotein A-1 binding to ABCA1 and preventing ABCA1 degradation. Atherosclerosis 2016; 248:149–159. [DOI] [PubMed] [Google Scholar]