

SUMMARY
Immunotherapy can reinvigorate dormant responses to cancer, but response rates remain low. Oncolytic viruses, which replicate in cancer cells, induce tumor lysis and immune priming, but their immune consequences are unclear. We profiled the infiltrate of aggressive melanomas induced by oncolytic Vaccinia virus using RNA sequencing and found substantial remodeling of the tumor microenvironment, dominated by effector T cell influx. However, responses to oncolytic viruses were incomplete due to metabolic insufficiencies induced by the tumor microenvironment. We identified the adipokine leptin as a potent metabolic reprogramming agent that supported antitumor responses. Leptin metabolically reprogrammed T cells in vitro, and melanoma cells expressing leptin were immunologically controlled in mice. Engineering oncolytic viruses to express leptin in tumor cells induced complete responses in tumor-bearing mice and supported memory development in the tumor infiltrate. Thus, leptin can provide metabolic support to tumor immunity and oncolytic viruses represent a platform to deliver metabolic therapy.
Graphical Abstract
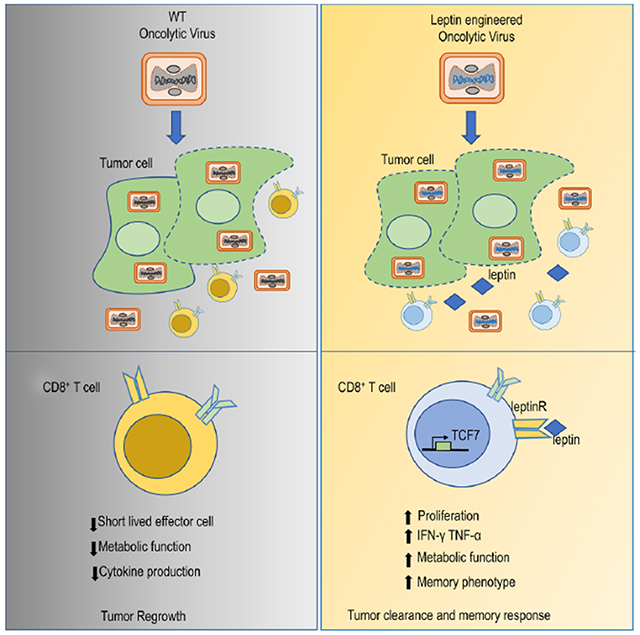
Blurb:
Metabolic insufficiency is a major barrier for anti-tumor immunity. Rivadeneira et al. demonstrate that engineering an oncolytic virus to express a metabolic modulator, in this case the adipokine leptin, improves T cell metabolic function in the tumor microenvironment allowing a superior antitumor response compared to a control oncolytic.
INTRODUCTION
The successes associated with immunotherapy as a cancer treatment have resulted in a major shift in both cancer research and clinical practice, with a dominant focus on understanding and modulating immune activity at the tumor site. In solid tumors, immunotherapies using monoclonal antibody mediated ‘checkpoint’ blockade of PD-1 and CTLA-4 have resulted in durable responses, leading to FDA approval in a variety of indications (La-Beck et al., 2015; Mahoney et al., 2015). However, the reality of single agent immunotherapies is that the majority of patients will not experience long-term durable benefits (Herbst et al., 2014; Weber et al., 2015). This resistance likely occurs for multiple reasons, but prominent among them are the failure to recruit T cells to the tumor and other, more dominant immunosuppressive mechanisms which limit T cell function in the tumor microenvironment (Sharma et al., 2017). Therefore, there is the need for new therapeutic modalities that could overcome these resistance mechanisms. Oncolytic virus immunotherapy is a class of therapeutic agent that has recently received more attention since the FDA approval in 2015 of a genetically modified herpes simplex virus, type 1 talimogene laherparepvec (T-VEC; Imlygic)(Andtbacka et al., 2015). Unlike checkpoint blockade approaches, oncolytic viruses have the ability to induce an immune response by triggering tumor-associated antigen and epitope spreading (Kanerva et al., 2013). Nonetheless, T-VEC’s approval was based on durable remission rate of 16% in melanoma patients (Andtbacka et al., 2015) highlighting the ample space for improvement of this class of therapy as well as further elucidate its mechanisms of action. The exact cell populations involved in oncolytic virus-induced immune response is poorly understood, and the functional status of newly infiltrating lymphocytes has not been well studied.
It is now appreciated that the metabolism of both T cells and tumor cells represent key mechanisms limiting immune function against cancer. Cancer cells become metabolically deregulated, depleting the local environment of essential nutrients and producing an excess of potentially toxic byproducts. In addition, tumor-infiltrating T cells acquire significant metabolic insufficiencies, including repressed glucose uptake and the loss of functional mitochondria. Thus, T cells are rendered insufficient in an environment that produces hypoxia and nutrient stress (Justus et al., 2015; Siska and Rathmell, 2015). Several groups including ours have demonstrated that metabolic reprogramming of T cells or modulation of the tumor microenvironment can result in increased antitumor immunity and response to immunotherapy (Rivadeneira and Delgoffe, 2018). Our group has shown this metabolic reprogramming can be achieved by retroviral expression of metabolic genes in tumor-specific T cells, co-stimulation via the 41BB receptor, and pharmacologic remodeling of the tumor microenvironment using the type II diabetes drug metformin (Menk et al., 2018; Scharping et al., 2016a; Scharping et al., 2017). However, the use of genetically encoded, protein-based soluble factors like adipokines for immunometabolic modulation remains unstudied.
Leptin is a canonical adipokine with potent metabolic reprogramming functions (Perez-Perez et al., 2017) such as the promotion of glucose and fatty oxidation as well as mitochondrial biogenesis (Saucillo et al., 2014; Steinberg et al., 2002). While discovered and typically studied in the context of obesity, T cells express the leptin receptor and leptin levels have been associated with inflammatory states (Abella et al., 2017). Indeed, T cells stimulated in the context of leptin can elaborate more cytokine and increase their proliferation (Dixit et al., 2004; Howard et al., 1999; Lord et al., 1998). Nonetheless, to date the study of leptin in immunity has been largely assessed in the context of obesity and not ascertained therapeutically.
In this study, we used single cell RNA sequencing to deeply profile the infiltrate of aggressive melanomas induced by oncolytic Vaccinia virus, revealing that oncolytic viruses promote the infiltration of a robust tumor infiltrate, but one that is ultimately ineffective at promoting complete responses, due in part to metabolic insufficiency. We next explored the utility of leptin as a tool to overcome the observed metabolic insufficiency by promoting the metabolic reprogramming of tumor-infiltrating T cells. Using a melanoma model in which leptin is locally elevated in the tumor microenvironment, we showed potent T cell activation and tumor control that was linked to metabolic reprogramming. Further, we re-designed the Vaccinia virus to genetically express and deliver leptin to the tumor microenvironment. This therapy resulted in complete therapeutic responses in mice compared to wild-type virus. Leptin expressing vaccinia virus simultaneously lysed tumor cells, leading to stimulation of new T cell infiltration, while also metabolically supporting the activity of that infiltrate through the local secretion of leptin.
Results
Oncolytic Vaccinia virus treatment of tumors resulted in the remodeling of the tumor immune microenvironment
While oncolytic viruses like T-VEC are FDA approved immunotherapies for cancer treatment, the immune consequences of these agents are unclear. We first sought to systematically profile the immune infiltrate induced by oncolytic virus infection. One major limitation of oncolytic virus therapy is that many viruses, including T-VEC, do not replicate efficiently in hypoxia (Friedman et al., 2012; Pipiya et al., 2005). Thus, we aimed to employ oncolytic Vaccinia virus, which is easily engineered, encodes its own polymerase, and, importantly, maintains replicative function in hypoxic tumor cells (Hiley et al., 2010; Moss, 2013). We utilized the Western Reserve laboratory strain Vaccinia virus which harbors a genetic deletion of thymidine kinase and Vaccinia growth factor genes generating a potent oncolytic viral agent (Buller et al., 1985; Puhlmann et al., 2000; Whitman et al., 1994). We employed a melanoma cell line termed clone 24 (CL24); generated in our lab from a single-cell of a Ptenf/fBrafLSL.V600ETyr2Cre.ER mouse that developed melanoma after tamoxifen administration (Dankort et al., 2009); recently reported (Najjar et al., 2019). This cell line is syngeneic to C57/BL6 mice, carries driver mutations common in human melanoma (as opposed to the often used B16). CL24 is poorly infiltrated and is completely insensitive to monoclonal antibody against Programmed Death 1( anti-PD1 monotherapy (Najjar et al., 2019). A single dose of Vaccinia virus was injected intratumorally when tumors reached 4 mm in size, which resulted in substantial tumor regression, but no complete responses (Fig. 1a). We thus sought to determine the character of the tumor infiltrate induced by oncolytic viruses, employing single cell RNA sequencing of the CD45+ tumor infiltrating leukocytes of PBS or Vaccinia infected clone 24 tumors. We first used unsupervised clustering data analysis to separate the CD45+ cells into distinct groups of immune populations (Fig. 1b). These immune populations were then classified based on the expression of known markers for each population (Fig S1a–b). Importantly, these analyses were conducted when tumors had not yet regressed. Our data identified known immune cell populations when analyzed in aggregate, however analysis based on treatment group revealed that oncolytic Vaccinia virus immunotherapy induced changes in the tumor immune microenvironment (Fig. 1c). Our data revealed Vaccinia infected tumors showed an influx of new, effector like CD8+ T cells, NK cells and monocytes, and a proportional loss of dysfunctional or suppressive cells like MDSC, and tumor-associated macrophages compared to PBS treated tumors. (Fig. 1c). Taken together, RNA sequencing analysis showed that oncolytic Vaccinia virus induced a remodeling of the tumor immune microenvironment, but one that ultimately succumbs to tumor-induced immune suppression and eventual outgrowth.
