

Abstract
Background
Muscle wasting from chronic kidney disease (CKD) or from defective insulin signalling results in morbidity and, ultimately, mortality. We have identified an endogenous mediator of insulin resistance, signal regulatory protein alpha (SIRPα), which leads to cachexia in mice and is associated with cachexia in patients with CKD.
Methods
We assessed insulin signalling and mechanisms causing muscle atrophy plus white adipose tissue (WAT) metabolism in mouse models of CKD or acute diabetes (streptozotocin treatment). We then examined these factors in mice with global knockout (KO) of SIRPα and sought mediators of metabolic responses in muscle and adipose tissues of mice with either muscle‐specific or adipose tissue‐specific KO of SIRPα. Metabolic responses were confirmed in primary cultures of adipose cells.
Results
In mice with CKD, SIRPα expression was increased in WAT (three‐fold, P < 0.05), and this was associated with precursors of cachexia: ‘pathologic browning', thermogenesis, and a two‐fold activation of protein kinase A (P < 0.05 vs. control mice) plus loss of adipose tissue mass. In contrast, mice with SIRPα global KO and CKD or acute diabetes experienced improved insulin signalling and activation of pAkt plus ‘physiologic browning' of WAT. These mice avoided losses of muscle and adipose tissues and experienced a 31% improvement in survival (P < 0.05) than did wild‐type mice with CKD. In muscle‐specific SIRPα KO mice with CKD, we uncovered that serum SIRPα levels (P < 0.05) were suppressed and were associated with improved insulin signalling both in skeletal muscles and in WAT. These changes were accompanied by physiologic WAT browning. However, in adipose‐specific SIRPα KO mice with CKD, levels of serum SIRPα were increased over two‐fold (P < 0.05), while muscle losses were minimally inhibited. Clinical implications of SIRPα signalling are suggested by our findings that include increased SIRPα expression in muscle and adipose tissues (P < 0.05 vs. healthy controls) plus higher SIRPα levels in the serum of patients with CKD (2.4‐fold, P=0.000017 vs. healthy controls).
Conclusions
Our results show that SIRPα plays an important role as an anti‐insulin mediator regulating pathways to cachexia. In muscle‐specific SIRPα KO, changes in SIRPα serum levels seem to improve insulin signalling in muscle and WAT, suggesting crosstalk between muscle and adipose tissue. Therefore, targeting SIRPα may prevent cachexia in patients with CKD or acute diabetes.
Keywords: SIRPα, Chronic kidney disease, Diabetes, Insulin resistance, Browning, UCP1, Cachexia
1. Background
Losses of muscle and adipose tissues occur commonly in patients with chronic kidney disease (CKD) and are associated with increased morbidity and mortality. For example, results from a National Institutes of Health (NIH)‐supported, multicentre, clinical trial of haemodialysis patients carried out over 2.5 years revealed that haemodialysis increased mortality by >46%.1 The authors concluded that decreases in muscle mass and adipocytes were linked to anthropometric evidence of cachexia with low values of body mass index, triceps skin‐fold thickness, and mid‐arm muscle circumference.1
These results prompted us to investigate mechanisms that could explain how CKD stimulates losses of both muscle and adipose tissues. In rodent models of CKD, several mechanisms have been linked to the development of protein catabolism, namely, inflammation, metabolic acidosis, insulin resistance, glucocorticoids, and myostatin (MSTN) expression.2, 3, 4, 5, 6, 7, 8 These catabolic conditions activate caspase‐3 and the ubiquitin–proteasome system (UPS), which develop loss of muscle proteins.9 In contrast, mechanisms causing loss of adipose tissues in CKD are not so well characterized. This prompted us to identify mechanisms responsible for losses of lipid stores in CKD because patients with advanced CKD lose adipose as well as protein stores.1 We also studied mechanisms causing loss of both adipocytes and muscle because they could lead to the design of therapies for combating the development of cachexia.
As insulin resistance is detectable early in the course of CKD and is associated with losses of muscle and adipose tissues, we investigated how impaired insulin signalling could affect CKD‐induced cachexia.10 We have identified a post‐insulin receptor mechanism that underlies the development of insulin resistance: it arises from dephosphorylation of phospho‐tyrosines in insulin receptors and insulin receptor substrate 1 (IRS1).11 In cultured muscle cells, dephosphorylation of tyrosines in the insulin receptor and IRS1 activates specific E3 ubiquitin ligases of the UPS resulting in IRS1 degradation and interruption of intracellular insulin signalling.2 In pursuing these findings, we identified a novel mediator of insulin resistance in mice with CKD, the signal regulatory protein alpha (SIRPα), a membrane glycoprotein that interacts with SHP‐1 and SHP‐2, tyrosine phosphatases, dephosphorylating insulin receptor and IRS1.12, 13, 14 This disruption of insulin signalling accelerates muscle proteolysis.12 In the present experiments, we evaluated the influence of SIRPα in vivo as a mediator of catabolic responses (i.e. sympathetic activation) that reduce adipose and muscle tissues and, hence, promote the development of cachexia.
2. Methods
2.1. Reagents and antibodies
Mouse Intact (1‐84) parathyroid hormone (PTH) enzyme‐linked immunosorbent assay (ELISA) assay kit was purchased from ALPCO (Salem, NH), calcium assay kit was from Cayman (Ann Arbor, MI), protein kinase A (PKA) kinase activity kit was from Enzo (Farmingdale, NY), Ultrasensitive Mouse Insulin ELISA Kit was from Crystal Chem (Elk Grove Village, IL), and https://www.promega.com/resources/protocols/technical-bulletins/0/tyrosine-phosphatase-assay-system-protocol/ was from Promega (Madison, WI). Mouse SIRPα ELISA kit was from EIAab Science Co. Ltd (Wuhan, China) and Human SIRPα ELISA kit from LifeSpan Biosciences (Seattle, WA). RNeasy Lipid Tissue Mini Kit, RNeasy Fibrous Tissue Mini Kit, and RNase‐Free DNase Set were from Qiagen (Valencia, CA); iScript cDNA Synthesis Kit was from Bio‐Rad (Hercules, CA). Phosphatase inhibitor and protease inhibitor were from Roche (Indianapolis, IN). TRIzol was from Life Technologies (Carlsbad, CA); RIPA lysis and extraction buffer was from G‐Biosciences (Louis, MO); and Pierce BCA Protein Assay was from Thermo Fisher Scientific (Rockford, IL). Streptozotocin (STZ), noradrenaline bitartrate (NE), forskolin (FSK), and insulin were from Sigma‐Aldrich (St. Louis, MO); and 666‐15 was from EMD Millipore (Burlington, MA), and H89 and LY294002 were from Tocris (Minneapolis, MN). Green fluorescent protein (GFP) plasmid was from Lonza (Allendale, NJ); SIRPα plasmid cDNA was from Open Biosystems (Lafayette, CO); DMEM/F12 and foetal bovine serum (FBS) were from Cellgro Mediatech (Manassas, VA). The antibodies against SIRPα (#13379), p‐Akt (Ser473, #4060), Akt (#2920), PPARγ (#2443), UCP‐1 (#14670), phosphorylated‐hormone sensitive lipase (pHSL; #4126), phosphorylated cAMP response element‐binding protein (pCREB; #9198), phosphorylated PKA (pPKA; #9624), GLUT4 (#2213), and GAPDH (#5174) were from Cell Signaling Technology (Beverly, MA). Antibody against phosphotyrosine (4G10, #05‐321) was from Millipore (Temecula, CA). IRS‐1 (sc‐7200) was from Santa Cruz Biotechnology (Dallas, TX). The antibody against UCP1 (#ab10983) for immunohistochemical staining was from Abcam (Cambridge, MA).
