

Abstract
Breast cancers (BC) with expression of estrogen receptor-alpha (ERα) occur in more than 70% of newly-diagnosed patients in the U.S. Endocrine therapy with antiestrogens or aromatase inhibitors is an important intervention for BCs that express ERα, and it remains one of the most effective targeted treatment strategies. However, a substantial proportion of patients with localized disease, and essentially all patients with metastatic BC, become resistant to current endocrine therapies. ERα is present in most resistant BCs, and in many of these its activity continues to regulate BC growth. Fulvestrant represents one class of ERα antagonists termed selective ER downregulators (SERDs). Treatment with fulvestrant causes ERα down-regulation, an event that helps overcome several resistance mechanisms. Unfortunately, full antitumor efficacy of fulvestrant is limited by its poor bioavailability in clinic. We have designed and tested a new generation of steroid-like SERDs. Using ERα-positive BC cells in vitro, we find that these compounds suppress ERα protein levels with efficacy similar to fulvestrant. Moreover, these new SERDs markedly inhibit ERα-positive BC cell transcription and proliferation in vitro even in the presence of estradiol-17β. In vivo, the SERD termed JD128 significantly inhibited tumor growth in MCF-7 xenograft models in a dose-dependent manner (P<0.001). Further, our findings indicate that these SERDs also interact with ER-positive immune cells in the tumor microenvironment such as myeloid-derived suppressor cells (MDSC), tumor infiltrating lymphocytes and other selected immune cell subpopulations. SERD-induced inhibition of MDSCs and concurrent actions on CD8+ and CD4+ T-cells promotes interaction of immune checkpoint inhibitors with BC cells in preclinical models, thereby leading to enhanced tumor killing even among highly aggressive BCs such as triple-negative BC that lack ERα expression. Since monotherapy with immune checkpoint inhibitors has not been effective for most BCs, combination therapies with SERDs that enhance immune recognition may increase immunotherapy responses in BC and improve patient survival. Hence, ERα antagonists that also promote ER downregulation may potentially benefit patients who are unresponsive to current endocrine therapies.
Keywords: Selective estrogen receptor downregulator (SERD), PD-L1, Myeloid-derived suppressor cells, Breast cancer, Immunotherapy, CD8+ T-cells
Graphical abstract
1. Introduction
Endocrine therapies that target the estrogen receptor (ER) in breast cancer (BC) have significant clinical benefit when used to treat ER-positive tumors and are often an effective targeted treatment for metastatic disease. However, a substantial number of patients with localized disease, and almost all patients with metastatic breast cancer, become resistant to endocrine therapies [1–3]. In the absence of options to current treatments such as antiestrogens (tamoxifen) or aromatase inhibitors (AI), cytotoxic chemotherapy is often the only alternative. Similarly, chemotherapies are often used for patients with triple-negative breast cancer (TNBC). The TNBC subtype occurs in 15–20% of BC patients and cannot be managed with endocrine or HER2-targeted therapies because TNBCs lack ERα and progesterone receptor (PR) expression and have no HER2 overexpression. However, recent clinical trials reveal that 20–30% of TNBC patients respond to immunotherapy such as immune checkpoint inhibitors (ICI) [4, 5]. Despite this advance, the great majority of patients with TNBC and other BC subtypes do not benefit from ICIs.
In the context of estrogen signaling in BC in vivo, it is important to note that estrogens do not only act directly on BC cells. Rather, it is known that estrogens also regulate the development and function of immune cells that occupy the tumor microenvironment (TME) [6–8]. Despite well-known sex-related differences in immune responses in various autoimmune diseases [9], little is known to date about the effect of estrogens or antiestrogens on tumor immune tolerance and immune checkpoint blockade in breast cancer. ERα, the major ER form, is known to exhibit high expression in early hematopoietic progenitors in bone marrow such as hematopoietic stem cells and common lymphoid and myeloid progenitors [6–8, 10, 11]. The programmed death-1 (PD-1) pathway is an immune checkpoint used by many tumor cells to evade detection and attack by tumor-directed T-cells [12–14] that are known to express ER [11]. PD-1 is expressed at the surface of activated T-cells where it interacts with its ligands, such as programmed death ligand-1 (PD-L1), to attenuate T-cell signaling, resulting in downregulation of T-cell proliferation, activation and the antitumor immune response. Although PD-L1 is rarely expressed in normal breast tissue, it is expressed in some BC cells and surrounding immune cells where it can mediate inhibition of tumor-infiltrating lymphocytes (TILs) which are a known prognostic indicator for benefit from ICIs [15, 16].
Among the subpopulations of immature myeloid cells that frequently arise during tumor progression and metastasis, myeloid-derived suppressor cells (MDSC) are known to express ER, and estrogen signaling is reported to promote MDSC expansion and activation in preclinical studies [7]. MDSCs are also identified in the TME of BC biopsies from the clinic [16, 17] and consist of two large groups of immune cells termed granulocytic or polymorphonuclear cells (G-MDSCs), which are phenotypically and morphologically similar to neutrophils, and monocytic cells (M-MDSCs) similar to monocytes. Immune suppression is a major property of MDSCs, with T-cells the main targets of MDSC action [16, 17] Accordingly, estrogen antagonists may disrupt BC progression by diminishing MDSC numbers and associated pro-tumorigenic functions potentially regardless of the ER status of the tumor. Among the challenges to make immuno-therapy a more effective intervention in BC management going forward, it is important to find ways to manipulate additional mechanisms of tumor immune tolerance and to enhance T-effector cell infiltration and access to the tumor. It is therefore reasonable to investigate the concept that BC escape from immune attack may be blocked by potent antiestrogens that exert antitumor activity in certain ER-positive immune cells, actions that should boost the action of ICIs.
It is well established that estrogens modulate BC gene transcription by binding ER with high affinity, thereby activating downstream signaling by use of genomic pathways that involve direct DNA binding of ligand-bound ER to estrogen-responsive elements in the promoter regions of responsive genes. In addition, nongenomic pathways often involve indirect modulation of transcription by ER interactions with components of other transcription or growth factor receptor kinase signaling complexes (such as MAPK, PI3K/AKT) via specific protein-protein interactions [18]. Current reports indicate that estrogen signaling in MDSCs occurs in part by the induced phosphorylation and activation of STAT3 which stimulates downstream signaling for the expansion of MDSCs [7]. STAT3 is required for MDSC survival and proliferation and also modulates expression of S100A8 and S100A9 proteins that are important for regulating MDSC expansion and migration to tumor sites [7, 8].
Antiestrogen therapy with tamoxifen has been widely used for more than 40 years, with evidence from clinical trials for significant reductions in BC mortality in ER-positive early BC [1, 19]. Although effective, tamoxifen has important drawbacks, including a limited period of activity before drug resistance; and an increased risk of endometrial cancer and thromboemboli due to its partial agonist activity as a selective ER modulator [2, 3, 20]. Use of AIs for postmenopausal patients has yielded better outcomes than the standard of 5 years tamoxifen [2, 19, 21]; but in patients with advanced breast cancer, only about 1/3 of ER-positive BCs respond to AIs, and resistance can evolve due to ER activation by different mechanisms such as ligand-independent activation [2, 3, 20–22] or emergence of ESR1 mutations [23, 24]. Consequently, a search is underway to discover new antiestrogens that lack agonist activity and override endocrine-resistance [20, 25]. As long as ER is present in breast tumors, growth may be stimulated by estrogen, partial agonists or estrogen-independent action. The first selective ER downregulator (SERD), fulvestrant, has no major agonist activity and good antitumor efficacy [20, 26, 27]. However, fulvestrant has very low bioavailability that is a significant liability in clinic [28]. Although fulvestrant has activity in ER-positive BCs that progress after AIs or tamoxifen including some patients with ESR1 mutations, discovery of improved SERDs with improved bioavailability and antitumor activity is a key goal. In 14–20% of metastatic ER-positive BCs from patients with multiple prior endocrine therapies, there is evidence for acquisition of functionally-aberrant ESR1 with point mutations often occurring in the ER ligand-binding domain, most commonly at D538G and Y537S [23, 24]. Some mutant ESR1 variants may continue to respond to fulvestrant, but higher doses of fulvestrant are required to achieve wild-type levels of tumor inhibition. Current data show that achievement of higher optimal doses of fulvestrant by intramuscular drug delivery is not feasible and underscore the need to develop more potent SERDs with enhanced bioavailability in advanced BC. A number of non-steroidal SERD candidates have been assessed, with many failing to advance beyond Phase I-II trials due to agonist activity in normal tissues, other off-target adverse side-effects or for unknown reasons [29, 30]. With this history, we elected to design estradiol-like SERDs targeting ER that differ from proposed nonsteroidal drugs. These new SERDs and fulvestrant were then assessed for antitumor activity in BCs as well as in ER-positive immune cells that occupy the TME and interactions with immune checkpoint inhibitors that may be beneficial to management of both ER-positive and potentially ER-negative BCs in the clinic.
2. Materials and Methods
2.1. Chemistry procedures for synthesis of 11β-aryloxy-estradiol derivatives
Reagents:
Tetrahydrofuran (THF) was distilled from benzoquinone ketyl radical under an argon atmosphere. Dichloromethane, toluene, benzene, and pyridine were distilled from calcium hydride under an argon atmosphere. Anhydrous N, N-dimethylformamide (DMF) was purchased from Sigma-Aldrich. All other solvents or reagents were purified according to standard procedures. (8S, 9S, 13S, 14S, 17S)-3,17-bis(Benzyloxy)-13-methyl-6, 7, 8, 9, 12, 13, 14, 15, 16, 17-decahydro-11H-cyclopenta[a]phenanthren-11-one (11-ketone) was prepared using established procedures [31–34].
Instrumentation:
1H NMR, 13C NMR, and 19F NMR spectra were obtained at 300 MHz, 400 MHz, or 500 MHz for proton, 75 MHz, 100 MHz, or 125 MHz for carbon, and 282 MHz, or 376 MHz for fluorine are so indicated. The chemical shifts are reported in parts per million (ppm, δ). The coupling constants are reported in Hertz (Hz) and the resonance patterns are reported with notations as the following: br (broad), s (singlet), d (double), t (triplet), q (quartet) and m (multiplet). High-resolution mass spectra were measured on a time-of-flight LC-MS. Thin-layer chromatography (TLC) was carried out using precoated silica gel sheets. Visual detection was performed with ultraviolet light, p-anisaldehyde stain, potassium permanganate stain or iodine. Flash chromatography was done using silica gel P60 (60 A, 40–63 μm) with compressed air.