Figure 1. Oncolytic Vaccinia virus has potent immunostimulatory activity.

(A) C57BL/6J mice were injected subdermally with CL24 cells. 5-7 days after tumor cell injection tumors were treated intratumorally with PBS, control Vaccinia virus (VVI) at 2.5x106 PFU and tumor growth monitored. Each line represents an individual mouse. (B) Single-cell RNA-seq data for 4000 cells CD45+ sorted cells treated as in (A). Cells were extracted on day 7. Data was generated by unsupervised clustering through Seurat. (C) UMAP analysis and quantification of PBS and VV treated mice. Data represents n=2 per condition. See also Figure S1.
Oncolytic Vaccinia virus promoted non-exhausted T cell infiltration with severe metabolic deficiencies
Flow cytometric analysis of the tumor infiltrating lymphocytes (TIL) from oncolytic virus treated mice confirmed that the influx of new immune cells appeared to be dominated by CD8+ T cells while we observe a decreased representation of regulatory T cells amongst the CD4+ compartment (Fig. 2a). Analysis of the co-inhibitory marker expression showed these ‘new’ CD8+ T cells have protein expression of T cell immunoglobulin and mucin containing 3 (Tim-3) alone (Fig.2b) as well as a low to mid expression of PD1 (Fig. 2c) suggesting these cells are not reinvigorated tumor residents but rather new immigrants that are not yet fully exhausted T cells. While co-inhibitory molecule expression is associated with T cell dysfunction, we and other have also shown that metabolic insufficiency, too, can predict T cell function (Scharping et al., 2016a). We further analyzed mitochondrial content as a marker for metabolic sufficiency, revealing that despite the ‘non-exhausted’ co-inhibitory molecule pattern of expression, TIL from oncolytic virus-treated tumors were still metabolically insufficient as evidenced by the lack of change in MitoTracker staining compared to untreated tumors (Fig. 2d). Overall, Vaccinia virus-induced oncolysis reprograms the immune microenvironment, promoting an influx of new T cells that ultimately succumb to metabolic insufficiency.
Figure 2. Tumor infiltrating lymphocyte (TIL) analysis of oncolytic Vaccinia virus treated tumors shows non-exhausted T cell infiltration with metabolic deficiencies.
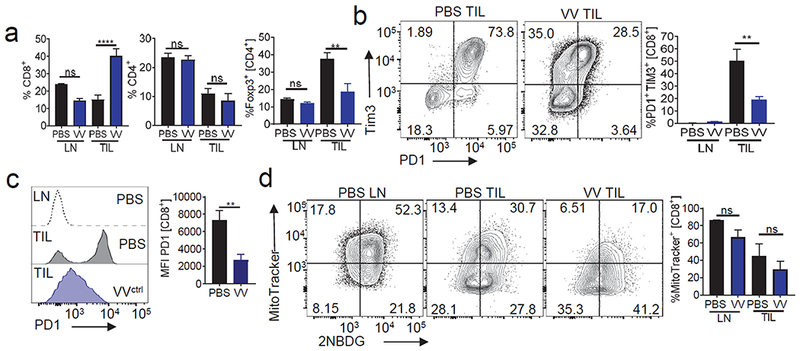
C57BL/6J mice were injected subdermally with CL24 cells. 5-7 days after tumor cell injection tumors were treated intratumorally with PBS, control Vaccinia virus (VVctrll). (A) CD8 and CD4 expression analysis on LN and TIL from mice treated as in (A). Representative flow cytogram of CD8 and CD4 staining in LN and TIL and tabulated flow cytometric data. (B) Expression of inhibitory molecules PD1 and TIM3 from mice treated as in (A). Representative flow cytogram of PD1 and Tim3 staining in LN and TIL and tabulated flow cytometric data. (C) Representative histogram PD1 expression on CD8+ T cells and tabulated data. (D) Mitochondrial content analyzed by mitotracker staining in CD8+ T cells from mice treated as in (A). Representative flow cytogram of mitotracker against 2NBDG staining in LN and TIL and tabulated flow cytometric data. Data represents at least 3 independent experiments *p <0.05, **p <0.01, ***p <0.001 by paired t-test. Error bars indicate s.e.m.
Leptin metabolically reprogramed activated T cells
Given that oncolytic viruses stimulated new immune infiltrate that still succumbed to metabolic dysfunction, we next sought to explore ways to bolster those new T cells such that they would be more competitive in the tumor microenvironment. We also wished to utilize genetically encoded payload, so that such an agent could be encoded in the viral vector. As previously discussed, leptin is a cytokine that modulates energy homeostasis as well as promotes an inflammatory response (La Cava and Matarese, 2004). We first sought to determine the metabolic reprogramming functions of leptin on T cells. We activated CD8+ T cells isolated from peripheral lymph nodes (LN) and cultured cells in increasing concentrations of leptin. Leptin induced increases in both basal oxygen consumption rate as well as spare respiratory capacity (a measure of mitochondrial reserve that defines long-lived memory T cells (Dixit et al., 2004)) of CD8+ T cells (Fig. 3a), but had little effect on activated T cells’ ability to perform glycolysis as measured by extracellular acidification (Fig. 3b). Flow cytometry analysis reinforced this data showing an increase in mitochondrial mass under leptin exposure indicative of higher oxidative phosphorylation, while observing no changes in glucose uptake (Fig. 3c). Treatment of activated T cells short-term with leptin revealed increased activation of phospho-p38 MAPK and consequent activation of the transcription factor ATF-2, which we and others have previously shown as critical for mitochondrial biogenesis (Akimoto et al., 2005; Menk et al., 2018) (Fig. 3d). Thus, leptin can stimulate T cells to increase their oxidative activity and capacity, a metabolic reprogramming event highly desirable in the tumor microenvironment.
Figure 3. The leptin receptor is upregulated in tumor infiltrating T cells and leptin is capable of metabolic reprogramming.

(A) Representative OCR trace and tabulated OCR and Spare Respiratory Capacity (SRC) of CD8+ T cells activated with 3ug/m: immobilized anti-CD3 in the presence of antiCD28 (2ug/mL) for 24h. Cells were treated with 0.0, 0.1, and 1.0nM of mouse recombinant leptin for 24h. (B) Representative ECAR trace for cells treated as (A). (C) Mitochondrial content analyzed by mitotracker staining and glucose uptake by 2NBDG staining in CD8+ T cells from mice treated as in (A). Representative flow cytogram of mitotracker against 2NBDG staining in LN and TIL and tabulated flow cytometric data. (D) T cells day 5 after activation treated with 1.0nM recombinant leptin and harvested at 10, 30 and 60 min. Immunoblot of p-p38MAPK and p-ATF2, actin was used as a loading control. (E) Leptin receptor staining of CD8+ T cells from mouse lymph nodes (LN) and tumor infiltrating CD8+ T cells (TIL) from tumor bearing mice. Leptin receptor staining of CD8+ T cells from mouse lymph nodes (LN) and tumor infiltrating CD8+ T cells (TIL) from tumor bearing mice. (F) Leptin receptor expression staining of PD1 and Tim3 in CD8+ T cells in LN and TIL. Data represents at least 3 independent experiments *p <0.05 by unpaired t-test. Error bars indicate s.e.m. Data represents at least 3 independent experiments *p <0.05, **p <0.01, ***p <0.001 by paired t-test. Error bars indicate s.e.m.
We assessed the expression of the leptin receptor in murine T cells, confirming expression of the leptin receptor in T cells as previously observed (Lord et al., 1998) (Fig. 3e). Furthermore, melanoma tumor infiltrating lymphocytes (TIL) expressed more leptin receptor compared to T cells in the lymph nodes (Fig. 3e). Categorizing the TIL according to expression of co-inhibitory molecules, we observed higher expression of the leptin receptor in activated or exhausted T cells with high expression of PD1 and Tim3 (Fig. 3f). Thus, leptin can promote metabolic reprogramming in T cells, and tumor-infiltrating T cells bear its receptor.