2.2. Animal studies
We studied 8‐ to 10‐week‐old male mice in protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine (BCM). Mice were maintained in 12 h light/dark cycles (6 a.m. to 6 p.m.) at 24°C and fed diets of standard or high‐protein (40%) rodent chow. C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). SIRPα Mt mice (Exons 7 and 8 were replaced with a neomycin selection cassette) sperm were obtained from Riken (Saitama, Japan) and backcrossed over five generations on C57BL/6 background. The BCM Genetically Engineered Mouse Core obtained the mouse from cryopreserved sperm. SIRPαfl/fl (Tm1C) mice were obtained in conjunction with the BaSH EUCOMM.15 Skeletal muscle‐specific SIRPα knockout (KO; mSIRPα−/−) or adipose‐specific mice (AD‐SIRPα−/−) with deletion of Exons 3 and 4 were generated using a Cre [muscle creatine kinase‐Cre or Adipoq‐Cre mice, respectively, from Jackson Laboratory (Bar Harbor, ME, USA)] recombinase:loxP system as previously described15; both are on C57BL/6 background. These mice appear to have grossly normal phenotype.
2.2.1. Chronic kidney disease model
To create mice with CKD, male C57BL/6 and SIRPα Mt mice at 8 weeks of age were anaesthetized, and a subtotal nephrectomy was performed.5 Wild‐type (WT) vs. SIRPα Mt ± CKD mice were provided either standard rodent chow or SIRPαfl/fl vs. mSIRPα−/− or AD‐SIRPα−/− ± CKD were treated with high‐protein diet (40%) to create mice with blood urea nitrogen (BUN) and PTH values similar to those of patients with advanced CKD. BUN was measured in blood collected from the tail vein. At post‐surgery Weeks 14 to 16, blood from anaesthetized mice was removed by perfusing phosphate‐buffered saline (PBS) into the left ventricle. The skeletal muscles [gastrocnemius, tibialis anterior (TA), extensor digitorum longus (EDL), and soleus] plus abdominal adipose tissues (inguinal and visceral adipose tissues) and other organs were removed, weighed, and immediately frozen in liquid nitrogen and maintained at −80°C until examined.
Lipids from livers were isolated from frozen tissues by extracting with CHCl3:CH3O:H2O, followed by drying of the organic phase under N2. Serum triglycerides and cholesterol were assessed by kit from Abcam (Cambridge, MA). https://www.promega.com/resources/protocols/technical-bulletins/0/tyrosine-phosphatase-assay-system-protocol/
2.2.2. Glucose and insulin tolerance tests
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed at 9 and 10 weeks post‐surgery using an Advanced Glucose Meter (https://www.google.com/search?q=Woonsocket+Rhode+Island&stick=H4sIAAAAAAAAAOPgE-LUz9U3MCqqMDRW4gAx47MNk7W0spOt9POL0hPzMqsSSzLz81A4VhmpiSmFpYlFJalFxQAcktlNQwAAAA&sa=X&ved=0ahUKEwilzvuCu8nZAhVJbKwKHSIcCJUQmxMI8gEoATAZ). For GTT, mice fasted for 16 h were injected intraperitoneally with 1.5 g/kg glucose; and tail vein blood was collected at 0, 30, 60, 90, and 120 min intervals to assess blood glucose concentration. For ITT, mice fasted for 4 h were injected intraperitoneally with 0.75 units/kg insulin; and blood was collected at 0, 15, 30, 60, and 90 min intervals to measure blood glucose concentration.
2.2.3. Whole‐body energy metabolism test
At post‐surgery Week 11, lean and fat mass of individual mice were quantified using an X‐ray Imager (PIXImus Body Composition, Lunar Corp). Whole‐body energy metabolism of the mice was assessed in the metabolic cages at post‐surgery Week 12 using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments) in the Mouse Phenotyping Core at BCM. CLAMS cages were housed in temperature‐controlled environmental chambers at 23°C on a standard 12 h light/dark cycles for the duration of the study. Animals were monitored for 24 h, and during that time, food and water were provided ad libitum. Parameters monitored include VO2, VCO2, respiratory exchange ratio (RER), heat, physical activity, and food/water consumption.
2.2.4. Streptozotocin model
To induce acute diabetes, 12‐week‐old C57BL/6 and SIRPα Mt male mice were injected intraperitoneally with two doses of 150 mg/kg/day STZ in PBS.8 Control mice received the vehicle (PBS) only. Mice were housed and fed a normal chow for 8 days. Basal insulin level (Ultrasensitive Mouse Insulin ELISA Kit) and blood glucose were checked using serum of tail vein blood after 4 h of fasting at Day 7. The mice were harvested at Day 8.
2.2.5. Grip strength
Forelimb grip strength of CKD mice was assessed (at Week 12) and STZ mice (at Day 6). Each mouse was pulled backwards away in a straight, horizontal line to display peak force obtained by a transducer (Model DFS II; Chatillon).
2.2.6. Noradrenaline bitartrate treatment on mice
Mice received intraperitoneal injections of NE (1 mg/kg). Control mice received the vehicle. At the end of the treatment, all mice were euthanized and harvested following heart perfusion with PBS.
2.3. Primary cultures of white adipocyte
Inguinal fat pads were obtained from 5‐ to 6‐month‐old male mice. Inguinal fat tissue was dissected gently from the skin and washed twice in PBS. The fat tissue was minced into 1 to 3 mm2 pieces followed by digestion, then filtered through a 100 and then 40 μm cell strainer, and resuspended in preadipocyte medium (DMEM/F12 containing 10% FBS and 1% penicillin/streptomycin). The mixture was plated in the six‐well format. Inguinal cells were grown to confluency and then treated by induction medium (1.5 μg/mL insulin, 1 μM dexamethasone, 500 μM IBMX, and 1 μM rosiglitazone in the preadipocyte medium). Three days after induction, cultured cells were maintained in medium (1.5 μg/mL insulin in the preadipocyte medium). At Day 8, the cells were treated after 2 h serum starvation with PI3K inhibitor LY294002 (50 μM for 30 min or 6 h). Activation of PKA by NE (100 nM for 30 min or 6 h), H89, a PKA inhibitor (50 μM for 30 min or 6 h), and insulin (1 nM for 10 min). After treatment, protein was extracted from cells and stored at −80°C.
2.4. 3T3‐L1 cell treatment
3T3‐L1 cells was purchased from American Type Culture Collection (Manassas, VA) and cultured in preadipocyte media in 5% CO2 in a humidified incubator at 37°C. At 80% confluence, the cells were serum starved for 2 h and treated with pCREB activator FSK (10 μM for 24 h) or pCREB inhibitor, 666‐15 (50 nM for 24 h). Subsequently, cellular proteins were extracted and stored at −80°C. Electroporation was performed with the Neon Transfection System (Invitrogen, Carlsbad, CA). 3T3‐L1 cells (106) were electroporated with 3 μg GFP control or 3 μg SIRPα plasmid. The cultured cells were maintained in DMEM/F12 containing 10% FBS for 24 h.