General chemistry procedures to prepare the several antiestrogen compounds described in this report are presented in detail in Supplementary Methods.
2.2. Cell culture
Cell lines were obtained from the American Type Culture Collection (ATCC) and cultured according to ATCC recommendations. Briefly, ERα-positive human BC cells MCF-7, T47D and ZR-75 were cultured in DMEM or RPMI-1640 media as before [35, 36], and MCF-7 cells with HER-2 overexpression [37] and MCF-7 cells with acquired tamoxifen resistance were established and cultivated as reported previously [38, 39]. Mouse triple-negative (ERα-/PR-/HER2-) 4T1 breast tumor cells were cultured in RPMI-1640 medium. Media were supplemented with 10% fetal bovine serum (FBS; Gemini Bio-Products), 100 units/ml penicillin, 100 μg/ml streptomycin sulfate and 2.5 μg/ml amphotericin B (Gemini Bio-Products). Cultures were maintained at 37°C in a 5% CO2 incubator. For steroid-free conditions, medium was changed 48 hrs. before studies to phenol red-free DMEM or phenol red-free RPMI-1640 with 5% dextran-coated, charcoal-treated (DCC-FBS) as before [35].
2.3. Cell Proliferation Assays
MCF-7 and other selected BC cells were seeded in 96-well plates at 3–5 × 105 cells/well in complete medium. After 24 hours, medium was switched to estrogen-free conditions as described above. After 48 hrs, cells were treated with indicated concentrations of antiestrogens for 72 hrs with or without estradiol-17β (E2). Cell number and viability were determined by either cell counts or by colorimetric assays using the CellTiter 96 Aqueous (Promega) assay or the cell proliferation ELISA BrdU assay (Roche) as per manufacturer’s instructions. Treatments were done in quadruplicate, and experiments were repeated at least three times. In selected experiments using the Incucyte™ Live Cell System (Essen Bioscience) as per the manufacturer’s instructions, the proliferation of 4T1 cells maintained in a tissue culture incubator was monitored by using the NucLight Rapid Red Reagent for cell labeling in 6-well plates. Images for cell confluence were obtained every 4–6 hrs; as cells proliferate, the confluence increases, and confluence is therefore a surrogate for proliferation. Images were analyzed using the Live-Cell Analysis System (Essen Bioscience).
2.4. Polyacrylamide gel electrophoresis and Western immunoblotting
MCF7 cells were plated in regular medium. After 24 hrs, cells were incubated in the presence of antiestrogens or fulvestrant for 4 hrs in phenol-red free medium without FBS. Cell lysates were prepared using RIPA buffer, and protein concentration was determined using the BCA Protein Assay Kit (PIERCE/ThermoFisher Scientific). Forty micrograms of total cell protein was resolved by 4–15% SDS-PAGE, transferred to a PVDF membrane and probed with antibody directed against ERα (1D5, 1:100, ThermoFisher cat# MA5–13191). Ribosomal protein L13A (RPL13A), an established housekeeping gene that is not regulated by estradiol-17β or tamoxifen was used as loading control (dilution 1:1000, Invitrogen/ThermoFisher cat# PA5–58528) [40].
2.5. Competition binding assays in ER-positive human breast tumor cells
Specific estradiol-17β (E2) binding and competition for binding by antiestrogen JD128 or fulvestrant was assessed in human MCF-7 breast cancer cells using methods as described before [36, 41]. In brief, MCF-7 cells were suspended in phenol red-free RPMI medium to a concentration of 1 ×107 cells/ml, and incubations for 60 min were begun with the addition of [2,4,6,7-3H (N)]-estradiol-17β (99 Ci/mmol; New England Nulcear/Perkin Elmer, Waltham, MA) at 37°C with shaking. A 100-fold molar excess of unlabeled estradiol-17β was present in paired samples to determine displaceable binding [41]. Competitive ligand binding to ER-positive MCF-7 cells is detected by the ability of a test compound to displace labeled estradiol-17β from the cells in vitro.
2.6. Estrogen receptor-dependent transcriptional activity
A stable ER-positive T47D ERE luciferase reporter cell line, in which the ERE and the reporter luciferase gene are consistently expressed in the cell line were used in this study (Signosis). The cell line was established by transfection of luciferase reporter vector along with neomycin expression vector followed by neomycin selection, with neomycin-resistant clones subsequently screened for E2 induced luciferase activity or for measurement of potential antiestrogenic activity. Early passages of cells were cultured in complete medium containing RPMI supplemented with penicillin (100 units/mL), streptomycin (100 μg/ml), 10% FBS and G418 (75μg/ml). At 24 hrs prior to assays, cells were trypsinized, washed and plated in each well of a 96-well plate with 5 ×104 cells in 100 μl with phenol-red-free medium containing 0.1% dextran-coated charcoal-treated FBS[42, 43]. Cells were then treated with 17β-estradiol alone or combined with fulvestrant or JD128 for 24 hrs. Thereafter, media was removed by aspiration and 100 μl of PBS was added to each well, followed by aspiration of medium and addition of 50 μl of lysis buffer to each well. Cells were incubated in lysis buffer for 30 min at room temperature. Lysate was mixed 1:1 with luciferase substrate (Promega), and luminescence was measured using a MLX microtiter plate luminometer (Dynex) and quantified as relative light units (RLU) according to established procedures [42, 43]. Total protein was quantified using BioRad Protein Assay (BioRad).
2.7. In vivo breast tumor models
Animals were housed in a pathogen-free environment with controlled light and humidity and received food and water ad libitum. All studies were approved by the UCLA Animal Research Protection Committee.
For experiments using human BC cells as subcutaneous xenografts, ovariectomized female nude mice at 6 weeks of age were obtained from Charles River. MCF-7 human BC cells (2 × 107) were implanted in the flanks of mice who had been primed three days before cell injections with estradiol-17β (0.36 mg, 60 days slow-release pellets, Innovative Research of America) as before [35, 36, 44]. When tumors grew to 50–100 mm3, animals were randomized to different treatment groups including a) vehicle control, b) JD128 at 15 mg/kg (by oral gavage daily for 28 days) and c) JD128 at 75 mg/kg (by oral gavage daily for 28 days). Tumor volumes for mice in experimental and control groups were measured every 3–4 days, with tumor volume calculated by (l × w × w) / 2, with tumor length l, and tumor width w in mm. Data were presented as the mean ± SEM for tumor volumes measured in cubic mm. Data were analyzed by use of ANOVA and student’s t-test statistical approaches as before [35, 36, 44].
To determine the potential effect of estrogen depletion on the progression of tumors in vivo, 4T1 murine TNBC cells (ATCC) were injected in the 4th mammary fat pad (2 × 105 cells) of either ovariectomized or sham-operated 6-week-old syngeneic female BALB/c mice (Jackson Laboratory). Tumors were measured every 3–4 days, and tumor volume was calculated as (l × w × w) / 2 as above.
In further studies to determine the effects of antiestrogen treatment alone or in combination with anti-PD-L1 antibody on murine tumor progression in vivo, ovariectomized 6-week-old female syngeneic BALB/c mice were used (Jackson Laboratory). Three days prior to tumor cell inoculation, mice were injected with estradiol-17β (0.36 mg, 60 days slow-release pellets, Innovative Research of America). 4T1 cells were inoculated in the 4th mammary fat pad (2 × 105 cells), and mice were randomized after tumors reached an average size of 200–250 mm3. For treatment, mice were divided into 6 groups: a) vehicle control or isotype IgG (IgG2b,κ, RTK4530, Biolegend), b) anti-PD-L1 antibody (Biolegend anti-CD274/B7-H1/PD-L1 clone 10F.9G2, 100 μg/mouse by intraperitoneal injection, every third day), c) fulvestrant (5mg/mouse subcutaneous, once a week), d) JD128 (50 mg/kg by oral gavage, daily) and e) combination treatment of fulvestrant and anti-PD-L1 antibody or f) JD128 and anti-PD-L1 antibody at doses as described for treatment as single agents. Tumors were measured every 3–4 days, and tumor volume was calculated as above. After 10–12 days, mice were anesthetized by established methods, with blood collected by cardiac puncture in BD vacutainer vials with EDTA (terminal procedure). An approved secondary method of euthanasia was then used to ensure animals were deceased. Tumors were harvested, with final tumor weights and sizes compared among groups. Mass cytometry studies to assess selected immune cell populations and biomarkers were performed as detailed below.
2.8. Mass cytometry for analyses of immune cell subpopulations, cytokines and selected biomarkers
Tumors from each mouse were harvested after 10–12 days of treatment as described above. Single cell suspensions were generated from tumors using the MACS mouse tumor dissociation kit (Miltenyi Biotech Cat. 130–096-730) following manufacturer’s instructions. One million cells per tumor were resuspended in PBS and labeled with Cell-ID Cisplatin (Fluidigm, Cat. 201064) to assess for live/dead cells. For antibody labeling, we used the recommended cell surface staining procedure (Fluidigm) followed by the FoxP3/Transcription Staining Buffer Set protocol (eBiosciences™). Cells were labeled with a panel of 28 metal-conjugated antibodies to determine different immune lineages in addition to memory, trafficking, activation, and exhaustion markers (see Supplementary Table 1 for list of antibodies). After washing and centrifugation, cells were fixed using MaxPar Fix and Perm buffer (Fluidigm, Cat. 201067) and labelled for single cell discrimination with Cell-ID Intercalator-Ir (Fluidigm, Cat. 201192A). Samples were resuspended with 10% EQ four-element calibration beads (Fluidigm, Cat. 201078), and filtered through a 40 μm mesh filter prior to acquisition on a Helios™ mass cytometer (Fluidigm), at a rate of 300–500 events/s.