Elevating local leptin protein concentrations in the tumor microenvironment enabled antitumor immunity
The therapeutic effects of leptin in the context of tumor infiltrating lymphocytes have not been previously investigated. We hypothesized that leptin can enhance the metabolic capacity of tumor infiltrating lymphocytes, consequently enhancing their function in the tumor. Initial studies treating tumor-bearing mice with recombinant leptin showed that systemic delivery, even at relatively high doses did not have any effects on tumor growth nor TIL makeup (Fig. S2a). Intratumoral administration of leptin showed small but not significant changes, suggesting that leptin may need to be more effectively delivered, locally, to bolster intratumoral immunity (Fig. S2a). To first test the metabolic reprogramming functions of leptin in isolation (outside of oncolytic virus infection), we instead engineered CL24 cells to express an empty vector (CL24hygro), or leptin (CL24leptin). CL24leptin cells expressed leptin intracellularly (Fig. S2b) and released it into culture supernatant (Fig. S2c). In vitro, CL24leptin showed a comparable growth kinetics with CL24-expressing a control plasmid (CL24hygro) (Fig. S3d). However, when CL24leptin cells were injected subdermally into C57BL/6J mice, they grew at a substantially slower rate compared to CL24hygro controls (Fig. 4a) and have significantly prolonged survival (Fig. 4b) suggesting leptin may stimulate host immunity. Indeed, depletion of CD8+ T cells (Fig. S2e) revealed that the controlled tumor growth observed in CL24leptin tumors required functional immunity (Fig. 4c). To determine whether this increased antitumor immunity was due to leptin acting on T cells, we injected control or leptin-overexpressing CL24 into mice bearing a T cell-restricted heterozygous deletion of the leptin receptor. Leptin receptor is haploinsufficient (Coleman, 1979), and T cells from these mice express lower protein expression of the leptin receptor (Fig. S2f). In these mice, leptin-overexpressing tumors were not controlled by the immune system, mirroring total CD8 T cell depletion (Fig. 4d). Thus, locally elevating leptin in the tumor microenvironment acted directly on T cells to induce immune-mediated tumor growth control. Analysis of the tumor infiltrating lymphocytes at day 10 (when tumors were of comparable size between groups) showed increased numbers of CD8+ T cells in tumors overexpressing leptin compared to control tumors (Fig. 4e) CL24 expresses gp100, so we directly tested whether leptin enhanced tumor’s ability to be infiltrated using an adoptive transfer of activated Pmel CD8+ T cells. These data show a significant increase in infiltration 72 h post adoptive transfer. (Fig S3a). We also noted leptin-overexpressing CL24 tumors had increased numbers of natural killer cells, but not other immune populations such as B cells (Fig. S3b). Thus, engineered overexpression of leptin in melanoma cells resulted in a tumor infiltrate more biased towards type 1 immunity, and localized expression of leptin controls tumor growth by enhancing a CD8+ T cell-dependent anti-tumor response.
Figure 4. Expression of leptin in cancer cells results in immune-mediated tumor control and metabolically improves the function of tumor infiltrating lymphocytes.

(A) CL24hygro and CL24leptin were injected subdermally on C57BL/6J mice and tumor growth monitored. Each line represents an individual mouse. (B) Survival plot of mice treated as in (A). (C) C57BL/6J mice were treated every other day with anti-CD8 (200ug). At day 6 mice were injected with either CL24hygro or CL24leptin and tumor growth was monitored. (D) Leptin receptor flox CD4creheterozygous mice (Leprf/wt CD4cre) and WT mice were injected with either CL24hygro or CL24leptin and tumor growth was monitored. (E) CD8 and CD4 expression analysis on LN and TIL from mice injected with CL24hygro and CL24leptin. (F) Representative flow cytograms of LN and TIL from mice injected with CL24hygro and CL24leptin cells were stimulated overnight with PMA and ionomycin for cytokine production analysis by staining for IFNγ and TNFα of CD8+ T cells. Tabulated flow cytometric data are shown. Tabulated flow cytometric data for CD8+ T cells from LN and TIL from mice injected with CL24hygro and CL24leptin analyzed for Ki67 expression (G) and metabolic markers Mitotracker FM staining and 2NBDG uptake (H). Data represents at least 3 independent experiments *p <0.05, **p <0.01, ***p <0.001 by paired t-test. Error bars indicate s.e.m. See also Figure S2 and Figure S3.
Leptin metabolically improved the function of tumor infiltrating lymphocytes
We next wanted to determine if leptin merely promoted increased infiltration of T cells or if T cells were functionally improved. CD8+ T cells infiltrating leptin-overexpressing tumors synthesized elevated protein expression of IFNγ and TNFα upon restimulation with PMA and ionomycin (Fig. 4f). Additionally, CD8+ T cells that infiltrated leptin expressing tumors were more proliferative in situ as measured by Ki67 staining (Fig. 4g). Consistent with our in vitro signaling analyses as well as prior work in other systems (Sanchez-Margalet and Martin-Romero, 2001), T cells infiltrating leptin-overexpressing tumors had higher steady-state phosphorylation of AKT, STAT3 and p38-MAPK (Fig. S3c). T cells from leptin-overexpressing tumors, consistent with our metabolic analyses in vitro, had increased mitochondrial mass, suggesting leptin was acting on the T cells to mediate metabolic reprogramming (Fig. 4h). Glucose uptake was unchanged in the TIL of CL24leptin, although this may be due to other inhibitory mechanisms in the TME (Chang et al., 2015; Ho et al., 2015). On the other hand, co-inhibitory receptor expression was unchanged in these cells (Fig. S3d). However, this is consistent with our previous studies employing various types of metabolic reprogramming (Scharping et al., 2016a; Scharping et al., 2017). Thus, while these cells may appear more phenotypically ‘exhausted’ (Fig. S3d) with similar high expression of PD1+ Tim3+ expression in both groups, leptin-induced metabolic support allowed cells to be polyfunctional, proliferative, and mediate tumor control.
Leptin expressing oncolytic Vaccinia virus induced superior antitumor responses
We next sought to locally elevate leptin through delivery by oncolytic virus. In order to generate a leptin expressing Vaccinia virus, the leptin gene (Lep) was cloned in the luciferase expressing pSC65 vector under the control of the Vaccinia p7.5 promoter. Leptin containing recombinant Vaccinia virus (VVleptin) and control luciferase expressing virus (VVctrl) were generated and used to infect CL24 cells. We analyzed expression of leptin in CL24 cells 24 and 48 hour post infection, (Fig. S4a) as well as the release of leptin in the media (Fig. S4b). Mice harboring CL24 tumors were treated with VVctrl or VVleptin with a dose of 2.5x106 PFU intratumorally, which was sufficient to induce luciferase expression specifically in the tumor (Fig. S4d) and detect free leptin in the tumor interstitial fluid; white adipose (WA) tissue interstitial fluid acted as a positive control (Fig. S4c). Importantly, leptin-engineered virus still retained oncolytic activity in vitro, inducing substantial cell death of CL24 cells after infection (Fig. S4e).
We treated CL24 melanoma bearing mice after an established tumor was formed, when these tumors could be reliably intratumorally injected (5 mm in one direction). Notably, in all of our studies mice received a single therapeutic dose of oncolytic virus. Consistent with our previous results, all mice injected with control virus experienced partial responses, leading to eventual tumor outgrowth. The partial response to control virus was especially exciting as this aggressive melanoma line is completely resistant to anti-PD1 immunotherapy (Najjar et al., 2019). In contrast to mice treated with VVctrl, those injected with the same dose of VVleptin had larger regressions, including a substantial proportion of complete responses (27%) (Fig. 5a). This resulted in a significant survival advantage for these mice (Fig. 5b). We observed no changes in bodyweight between all three treatments (Fig. S4f). More immunogenic tumor models, like the anti-PD1 sensitive model MC38, received no added benefit from leptin-engineered virus (Fig. S4g), suggesting leptin-mediated metabolic support may be especially important in immunologically harsh environments. Thus, we also employed the aggressive pancreatic tumor model PanC02, which is characterized by very poor therapeutic response to current therapies. A single intratumoral dose of VVleptin mediated a substantially improved response and survival compared to VVctrl, which afforded essentially no measurable benefit (Fig. 5c).
Figure 5. Leptin-engineered Vaccinia virus promotes improved antitumor immune response through metabolic reprogramming.