2.5. Reverse transcription–quantitative polymerase chain reaction
Total RNA of fat and muscle was extracted using TRIzol and purified with RNeasy Lipid Tissue Mini Kit and RNeasy Fibrous Tissue Mini Kit, respectively. First‐strand cDNA was synthesized from 1 μg DNase‐treated total RNA using Reverse Transcription Supermix for reverse transcription–quantitative polymerase chain reaction (RT–qPCR). The mRNA levels were evaluated by qPCR using SYBR Green Supermix. Reactions of qPCR were performed in 96‐well format using a CFX96™ Real‐Time System (Bio‐Rad, Hercules, CA). The reaction volume was 10 μL, including 5 μL SYBR Green Supermix, 2 μL 2.5 μM primer, and 1 μL cDNA. The following thermal cycling profile was used: 95°C 3 min; 40 cycles of 95°C for 15 s, 60°C for 30s; followed by 55°C to 95°C increment for dissociation curve analysis. The relative mRNA levels were calculated using the comparative CT Method (Livak and Schmittgen, 2001) and normalized to GAPDH mRNA. Sequence information of mouse primers is as follows: GAPDH‐F: 5′‐TGTGATGGGTGTGAACCACGAGAA‐3′, R: 5′‐CATGAGCCCTTCCACAATGCCAAA‐3′; SIRPα‐F: 5′‐CTCTGTGGACGCCTGTAA‐3′, R: 5′‐GATGCTGCGTCGTTTGTT‐3′; IL‐6 F: 5′‐GAGGATACCACTCCCAACAGACC‐3′, R: 5′‐AAGTGCATCATCGTTGTTCATACA‐3′; MuRF1 F: 5′‐GGGTAAAGAAGAACACCAATG‐3′, R: 5′‐GAAGACACACTTCCCTATTG‐3′; Atrogin‐1 F: 5′‐CTGAAAGTTCTTGAAGACCAG‐3′, R: 5′‐GTGTGCATAAGGATGTGTAG‐3′; MSTN F: 5′‐CTATAAGACAACTTCTGCCAAG‐3′, R: 5′‐AGAAAGTCAGACTCTGTAGG‐3′; UCP1F: 5′‐CTTTTTCAAAGGGTTTGTGG‐3′, R: 5′‐CTTATGTGGTACAATCCACTG‐3′; PGC1α F: 5′‐CACATACAAGGGAGAATTGC‐3′, R: 5′‐TCCTCTTCAAGATCCTGTTAC‐3′; CIDEA F: 5′‐CTATAACAGAGAGCAGGGTC‐3′, R: 5′‐GTGTTAAGGAATCTGCTGAG‐3′; ATG12 F: 5′‐CTCTATATGAGTGTTTTGGCAG‐3′, R: 5′‐TTGATAGTAAGTCTCTTCCCAC‐3′; BECN1 F: 5′‐CAATAATTTCAGACTGGGTCG‐3′, R: 5′‐ATTTGTCTGTCAGAGACTCC‐3′; LC3 F: 5′‐TGAACAAAGAGTGGAAGATG‐3′, R: 5′‐GCCGTCTGATTATCTTGATG‐3′; P62 F: 5′‐AATGTGATCTGTGATGGTTG‐3′, R: 5′‐GAGAGAAGCTATCAGAGAGG‐3′. Sequence information of human primers is as follows: GAPDH‐F: 5′‐CTTTTGCGTCGCCAG‐3′, R: 5′‐TTGATGGCAACAATATCCAC‐3′; SIRPα F: 5′‐GAACGGAACATCTATATTGTGG‐3′, R: 5′‐CATGCAACCTTGTAGAAGAAG‐3′; UCP1 F: 5′‐ACAGCACCTAGTTTAGGAAG‐3′, R: 5′‐CTGTACGCATTATAAGTCCC‐3′; IL‐6 F: 5′‐GCAGAAAAAGGCAAAGAATC‐3′, R: 5′‐CTACATTTGCCGAAGAGC‐3′; MuRF1 F: 5′‐GACAGATGAGGAAGAGGAAG‐3′, R: 5′‐TCATTCATCCAGCTCCTTAC‐3′; Atrogin‐1 F: 5′‐AACTCAGTATTTCCACCAAG‐3′, R: 5′‐GAAGTCCAGTCTGTTGAAAG‐3′.
2.6. Western blots
We homogenized 100 mg fat tissue (or 30 mg muscle tissue) for 1 min in 300 μL cold RIPA buffer supplemented with protease and phosphatase inhibitor cocktails. The homogenates were incubated on ice for 10 min and then centrifuged (15,600 g) at 4°C for 15 min. The supernatants were used as whole cell lysates. Protein concentration was determined by Pierce BCA Protein Assay. An equal amount of protein (50–80 μg) was separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis in tris/glycine buffer, transferred to nitrocellulose blotting membrane, blocked for 20 min, and blotted with all primary antibodies diluted (1:1000) except GAPDH (1:2000) in blocking buffer overnight at 4°C. After 3 times of washing in TBS containing 0.1% Tween20, the membrane was incubated in secondary antibody diluted with TBS containing 0.1% Tween20 for 1 h and washed mentioned earlier above, and the protein was detected using the ChemiDoc MP Imaging System (Bio‐Rad, Hercules, CA). The proteins of interest were analysed using Image Lab 6.0 (Bio‐Rad, Hercules, CA), and the protein intensities were quantified using NIH ImageJ software. GAPDH was used as an internal standard unless otherwise specified to quantify western blot bands.
2.7. Section and staining
For https://vectorlabs.com/products/immunohistochemistry staining, tissues were fixed in 10% formalin, embedded in paraffin, and cut into 6 μm sections on slides. The slides were incubated with the 1:250 diluted anti‐UCP1 antibody and the 1:50 diluted anti‐SIRPα antibody. Frozen cryosections (8 μm) of TA from control and CKD mice were mounted on glass slides (Zhang et al., 2010). After the immunostaining with anti‐laminin, the myofiber sizes were analysed using NIS‐Elements Br 3.0 software (Nikon). For Oil Red O staining, adipocyte cells were washed with PBS and fixed in 4% paraformaldehyde for 1 h at room temperature. After being washed with PBS, the cells were stained with a 0.2% Oil Red O solution for 15 min. Images were processed using a Nikon 80i microscope (Melville, NY).
2.8. Human study
Biopsies of fat and muscle were obtained from healthy subjects during abdominal hernia surgeries. Serum samples from healthy controls were obtained from blood donors. Serum samples and biopsies of fat and muscle from CKD patients were obtained prior to peritoneal dialysis catheter placement. The rectus abdominis muscle and periumbilical fat were biopsied, and cDNA isolated or biopsy samples were frozen at −80°C or fixed in 10% formalin, embedded in paraffin, and stored until analysed. Informed consent was obtained and possible consequences of the studies were explained to all participants. The procedures were approved by the Ethics Committee of the Department of Internal Medicine of the University of Genoa, in accordance with the Declaration of Helsinki regarding ethics of human research.
2.9. Statistical analysis
Values are expressed as mean ± SEM. Significant analysis was performed using two‐tailed unpaired t‐test for single variables and for multiple variables by analysis of variance followed by Bonferroni test. P < 0.05 was considered statistically significant. GraphPad Prism software was used for statistical analysis.
3. Results
3.1. Signal regulatory protein alpha deletion prevents chronic kidney disease‐induced muscle protein wasting
In the present experiments, we explored mechanisms by which SIRPα contributes to the development of cachexia by characterizing WT mice with CKD. These mice had BUN values and body weight losses similar to those of responses that occur in patients with advanced CKD (Figure 1A and 1B). Mice with CKD lost lean body and muscle mass plus reduced grip strength vs. results from WT, sham‐operated, control mice (Figure 1C–E). In contrast, global KO of SIRPα mutant (Mt) mice with CKD exhibited improvements in body weight, lean body mass, and muscle mass despite a similar level of azotemia. The SIRPα Mt mice with CKD also had significantly stronger grip strength (P < 0.05) than WT mice with CKD (Figure 1D). The responses were confirmed when we analysed myofibers: there was a rightward shift of cross‐sectional areas of myofibers in TA muscles (Figure 1F). SIRPα absence also improved intracellular insulin signalling in muscles (Figure S1A) and there was no enhancement of the expression of IL‐6 or atrophy‐related genes (e.g. MuRF1, Atrogin‐1, and MSTN; Figure 1G). In contrast, WT mice with CKD developed a five‐fold increase in SIRPα expression and increased the expressions of atrophy‐related genes (Figure 1G). In the present experiments, these changes in muscle metabolism were associated with a 31% survival advantage of the SIRPα Mt mice with CKD vs. the survival of WT mice with CKD (Figure 1H).
Figure 1.