2.9. Dimensionality Reduction, Cluster Analysis and Visualization
Collected mass cytometry data was analyzed as previously described [45]. Briefly, samples were normalized utilizing a bead standard. First, each cytometry file was processed in FlowJo (v10.3), then manually gated for stability of signal over time, followed by exclusion of normalization beads, ratio of DNA intercalators (191Ir+ vs 193Ir+), with finally single cell events (Ir193 vs event length)(Fig. 9A). After that, viable (195Pt−)CD45+ events were exported and uploaded into the X-shift (VorteX) clustering environment to obtain the k -nearest-neighbor density estimation as described before [45, 46]. Dimensionality reduction of unclustered data was performed using the t-stochastic neighborhood embedding (t-SNE) and PhenoGraph algorithms implemented in the Cytofkit library [47], supplied by Bioconductor v.3.4 and run in RStudio v.1.1.463. A fixed number of 10,000 cells were sampled without replacement from each file and combined for analysis. Resulting t-SNE plots were subsequently filtered by marker expression to visualize differences between different treatment groups. Heatmaps were generated using Z-scores based on median marker expression (excel and Prism v7). Then, we used Wei et al. [48] criteria to exclude clusters from analyses that had an expression level lower than 0.5%.
Fig. 9.

High-dimensional analysis of mass cytometry data shows antiestrogens decrease the amount of myeloid derived suppressor cells present in 4T1 tumors. Single cells were purified from 4T1 tumors grown in BALB/c mice, stained with a panel of 28 markers by mass cytometry. A) Sequential gating strategy to analyze tumor CD45+ cell subsets present in the TME. B) Phenograph example of different cell populations identified by single cell analysis using Cytofkit. C) tSNE plots show cluster expression of markers for both populations of myeloid cells G MDSC and M-MDSC. D) Representative plots of G-MDSC (CD11b+Ly6Ghi, Ly6Clo) and M-MDSC (CD11b+Ly6Chi, Ly6Glo) as percentage of CD45+ cells. E) Quantification of G-MDSC and M-MDSC present in the tumor bed of BALB/c mice with 4T1 tumors. *P < 0.05, ** P < 0.01. n = 6–11. F) ERα expression in total MDSC, G-MDSC and M-MDSC.
2.10. Flow cytometry and bone marrow cell analysis
Human myeloid-derived suppressor cells were expanded from bone marrow (BM) specimens of BC patients after standard Ficoll gradient purification and red blood cell lysis. Briefly, 2 × 106 BM cells were cultured in the presence of 1000 IU/ml of GM-CSF and 40 ng/ml IL-6 in different media conditions including regular RPMI-1640 with 15% FBS or phenol red-free medium with 15% DCC-FBS with or without 100 nM E2 (7). After 6 days of culture, cells were harvested, stained with a 14 antibody panel including anti-phospho-STAT3 (pSTAT3) and analyzed by flow cytometry with an LSRII with a 5 lasers (UV, violet, blue, green-yellow and red). Data was processed using FlowJo (v10.3). De-identified BM specimens were retrospectively-collected and deposited in the UCLA Pathology Tumor Bank according to Human Subject Protection Committee guidelines at our institution.
2.11. Immunohistochemistry
Paraffin-embedded sections from 4T1 tumors were cut at 4 μm thickness and paraffin removed with xylene and rehydrated through graded ethanol. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 10 min. Heat-induced antigen retrieval (HIER) was carried out for all sections in 0.001M EDTA buffer, pH = 8.00 using a vegetable steamer at 95°C for 25 min. Sections were incubated with anti mouse CD8a (eBioscience, 14–0808-82) at 1:100 dilution for 1 hour at room temperature. After primary antibody incubation, tissues were then incubated with secondary rabbit anti-rat immunoglobulin for 30 min at 1:200 dilution (Vector, AI-4001) followed by a 30 min incubation with Dakocytomation EnvisionÅ System Labelled Polymer HRP anti rabbit (Agilent, K4003). All sections were visualized with the diaminobenzidine reaction and counterstained with hematoxylin. The number of immune-positive cells were counted in five randomly chosen fields per tumor at 100-fold magnification. 4–6 mice tumors per condition were used for analysis. Results from the five areas/mouse were averaged and used in the statistical analysis.
2.12. Statistics
For in vitro studies, triplicates of experiments were done to verify results. ANOVA or t-tests were used as appropriate to compare interventions. Analyses of cells were evaluated using bar and scatter graphs with mean, standard deviation (SD) and standard error (SE). Repeated measures ANOVA was used as appropriate to assess time, condition, and time by condition interaction effects. For in vivo studies, mice with similar tumor size were randomized to different treatment groups with controls for up to 28 days. Data analyses by appropriate parametric or nonparametric methods were applied [22, 35–37]. Briefly, these analyses use mixed-effects models with tumor size as outcome measure (transformed as needed). Analyses of mass and flow cytometry data were performed using GraphPad Prism version 7.0 (GraphPad, San Diego, CA) using one-way ANOVA followed Bonferroni’s multiple comparisons test or two-tailed unpaired Student’s t-test approaches as described before [45, 46, 49, 50]. Differences were considered significant for P values less than 0.05.
3. Results
3.1. SERD Synthesis and Properties
As reported earlier [51], we designed, synthesized and screened more than 65 new SERD candidates, all of which have the general structure shown in 1, namely 11β-aryloxy estradiols, with a basic amine positioned at the 4-position of the aryl ring (Fig. 1). The basic amine is connected to the aryl unit either directly or via a spacer that varies from 3–6 atoms. In some of the more active compounds, we also attached an electron-withdrawing group, e.g. a trifluoromethyl unit or a fluoride atom, in the 3-positon (ortho to the amino chain). Of these compounds, several had activity comparable to fulvestrant 2 but JD128, 4, in particular was more potent than fulvestrant in a number of antitumor assays as shown below. We note that this new class of steroid-like SERDs lack the prototypical side chain (as in fulvestrant) widely used to design other drugs with ERα antagonism, but these SERD candidates generate a full antagonist profile and induce significant ERα down-regulation, likely similar to significant ‘indirect’ receptor antagonism as reported in previous independent studies of 11β-substitutions in ERα [52, 53] and other structural changes as in an independent report [54].
Fig 1.

New substituted estradiols. See text for details.
This new series of estradiol analogues, namely 11β-(4-aminoalkyl) aryloxy-estradiols, are expected to bind the ER ligand-binding domain since they are close structural analogues of estradiol (cf. Hansen et al. [53]). The 11β-aryloxy group, bearing a variable length chain ending in a basic dialkylamino group, would be expected to block the folding of helix-12 by potentially both steric hindrance and a salt bridge formation between the protonated amine and an acidic side chain on helix 12. Thus, these ER antagonists should bind to ER in such a way as to prevent the folding of helix-12 and thereby potentially inhibit BC proliferation.
The synthesis of the new analogues (Fig. 2) started with estradiol 7 which was converted into the bis (benzyloxy)ketone 8 by a known route [31–34] (protection, benzylic oxidation to the 9, 11-alkene, hydroboration-oxidation, and final oxidation to the ketone). Reduction of this protected ketone 8 with sodium borohydride afforded the expected 11β-alcohol 9 by attack of the hydride on the less hindered α– face, away from the hindering 13β–methyl group. Formation of the 11β-alkoxide anion of 9 using potassium hydride in THF/DMF followed by addition of 4-fluoronitrobenzene 10 effected a clean SNAr reaction to afford the 4-nitrophenyl ether 11. Nickel boride reduction [55, 56] of the nitro group (sodium borohydride with NiCl2 6H2O in methanol) gave the aminophenyl ether 12 in good yield. Removal of the two benzyl ethers from 12 by catalytic hydrogenolysis using Pd(OH)2 in methanol gave the first analogue, the simple aniline 13 (JD105), namely 11β-(4-aminophenyloxy) estradiol. For nearly all of the other analogues, the crude aniline 12 was not isolated but rather treated directly with an acid chloride. The analogues having a three-atom linker between the aryl ring and the basic amine were all prepared by the same route. Thus, treatment of 12 with chloroacetyl chloride and catalytic DMAP in triethylamine afforded the intermediate chloroacetamide, which was immediately reacted with one of four secondary amines, e.g., piperidine, pyrrolidine, morpholine, and dimethylamine, to give the amides.
FIG. 2.

Synthesis pathways for analogues. Analogues 13–16. See text for more details.
Again hydrogenolysis of the benzyl ethers using hydrogen and a palladium catalyst gave the desired analogues, 14a-d (JD101-JD104). After coupling of 12 with the acid chloride to give the amide, hydride reduction afforded the 2-(dialkylamino)ethyl amines, the benzyl ethers of which were hydrogenolyzed to give another set of analogues 15a-d, namely the N-(2-aminoethyl)anilines. In addition the 4-amino group was completely removed to give the simple 11β-phenyl ether 16.
The next set of analogues each had a 3-carbon chain between the aniline and the secondary amine (see Fig. 3). Thus treatment of the crude aniline 12 with 3-chloropropionyl chloride furnished the 3-chloropropanamide and displacement of the chloride with the secondary amines and subsequent hydrogenolysis afforded the analogues with a 5-atom side chain ending in the basic amine, 17a-d (JD106–109).
Fig. 3.

Synthesis pathways for analogues. Analogues 17–20. See text for more details.
Likewise using 4-chlorobutanoyl chloride, after displacement of the chloride with the secondary amines and subsequent hydrogenolysis, one obtained the analogues with a 6-atom side chain ending in the basic amine, 18a-d (JD110–112, JD116). Finally, following the same route starting with 5-chloropentanoyl chloride gave the analogues with a 7-atom side chain, 19a-d. Again after coupling of 12 with the 3-carbon acid chloride to give the amide, hydride reduction afforded the 2-(dialkylamino)ethyl amines, the benzyl ethers of which were hydrogenolyzed to give another set of analogues 20a-d, namely the N-(3-aminopropyl) anilines. By substituting the 4-fluoronitrobenzene unit for other aryl fluorides, one could prepare several other sets of analogues (see Fig. 4). Thus, alkylation of the 11β-alcohol 9 with 2, 4-difluoronitro-benzene led to the 3-fluoro-4-nitrophenyl ether (which after hydrogenolysis gave the analogue 21). From that compound were prepared the 16 analogues, 23a-d, 24a-d, 25a-d, and 26a-d and the unsubstituted aniline 22. In a similar manner, using 4-fluoro-2-(trifluoromethyl) nitrobenzene to alkylate the anion of 12 resulted in the 3-trifluoromethyl-4-nitrophenyl ether (which after hydrogenolysis gave the analogue 27) and thus the 16 additional analogues, 29a-d, 30a-d, 31a-d, and 32a-d and the unsubstituted aniline 28.