(A) C57BL/6J mice were injected subdermally with CL24 cells. 5-7 days after tumor cell injection tumors were treated intratumorally with PBS (n=8), VVcontrol (n=8), or VVleptin (n=10), at 2.5x106 PFU and tumor growth monitored. Each line represents an individual mouse. (B) Mice were monitored for survival. (C) C57BL/6J mice were injected subdermally with PanCO2 cells 7-10 days after tumor injection tumors were treated as in (A). (D) Mice were monitored for survival. On day 10 after treatment lymphocytes were isolated from TIL and LN. (E) Tabulated flow cytometric data for CD8 and CD4 expression. (F) Isolated lymphocytes were stimulated overnight with PMA and ionomycin. Tabulated flow cytometric data for cytokine production analysis by staining for IFNγ and TNFα. (G) Tabulated flow cytometric data for CD8+ T cells from LN and TIL from mice treated as in (A) analyzed for Ki67 expression. (H) Representative histograms and tabulated flow cytometric data of CD8+ T cells isolated from LN and TIL were analyzed for mitochondrial protein VDAC. Data represents at least 3 independent experiments *p <0.05, **p <0.01, ***p <0.001 by two-way ANOVA. Error bars indicate s.e.m. See also Figure S4 and S5.
Work from other groups suggests obesity may be associated with better responses to anti-PD1 based immunotherapy (Wang et al., 2019). Anti-PD1 likely functions through mechanisms distinct from oncolytic viruses, but nevertheless we sought to determine how obesity (in which leptin is systemically elevated) may impact VV therapy, with or without our engineered construct. Mice treated with VVctrl had the same tumor growth kinetics when comparing lean mice with obese mice. Leptin-engineered virus provided no additional benefit to obese mice (Fig. S4h), which we hypothesize may be due to the fact that obese mice are rendered insensitive to leptin (Wang et al., 2019). Furthermore, anti-PD1 does not provide further therapeutic benefit to VVleptin in lean mice (Fig. S4i). Altogether, the greatest therapeutic benefit was observed when lean mice where treated with VVleptin, inducing complete responses in anti-PD1 insensitive tumor models.
Leptin-engineered Vaccinia virus therapy promotes a memory response to secondary tumor challenge
Consistent with our prior scRNAseq data, analysis of immune infiltrate in tumors treated with VVctrl and VVleptin showed that both oncolytic viruses induced an increase in T cell infiltration at the tumor site (Fig. 5d). Furthermore, analysis of CD8+ T cells infiltrating treated tumors revealed VVleptin induced a qualitatively superior tumor infiltrate: increased T cell activity at the tumor site shown by an increase in cytokine competency (Fig. 5e), as well as increased proliferative capacity (Fig. 5f). Furthermore, we observed no difference in expression of PD1 and Tim3 between VVctrl and VVleptin treatment (Fig. S4j). Consistent with our data in T cells infiltrating leptin-overexpressing tumors, CD8+ T cells in tumors treated with VVleptin exhibited an increase in mitochondrial mass as measured by the mitochondrial protein VDAC (Fig. 5g) as well as MitoTracker staining (Fig. S5a), suggesting T cells infiltrating this leptin-reprogrammed microenvironment are more metabolically sufficient.
Unsupervised clustering of T lymphocytes from scRNA-seq in VVleptin tumors confirmed our flow cytometric findings: T cells in tumors responding to leptin-engineered virus treatment were not in greater number (compared to control virus treatment) but their phenotype was changed (Fig. 6a). Notably, we saw increases in effector-memory and memory signature (Fig. 6b). We also saw changes in the macrophage compartment, suggesting a potential switch to a more inflammatory state (Fig. S6a,b). Leptin has been shown to inhibit regulatory T cells (Treg) and modulate the inflammatory response in autoimmune diseases (De Rosa et al., 2007; Klingenberg et al., 2010; Matarese et al., 2001). Furthermore, previous work have shown that oncolytic virus therapy can reduce the infiltration of Treg cells (Barve et al., 2008; Ricca et al., 2018). Consistently, after oncolytic virus treatment of VVctrl and VVleptin we observed a decrease in percentage of the Treg population compared to PBS treatment with comparable percent population between VVctrl and VVleptin (Fig. S5d) suggesting that leptin was not necessarily acting at the level of Treg cell modulation.
Figure 6. Leptin promotes memory responses in the oncolytic virus-induced immune infiltrate.

C57BL/6J mice were injected subdermally with CL24 cells. 5-7 days after tumor cell injection tumors were treated intratumorally with PBS, VVcontrol, or VVleptin. (A) Unsupervised clustering within T cells (scRNA-seq) from PBS, VVctrl, and VVleptin treated tumor infiltrate. (B) Comparison of T cell population between PBS, VVctrl and VVleptin treatment. Quantification of the proportion of each cell type population based on treatment. On day 10 after treatment lymphocytes were isolated from TIL and LN. (C) TCR sequencing of genomic DNA extracted from CL24 bearing mice treated intratumorally with PBS, VVcontrol, or VVleptin at 2.5x106 PFU. (n=5 each treatment) Analysis of Total templates and productive rearrangements. (D) Analysis of sample clonality and mean frequency. (E) Mice were treated as in (A). Representative histograms and tabulated flow cytometric data of CD8+ T cells stained for CD127 expression, CD127 with KLRG1 expression (F) and TCF7 expression in KLRG1+ population (G). (H) Mice were treated as in (A). Complete responders from VVleptin treatment of CL24 tumors (n=11) and naïve mice (n=10) received an injection of CL24 cells and monitored for tumor growth. Data represents at least 3 independent experiments *p <0.05, **p <0.01, ***p <0.001 by two-way ANOVA. Error bars indicate s.e.m. See also Figure S5 and S6.
As oncolytic viruses have been purported to induce new T cell priming to viral and tumor antigens (Brown et al., 2017; Russell and Barber, 2018), we next wanted to ascertain the effects of our treatments on the T cell repertoire at the tumor site. TCR sequencing revealed that while PBS treated tumors had few infiltrating T cells dominated by an oligoclonal population, treatment with Vaccinia resulted in a substantial influx of new T cells with a polyclonal repertoire (Fig. 6c). Leptin-engineered Vaccinia had a slightly less clonal population, suggesting at this time point (7 days after viral treatment) some clones were preferentially expanding (Fig. 6d). TRP2 tetramer-binding cells were similar or underrepresented in the virus-treated tumors (Fig. S5b), suggesting the majority of this tumor infiltrate was specific to previously hidden antigens that were revealed by oncolysis. The clonal expansion could be indicative of expansion of some memory precursors, and indeed leptin-engineered VV induced a greater percentage of CD127hi memory precursors (Fig. 6e). Furthermore, while we observed a trend (but not statistically significant) increase in KLRG1hiCD127+ memory precursors (Fig. 6f), TCF7, a transcription factor responsible for the formation of central CD8 T cells memory response (Zhou et al., 2010) was upregulated even in KLRG1+ effector T cells (Fig. 6g).
A concern with oncolytic viruses is that the immunity they may elicit may be dominated by virus-specific cells. Elevated percentage of Vaccinia-derived B8R tetramer binding cells were evident in the infiltrate of leptin and control virus-infected tumors (Fig. S5c). However, when the survivors of VVleptin treated tumor-bearing mice were rechallenged with uninfected, wild-type CL24 cells after complete responses, most mice completely rejected their tumors, while the minority that grew out were substantially slower compared to naïve mice (Fig. 6g). Thus, while anti-viral immunity is certainly primed in oncolytic VV-treated mice, the leptin-engineered virus was able to promote a strong memory response to subsequent tumor challenge. As memory T cells have increased mitochondrial reserve and depend on that reserve for their memory function (van der Windt et al., 2013), leptin may preferentially support the memory phenotype in the face of inflammation and oncolysis in the tumor microenvironment. Our data suggest that by providing metabolic support to newly infiltrated T cells induced by oncolytic virus treatment, memory precursor populations with superior anti-tumor capabilities can preferentially expand and mediate complete responses.
DISCUSSION
Among the many challenges encountered by the immune response in solid tumors is the poor capacity to infiltrate as well as being able to carry out their effector function appropriately in a hostile microenvironment. Our study shows that we can overcome both obstacles by engineering an oncolytic virus that can deliver metabolic modulation (in the form of the adipokine leptin) directly to the microenvironment, consequently improving therapeutic efficacy. Recent studies have started to explore the genetic signature defined by oncolytic viruses in the tumor and determining targets that can be expressed in oncolytic viruses (Zamarin et al., 2017). The present study detailed the changes in the immune landscape after oncolytic viral treatments utilizing single cell RNA-seq analysis. Our findings reveal changes in tumor infiltrate at an early time-point when tumors are not yet regressing. These data suggest that oncolytic viruses do not simply lyse a portion of tumor cells and promote some immunogenic cell death, but rather have the capacity to completely remodel the tumor immune microenvironment. This remodeling promotes the infiltration of new T cells that are sensitive to signals that may influence their fate towards dysfunction or memory. Our data show not only an increased infiltration in the T cell compartment, which is likely central to the antitumor immunity we observe, but a wide array of changes in the myeloid population particularly in the macrophage compartment. A better understanding of macrophage classification and their function would be critical to dissect their role in the response to oncolytic viruses. What is unclear is whether these are a consequence of new T cell immunity, viral infection, or the tissue damage induced by oncolysis. Our study sheds light on the potent immunity induced by oncolytic viruses and suggests that this immune response can be bolstered in specific ways to promote more durable responses.