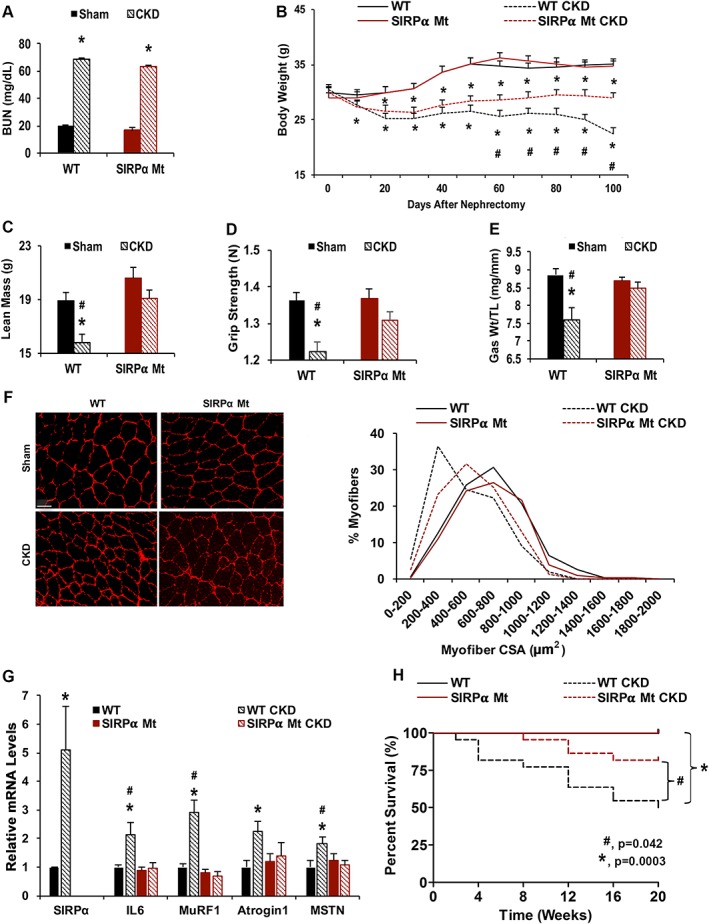
Absence of SIRPα suppresses CKD‐induced muscle wasting. At 12–16 weeks after subtotal nephrectomy (A–G: n = 4–6 mice/group): (A) blood urea nitrogen (BUN) and body weight (B) were measured. (C) Lean mass was measured by dual‐energy X‐ray absorptiometry (DEXA) and (D) grip strength in Newton (N) was determined. (E) Weight (Wt) of gastrocnemius (Gas) muscles was determined and normalized to tibia length (TL). (F) Cryosections of TA muscles were immunostained with anti‐laminin (red) to assess myofiber sizes (scale bar = 100 μm). Myofiber areas were measured, and the distribution of areas of 500 myofibers was assessed by a blinded observer. (G) mRNAs from gas skeletal muscles were determined by RT–qPCR. (A–G) Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, WT vs. SIRPα Mt mice. (H) Survival was calculated from the percentage of mice surviving at 2 weeks after creating CKD [n = 5–7 sham, n = 20–22 mice per CKD group, log‐rank (Mantel–Cox) test, P values illustrated]. See also Figure S1.
3.2. Signal regulatory protein alpha knockout improves chronic kidney disease‐impaired insulin signalling in adipose tissues
In WT mice, we confirmed that CKD reduces glucose and insulin tolerances (Figure 2A and 2B) and impairs the metabolism of adipose tissue. Specifically, in WT mice with CKD, epididymal white adipose tissue (eWAT; Figure 2C) and inguinal WAT (iWAT; Figure S2A) exhibited tyrosine dephosphorylation of IRS1. These responses were associated with impaired insulin signalling and decreased pAkt (Figures 2C and S2A). In contrast, SIRPα Mt mice had improved insulin and glucose tolerance vs. WT mice even when the mice had CKD (Figure 2A and 2B). Moreover, WAT of SIRPα Mt mice with CKD exhibited improved insulin signalling, with an increase in levels of pAkt plus phosphorylation of tyrosines in IRS1 (Figures 2C and S2A). The striking differences in responses to WT and SIRPα Mt mice included a significantly higher level of tyrosine phosphatase activities in WT mice with CKD (Figure 2D and 2E). While SIRPα Mt mice with CKD exhibited blunting of tyrosine phosphatase activity despite CKD (Figure 2D and 2E).
Figure 2.
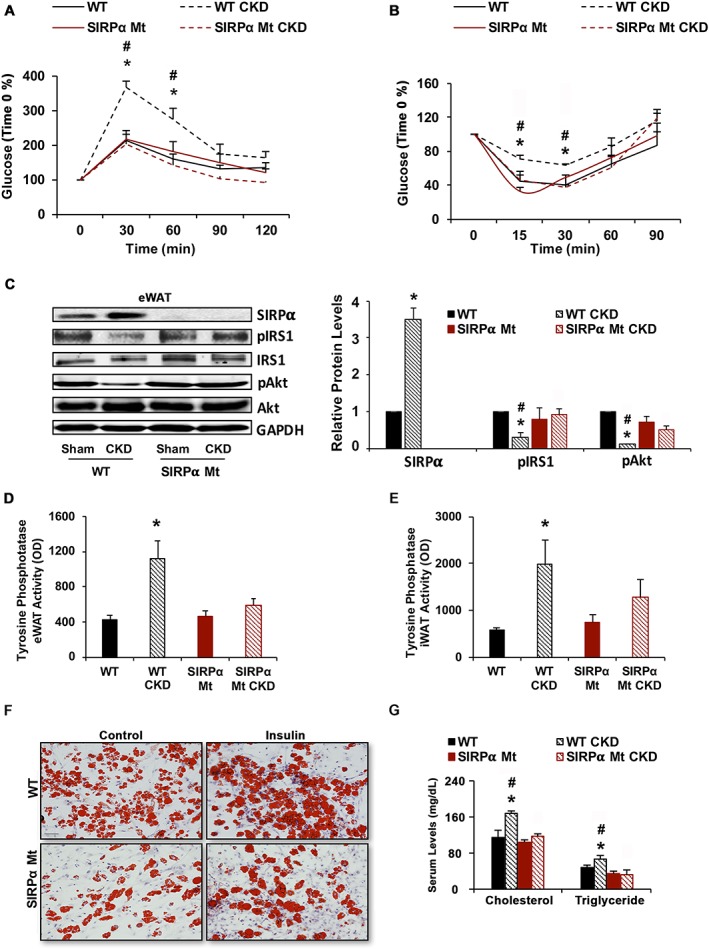
Suppressing SIRPα improves insulin signalling in white adipose tissue (WAT) and lipid deposition. Insulin sensitivity in mice after a 16 h fast was determined by (A) following intraperitoneal glucose (1.5 g/kg). (B) Intraperitoneal insulin (0.75 U/kg) tolerance was measured after a 4 h fast. (C) Protein lysates of epididymal white adipose tissue (eWAT) were immunoblotted to detect SIRPα, pIRS1, IRS1, and pAkt (Ser473), Akt and GAPDH (left panel) and the relative densities to GAPDH or pAkt to Akt ratios are shown (right panel). (D and E) Tyrosine phosphatase activity was measured in eWAT or inguinal white adipose tissue (iWAT). (F) Primary cultures of inguinal adipocytes from WT vs. SIRPα Mt mice were treated with or without insulin and stained for Oil Red O (scale bar = 100 μm). (G) Serum cholesterol and triglyceride levels were measured (A–G; n = 4–6 mice/group). Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, WT vs. SIRPα Mt mice. See also Figure S2.
We also studied primary cultures of white adipocytes obtained from WT or SIRPα Mt mice. Lipid accumulation in cells from SIRPα Mt mice was reduced compared with results from WT mice. Insulin administration stimulated lipid accumulation in WT adipocytes; this response was blunted in adipocytes of SIRPα Mt mice treated with insulin (Figure 2F). Besides these results from primary cultures of adipocytes, there was lipid accumulation in livers of WT mice with CKD vs. results from cells of SIRPα Mt mice with CKD (Figure S2B). In addition, serum levels of cholesterol and triglycerides in WT mice with CKD were increased vs. results from SIRPα Mt mice with CKD (Figure 2G). Thus, SIRPα elimination in mice with CKD prevents the development of insulin resistance and changes in lipid metabolism that occurs in patients with metabolic syndrome.
3.3. Suppressed signal regulatory protein alpha promotes white adipose tissue browning independently of parathyroid hormone
We hypothesized that the reduction in lipid accumulation in SIRPα Mt mice may be related to browning of WAT. Based on indirect calorimetry, VO2 consumption was higher in WT mice with CKD vs. SIRPα Mt mice with CKD (Figure S3A). In addition, the CO2 production, the RER, and heat generation were all significantly higher in WT mice with CKD vs. SIRPα Mt mice with CKD (Figure S3B–D). These responses were not due to differences in physical activity (Figure S3E).