Fig. 4.

Synthesis pathways for analogues. Analogues 21–32. See text for more details.
3.2. Selected steroid-like SERD candidates promote ER downregulation, bind ER-positive breast tumor cells and block ER-dependent transcription in vitro.
We used different assays to screen antiestrogen/SERD candidates (see Figs. 1–4), including determination of the effect of antiestrogens on downregulation of ERα protein using PAGE and Western immunoblots (Fig. 5). As shown in the figure, SERD candidates 128 and 140 were most effective in reducing ER protein levels in ER-positive MCF-7 BC cells in vitro, with the effect of 128 comparable to that of fulvestrant. Additional studies were also done to assess competitive binding of SERD 128 in MCF-7 cells (Fig. 5B) and inhibition of ER-dependent transcription in ER-positive T47D BC cells stably transfected with an ER-dependent luciferase reporter gene (Fig. 5C).
Fig. 5.

Biologic activity of selected SERD candidates. A) Downregulation of ER protein. ER-positive MCF-7 cells were treated in phenol-red free RPMI 1640 without FBS and containing vehicle control (CON) or 100 nM concentrations of either fulvestrant (FX) or antiestrogens 105, 109, 121, 140, 151, 160 or JD128 in vitro. After 4 hours, cells were harvested and processed for PAGE and Western immunoblots using ERα antibody (1D5, Thermofisher Scientific). RPL13A was used as a loading control. B) Specific [3H]estradiol-17β (E2) binding and competition for binding by antiestrogen JD128 or fulvestrant (FX) at 10 nM was assessed in human MCF-7 breast cancer cells using methods as described before [36, 41]. C) Response of the ERE-luciferase T47D reporter construct to estrogen antagonists fulvestrant (10 nM) or JD128 (10 nM) in combination with 2nM 17β-estradiol as compared to treatment with 17β-estradiol alone (E2; 2 nM). Cells were dosed with either E2 alone or with SERDs combined with E2 in phenol red-free medium with 0.1% dextran-coated charcoal-treated FBS in luminometer plates. Data are presented as relative light units (RLU) relative to that of E2 alone in three replicate assays (4 wells per replicate) + SEM. Treatment with E2 alone induced a 12-fold induction of ER-dependent luciferase activity quantified as RLU relative to vehicle control-treated samples.
The combined results of these studies indicate that JD128 is a promising SERD with ER antagonist activity in ER downregulation, target cell binding and ER-dependent transcription comparable to that of the pure antiestrogen fulvestrant.
3.3. Steroid-like SERD JD128 inhibits human BC progression in vitro and in xenograft models in vivo.
Investigations of the properties of SERD JD128 in blocking the progression of human breast tumors in vitro and in vivo. As shown in Fig. 6A, the E2-induced proliferation of several ER-positive BC cells including MCF-7, T47D and ZR75 cells was significantly inhibited by treatment with 10 nM JD128 (all at P<0.001). This antiproliferative action of JD128 was also found with different MCF-7 cell populations that included cells with no HER2-overexpression (MCF-7/PAR), cells with HER2-overexpression (MCF-7/HER2) and MCF-7 cells with tamoxifen resistance (MCF-7/TMR).
Fig. 6.

Steroid-like SERD 128 inhibits estrogen-induced BC cell proliferation in vitro and in vivo. A) ER-positive MCF-7, T47D and ZR75 cells were grown in phenol red-free media with 1% DCC-FBS for 48 hr., then treated 48 hr. with 2 nM estradiol-17β alone (control) or in combination with 10 nM doses of JD128. Note that MCF-7 cell populations included cells with no HER2-overexpression (MCF-7/PAR), cells with HER2-overexpression (MCF-7/HER2) and MCF-7 cells with tamoxifen resistance (MCF-7/TMR). Cell proliferation is shown as % of that in estradiol-treated controls (n=3 experiments). Inhibition of cell proliferation in MCF-7/PAR, MCF-7/HER2, MCF-7/TMR, T47D and ZR75 cells averaged 98%, 85%, 94%, 97% and 98% as compared to estradiol-treated controls. JD128 significantly blocked proliferation in all BC cell models in vitro (P<0.001). Of note, E2 alone stimulated cell proliferation several-fold in each cell line as compared to cells treated only with vehicle (not shown). B) JD128 inhibits growth of human breast tumor xenografts in vivo. MCF-7 human breast cancer cells were subcutaneously inoculated in nude mice previously primed with estradiol pellets. When animals developed tumors of comparable size they were randomized to treatment with vehicle control (control) or JD128 at 15 and 75 mg/kg once a day by oral gavage for 28 days. Tumors were measured every 3 days, and tumor volume was calculated as V= (l ×w × w)/2). Results are expressed as mean ± SEM. *** P < 0.001 as compared to control group.
In Fig. 6B, orally administered JD128 is shown to inhibit the growth of human MCF-7 breast tumor xenografts in vivo in a dose-dependent manner. MCF-7 cells were subcutaneously inoculated in nude mice previously primed with estradiol pellets. When animals developed tumors of comparable size, they were randomized to treatment with vehicle control (vehicle) or JD128 at 15 and 75 mg/kg once a day by oral gavage. It is important to note that JD128, in contrast to fulvestrant [28], has potent biologic action in blocking the progression of breast tumors in vivo via an oral route of administration.
3.4. Effects of estrogen and antiestrogens on expansion and activation of human immune MDSCs
Emerging findings indicate that E2 can modulate expansion/activity of MDSCs [7, 8, 17]. Since MDSCs that often occur in the TME reportedly play a critical role in tumor immune tolerance and cancer progression, we assessed effects of E2 and potential antagonist effects of fulvestrant and JD128 (Fig. 7).
Fig. 7. Expansion of MDSCs is dependent on estrogen signaling and reversed by antiestrogens.

E2 increases total numbers of MDSCs, with total numbers of human MDSCs derived from bone marrow (BM) of BC patients. Retrospectively-collected BM cells from de-identified BC patients were purified by established methods including red blood cell lysis and Ficoll gradients and then incubated with GM-CSF and IL-6 for 6 days in either regular RPMI medium + 15% FBS (NM) (contains estrogens), NM with antiestrogens (FULV or JD128) or in phenol red-free medium with 15% charcoal coated-dextran treated FBS (EFM) (estrogen-depleted) with or without the addition of 100 nM estradiol-17β (EFM+E2). Normal cell culture medium (NM) drives E2-dependent signaling due to the presence of various estrogens in FBS as well as estrogenic properties of phenol red. These effects were significantly inhibited by fulvestrant (FULV) and JD128 at 1μM concentrations in normal medium. A) The gating strategy used to identify MDSCs is shown. The figure shows total MDSC populations (CD45+CD3−CD19−CD20−CD56−CD11b+) identified by following the gating strategy of Ruffell et al. [16] and modified by Svoronos et al. [7]. B) Top panel: graph showing quantification of total MDSC cultivated as described. Lower panel: JD128 blocks phosphorylation/activation of STAT3 in G-MDSC subpopulations under the same conditions described in A. Results show median fluorescence intensity for p-STAT3 in G-MDSC subsets (CD45+HLA-DR−CD11b+CD14-CD15+) after expansion of human total MDSC. Of note, the effect of JD128 is similar to that achieved with E2 depletion (EFM).
In these studies, we used archival retrospectively-collected bone marrow (BM) cells from de-identified BC patients. The BM cells were purified by established methods and then stimulated with cytokines under conditions specified in Fig. 7. Thereafter, MDSC cells were detected using established gating strategies by flow cytometry. When compared to MDSCs derived from BM cultivated in normal medium containing E2 and cytokines, several findings are apparent: a) MDSC levels are markedly reduced in E2-free medium; b) addition of E2 to E2-depleted medium stimulates significant expansion of MDSC numbers; c) JD128 and fulvestrant each block E2-induced expansion of MDSCs, and the effect of JD128 exceeds that of fulvestrant at equivalent doses (all at P<0.05; Fig. 7B, top panel). Furthermore, the accumulation of MDSCs is known to involve the expansion of immature myeloid cells and activation/conversion of immature cells to MDSCs, a process that appears to be driven at least in part by STAT3 signaling [17]. Importantly, estrogen is reported to activate such signaling pathways in MDSCs via the phosphorylation of STAT3 [7]. Accordingly, antiestrogen JD128 is especially effective in blocking the phosphorylation and activation of STAT3 in G-MDSC subsets, an action that may be crucial for blocking the enhanced immunosuppressive activity of MDSCs in BC (Fig. 7B, lower panel).
3.5. Effects of estrogens and antiestrogens on ERα-negative tumor growth in vitro and in vivo.
TNBC cells that lack expression of ERα, PR and HER2 amplification were selected for use in experiments to investigate the potential actions of antiestrogens primarily on immune cells in the TME. In mice with implants of E2-insensitive orthotopic tumors, Svoronos et al. [7] reported a significant survival benefit associated with ovariectomy (OVX; estrogen depletion) when compared to non-OVX controls (normal estrogen levels), while treatment of OVX mice with E2 reversed the protective effect of OVX. Further, the survival benefit of OVX was not observed in immune-deficient as compared to wild-type mice, suggesting that immune activity is critical in the antitumor effect of E2 depletion [7]. We confirm in our experiments that OVX reduces the progression of 4T1 TNBCs as compared to that of intact animals in a murine model, thus suggesting that ovarian E2 may play a role in stimulating TNBC growth in vivo (FIG. 8A) P < 0.0001. To determine if estrogens have a direct effect on 4T1 TNBC (ERα-negative) cell proliferation, we used the Incucyte™ system as described in methods to investigate 4T1 cell progression in vitro. No growth stimulation of cells as monitored by cell confluence was observed when tumor cells were grown in the presence of E2 as compared to control-treated 4T1 cells over a time course of 5 days (Fig. 8B). Furthermore, treatment with SERD JD128 at doses ranging from 10–1000 nM did not elicit any significant effect on cell growth in vitro as shown in Figure 8B. Together, these data indicate that effects of sex steroids on progression of 4T1 tumors in vivo are likely due to interactions with cells in the TME.
Fig. 8. Estrogen effects on ERα negative tumor growth in vitro and in vivo.