There is increasing evidence showing that improving T cell metabolic function in the tumor microenvironment allows for a better therapeutic response (Rivadeneira and Delgoffe, 2018). This study employed leptin therapeutically as a metabolic modulator of the immune response, especially in cancer. Looking closely at the T cell compartment by scRNAseq or by flow cytometry analysis we observed clear differences between the two viruses presented here, most notably the concept that providing induced increased metabolic capacity in T cells, resulting in improved function and differentiation toward a memory-like phenotype, essential for a durable responses. These complete responders develop a full memory response, preventing tumor regression when encountering a second tumor cell challenge. Thus, this new immunity induced in response to oncolytic viruses is not dominated by virus-specific clones but rather contains potent, tumor-specific T cells that can prevent future tumor encounter. Furthermore, we highlight the potential for utilizing oncolytic viruses as an effective delivery system for molecules that can modulate specifically the tumor microenvironment and improve therapeutic response.
Previous studies have shown that immune cells express the leptin receptor (Procaccini et al., 2012) and that leptin as a cytokine can have pro-inflammatory functions in innate and adaptive immune responses (La Cava and Matarese, 2004; Loffreda et al., 1998; Santos-Alvarez et al., 1999). Regarding the adaptive immune response, leptin can activate and enhance proliferation of human T lymphocytes (Martin-Romero et al., 2000). There are some observations that leptin might inhibit regulatory T cell proliferation and function in models of inflammation and autoimmunity (Feuerer et al., 2009; Matarese et al., 2001), our data using oncolytics suggest that Treg cells are certainly not stimulated in a leptin-rich tumor environment, although it remains unclear whether they are functionally inhibited when leptin is overexpressed.
Little is known about the role of leptin or the leptin receptor in cancer, particularly in the tumor microenvironment. Our findings demonstrate that there is an increase leptin receptor expression in T cells in the tumor microenvironment compared to those in the secondary lymphoid organs. Leptin can metabolically enhance tumor infiltrating T cell effector function through the persistence of mitochondrial function and an increase in oxidative phosphorylation. Previous studies have introduced the concept that leptin can have a direct metabolic effect by promoting fatty acid oxidation in skeletal muscle (Steinberg et al., 2002). In the context of immune cells, CD4+ T cells from leptin deficient mice show a reduction in glucose uptake along with decreased proliferation and cytokine production (Saucillo et al., 2014). It is important to note that previous studies of leptin-induced changes in T cell metabolism are conducted in the context of obesity or fasting (Gerriets et al., 2016; Saucillo et al., 2014). Our data suggest that T cells that are ‘starved’ in the nutrient dearth tumor microenvironment may be ideal targets for metabolic mediators like leptin. Our data reinforces previous studies showing that leptin signals through the activation of important signaling pathways like p38-MAPK and STAT3 (Ghilardi and Skoda, 1997; Niswender et al., 2001; Papathanassoglou et al., 2006), and can increase mitochondrial content and quality. Leptin can promote PGC1α activation and promote oxidative phosphorylation as well as promote mitochondrial fusion through the expression of mitofusin 1 (Hsu et al., 2015; Roman et al., 2010). As we have previously shown that tumor infiltrating T cells repress the expression of PGC1α (Scharping et al., 2016a), leptin may support TIL function through maintenance of that axis.
Our analysis of the T cell infiltrate of both wild-type and leptin-engineered oncolytic Vaccinia shed light on the immune populations that were more predominant in the tumors treated with leptin-expressing Vaccinia virus. We found an increase in proportions of memory T cells which can explain the sustained therapeutic response observed. Memory T cells are superior antitumor T cells, have a higher mitochondrial content and oxidative phosphorylation capacity (Sukumar et al., 2016; van der Windt et al., 2012) in accordance with our data showing an increase in mitochondrial content. TCR sequencing analysis further expanded our understanding on the effects of oncolytic viruses on tumor infiltrating lymphocytes. While oncolytics predictably induced new T cell clones to infiltrate the tumor, we observed a T cell clonal expansion in tumors treated with leptin-expressing Vaccinia virus. Our TRP2-tetramer data highlights the notion that the new T cell infiltrate is potentially recognizing new antigens and therefore the TRP2 specific population is underrepresented. While our data employing tumor rechallenge suggests at least a portion of these T cells are tumor specific, further studies could determine the antigen specificity of these clones and how important these clones are for therapeutic response.
Our work and others have shown the benefits of metabolically enhancing mitochondrial function in tumor infiltrating lymphocytes. Our study opens up the possibilities to further expand the repertoire of metabolic modulators, among the myriad encoded in the genome that can be delivered directly into the tumor. An attractive method of therapeutic delivery of these metabolic modulators is the utilization of oncolytic viruses, which can deliver genetically encoded payload directly to the tumor microenvironment. Until now, the majority of oncolytic-delivered genes have been immunologic in nature: cytokines, costimulatory molecules, etc. However, our study represents a proof of concept that metabolic modulators can be delivered by oncolytic viruses. While we chose Vaccinia for its distinct characteristics, our data suggest these modalities may be broadly applicable and encoded in other oncolytics like herpes simplex virus (HSV), Newcastle Disease Virus, adenovirus (VND), and vesicular stomatitis virsus (VSV). Furthermore, testing Vaccinia and other oncolytics model in systemic delivery would be of great interest for future studies, particularly in tumor models where intratumoral injections are not viable or in the treatment of distant metastases. While our scRNA-seq revealed that oncolytics have potent immune-stimulatory potential early after infection, it is clear that to achieve durable, complete responses, metabolic support is crucial, and may help guide the strong early effector response into long-lived memory capable of mediating robust antitumor effects.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for materials and resource will be fulfilled by the Lead Contact, Greg Delgoffe (gdelgoffe@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57/BL6 mice and Ptenf/fBrafV600ETyrCre.ER mice were obtained from Jackson Laboratories and bred in house. Leptin receptor flox mice (ObRFlox) were obtained from The Jackson Laboratory. These mice were crossed to CD4 Cre expressing mice. These mice when used in experimental procedure were males and females between of 6-7 weeks old. Diet induced obesity (DIO) mice were obtained from The Jackson Laboratory and were all males. All animal work and protocols in the current study were approved by the University of Pittsburgh Institutional Animal Care and Use Committee, accredited by the AAALAC.
Cell culture
Tumor experiments were conducted using a single-cell clone derived from a melanoma tumor formed from a female Ptenf/fBrafV600ETyrCre.ERmouse painted with tamoxifen (clone 24, described in (Najjar et al., 2019)). Clone 24 is a male cell line. PanCO2, a male mouse pancreatic ductal adenocarcinoma cell line, was a kind gift from Dr. Michael Lotze’s lab. MC38 is a male mouse colon adenocarcinoma cell line was a kind gift from Dr. Dario Vignali. CL24, PanCO2 and MC38 were all cultured in DMEM, supplemented with 10% FBS (v/v) at 37°C with 5% CO2.The cDNA for leptin was obtained from OriGene and transfected into clone 24 followed by hygromycin selection (an empty vector plasmid was used as a control). Single cell clones were selected and grown as cell line CL24hygro for control plasmid and CL24leptin for leptin expressing cell line.
METHOD DETAILS
Tumor models
C57BL/6J mice were injected with CL24hygro or CL24leptin melanoma cell line (250,000 cells intradermally) on day 0 and followed until tumors reach 15 mm in any direction. Tumors were measured every other day with digital calipers and tumor size was calculated by LxW.
Tumors (CL24, MC38 and PanCO2) were treated with PBS, VVctrl or VVleptin (2.5x106 PFU) intratumorally when tumors reached approximately a 20mm2 and tumor growth was monitored until tumors treated with PBS reached 15mm in any direction. For CD8 depletion experiments mice were injected every other day starting at day 0 with anti-CD8 (YTS) at 200ug per mouse. On day 7 mice were injected with CL24hygro or CL24leptin melanoma cell line (250,000 cells intradermally) and followed until tumor reach 15mm in any direction.
On day 7 when tumors reached 5mm diameter, mice were treated with Vaccinia virus as previously described and were started on either 0.2mg anti– PD1 or hamster IgG isotype control (Bio X Cell), injected every other day days intraperitoneally. Cohorts were sacrificed when control mouse tumors reached 15 mm in any direction measured.
Oncolytic virus production
The wild-type Vaccinia virus Western Reserve (WR) strain was obtained from the American Type Culture Collection (BEI Resources). WR.TK–.Luc+ were described previously (Kirn et al., 2007) and were constructed for this work, with the pSC65 plasmid (from Prof. Bernie Moss, NIH) cloned to express firefly luciferase from the viral pSE/L promoter and mouse leptin (Lep) from the p7.5 promoter. This was recombined into the viral TK gene. Vaccinia virus expressing leptin was generated by cloning in the leptin gene using Gibson Cloning (New England BioLabs) into the Vaccinia plasmid. Leptin gene was cloned from a mouse leptin ORF mammalian expression plasmid (Sino Biological Inc.).