It has been reported that WAT browning plays an important role in causing muscle protein wasting. Kir et al. noted that increased levels of PTH in WT CKD mice were associated with browning of WAT, and these in turn led to activation of muscle protein degradation.16 We evaluated the role of PTH in the mice we studied. We found that plasma levels of PTH were increased in WT mice with CKD but suppressed in SIRPα Mt mice with CKD (Figure 3A). These responses were not associated with changes in serum calcium (Figure 3B). However, in SIRPα Mt mice with or without CKD, both eWAT and iWAT underwent browning (Figure 3C and 3D). In SIRPα Mt mice, fat mass was preserved compared with values in WT mice with CKD (Figure 3E and 3F). Interestingly, WT mice with CKD or SIRPα Mt mice with or without CKD exhibited increases in thermogenic genes (e.g. UCP1, PGC1α, and CIDEA) (Figure 3G and 3H). Notably, mRNAs of these genes were associated with up‐regulation of UCP1 and PPARγ proteins in both eWAT or iWAT of WT mice with CKD. Similar responses occurred in SIRPα Mt mice with or without CKD (Figures 3I–L and S3F–G). Thus, browning of WAT isolated from SIRPα Mt mice with CKD was not stimulated by PTH and did not increase VO2 consumption or heat generation. There also was no evidence indicating ubiquitin proteasome system (UPS) activation nor loss of muscle mass (Figure 1C–G).
Figure 3.
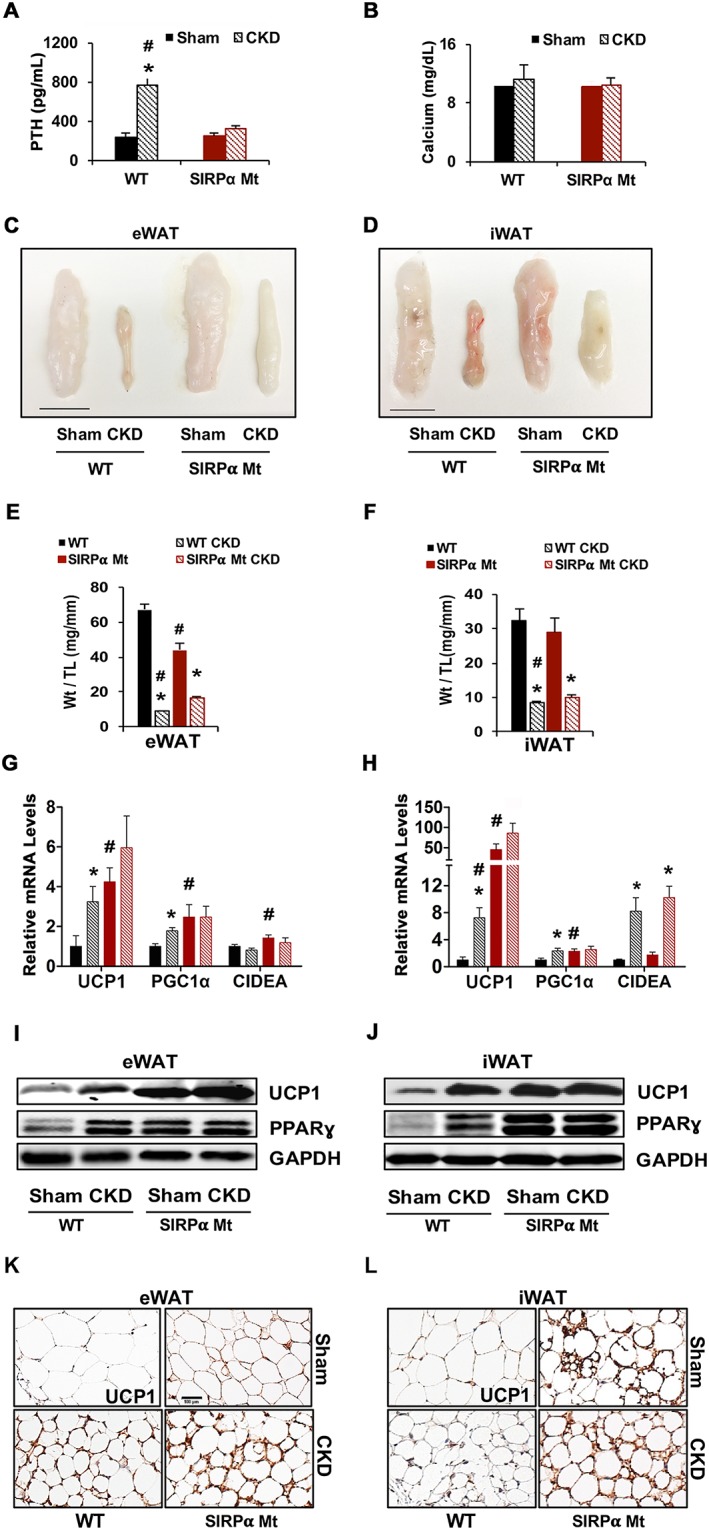
The absence of SIRPα promotes thermogenic genes, independently of parathyroid hormone (PTH). (A) Plasma PTH and (B) serum calcium levels were measured. (C) eWAT and (D) iWAT are pictured (scale = 1 cm). Tissue weight from (E) eWAT and (F) iWAT is illustrated. mRNA levels of (G) eWAT and (H) iWAT were determined by RT–qPCR analysis. (I) Protein lysates from eWAT and (J) iWAT samples were immunoblotted for UCP1, PPARγ, and GAPDH. Immunohistochemical staining for UCP1 protein was performed on (K) eWAT and (L) iWAT (scale bar = 100 μm) (A–L: n = 4–6 mice/group). Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, WT vs. SIRPα Mt mice. See also Figure S3.
3.4. In chronic kidney disease, signal regulatory protein alpha suppression promotes protein kinase A‐independent adipose tissue browning
To determine if WAT browning is controlled by sympathetic activation of PKA, we examined the influence of PKA on SIRPα Mt mice with CKD. Potential stimulators of PKA include an increase in PTH.16 However, our measurements in SIRPα Mt mice with CKD show that PTH is not elevated (Figure 3A). Another mechanism by which CKD could stimulate PKA would occur with activation of the sympathetic nervous system.16, 17, 18 We found that pPKA was increased in both iWAT and eWAT of WT mice with CKD vs. results obtained in SIRPα Mt mice with CKD (Figure 4A and 4B). Moreover, adipocytes from WT mice with CKD displayed increased expression of mediators of browning, namely, pHSL and pCREB. The same pathways were significantly reduced in SIRPα Mt mice with CKD. Notably, in WT mice with CKD, PKA activity in eWAT was significantly higher vs. the level in SIRPα Mt mice with CKD (Figure 4C). We conclude that WAT browning in WT mice with CKD develops as a result of a PKA‐dependent response. In contrast, browning of WAT in SIRPα Mt mice did not depend on activation of PKA or its mediators.
Figure 4.
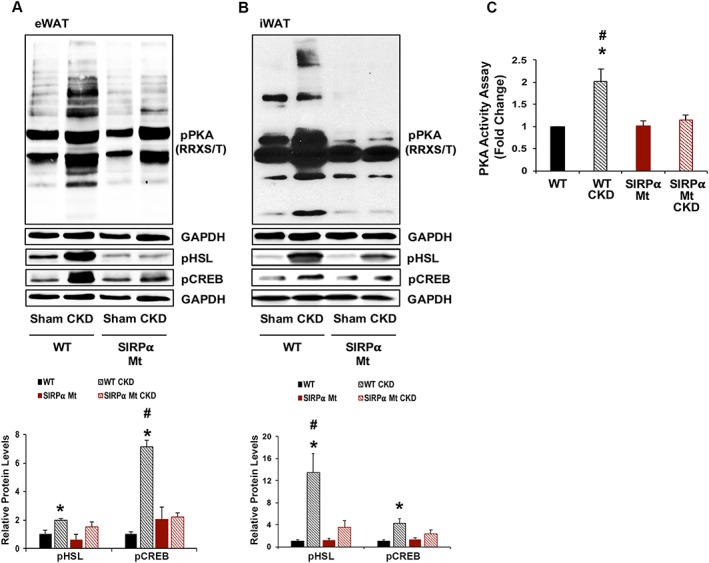
PKA activity and its mediators are suppressed in SIRPα Mt Mice with CKD. (A–B) eWAT and iWAT protein lysates were immunoblotted to detect phosphorylated protein kinase A [pPKA (RRXS/T)], pHSL, pCREB, and GAPDH (top panel); and the relative densities to the level of GAPDH are shown (bottom panel). (C) PKA activity assay was determined in eWAT (A–C: n = 4–6 mice/group). Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, WT vs. SIRPα Mt mice.