A) Ovariectomy reduces progression of 4T1 TNBC in syngeneic mice in vivo. Female 6-wk-old BALB/c mice, either ovariectomized (ovx) or sham-operated (intact), were inoculated s.c. with 2 ×105 4T1 TNBC cells. Tumor growth was then assessed every 3 days, with tumor volume calculated as V= (l × w × w)/2. **** P ≤ 0.0001. B) 4T1 triple negative breast cancer cells do not respond to estrogen or antiestrogens in vitro. 4T1 cells were grown in the presence (+E2) or absence (−E2) of estradiol-17β and increasing concentrations of JD128 at 10 nM (128–10), 100 nM (128–100) or 1000 nM (128–1000). Cell proliferation was assessed using the Incucyte S3 Live-Cell Analysis system with pictures obtained every 4–6 hours. Graph shows average cell proliferation expressed as phase object confluence measured for 5 days. No significant differences were observed in cell proliferation. C) Antiestrogens combined with anti-PD-L1 checkpoint antibody inhibit TNBC cell growth. Female BALC/c mice (6-wk-old) received intra-mammary implants with 2 × 105 murine 4T1 TNBC cells. After tumors reached approx. 200 mm3 mice were randomized and treated with vehicle (CON), 100 mg of anti-PD-L1 antibody every 3rd day (Ab), 50 mg/kg SERD128 via oral gavage (JD128), 5 mg fulvestrant s.c. (FULV) and combinations of antibody and fulvestrant (FULV+Ab) or antibody and JD128 (JD128+Ab). Tumors were measured every 3 days, and tumor volume was calculated as V= (l × w × w)/2. ** P < 0.01, **** P < 0.0001 as compared to control group.
Furthermore, in this in vivo study, we assessed antitumor efficacy of JD128 alone and combined with an anti-PD-L1 checkpoint antibody. The 4T1 tumor cells implanted in mammary glands exhibit highly aggressive behavior and are generally found to metastasize widely to cause early mortality. In contrast to a lack of effects of either estrogens or antiestrogens on 4T1 tumor progression in vitro, we find that the antiestrogens fulvestrant and JD128 are each effective in inhibition of 4T1 tumor growth in vivo in syngeneic BALB/c mouse models (Fig. 8C). Since these mice are immune-intact, we next assessed the effect of treatment with an anti-PD-L1 checkpoint inhibitor alone and in combination with either fulvestrant or JD128. As shown in Fig. 8C, anti-PD-L1 antibody alone elicited no significant effect on 4T1 tumor progression, while JD128 and fulvestrant were each able to induce significant suppression of 4T1 tumor progression in vivo. These results appear to be consistent with the notion that antiestrogens interact with immune cells in the TME and may play an important role in stopping tumor progression in vivo. To investigate effects of antiestrogens and immune checkpoint inhibitors when administered in combination, we next used mass cytometry to study the immune cell subpopulations present in the TME in vivo.
3.7. Mass cytometry analyses show that antiestrogens reduce MDSCs in murine 4T1 tumors in syngeneic mice.
To explore mechanistic pathways that underlie the antitumor effects of antiestrogens alone and combined with an ICI (Fig. 8C), we used mass cytometry by time of flight analyses (cyTOF) with a panel of selected labeled antibodies to track the levels and activities of immune cell subsets in the TME.
Single cell suspensions were prepared from 4T1 tumors grown in BALB/c mice that were treated for 12 days as detailed in FIG. 8C. Cells were then labeled and analyzed by cyTOF. Results of these analyses are summarized in FIG. 9. Of the two major MDSC subsets that have been described in humans and mice based on their phenotypic, morphological and functional characteristics (e.g. G-MDSC and M-MDSC), both G-MDSC and M-MDSC subsets are significantly reduced on treatments with either antiestrogens alone or when given in combination with anti-PD-L1 antibody as compared to appropriate controls (FIG. 9D, E), with a somewhat enhanced effect on the GMDSC population. The results indicate that this biologic effect of antiestrogens may be due to expression of ERα in both G-MDSC and M-MDSC subsets (Fig. 9F).
3.8. Effects of antiestrogens on tumor-infiltrating lymphocytes and cytokines in 4T1 tumors in vivo.
In order to gain a better understanding of all tumor infiltrating leukocytes, we analyzed single cell suspensions from tumors (Fig. 8C and 9) by looking at CD8+ and CD4+ TILs. An adaptive T-cell response, which requires antigen recognition, is composed of both cytotoxic CD8+ T cells and CD4+ T cells [57]. Animal models have shown that in vivo eradication of tumors is for the most part mediated by cytotoxic T-cells. The presence of intratumoral T-cells is an independent predictor of improved survival and has also been associated with increased secretion of interferon-gamma (IFNα), interleukin-2 (IL-2) and TNFα [16, 58, 59]. As in Fig. 8C, groups included mice treated with control vehicle, anti-PDL1 antibody, fulvestrant, JD128 or the combination of fulvestrant with anti-PD-L1 antibody or JD128 and anti-PD-L1 antibody (Fig. 10).
Fig. 10.

Tumor infiltrating lymphocytes (TILs) in 4T1 tumors from BALB/c mice, with CD8+ and CD4+ TILs shown. Single cell suspensions were purified, stained and analyzed by cyTOF. Groups include mice treated with control vehicle (CON), anti-PD-L1 antibody (Ab), fulvestrant (Fulv), JD128 or the combination of fulvestrant with anti-PD-L1 antibody (Fulv+Ab) or JD128 and anti-PD-L1 antibody (JD128 + Ab). A) Sequential gating strategy to analyze tumor CD3+ cell subsets. B) Z-scores of median intensity of distinct protein markers are show in heatmap for all clusters analyzed by Cytofkit. C) tSNE scatter plot visualization of CD3+ cells showing clusters of CD8+, CD4+ and Tregs (CD4+CD25+FoxP3+) cells are observed (upper left). Right: t-SNE plots with arcsinh transformed signal intensity of different activation markers. D) Percentage of different type of CD8+ and CD4+ T cells, naïve (nT) (CD44−CD62L+CD69−), effector (effT) (CD44+CD69+Tbethieomes−) and effector memory (TEM) (CD44+CD62L−). E) Increased expression of activation cytokines in CD8+ and CD4+TIL population are shown. F) CD4+ CD25+FoxP3+ Tregs are significantly decreased by antiestrogen therapy. G) CD8+ TIL in same tumor tissues used for cytof detected by IHC, antiestrogens and combination treatment increase CD8 infiltration in the tumor bed. *P < 0.05, **P < 0.01, *** P < 0.001. n=6–11.
A sequential gating strategy to analyze tumor CD3+ cell subsets is shown in Fig. 10A, while the normalized median intensity of distinct protein markers are show in a heatmap for all clusters analyzed by Cytofkit in Fig. 10B [45, 46]. tSNE scatter plots for visualization of CD3+ cells that show clusters of CD8+, CD4+ and Tregs cells are presented in Fig. 10C. Importantly, the results show that both effector and effector memory CD8+ and CD4+ T-cells in tumors are several-fold higher in mice treated with either fulvestrant or JD128 antiestrogens when combined with PD-L1 antibody as compared to controls (P<0.05) (Fig. 10D). In addition, we find increased expression of known activation cytokines IFNγ, IL-2 and TNFα in CD8+ and CD4+ TIL subpopulations (Fig. 10E). These data appear to complement reports on estrogen-specific alterations of these cytokines in independent murine models [60]. Furthermore, antiestrogen treatments evoke a significant reduction of T-regulatory T-cells (Tregs) (Fig. 10F) which are known to play an important role in the maintenance of tumor immune tolerance [61]. In the process of tumor progression, Treg cells tend to accumulate in tumors and suppress T-cell responses at the tumor site. The number of tumor-infiltrating CD25+FoxP3+ Tregs is associated with poor prognosis and is identified as a significant predictor of poor outcome [62]. In addition, to corroborate mass cytometry data we performed immunohistochemistry to detect CD8+ TILs presence in the tumor bed. IHC results shown in Fig. 8G confirmed data obtained by cytof, antiestrogens and combination treatment significantly increase CD8+ TIL infiltration (P < 0.0001).
3.9. Effects of antiestrogens combined with ICIs on macrophage and dendritic cell subsets in 4T1 tumors in vivo.
Since recent findings suggest that cells of the innate immune system play an important role in the decision between an effective immune response versus induction of immune tolerance, we also investigated levels of dendritic cells (DC) that have a special function linking the innate immune response with the induction of adaptive immunity. These cells play a major role by processing and presenting antigens to T and B cells to generate an immune response. Stimulatory DCs promote effective immune responses by stimulating T-cell proliferation and shaping specific T-cell response phenotypes [63]. Importantly, treatment with both antiestrogens fulvestrant and JD128 alone as well as combined with anti-PD-L1 antibody (as in Fig. 8C) increased the population of DCs in 4T1 tumors (Fig. 11A).
Fig. 11.

Combined therapy with JD128 and anti-PD-L1 antibody enhance tumor infiltrating dendritic cells and M1 macrophages. Single cell suspensions were purified, stained and analyzed by cyTOF as described above. Groups include mice treated with control vehicle (CON), anti-PD-L1 antibody (Ab), fulvestrant (Fulv), JD128 or the combination of fulvestrant with anti-PD-L1 antibody (Fulv+Ab) or JD128 and anti-PD-L1 antibody (JD128 + Ab). A) Subset of dendritic cells present in the tumor bed show a significant increase in the total number of DC gated as (CD45+CD11c+MHCII+) when SERD JD128 was added to ICI therapy. B) M1 tumor infiltrating macrophages were significantly increased by combination therapy of SERD JD128 and anti-PD-L1 antibody. Macrophages were gated as (CD45+CD11b+F4/80+ M1: MHCIIhi M2: MHCIIlo). *P ≤ 0.05, **** P < 0.0001, n= 6–11.
Further, it is well documented that the highly inflammatory microenvironment of tumors tends to recruit macrophages and peripheral blood monocytes [16]. These myeloid cells receive tumor-derived signals that alter gene expression and phenotype. A prominent myeloid cell subset that develops in the breast TME is the tumor-associated macrophage (TAM). Macrophages are key modulators and effector cells in the immune response that exhibit high plasticity in response to various external signals (61). Depending on TME signals, macrophages occur as M1 macrophages associated with ‘tumoricidal’ activity with high production of reactive nitrogen and oxygen intermediates and pro-inflammatory cytokines or M2 macrophages involved in tumor progression and immunoregulatory functions [64]. The M2 phenotype predominates among TAMs, and a high density of TAMs correlates with poor prognosis in BC [65]. CyTOF analyses based on experiments noted in Fig. 8C reveal that therapy with SERD128 combined with anti-PD-L1 antibody elicits a significant increase in the M1 tumoricidal subset of macrophages in the TME (P<0.05) while simultaneously trending toward a reduction in the M2 macrophage subset (P= 0.05) (Fig. 11B), thus contributing to overall antitumor actions of dual antiestrogen-ICI therapy.