T cell isolations from lymph node, tumor and adoptive transfer
Spleen and lymph node CD8+ T cells were isolated from mice. Tissue was harvested, mechanically disrupted, and incubated with a biotinylated antibody cocktail consisting of antibodies (BioLegend) to B220, CD11b, CD11c, CD16/32, CD19, CD25, CD105, NK1.1, TCRγδ, and CD4. After a wash step, cells were incubated with streptavidin-coated magnetic nanoparticles (BioLegend). After washing, CD8+ cells were isolated by applying a magnetic field and removing untouched cells. For adoptive transfer, T cells were generated by activating Pmel-1 x Thy1.1 mice with peptide and expanding cells for 5 days in IL-2. Pmel CD8+ T cells were intravenously delivered (107) to mice bearing CL24hygro and CL24leptin tumors. Mice were then sacrificed and TIL analyzed.
To obtain single-cell suspensions of tumor infiltrating lymphocytes, tumor bearing mice were sacrificed and tumors were harvested. Excised, whole tumors were injected repeatedly using 20G needles with 2mg/mL collagenase type VI, 2U/mL hyluronidase (Dispase), and 10U/mL DNAse I (Sigma) in buffered RPMI with 10% FBS and incubated for 30 min at 37°C. Tumors were then mechanically disrupted between frosted glass slides and filtered to remove particulates, then vortexed for 2 minutes. In many experiments (especially prior to sorting), tumor homogenates were debulked of tumor cells using CD105-biotin mediated magnetic depletion.
Metabolic Assays
T cell metabolic output was measured by Seahorse technology as previously described (Scharping et al., 2016b). Briefly, 100,000 T cells were seeded into Cell-Tak-coated XFe96 plates in minimal unbuffered assay media containing 25 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate. Cells received sequential injections of 2 μM oligmycin, 2 μM FCCP, 10 mM 2-deoxyglucose, and 0.5μM rotenone/antimycin A.
We assayed single-cell metabolic capacity by flow cytometry. Specifically, we utilized 2-NBD-glucose (Cayman Chemical) and MitoTracker FM dyes (ThermoFisher) to assay the propensity of cells to take up glucose or generate intermediates via their mitochondria. Nondraining and draining lymph node or tumor preparations were pulsed with 20 μM 2-NBDG in 5% FBS-containing media for 30 min at 37°0. Cells were surface stained and loaded with MitoTracker FM dyes to measure mitochondrial mass and function.
Immunoblotting
Immunoblotting was performed as previously described (Delgoffe et al., 2009). Briefly cells were lysed in 1% NP-40 lysis buffer. Cell lysates were then separated by SDS-PAGE using a 4-12% Bio-Rad gels. Gels were then transferred to a polyvinylidene difluoride membrane and blocked in 5% milk in Tris-buffered saline 0.1%Tween-20 (TBST). Membrane was then incubated overnight at 4°C with primary antibodies diluted in blocking buffer. Membrane was incubated with secondary antibody (anti-mouse horseradish peroxidase, Jackson ImmunoResearch) in blocking buffer for 1 hour at room temperature and subsequently washed 3 times for 10 minutes with TBST. Protein was visualized by chemiluminescence by using Western Lightning (PerkinElmer). Mouse Leptin/OB antibody (R&D system BAF498), p-ATF2 (Cell Signaling), β-actin (Santa Cruz) and p38 MAPK (Cell Signaling).
ELISA
ELISA plate was coated with 50uL capture antibody (1:1000 in PBS) and put at 4°C overnight. Next day plate was washed 3 times with Wash Buffer (1L PBS + 0.05% Tween 20). Plate was Blocked with 200uL blocking buffer (200mL PBS + 1% BSA) for 1 hour at room temperature. Samples were added (50ul) in blocking buffer to the wells together with Standard Curve samples. Plate was incubated at room temperature for 2 hours. Secondary antibody was added (1:2000 in blocking buffer) and incubated at room temperature for 1 hour. After one hour HRP streptavidin (1:2000 in blocking buffer) was added and incubated at room temperature for 30min. We added 40uL TMB substrate A and 40uL TMB substrate B to develop samples. Plate was read at 450nm in a plate reader. Antibodies used for leptin Elisa experiment: Capture Mouse Leptin/OB antibody (R&D systems AF498) and detection antibody Mouse Leptin/OB antibody (R&D system BAF498).
TCR Sequencing
CL24 tumors treated with PBS, VVctrl or VVleptin were excised and processed for genomic DNA extraction (DNeasy QIAGEN kit). TCR sequencing was then performed following the immunoSEQ assay (Adaptive Biotechnologies) for immunosequencing of the complementaritydetermining region 3 (CDR3) variable regions of T cell receptor-β chains (TCRβ) (Robins et al., 2009). Sequences were then filtered for the identification and quantification of abundance of unique TCRβCDR3 regions and compared across samples.
Single cell RNA sequencing analysis
CL24 tumors were treated with PBS, VVctrl or VVleptin (2.5x106 PFU) intratumorally for 7 days. Tumor infiltrating lymphocytes were isolated and sorted for CD45+ lymphocytes. CD45+ cell were loaded into the Chromium instrument (10X Genomics, Pleasanton, CA), and the resulting barcoded cDNAs were used to construct libraries. The libraries from each sample were then RNA-sequenced. Cell-gene unique molecular identifier counting matrices were generated usigng the 10x Genomics CellRanger (v.2.1.1) pipeline. Quality control and normalization and analysis were done using Scanpy package (Wolf et al., 2018). After QC and normalization, the Louvain algorithm which is a part of the Scanpy package was used to cluster cells. Identities were assigned to cell types by using a combination of top expressed genes and canonical cell type marker genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using Prism 7 (GraphPad Software). Data were analyzed with the unpaired t test or one-way ANOVA with Tukey correction. Data are presented as mean ± SD. * p< 0.05; ** p<0.01; **** p<0.0001; ns, not significant.
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE133699. All software used in the analysis is listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B220-PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 103236; RRID:AB_893354 |
| TNFα PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 506322; RRID:AB_961434 |
| CD4 PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 100433; RRID:AB_893330 |
| VDAC AF488 (monoclonal) | Abcam | Cat# ab179839 |
| IFNγ BV510 (monoclonal) | Biolegend | Cat# 505842; RRID:AB_2734494 |
| PD1 BV786 (monoclonal) | Biolegend | Cat#135225; RRID:AB_2563680 |
| NK1.1 APC (monoclonal) | BD pharmigen | Cat# 561117; RRID:AB_10563422 |
| Tim3 PE (monoclonal) | Biolegend | Cat# 119703; RRID:AB_345377 |
| Ki67 BV605 (monoclonal) | Biolegend | Cat# 652413; RRID:AB_2562664 |
| p-p38MAPK (T180/Y182) – PE (monoclonal) | Cell Signaling | Cat# 6908S; RRID:AB_10839411 |
| p-NF-kappaB p65 (S536) - A647 (monoclonal) | Cell Signaling | Cat# 4887S; RRID:AB_561198 |
| pSTAT3 (Tyr705) BV421 (monoclonal) | Biolegend | Cat# 651009; RRID:AB_2572087 |
| CD127 (IL7Ra) PE- Cy7 (monoclonal) | Biolegend | Cat# 135014; RRID:AB_1937265 |
| CD62L BV786 (monoclonal) | BD Horizon | Cat# 564109; RRID:AB_2738598 |
| KLRG1 APC-Cy7 (monoclonal) | Biolegend | Cat# 138426; RRID:AB_2566554 |
| TCF7/TCF1 FITC (monoclonal) | R&D systems | Cat# IC8224G |
| Foxp3 AxF700 (monoclonal) | Thermo Fisher | Cat# 56-5773-82; RRID:AB_1210557 |
| Leptin R biotinylated (mouse polyclonal) | R&D systems | Cat# BAF497; RRID:AB_2296953 |
| Mouse Leptin (polyclonal Goat) | R&D systems | Cat # AF498; RRID:AB_355394 |
| Phospho- ATF-2 (Thr71) (rabbit monoclonal) | Cell Signaling | Cat# 24329 |
| P38 MAPK (rabbit monoclonal) | Cell Signaling | Cat# 9212S; RRID:AB_330713 |
| InVivo Mab Anti-mouse CD8α(YTS 169.4) | BioXCell | BE0117; RRID:AB_10950145 |
| InVivoMAb rat IgG1 Isotype control | BioXCell | BE0290; RRID:AB_2687813 |
| β-Actin (C4) (mouse monoclonal) | Santa Cruz | Cat# SC-47778; RRID:AB_2714189 |
| Bacterial and Virus Strains | ||
| wild-type Vaccinia virus Western Reserve (WR) | American Type Culture Collection (BEI Resources) | Cat# NR2639 |
| One Shot® Stbl3™ Chemically Competent E. coli | Fisher | Cat# C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2NBDG | Cayman Chemical | Cat# 186689-07-6 |
| Mitotracker™ Deep Red FM | Invitrogen | Cat# M22426 |
| Tetramer H-2Kb TRP-2 PE | MBL International | Cat# TB-5004-1 |
| Tetramer VACV B8R BV421 | MBL International | Cat# BTB-M538 |
| Recombinant mouse Leptin | R&D systems | Cat# 498-OB-01M |
| L-glutamine | Fisher | Cat# MT25005CI |
| Oligomycin Complex | Cayman Chemical | Cat# 11341 |
| FCCP | Cayman Chemical | Cat# 15218 |
| 2-deoxy-D-glucose (2DG) | Sigma | Cat# D8375-5G |
| Rotenone | Sigma | Cat# R8875-5G |
| Antimycin A | Sigma | Cat# A8674-25MG |