3.5. Protein kinase A activation of cAMP response element‐binding protein to signal regulatory protein alpha interferes with insulin signalling
CREB binding sites are found in the SIRPα promoter (http://natural.salk.edu/CREB/), whether CREB activation regulates SIRPα functions is largely unknown. We activated CREB by adding forskolin (FSK) to 3T3‐L1 adipocytes and found that the expressions of pCREB, SIRPα, pHSL, and UCP1 increased while pAkt was reduced (Figure 5A). When we treated 3T3‐L1 adipocytes with the CREB inhibitor, 666‐15, we found reduced expressions of pCREB, SIRPα, and UCP1 but no change in the level of pHSL or pAkt (Figure 5B). These results raise the possibility that PKA activates CREB, which stimulates SIRPα expression. To determine how SIRPα expression occurs, we injected WT and SIRPα Mt mice with 1 mg/kg of norepinephrine (NE). Western blotting of NE‐stimulated WAT from WT mice revealed significant increases in the expressions of SIRPα, pCREB, and pHSL plus the thermogenin gene (Figure 5C). In addition, we found decreased expression of pAkt at 6 h after NE treatment but not at 2 h (Figures S4A and 5C). Interestingly, treatment of SIRPα Mt mice with NE not only suppressed pAkt, pHSL, and pCREB but also blocked UCP1 activation (Figures 5C and S4B). We conclude that browning was reduced in SIRPα Mt mice treated with NE; the mechanism may be due to a reduction in pAkt. Moreover, WAT treatment with NE led to increases in PKA activity in primary cell cultures of WAT from WT mice. In contrast, PKA activity was suppressed in primary cell cultures of WAT from SIRPα Mt mice that were treated with NE (Figure S4B).
Figure 5.
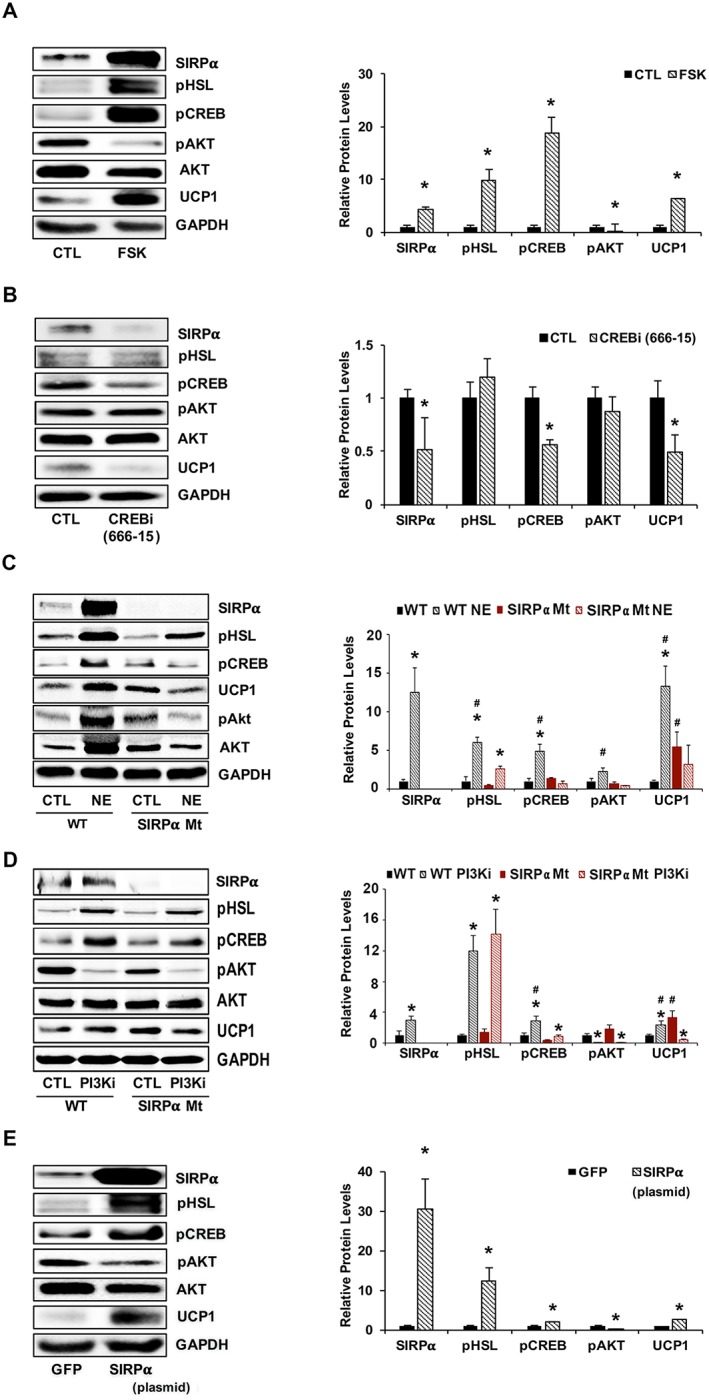
PKA activates CREB, stimulating SIRPα and interfering with insulin signalling. (A) 3T3‐L1 adipocytes were treated with forskolin (FSK, 10 μM) for 24 h, and lysates immunoblotted (left panel) for relative densities to GAPDH or pAkt to AKT are shown (right panel) for SIRPα, pHSL, pCREB, pAkt (Ser473), Akt, and UCP1 (n = 4 independent experiments). (B) 3T3‐L1 adipocytes were treated for 24 h with 50 nM CREB inhibitor (compound 666‐15) and lysates were immunoblotted (left panel) for SIRPα, pHSL, pCREB, pAkt (Ser473), Akt, UCP1, and GAPDH, and relative densities to GAPDH or pAkt to AKT are shown ( right panel; n = 4 independent experiments). (C) Mice treated with 1 mg/kg noradrenaline bitartrate (NE) for 2 h were compared with vehicle‐treated mice. iWAT was homogenized and immunoblotted (left panel) for SIRPα, pHSL, pCREB, pAkt (Ser473), Akt, and UCP1, and relative densities that obtained to GAPDH or pAkt to AKT are shown (right panel; n = 4–6 mice/group). (D) Primary cultures of iWAT from WT or SIRPα Mt mice were treated with or without a PI3K inhibitor (PI3Ki) at 50 μM for 6 h. Lysates from these cells were immunoblotted (left panel) for SIRPα, pHSL, pCREB, pAkt (Ser473), Akt, and UCP1; and relative densities obtained to GAPDH or pAkt to AKT are shown (right panel; n = 4 independent experiments). (E) 3T3‐L1 adipocytes were transfected with a plasmid stimulating SIRPα expression and compared with results from a plasmid that expresses green fluorescent protein (GFP). Lysates were immunoblotted (left panel) for SIRPα, pHSL, pCREB, pAkt (Ser473), Akt, and UCP1; and relative densities obtained to GAPDH or pAkt to AKT are shown (right panel; n = 4 independent experiments). Values are a mean ± SEM. (A–B, E) *P < 0.05, control vs. treatment. (C–D) *P < 0.05, control vs. treatment and #P < 0.05, WT vs. SIRPα Mt mice. See also Figure S4.
To determine if similar events regulate UCP1 expression, we studied primary cell cultures of WAT obtained from WT or from SIRPα Mt mice. When either of the primary cells were treated with the PI3K inhibitor, LY294002, pAkt was suppressed, but the expressions of SIRPα, pHSL, pCREB, and UCP1 were increased (Figure 5D). Adipocytes from SIRPα Mt mice treated with the PI3K inhibitor led to suppression of UCP1 expression. The results suggest that reduced pAkt contributes in a major way to the browning of WAT obtained from SIRPα Mt mice (Figure 5D). When we treated cultured adipocytes from WT or SIRPα Mt mice with the PI3K inhibitor, we found an increase in the activation of PKA in WAT from WT adipocytes (Figure S4C). Next, we transfected 3T3‐L1 adipocytes with a plasmid that overexpresses SIRPα. In this case, levels of pHSL, pCREB, and UCP1 were up‐regulated which was associated with a suppression of pAkt (Figure 5E). Additionally, when SIRPα was overexpressed, PKA activation was stimulated (Figure S4D). These responses cause an increase in UCP1 and pathologic browning of WAT.