Discussion
The role of estrogen signaling in the progression of BCs with ERα expression is well-established by the successful use of ER antagonists in the clinic [2, 3, 19]. In addition, the present findings indicate that antiestrogens also have a significant effect on antitumor immunity independent of their direct activity on BC cells. Although ICIs have been shown to improve overall survival for subsets of patients with advanced melanoma, lung and TNBC [4, 13], the bulk of patients with BC, particularly ER-positive disease, do not have significant benefit from this promising therapeutic approach [4, 14]. Despite known sex-related differences in immune responses [9, 66, 67], little is known about the effect of sex hormones on immunotherapy in malignancy. An important question regarding the use of targeted therapies is whether these agents may positively or negatively affect immune cells. There is increasing awareness of the role of nonmalignant cells in the TME in regulating the tumor response to therapies. As indicated in the present report, the TME plays a critical role in modulating cancer progression and therapeutic responses. The presence of tumor-infiltrating lymphocytes in the TME is a prognostic indicator for benefit from ICI in TNBC, and T-cell inhibitory pathways in the TME such as MDSCs are identified [7, 8, 17]. Most immune cells including MDSC and CD8+ T-cells express estrogen receptors, ERα and ERβ, with ERα the predominant receptor type [7, 8, 10, 11, 17]. The accumulation of MDSCs is a complex process involving expansion of immature myeloid cells and pathologic activation and conversion of immature cells to MDSCs. Mechanistically, E2 signaling via JAK/STAT pathways may accelerate progression of E2-responsive and -unresponsive tumors by driving the expansion of MDSCs and enhancing their immunosuppressive activity in vivo as reported here and in previous work [7]. In contrast, blockade of E2 action appears to delay tumor progression due to a decrease in MDSC numbers and immunosuppressive activity that promotes T-cell-dependent antitumor immunity. Our findings suggest that antiestrogens particularly when administered in combination with anti-PD-L1 antibodies act to inhibit BC progression in part by blocking the expansion and mobilization of MDSCs that would otherwise promote tumor immune tolerance. In addition, emerging findings show that serine/threonine protein kinase casein kinase 2 that is overexpressed in BC plays a critical role in myeloid cell differentiation. Importantly, inhibition of casein kinase 2 disrupts the myeloid cell differentiation in BCs and enhances the efficacy of immunotherapy in mice [68]. This report is relevant to the present study because ERα signaling is known to activate transcription of casein kinase 2 [69], and ER antagonists block this action.
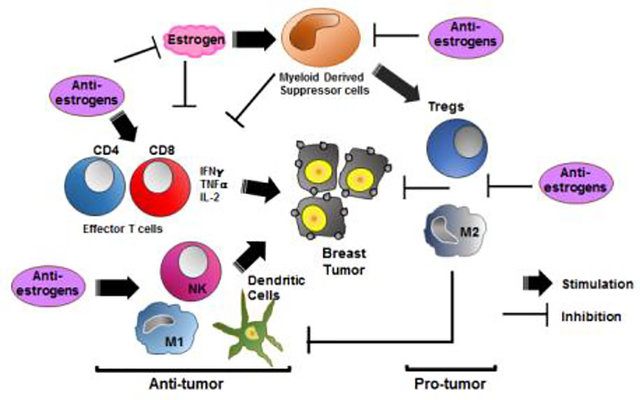
In general, MDSCs are not present in healthy individuals but occur in pathologic states associated with chronic inflammation and cancer [17]. For example, BC biopsies from patients with residual disease after chemotherapy contain relatively high levels of infiltrating myeloid-derived cells [16]. Of note, these mechanisms may also be important during pregnancy, where E2 may drive the expansion/activation of MDSCs to promote maternal-fetal immune tolerance [70, 71]. Importantly, the current findings provide evidence in preclinical human and murine models that blockade of E2 signaling acts to inhibit the expansion of MDSCs that are major contributors to pathologic myelopoiesis and immune tolerance in BC [7, 17]. In addition, ovariectomized mice with E2 depletion have significantly reduced progression of murine E2-insensitive TNBCs when grown as implants in syngeneic immune-intact mice. These results are consistent with earlier reports on the crucial role of MDSCs and TILs on modulating antitumor immunity [7]. Antitumor immunity includes several functional steps required for an immune response to eliminate tumors, such as blockade of immunosuppression, promotion of immune infiltration, activation of antigen-presenting cells and enhancement of effector cell activity [72]. The presence of TILs in the TME is predictive of patient survival. Several types of CD45+ leukocytes infiltrate the TME including CD4+ and CD8+ T-cells identified by specific phenotypic markers. It is recognized that effective antitumor immune responses require the involvement of both CD4+ and CD8+ T cells, with CD4+ T cells critical for priming of tumor-specific CD8+ T cells and for the secondary expansion and memory of CD8+ T cells [73]. However, CD4+FoxP3+ Treg cell-induced immune suppression represents a major obstacle for successful antitumor immunity. Accordingly, our data show that antiestrogens stimulate increments in the levels of effector and effector memory CD8+ and CD4+ T cells, while simultaneously suppressing the levels of immunosuppressive CD4+FoxP3+Treg cells. Furthermore, MDSCs are reported in turn to suppress antitumor activities of effector and memory effector CD8+ T-cells in vivo [17] and other natural immune cells such as macrophages and dendritic cells [70, 74], actions that appear to be reversed on treatment with antiestrogens combined with ICIs in murine models in vivo. As suggested from our findings, cytokine secretion modulated by antiestrogen therapy may also play a role as functional chemoattractants for selected immune cells. Hence, the current data provide evidence that beneficial antitumor effects occur on treatment of murine TNBCs with antiestrogens combined with ICIs in syngeneic, immune-intact mice, including promotion of effector and memory effector T-cells in the TME and modulation of macrophage and dendritic cell subsets. Thus, SERDs that enhance and/or maintain the activation status of effector T-cells may be used in dual therapies to enhance the effects of ICIs. A schematic representation of postulated effects of E2 and antiestrogen signaling on immune cells in the TME is shown in Fig. 12.
Fig. 12.

Estrogen (E2) (left panel) orchestrates a number of effects on immune cells in the TME. Evidence suggests that E2 promotes tumor immune tolerance through inhibition of CD8+ and CD4+ T cell effector responses, as well as antigen-presenting cells such as M1 macrophages and dendritic cells (DC). In addition, E2 signaling also stimulates suppressive actions of MDSC that can increase Tregs and M2 macrophages for tumor-promotion. In contrast, antiestrogen therapy with SERDs (right panel), particularly when used in combination with immune checkpoint inhibitors, helps to reverse the several actions of E2 and may represent a novel option in combination with immune checkpoint inhibitors to overcome an immunosuppressive BC microenvironment and stimulate more effective anti-tumor responses.
Tumor mutational burden (TMB) and the expression of immune checkpoints such as PD-L1 also play an important role in determining tumor sensitivity to ICIs [4, 13, 75]. Reduced TMB and low expression of PD-L1 may be important factors that explain the relative resistance of most BCs to ICI therapies [4, 14]. In this regard, recent reports indicate that immunotherapeutic target expression on BCs such as α-lactalbumin, a lactation protein negatively regulated by E2, can be amplified several-fold by antiestrogen therapy and thereby potentially enhance the efficacy of ICIs if combined with antiestrogens [76]. In addition, estrogens are also found to modulate the expression of PD-L1 in endometrial tissues [77, 78], in immune cells from reproductive tract and in ER-positive BC cells in vitro [79, 80]. The latter indicates that E2 may upregulate PD-L1 expression in ERα-positive BC cells to potentially suppress immune functions of T-cells in the TME and drive cancer progression. Of note, only 19.4% of patients with ER-positive/HER2-negative BCs were found to be PD-L1 positive in recent clinical trials, while 58.6% of TNBC patients screened in trials were PD-L1 positive [14, 81]. This difference in PD-L1 expression may account in part for a corresponding difference in clinical responses to ICI treatment. These reports raise the possibility of using antiestrogens as a priming approach to reverse immune-resistant ‘cold’ BCs to immune-sensitive ‘hot’ tumors more likely to respond to ICIs.
The current results also have implications for understanding potential gender-and/or age-dependent differences in tumor initiation and malignant progression. Humans show strong sex differences in immunity to infection and autoimmunity, suggesting sex hormones play a role in regulating immune responses. Indeed, receptors for E2 regulate cells and pathways in the innate and adaptive immune system, as well as immune cell development [82] and T cell functions [11, 79].
We note that ATP-competitive inhibitors of cyclin-dependent kinases 4/6 (CDK 4/6) such as abemaciclib were also reported to enhance the action of ICIs. The mechanism for this effect appears to involve modulation of T-cell activation and down-regulation of immunosuppressive myeloid populations [83]. This action may be dependent in part on the activity of E2, since E2 is well-known to stimulate expression/activity of cyclin D which is a requisite partner of CDK 4/6 to induce hyper-phosphorylation of Rb, thereby promoting cell proliferation and regulation of the cell cycle [84, 85].
Results of this translational research indicate that SERDs with strong antiestrogen activity such as JD128 and fulvestrant and potentially other antiestrogens [86–89] can augment the action of immune checkpoint inhibitors to inhibit BC progression. This work provides a preclinical rationale for considering treatment combinations and schedules that include antiestrogens. Thus, use of antiestrogens together with ICIs could lead to timely introduction of this dual treatment strategy in both ER-positive and potentially ER-negative or treatment-resistant breast cancers, thus significantly expanding the application and life-extending benefits of these drugs in the clinic to promote patient survival.
Supplementary Material
Highlights.