| Recombinant Murine IL-2 | Peprotech | Cat# 212-12 |
| SIINFEKL peptide | AnaSpec | Cat# AS-60193-1 |
| Deposited Data | ||
| Raw and analyzed data SC-RNAseq | This paper | GEO: GSE133699 |
| Experimental Models: Cell Lines | ||
| CL24 | Najjar et al., 2019 | N/A |
| MC38 | Gift from Dr. Dario Vignali (University of Pittsburgh) | N/A |
| PanCO2 | Gift from Dr. Michael Lotze (University of Pittsburgh) | N/A |
| Experimental Models: Organisms/Strains | ||
| B6.129P2-Leprtm1Rck | Jackson Laboratory | Stock# 008327 |
| Ptenf/fBrafV600ETyrCre.ER | Jackson Laboratory | Stock # 013590 |
| B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | Jackson Laboratory | Stock # 022071 |
| Recombinant DNA | ||
| Mouse Leptin natural ORF mammalian (pPCMV3-mLEP) | Sino Biological Inc. | Cat# MG50442-UT |
| PCMV3-untagged Negative Control Vector | Sino Biological Inc. | Cat# CV011 |
| pSC65 plasmid | Kind gift from Prof. Bernie Moss, NIH | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc | https://www.graphpad.com/demos/ |
| BD FACS Diva Sotware | BD Biosciences | |
| FlowJo version 10 | TreeStar, Ahsland Oregon | https://www.flowjo.com |
| Seahorse Wave Desktop Software | Agilent | https://www.graphpad.com/demos/ |
| Cell Ranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy | Wolf et al., 2018 | https://github.com/theislab/scanpy |
| Critical Commercial Assays | ||
| 10X Genomics | 10X Genomics, Pleasanton, CA | https://www.10xgenomics.com/ |
| Seahorse XFe96 Analyzers | Agilent | https://www.agilent.com/en/products/cell-analysis/seahorse-analyzers/seahorse-xfe96-analyzer |
Supplementary Material
Highlights:
Single cell sequencing reveals immune remodeling in Vaccinia virus treated tumors
The adipokine leptin enhances T cell metabolic function in vitro and in vivo
Vaccinia engineered to express leptin metabolically enhances antitumor responses
Leptin-engineered Vaccinia promotes a memory response upon tumor rechallenge
ACKNOWLEDGEMENTS
The authors thank Nicole Scharping for technical and mentoring assistance as well as Dr. Chandranath Roy for assistance in virus production.
This work was primarily supported by a Stand Up 2 Cancer Innovative Research Grant (SU2C-AACR-IRG-04-16) to G.M.D., and additionally supported by the NIH Director’s New Innovator Award (DP2AI136598 to G.M.D.) the UPMC Hillman Cancer Center Melanoma and Skin Cancer (P50CA121973-09) and Head and Neck Cancer SPOREs (P50CA097190), to R.L.F. and G.M.D.), the Alliance for Cancer Gene Therapy/Swim Across America (to G.M.D.), and the Sy Holzer Endowed Immunotherapy Fund (to G.M.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The use of metabolic-reprogramming oncolytic viruses is the subject of a patent application on which D.B.R., P.S., S.H.T., and G.M.D. are listed as inventors. P.S. and S.H.T. are employees of Western Oncolytics.
REFERENCES
- Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, Mera A, Lago F, Gomez R, and Gualillo O (2017). Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol 13, 100–109. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, and Yan Z (2005). Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 280, 19587–19593. [DOI] [PubMed] [Google Scholar]
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. (2015). Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 33, 2780–2788. [DOI] [PubMed] [Google Scholar]
- Barve M, Bender J, Senzer N, Cunningham C, Greco FA, McCune D, Steis R, Khong H, Richards D, Stephenson J, et al. (2008). Induction of immune responses and clinical efficacy in a phase II trial of IDM-2101, a 10-epitope cytotoxic T-lymphocyte vaccine, in metastatic non-small-cell lung cancer. J Clin Oncol 26, 4418–4425. [DOI] [PubMed] [Google Scholar]
- Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, Bigner DD, Gromeier M, and Nair SK (2017). Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller RM, Smith GL, Cremer K, Notkins AL, and Moss B (1985). Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature 317, 813–815. [DOI] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman DL (1979). Obesity genes: beneficial effects in heterozygous mice. Science 203, 663–665. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, and Bosenberg M (2009). Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 41, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, and Matarese G (2007). A key role of leptin in the control of regulatory T cell proliferation. Immunity 26, 241–255. [DOI] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Cotter RJ, and Powell JD (2009). Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Molecular immunology 46, 2694–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW Jr., and Taub DD (2004). Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest 114, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman GK, Haas MC, Kelly VM, Markert JM, Gillespie GY, and Cassady KA (2012). Hypoxia Moderates gamma(1)34.5-Deleted Herpes Simplex Virus Oncolytic Activity in Human Glioma Xenoline Primary Cultures. Transl Oncol 5, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Danzaki K, Kishton RJ, Eisner W, Nichols AG, Saucillo DC, Shinohara ML, and MacIver NJ (2016). Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol 46, 1970–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi N, and Skoda RC (1997). The leptin receptor activates janus kinase 2 and signals for proliferation in a factor-dependent cell line. Mol Endocrinol 11, 393–399. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiley CT, Yuan M, Lemoine NR, and Wang Y (2010). Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther 17, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Antitumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, Lechler RI, and Bloom SR (1999). Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest 104, 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu WH, Lee BH, and Pan TM (2015). Leptin-induced mitochondrial fusion mediates hepatic lipid accumulation. Int J Obes (Lond) 39, 1750–1756. [DOI] [PubMed] [Google Scholar]
- Justus CR, Sanderlin EJ, and Yang LV (2015). Molecular Connections between Cancer Cell Metabolism and the Tumor Microenvironment. Int J Mol Sci 16, 11055–11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tahtinen S, Oksanen M, Heiskanen R, Pesonen S, et al. (2013). Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res 19, 2734–2744. [DOI] [PubMed] [Google Scholar]
- Kirn DH, Wang Y, Le Boeuf F, Bell J, and Thorne SH (2007). Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med 4, e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg R, Lebens M, Hermansson A, Fredrikson GN, Strodthoff D, Rudling M, Ketelhuth DF, Gerdes N, Holmgren J, Nilsson J, et al. (2010). Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 30, 946–952. [DOI] [PubMed] [Google Scholar]
- La-Beck NM, Jean GW, Huynh C, Alzghari SK, and Lowe DB (2015). Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 35, 963–976. [DOI] [PubMed] [Google Scholar]
- La Cava A, and Matarese G (2004). The weight of leptin in immunity. Nat Rev Immunol 4, 371–379. [DOI] [PubMed] [Google Scholar]
- Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, et al. (1998). Leptin regulates proinflammatory immune responses. FASEB J 12, 57–65. [PubMed] [Google Scholar]
- Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, and Lechler RI (1998). Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901. [DOI] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD, and Freeman GJ (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nature Reviews Drug Discovery 14, 561. [DOI] [PubMed] [Google Scholar]
- Martin-Romero C, Santos-Alvarez J, Goberna R, and Sanchez-Margalet V (2000). Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol 199, 15–24. [DOI] [PubMed] [Google Scholar]
- Matarese G, Di Giacomo A, Sanna V, Lord GM, Howard JK, Di Tuoro A, Bloom SR, Lechler RI, Zappacosta S, and Fontana S (2001). Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol 166, 5909–5916. [DOI] [PubMed] [Google Scholar]
- Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, and Delgoffe GM (2018). 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med 215, 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B (2013). Poxvirus DNA replication. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, Zhai S, Kirkwood JM, and Delgoffe GM (2019). Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr., and Schwartz MW (2001). Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 413, 794–795. [DOI] [PubMed] [Google Scholar]
- Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, and Mantzoros C (2006). Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J Immunol 176, 7745–7752. [DOI] [PubMed] [Google Scholar]