3.6. In the cachexia of acute diabetes, white adipose tissue browning is protein kinase A dependent
To test if the responses we identified were suggestive of increased insulin levels alone, we utilized another model of cachexia, STZ treatment. STZ is characterized by loss of gastrocnemius muscle and WAT adipocytes.19 Metabolic responses to STZ treatment included the development of insulinopenia and hyperglycaemia in WT and SIRPα Mt mice (Figure S5A and B). In SIRPα Mt mice, the loss of gastrocnemius and WAT weights was ameliorated despite the presence of acute diabetes and a suppressed level of insulin (Figure S5C). In addition, grip strength in acutely diabetic WT mice exhibited significant impairment vs. results from the acutely diabetic SIRPα Mt mice (Figure S5D; P < 0.05). There also were increases in the expressions of autophagy genes (e.g. ATG12, BECN1, LC3, and p62) in WT, STZ‐treated mice (Figure S5E). Acutely diabetic SIRPα Mt mice did not express the same autophagy genes that were significantly suppressed vs. results from acutely diabetic WT mice (Figure S5E; P < 0.05). Notably, SIRPα was increased in both eWAT and iWAT of acutely diabetic WT mice, and there was evidence for impairment in intracellular insulin signalling plus activation of UCP1 (Figure S5F and G). In gastrocnemius muscles from acutely diabetic WT mice, pAkt was reduced but SIRPα expression was increased (Figure S5H). We also found activation of pPKA, pHSL, and pCREB in eWAT and iWAT (Figure S5I and J). Conversely, in acutely diabetic SIRPα Mt mice, pAkt levels in WAT were similar to those in control mice. UCP1 remained high in both groups. Notably, the levels of pPKA, pHSL, and pCREB were suppressed in acutely diabetic SIRPα Mt mice in comparison with acutely diabetic WT mice (Figure S5I and J). On the basis of these results, we conclude that blocking SIRPα improves the metabolism of muscle protein and adipocytes in acute diabetes. This promotes physiologic browning via a pathway that does not require activation of PKA or PKA mediators.
3.7. Muscle‐specific suppression of signal regulatory protein alpha improves insulin signalling and prevents cachexia in chronic kidney disease
Skeletal muscle‐specific SIRPα KO (mSIRPα−/−) mice were created by crossing Mck‐Cre mice with mice bearing floxed exons 3–4 of SIRPα. Both SIRPα flox/flox(fl/fl) and mSIRPα−/− mice were treated to produce CKD and compared. The two groups of mice displayed significant (P < 0.05) elevations in BUN (Figure 6A). Additionally, SIRPαfl/fl mice with CKD exhibited reduced grip strength compared with results from mSIRPα−/− mice that also had CKD (Figure 6B). Glucose tolerance was improved in mSIRPα−/− compared with SIRPαfl/fl mice (Figure 6C). Importantly, SIRPαfl/fl mice with CKD had evidence of hyperinsulinaemia (5.2‐fold higher) as compared with normal insulin levels in mSIRPα−/− mice with CKD (Figure 6D). Weights of WAT and gastrocnemius, TA, EDL, and soleus skeletal muscles were all decreased in SIRPαfl/fl mice with CKD vs. measurements from mSIRPα−/− mice with CKD (Figure 6E). Thus, mSIRPα−/− mice with CKD have preservation of the weight of WAT and skeletal muscles compared with results from SIRPαfl/fl mice with CKD. We explored the mechanisms for these responses by measuring insulin signalling in both WAT and skeletal muscles. In skeletal muscle, mSIRPα−/− mice with CKD exhibited improved intracellular insulin signalling with increased levels of pAkt. In contrast, WAT displayed increased expression of SIRPα. In this case, we found that intracellular insulin signalling in skeletal muscle and WAT improved in mSIRPα−/− mice, despite the presence of CKD (Figure 6F and 6G). Notably, pathologic browning was present in WAT of SIRPαfl/fl mice with CKD. In the mSIRPα−/− mice with CKD, we found up‐regulation of UCP1 plus suppression of activated pPKA and its mediators (Figure 6G). These responses suggest the presence of physiologic browning. Consistent with this formulation, we find evidence of increased circulating levels of SIRPα in serum of SIRPαfl/fl mice with CKD (Figure 6H). This result suggests that SIRPα may be a mediator of inter‐organ communication. Therefore, crosstalk between skeletal muscles and WAT includes our finding that elimination of SIRPα in muscle unexpectedly improved insulin sensitivity and led to physiologic browning in WAT without loss of skeletal muscles plus diminished loss of WAT.
Figure 6.
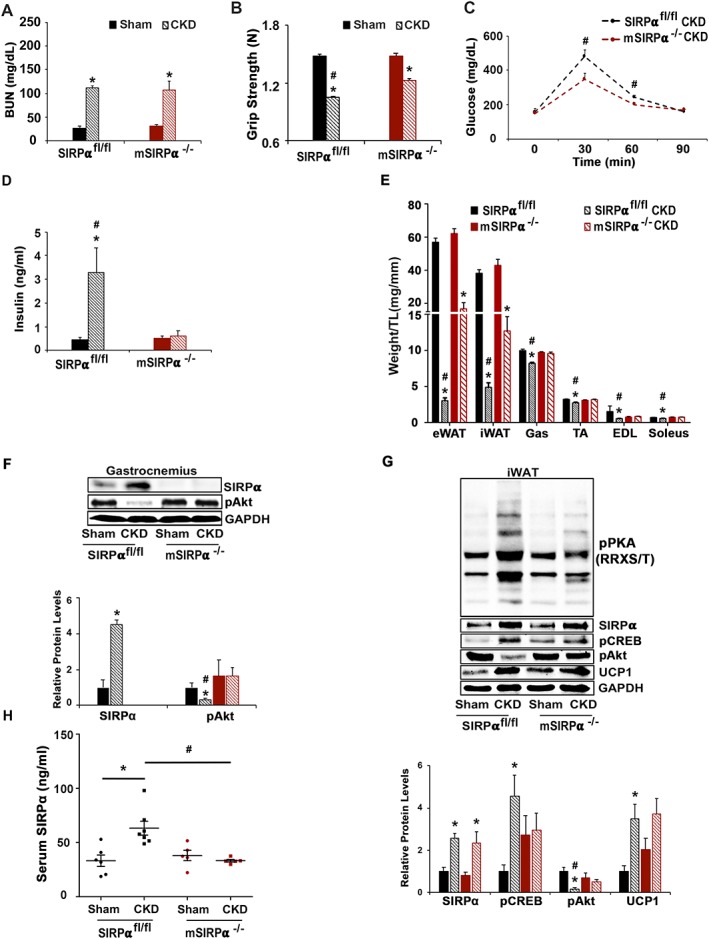
Blocking SIRPα in muscle facilitates inter‐organ communication and prevents cachexia in CKD. At 8–12 weeks after subtotal nephrectomy (A–G: n = 4–6 mice/group): (A) blood urea nitrogen (BUN), (B) grip strength, (C) glucose tolerance, and (D) insulin levels were measured by ELISA. (E) Organ harvest weight normalize to tibia length (TL) was measured. (F) Protein lysates of gastrocnemius (Gas) skeletal muscle were immunoblotted (top panel) to detect SIRPα, pAkt (Ser473), and GAPDH; and relative densities were measured (bottom panel). (G) Inguinal white adipose tissue (iWAT) was immunoblotted (top panel) to detect SIRPα, pCREB, pAkt (Ser473), UCP1, phosphorylated protein kinase A [pPKA (RRXS/T)], and relative densities obtained to GAPDH are shown (bottom panel). (H) ELISA was performed to detect circulating serum levels of SIRPα. Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, SIRPαfl/fl vs. mSIRPα−/−.