Select 11β-aryloxy-estradiol analogs downregulate ERα and are potent antiestrogens
New antiestrogens (AE) block ER+ breast tumor progression in vitro and in vivo
Estrogen depletion by ovariectomy reduces progression of murine TNBC in vivo
AEs alter immune cells in the tumor microenvironment to reduce immune tolerance
AEs augment anti-PD-L1 checkpoint inhibitors to block tumor progression in vivo
Acknowledgments.
This work was funded by the Tower Cancer Research Foundation-Jessica M. Berman Breast Cancer Fund, NIH/NCI U54 CA143930 Charles Drew University School of Medicine-UCLA Jonsson Cancer Center Partnership, California Breast Cancer Research Program and a CDMRP DOD BCRP Level 2 Award. We thank Dr. Hermes J. Garbán for thoughtful discussions, Dr. Antoni Ribas for use of research resources for this project, Mr. Colin Sterling, Jr. for laboratory contributions and Dr. Dinesh Rao for guidance in hematopathology issues.
Footnotes
Conflict of Interest. RJP has consulted previously with Genentech, Pfizer and Astra-Zeneca Pharmaceuticals. BCA has consulted with Advarro IBC and Kite Pharma.
References Cited
- [1].Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials., Lancet 351(9114) (1998) 1451–1467. [PubMed] [Google Scholar]
- [2].Hurvitz SA, Pietras RJ, Rational management of endocrine resistance in breast cancer: a comprehensive review of estrogen receptor biology, treatment options, and future directions, Cancer 113(9) (2008) 2385–2397. [DOI] [PubMed] [Google Scholar]
- [3].Osborne CK, Schiff R, Mechanisms of endocrine resistance in breast cancer, Annu Rev Med 62 (2011) 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, Kuter I, Nanda R, Cassier PA, Delord JP, Gordon MS, ElGabry E, Chang CW, Sarkar I, Grossman W, O’Hear C, Fasso M, Molinero L, Schmid P, Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study, JAMA Oncol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, Investigators IMT, Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer, N Engl J Med 379(22) (2018) 2108–2121. [DOI] [PubMed] [Google Scholar]
- [6].Jiang X, Shapiro DJ, The immune system and inflammation in breast cancer, Mol Cell Endocrinol 382(1) (2014) 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Svoronos N, Perales-Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, Nguyen JM, Curiel TJ, Cadungog MG, Singhal S, Eruslanov EB, Zhang P, Tchou J, Zhang R, Conejo-Garcia JR, Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells, Cancer Discov 7(1) (2017) 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Welte T, Zhang XH, Rosen JM, Repurposing Antiestrogens for Tumor Immunotherapy, Cancer Discov 7(1) (2017) 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moulton VR, Sex Hormones in Acquired Immunity and Autoimmune Disease, Front Immunol 9 (2018) 2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lambert KC, Curran EM, Judy BM, Milligan GN, Lubahn DB, Estes DM, Estrogen receptor alpha (ERalpha) deficiency in macrophages results in increased stimulation of CD4+ T cells while 17beta-estradiol acts through ERalpha to increase IL-4 and GATA-3 expression in CD4+ T cells independent of antigen presentation, J Immunol 175(9) (2005) 5716–5723. [DOI] [PubMed] [Google Scholar]
- [11].Mohammad I, Starskaia I, Nagy T, Guo J, Yatkin E, Vaananen K, Watford WT, Chen Z, Estrogen receptor alpha contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation, Sci Signal 11(526) (2018). [DOI] [PubMed] [Google Scholar]
- [12].Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, Siemens DR, Koti M, Craig AW, Graham CH, Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis, Oncotarget 7(9) (2016) 10557–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade, Science 359(6382) (2018) 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Tourneau CL, van Brummelen EMJ, Varga A, Salgado R, Loi S, Saraf S, Pietrangelo D, Karantza V, Tan AR, Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer, Clin Cancer Res 24(12) (2018) 2804–2811. [DOI] [PubMed] [Google Scholar]
- [15].Ali HR, Glont SE, Blows FM, Provenzano E, Dawson SJ, Liu B, Hiller L, Dunn J, Poole CJ, Bowden S, Earl HM, Pharoah PD, Caldas C, PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes, Ann Oncol 26(7) (2015) 1488–1493. [DOI] [PubMed] [Google Scholar]
- [16].Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM, Leukocyte composition of human breast cancer, Proc Natl Acad Sci U S A 109(8) (2012) 2796–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gabrilovich DI, Myeloid-Derived Suppressor Cells, Cancer Immunol Res 5(1) (2017) 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boonyaratanakornkit V, Hamilton N, Marquez-Garban DC, Pateetin P, McGowan EM, Pietras RJ, Extranuclear signaling by sex steroid receptors and clinical implications in breast cancer, Mol Cell Endocrinol 466 (2018) 51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Group EBCTC, Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials, Lancet 365(9472) (2005) 1687–1717. [DOI] [PubMed] [Google Scholar]
- [20].Robertson JF, Lindemann J, Garnett S, Anderson E, Nicholson RI, Kuter I, Gee JM, A good drug made better: the fulvestrant dose-response story, Clin Breast Cancer 14(6) (2014) 381–389. [DOI] [PubMed] [Google Scholar]
- [21].Howell A, Cuzick J, Baum M, Buzdar A, Dowsett M, Forbes JF, Hoctin-Boes G, Houghton J, Locker GY, Tobias JS, Group AT, Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer, Lancet 365(9453) (2005) 60–62. [DOI] [PubMed] [Google Scholar]
- [22].Pietras RJ, Marquez-Garban DC, Membrane-associated estrogen receptor signaling pathways in human cancers, Clin Cancer Res 13(16) (2007) 4672–4676. [DOI] [PubMed] [Google Scholar]
- [23].Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S, ESR1 ligand-binding domain mutations in hormone-resistant breast cancer, Nat Genet 45(12) (2013) 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA, An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer, Cancer Res 57(7) (1997) 1244–1249. [PubMed] [Google Scholar]
- [25].Kieser KJ, Kim DW, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA, Characterization of the pharmacophore properties of novel selective estrogen receptor downregulators (SERDs), J Med Chem 53(8) (2010) 3320–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wakeling AE, O’Connor KM, Newboult E, Comparison of the biological effects of tamoxifen and a new antioestrogen (LY 117018) on the immature rat uterus, J Endocrinol 99(3) (1983) 447–453. [DOI] [PubMed] [Google Scholar]
- [27].Wijayaratne AL, McDonnell DP, The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators, J Biol Chem 276(38) (2001) 35684–35692. [DOI] [PubMed] [Google Scholar]
- [28].van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, Brown M, Schroder CP, de Vries EF, Hospers GA, Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer, Cancer Discov 5(1) (2015) 72–81. [DOI] [PubMed] [Google Scholar]
- [29].Evaluating an ER Degrader for Breast Cancer., Cancer Discov 5(7) (2015) OF15. [DOI] [PubMed] [Google Scholar]
- [30].Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, Douglas K, Sensintaffar J, Lu N, Lee KJ, Aparicio A, Kaufman J, Qian J, Shao G, Prudente R, Moon MJ, Joseph JD, Darimont B, Brigham D, Grillot K, Heyman R, Rix PJ, Hager JH, Smith ND, Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts, J Med Chem 58(12) (2015) 4888–4904. [DOI] [PubMed] [Google Scholar]
- [31].Kurti L, Czako B, Corey EJ, A short, scalable synthesis of the carbocyclic core of the anti-angiogenic cortistatins from (+)-estrone by B-ring expansion, Org Lett 10(22) (2008) 5247–5250. [DOI] [PubMed] [Google Scholar]
- [32].Labaree DC, Zhang JX, Harris HA, O’Connor C, Reynolds TY, Hochberg RB, Synthesis and evaluation of B-, C-, and D-ring-substituted estradiol carboxylic acid esters as locally active estrogens, J Med Chem 46(10) (2003) 1886–1904. [DOI] [PubMed] [Google Scholar]
- [33].E. G Lim C, Perrault WR, Pearlman BA, An environmentally friendly and cost effective synthesis of estradiol featuring two novel reagents: Si(0)/KF and PMHS/hexamethyldisiloxane/pTSA, Tetrahedron Letters 47(36) (2006) 6417–6420. [Google Scholar]
- [34].F. R Tedesco R, Napolitano E, Novel Stereoselective Synthesis of 11.beta.-Carbon-Substituted Estradiol Derivatives, J. Org. Chem 60(16) (1995) 5316–5318. [Google Scholar]
- [35].Marquez DC, Chen HW, Curran EM, Welshons WV, Pietras RJ, Estrogen receptors in membrane lipid rafts and signal transduction in breast cancer, Mol Cell Endocrinol 246(1–2) (2006) 91–100. [DOI] [PubMed] [Google Scholar]
- [36].Marquez DC, Pietras RJ, Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells, Oncogene 20(39) (2001) 5420–5430. [DOI] [PubMed] [Google Scholar]
- [37].Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ, HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells, Oncogene 10(12) (1995) 2435–2446. [PubMed] [Google Scholar]
- [38].Berstein LM, Yue W, Wang JP, Santen RJ, Isolated and combined action of tamoxifen and metformin in wild-type, tamoxifen-resistant, and estrogen-deprived MCF-7 cells, Breast Cancer Res Treat 128(1) (2011) 109–117. [DOI] [PubMed] [Google Scholar]
- [39].Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI, Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells, Endocrinology 144(3) (2003) 1032–1044. [DOI] [PubMed] [Google Scholar]
- [40].Schroder AL, Pelch KE, Nagel SC, Estrogen modulates expression of putative housekeeping genes in the mouse uterus, Endocrine 35(2) (2009) 211–219. [DOI] [PubMed] [Google Scholar]
- [41].Williams D, Gorski J, Preparation and characterization of free cell suspensions from the immature rat uterus, Biochemistry 12(2) (1973) 297–306. [DOI] [PubMed] [Google Scholar]
- [42].Shanle EK, Hawse JR, Xu W, Generation of stable reporter breast cancer cell lines for the identification of ER subtype selective ligands, Biochem Pharmacol 82(12) (2011) 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wilson VS, Bobseine K, Gray LE Jr., Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists, Toxicol Sci 81(1) (2004) 69–77. [DOI] [PubMed] [Google Scholar]