- Perez-Perez A, Vilarino-Garcia T, Fernandez-Riejos P, Martin-Gonzalez J, Segura-Egea JJ, and Sanchez-Margalet V (2017). Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev 35, 71–84. [DOI] [PubMed] [Google Scholar]
- Pipiya T, Sauthoff H, Huang YQ, Chang B, Cheng J, Heitner S, Chen S, Rom WN, and Hay JG (2005). Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther 12, 911–917. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Jirillo E, and Matarese G (2012). Leptin as an immunomodulator. Mol Aspects Med 33, 35–45. [DOI] [PubMed] [Google Scholar]
- Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, and Bartlett DL (2000). Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther 7, 66–73. [DOI] [PubMed] [Google Scholar]
- Ricca JM, Oseledchyk A, Walther T, Liu C, Mangarin L, Merghoub T, Wolchok JD, and Zamarin D (2018). Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol Ther 26, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivadeneira DB, and Delgoffe GM (2018). Antitumor T-cell Reconditioning: Improving Metabolic Fitness for Optimal Cancer Immunotherapy. Clin Cancer Res 24, 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, and Carlson CS (2009). Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114, 4099–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman EA, Reis D, Romanatto T, Maimoni D, Ferreira EA, Santos GA, Torsoni AS, Velloso LA, and Torsoni MA (2010). Central leptin action improves skeletal muscle AKT, AMPK, and PGC1 alpha activation by hypothalamic PI3K-dependent mechanism. Mol Cell Endocrinol 314, 62–69. [DOI] [PubMed] [Google Scholar]
- Russell SJ, and Barber GN (2018). Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 33, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Margalet V, and Martin-Romero C (2001). Human leptin signaling in human peripheral blood mononuclear cells: activation of the JAK-STAT pathway. Cell Immunol 211, 30–36. [DOI] [PubMed] [Google Scholar]
- Santos-Alvarez J, Goberna R, and Sanchez-Margalet V (1999). Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol 194, 6–11. [DOI] [PubMed] [Google Scholar]
- Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, and Maciver NJ (2014). Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol 192, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016a). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 701–703. [DOI] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Whetstone RD, Zeng X, and Delgoffe GM (2016b). Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer immunology research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Whetstone RD, Zeng X, and Delgoffe GM (2017). Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res 5, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, and Rathmell JC (2015). T cell metabolic fitness in antitumor immunity. Trends Immunol 36, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Parolin ML, Heigenhauser GJ, and Dyck DJ (2002). Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab 283, E187–192. [DOI] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab 23, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL (2012). Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJW, O’Sullivan D, Everts B, Huang SC-C, Buck MD, Curtis JD, Chang C-H, Smith AM, Ai T, Faubert B, et al. (2013). CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proceedings of the National Academy of Sciences 110, 14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, Mirsoian A, Minnar CM, Stoffel KM, Sturgill IR, et al. (2019). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 25, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr., Lao CD, et al. (2015). Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16, 375–384. [DOI] [PubMed] [Google Scholar]
- Whitman ED, Tsung K, Paxson J, and Norton JA (1994). In vitro and in vivo kinetics of recombinant vaccinia virus cancer-gene therapy. Surgery 116, 183–188. [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, Wolchok JD, and Allison JP (2017). Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun 8, 14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH (2010). Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE133699. All software used in the analysis is listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B220-PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 103236; RRID:AB_893354 |
| TNFα PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 506322; RRID:AB_961434 |
| CD4 PerCP Cy5.5 (monoclonal) | Biolegend | Cat# 100433; RRID:AB_893330 |
| VDAC AF488 (monoclonal) | Abcam | Cat# ab179839 |
| IFNγ BV510 (monoclonal) | Biolegend | Cat# 505842; RRID:AB_2734494 |
| PD1 BV786 (monoclonal) | Biolegend | Cat#135225; RRID:AB_2563680 |
| NK1.1 APC (monoclonal) | BD pharmigen | Cat# 561117; RRID:AB_10563422 |
| Tim3 PE (monoclonal) | Biolegend | Cat# 119703; RRID:AB_345377 |
| Ki67 BV605 (monoclonal) | Biolegend | Cat# 652413; RRID:AB_2562664 |
| p-p38MAPK (T180/Y182) – PE (monoclonal) | Cell Signaling | Cat# 6908S; RRID:AB_10839411 |
| p-NF-kappaB p65 (S536) - A647 (monoclonal) | Cell Signaling | Cat# 4887S; RRID:AB_561198 |
| pSTAT3 (Tyr705) BV421 (monoclonal) | Biolegend | Cat# 651009; RRID:AB_2572087 |
| CD127 (IL7Ra) PE- Cy7 (monoclonal) | Biolegend | Cat# 135014; RRID:AB_1937265 |
| CD62L BV786 (monoclonal) | BD Horizon | Cat# 564109; RRID:AB_2738598 |
| KLRG1 APC-Cy7 (monoclonal) | Biolegend | Cat# 138426; RRID:AB_2566554 |
| TCF7/TCF1 FITC (monoclonal) | R&D systems | Cat# IC8224G |
| Foxp3 AxF700 (monoclonal) | Thermo Fisher | Cat# 56-5773-82; RRID:AB_1210557 |
| Leptin R biotinylated (mouse polyclonal) | R&D systems | Cat# BAF497; RRID:AB_2296953 |
| Mouse Leptin (polyclonal Goat) | R&D systems | Cat # AF498; RRID:AB_355394 |
| Phospho- ATF-2 (Thr71) (rabbit monoclonal) | Cell Signaling | Cat# 24329 |
| P38 MAPK (rabbit monoclonal) | Cell Signaling | Cat# 9212S; RRID:AB_330713 |
| InVivo Mab Anti-mouse CD8α(YTS 169.4) | BioXCell | BE0117; RRID:AB_10950145 |
| InVivoMAb rat IgG1 Isotype control | BioXCell | BE0290; RRID:AB_2687813 |
| β-Actin (C4) (mouse monoclonal) | Santa Cruz | Cat# SC-47778; RRID:AB_2714189 |
| Bacterial and Virus Strains | ||
| wild-type Vaccinia virus Western Reserve (WR) | American Type Culture Collection (BEI Resources) | Cat# NR2639 |
| One Shot® Stbl3™ Chemically Competent E. coli | Fisher | Cat# C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2NBDG | Cayman Chemical | Cat# 186689-07-6 |
| Mitotracker™ Deep Red FM | Invitrogen | Cat# M22426 |
| Tetramer H-2Kb TRP-2 PE | MBL International | Cat# TB-5004-1 |
| Tetramer VACV B8R BV421 | MBL International | Cat# BTB-M538 |
| Recombinant mouse Leptin | R&D systems | Cat# 498-OB-01M |
| L-glutamine | Fisher | Cat# MT25005CI |
| Oligomycin Complex | Cayman Chemical | Cat# 11341 |
| FCCP | Cayman Chemical | Cat# 15218 |
| 2-deoxy-D-glucose (2DG) | Sigma | Cat# D8375-5G |
| Rotenone | Sigma | Cat# R8875-5G |
| Antimycin A | Sigma | Cat# A8674-25MG |
| Recombinant Murine IL-2 | Peprotech | Cat# 212-12 |
| SIINFEKL peptide | AnaSpec | Cat# AS-60193-1 |
| Deposited Data | ||
| Raw and analyzed data SC-RNAseq | This paper | GEO: GSE133699 |
| Experimental Models: Cell Lines | ||
| CL24 | Najjar et al., 2019 | N/A |
| MC38 | Gift from Dr. Dario Vignali (University of Pittsburgh) | N/A |
| PanCO2 | Gift from Dr. Michael Lotze (University of Pittsburgh) | N/A |
| Experimental Models: Organisms/Strains | ||
| B6.129P2-Leprtm1Rck | Jackson Laboratory | Stock# 008327 |
| Ptenf/fBrafV600ETyrCre.ER | Jackson Laboratory | Stock # 013590 |
| B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | Jackson Laboratory | Stock # 022071 |
| Recombinant DNA | ||
| Mouse Leptin natural ORF mammalian (pPCMV3-mLEP) | Sino Biological Inc. | Cat# MG50442-UT |
| PCMV3-untagged Negative Control Vector | Sino Biological Inc. | Cat# CV011 |
| pSC65 plasmid | Kind gift from Prof. Bernie Moss, NIH | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc | https://www.graphpad.com/demos/ |
| BD FACS Diva Sotware | BD Biosciences | |
| FlowJo version 10 | TreeStar, Ahsland Oregon | https://www.flowjo.com |
| Seahorse Wave Desktop Software | Agilent | https://www.graphpad.com/demos/ |
| Cell Ranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy | Wolf et al., 2018 | https://github.com/theislab/scanpy |
| Critical Commercial Assays | ||
| 10X Genomics | 10X Genomics, Pleasanton, CA | https://www.10xgenomics.com/ |
| Seahorse XFe96 Analyzers | Agilent | https://www.agilent.com/en/products/cell-analysis/seahorse-analyzers/seahorse-xfe96-analyzer |