- [44].Weinberg OK, Marquez-Garban DC, Fishbein MC, Goodglick L, Garban HJ, Dubinett SM, Pietras RJ, Aromatase inhibitors in human lung cancer therapy, Cancer Res 65(24) (2005) 11287–11291. [DOI] [PubMed] [Google Scholar]
- [45].Kimball AK, Oko LM, Bullock BL, Nemenoff RA, van Dyk LF, Clambey ET, A Beginner’s Guide to Analyzing and Visualizing Mass Cytometry Data, J Immunol 200(1) (2018) 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Samusik N, Good Z, Spitzer MH, Davis KL, Nolan GP, Automated mapping of phenotype space with single-cell data, Nat Methods 13(6) (2016) 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, Chen J, Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline, PLoS Comput Biol 12(9) (2016) e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, Allison JP, Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade, Cell 170(6) (2017) 1120–1133 e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Comin-Anduix B, Sazegar H, Chodon T, Matsunaga D, Jalil J, von Euw E, Escuin-Ordinas H, Balderas R, Chmielowski B, Gomez-Navarro J, Koya RC, Ribas A, Modulation of cell signaling networks after CTLA4 blockade in patients with metastatic melanoma, PLoS One 5(9) (2010) e12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, Ribas A, Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma, Sci Transl Med 7(279) (2015) 279ra241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].M.-G. D Bert B, Deng G, Diers E, Jung ME, Pietras RJ, New Estrogen Receptor Down-regulators to Treat Human Breast Cancer, Proceedings of the American Association for the Advancement of Science, Pacific Division 31(Part I) (2016) 110. [Google Scholar]
- [52].N. L Claussner A, Nique F, Philibert D, Teutsch G, Van de Velde P., 11 beta-amidoalkyl estradiols, a new series of pure antiestrogens., J Steroid Biochem Mol Biol 41(3–8) (1992) 609–614. [DOI] [PubMed] [Google Scholar]
- [53].H. E Hanson RN, Hendricks JA, Labaree D, Hochberg RB, Synthesis and evaluation of 11β-(4-substituted phenyl) estradiol analogs: transition from estrogen receptor agonists to antagonists., Bioorg Med Chem 20(12) (2012) 3768–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Srinivasan S, Nwachukwu JC, Bruno NE, Dharmarajan V, Goswami D, Kastrati I, Novick S, Nowak J, Cavett V, Zhou HB, Boonmuen N, Zhao Y, Min J, Frasor J, Katzenellenbogen BS, Griffin PR, Katzenellenbogen JA, Nettles KW, Corrigendum: Full antagonism of the estrogen receptor without a prototypical ligand side chain, Nat Chem Biol 13(6) (2017) 691. [DOI] [PubMed] [Google Scholar]
- [55].K. T Nose A, Studies of Reduction with the Sodium Borohydride-Transition Metal Boride System. I.: Reduction of Nitro and the Other Functional Groups with the Sodium Borohydride-Nickel Boride System., Chem. Pharm. Bull(36) (1988) 1529–1533. [Google Scholar]
- [56].Osby JO GB, Rapid and efficient reduction of aliphatic nitro compounds to amines., Tetrahedron Lett(26) (1985) 6413–6416. [Google Scholar]
- [57].Ostroumov D, Fekete-Drimusz N, Saborowski M, Kuhnel F, Woller N, CD4 and CD8 T lymphocyte interplay in controlling tumor growth, Cell Mol Life Sci 75(4) (2018) 689–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF, Understanding the tumor immune microenvironment (TIME) for effective therapy, Nat Med 24(5) (2018) 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Disis ML, Immune regulation of cancer, J Clin Oncol 28(29) (2010) 4531–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dai R, Edwards MR, Heid B, Ahmed SA, 17beta-Estradiol and 17alpha-Ethinyl Estradiol Exhibit Immunologic and Epigenetic Regulatory Effects in NZB/WF1 Female Mice, Endocrinology 160(1) (2019) 101–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sakaguchi S, Yamaguchi T, Nomura T, Ono M, Regulatory T cells and immune tolerance, Cell 133(5) (2008) 775–787. [DOI] [PubMed] [Google Scholar]
- [62].Chang LY, Lin YC, Kang CW, Hsu CY, Chu YY, Huang CT, Day YJ, Chen TC, Yeh CT, Lin CY, The indispensable role of CCR5 for in vivo suppressor function of tumor-derived CD103+ effector/memory regulatory T cells, J Immunol 189(2) (2012) 567–574. [DOI] [PubMed] [Google Scholar]
- [63].Schmidt SV, Nino-Castro AC, Schultze JL, Regulatory dendritic cells: there is more than just immune activation, Front Immunol 3 (2012) 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sica A, Mantovani A, Macrophage plasticity and polarization: in vivo veritas, J Clin Invest 122(3) (2012) 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Martinez FO, Gordon S, The M1 and M2 paradigm of macrophage activation: time for reassessment, F1000Prime Rep 6 (2014) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Capone I, Marchetti P, Ascierto PA, Malorni W, Gabriele L, Sexual Dimorphism of Immune Responses: A New Perspective in Cancer Immunotherapy, Front Immunol 9 (2018) 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mirandola L, Wade R, Verma R, Pena C, Hosiriluck N, Figueroa JA, Cobos E, Jenkins MR, Chiriva-Internati M, Sex-driven differences in immunological responses: challenges and opportunities for the immunotherapies of the third millennium, Int Rev Immunol 34(2) (2015) 134–142. [DOI] [PubMed] [Google Scholar]
- [68].Hashimoto A, Gao C, Mastio J, Kossenkov A, Abrams SI, Purandare AV, Desilva H, Wee S, Hunt J, Jure-Kunkel M, Gabrilovich DI, Inhibition of Casein Kinase 2 Disrupts Differentiation of Myeloid Cells in Cancer and Enhances the Efficacy of Immunotherapy in Mice, Cancer Res 78(19) (2018) 5644–5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Das N, Datta N, Chatterjee U, Ghosh MK, Estrogen receptor alpha transcriptionally activates casein kinase 2 alpha: A pivotal regulator of promyelocytic leukaemia protein (PML) and AKT in oncogenesis, Cell Signal 28(6) (2016) 675–687. [DOI] [PubMed] [Google Scholar]
- [70].Ostrand-Rosenberg S, Sinha P, Figley C, Long R, Park D, Carter D, Clements VK, Frontline Science: Myeloid-derived suppressor cells (MDSCs) facilitate maternal-fetal tolerance in mice, J Leukoc Biol 101(5) (2017) 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pan T, Liu Y, Zhong LM, Shi MH, Duan XB, Wu K, Yang Q, Liu C, Wei JY, Ma XR, Shi K, Zhang H, Zhou J, Myeloid-derived suppressor cells are essential for maintaining feto-maternal immunotolerance via STAT3 signaling in mice, J Leukoc Biol 100(3) (2016) 499–511. [DOI] [PubMed] [Google Scholar]
- [72].Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, Immunity 39(1) (2013) 1–10. [DOI] [PubMed] [Google Scholar]
- [73].Huang Y, Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Hunborg P, Varvares MA, Hoft DF, Hsueh EC, Peng G, CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome, Oncotarget 6(19) (2015) 17462–17478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK, Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression, Semin Cancer Biol 22(4) (2012) 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science 348(6230) (2015) 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jaini R, Loya MG, Eng C, Immunotherapeutic target expression on breast tumors can be amplified by hormone receptor antagonism: a novel strategy for enhancing efficacy of targeted immunotherapy, Oncotarget 8(20) (2017) 32536–32549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wu L, Lv C, Su Y, Li C, Zhang H, Zhao X, Li M, Expression of programed death-1 (PD-1) and its ligand PD-L1 is upregulated in endometriosis and promoted by 17beta-estradiol, Gynecol Endocrinol (2018) 1–6. [DOI] [PubMed] [Google Scholar]
- [78].Yang L, Huang F, Mei J, Wang X, Zhang Q, Wang H, Xi M, You Z, Posttranscriptional Control of PD-L1 Expression by 17beta-Estradiol via PI3K/Akt Signaling Pathway in ERalpha-Positive Cancer Cell Lines, Int J Gynecol Cancer 27(2) (2017) 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Polanczyk MJ, Hopke C, Vandenbark AA, Offner H, Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1), Int Immunol 19(3) (2007) 337–343. [DOI] [PubMed] [Google Scholar]
- [80].Shen Z, Rodriguez-Garcia M, Patel MV, Barr FD, Wira CR, Menopausal status influences the expression of programmed death (PD)-1 and its ligand PD-L1 on immune cells from the human female reproductive tract, Am J Reprod Immunol 76(2) (2016) 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L, Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study, J Clin Oncol 34(21) (2016) 2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kovats S, Estrogen receptors regulate innate immune cells and signaling pathways, Cell Immunol 294(2) (2015) 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Teh JLF, Aplin AE, Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment, Clin Cancer Res (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ, PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro, Breast Cancer Res 11(5) (2009) R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Musgrove EA, Sutherland RL, Cell cycle control by steroid hormones, Semin Cancer Biol 5(5) (1994) 381–389. [PubMed] [Google Scholar]
- [86].Guo S, Zhang C, Bratton M, Mottamal M, Liu J, Ma P, Zheng S, Zhong Q, Yang L, Wiese TE, Wu Y, Ellis MJ, Matossian M, Burow ME, Miele L, Houtman R, Wang G, ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models, Oncotarget 9(6) (2018) 6924–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, Dann SG, Arndt KT, Primeau T, Griffin E, Shao J, Crowder R, Lai JP, Norris JD, McDonnell DP, Li S, Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer, Clin Cancer Res 21(22) (2015) 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, Curwen JO, de Almeida C, Ballard P, Hulse M, Donald CS, Feron LJ, Karoutchi G, MacFaul P, Moss T, Norman RA, Pearson SE, Tonge M, Davies G, Walker GE, Wilson Z, Rowlinson R, Powell S, Sadler C, Richmond G, Ladd B, Pazolli E, Mazzola AM, D’Cruz C, De Savi C, AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models, Cancer Res 76(11) (2016) 3307–3318. [DOI] [PubMed] [Google Scholar]
- [89].Zhao L, Huang S, Mei S, Yang Z, Xu L, Zhou N, Yang Q, Shen Q, Wang W, Le X, Lau WB, Lau B, Wang X, Yi T, Zhao X, Wei Y, Warner M, Gustafsson JA, Zhou S, Pharmacological activation of estrogen receptor beta augments innate immunity to suppress cancer metastasis, Proc Natl Acad Sci U S A 115(16) (2018) E3673–E3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.