3.8. Adipose‐specific signal regulatory protein alpha knockout with chronic kidney disease displayed improved insulin signalling but did not prevent dyslipidaemia or losses of skeletal muscle
We evaluated whether there is inter‐organ communication between adipose tissue and skeletal muscle by creating adipose‐specific SIRPα KO (AD‐SIRPα−/−) mice on the basis of methods similar to those we used in obtaining mSIRPα−/− mice. We used adiponectin‐Cre to delete SIRPα floxed exons 3–4 specifically in adipose tissues. Initially, we compared results of glucose tolerance testing in SIRPαfl/fl and AD‐SIRPα−/− mice with CKD (Figure 7A). There were similar levels of circulating insulin in SIRPαfl/fl mice with CKD and AD‐SIRPα−/− mice with CKD (Figure 7B); both insulin values were higher than those present in mSIRPα−/− mice with CKD (Figure 6D). With CKD, serum cholesterol in both SIRPαfl/fl mice and AD‐SIRPα−/− mice was not different (Figure 7C). However, grip strength of AD‐SIRPα−/− mice with CKD was greater compared with results from SIRPαfl/fl mice with CKD (Figure 7D). Upon harvest, AD‐SIRPα−/− mice with CKD exhibited preservation of WAT weight plus improved insulin signalling and physiologic browning of WAT (Figure 7E and 7F). These responses occurred without evidence of PKA activation (Figure 7F). In skeletal muscles, AD‐SIRPα−/− mice with CKD exhibited no statistically significant improvement in weight of the gastrocnemius, TA, and soleus (Figure 7E) with evidence of SIRPα up‐regulation in gastrocnemius skeletal muscle despite improvements in activation of pAkt (Figure 7F). Finally, serum SIRPα levels in SIRPαfl/fl mice with CKD were increased and remained elevated in AD‐SIRPα−/− mice with CKD (Figure 7G). These results are consistent with a communication between skeletal muscles and adipose tissues regulated by a novel mediator of insulin resistance, activated by SIRPα.
Figure 7.
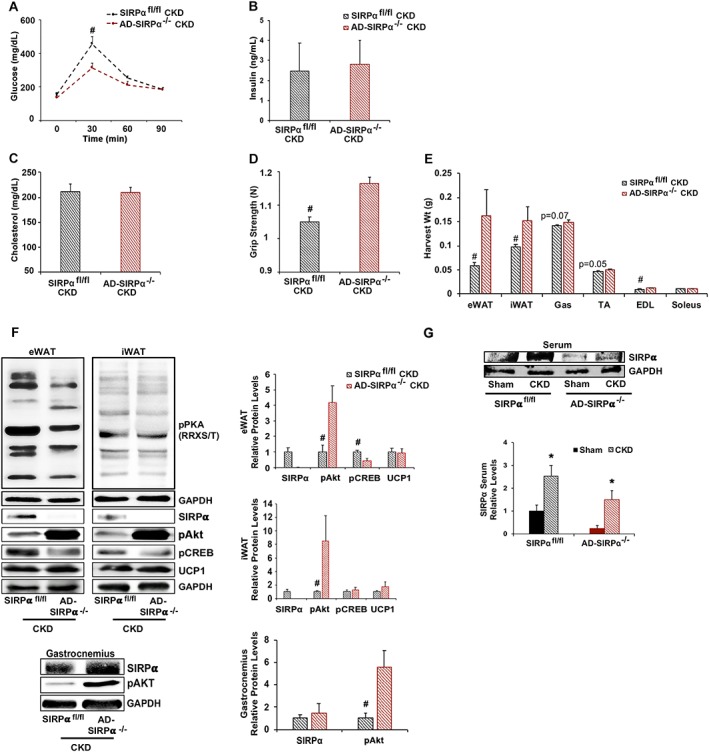
Blocking SIRPα in adipose tissue does not lead to significant inter‐organ communication or prevention of cachexia (A–G: n = 4–6 mice/group). (A) Glucose tolerance, (B) insulin levels, (C) serum cholesterol, and (D) grip strength were measured. (E) Organ harvest weight was measured. (F) Protein lysates of inguinal and epididymal white adipose tissue (iWAT/eWAT) were immunoblotted (left panel) to detect phosphorylated protein kinase A [pPKA (RRXS/T)], SIRPα, pAKT (Ser473), pCREB, UCP1, and GAPDH. Protein lysates of gastrocnemius skeletal muscle were immunoblotted (left panel) to detect either SIRPα or pAkt (Ser473), and relative densities obtained to GAPDH are shown (right panel). (G) Serum SIRPα was measured by immunoblot. Values are expressed as mean ± SEM; *P < 0.05, sham vs. CKD and #P < 0.05, SIRPαfl/fl vs. AD‐SIRPα−/−.
3.9. Signal regulatory protein alpha and white adipose tissue browning are increased in patients with chronic kidney disease
We examined biopsies of rectus abdominis muscles and periumbilical WAT obtained from control adults and patients with advanced CKD, characterized by increased C‐reactive protein and serum creatinine (Table S1A). In muscles of CKD patients, we detected increased mRNAs of SIRPα, IL‐6, and atrophy genes (Atrogin‐1 and MuRF1): these values were compared with those present in control adults (Figure 8A). Likewise, skeletal muscle SIRPα levels in patients with CKD were increased vs. values in control subjects (Figure 8B and 8C). When we examined measurements in those subjects with CKD, we found that there were higher mRNA levels of SIRPα and UCP1, indicating WAT browning vs. levels in control adults (Figure 8D). Immunohistochemical staining of WAT from CKD patients uncovered higher levels of SIRPα plus UCP1 proteins (Figure 8E‐F). These increases in SIRPα expression and markers of browning in WAT of CKD patients were associated with weight loss (Table S1A). Finally, serum SIRPα was increased 2.4‐fold in patients with advanced CKD when compared with healthy controls (Figure 8G, Table S1B).
Figure 8.
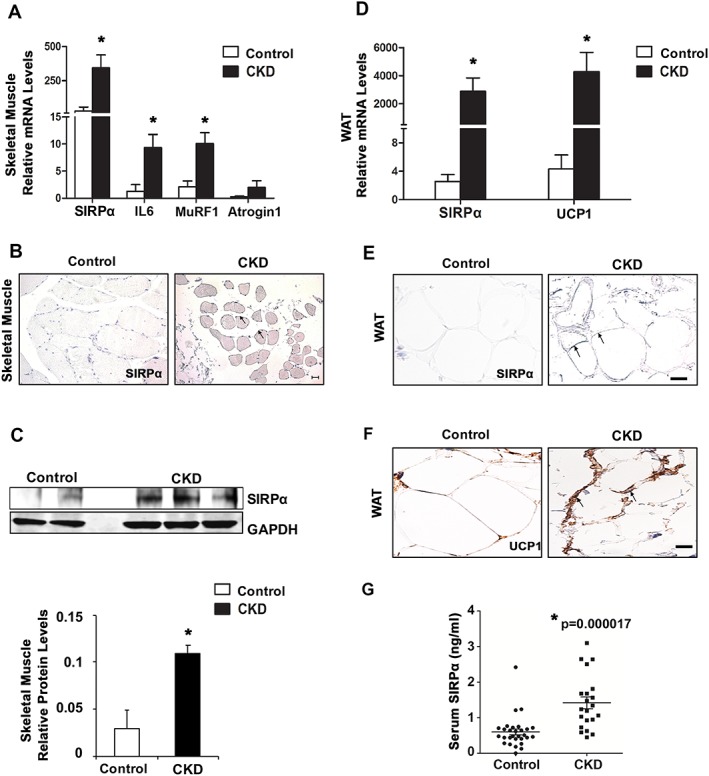
Patients with CKD expressed up‐regulation of SIRPα, atrophy‐related genes and browning of WAT (n = 7–9 patients/group). (A) mRNAs of skeletal muscles were determined on the basis of RT–qPCR analysis. (B) Immunostaining of muscle sections for SIRPα (see arrows) from biopsies of age‐matched, healthy control subjects and CKD patients. (C) Skeletal muscle lysates were immunoblotted (top panel) for SIRPα; and relative densities obtained to GAPDH are shown (bottom panel). (D) mRNAs of white adipose tissue (WAT) samples were determined on the basis of RT–qPCR analysis. Immunostaining of WAT sections for (E) SIRPα (see arrows) and (F) UCP1 (see arrows) was performed on healthy control subjects and CKD patients (scale bar = 100 μm). (G) Serum was obtained from patients with CKD vs. healthy controls, and ELISA was performed to detect circulating serum levels of SIRPα (n = 21–29 patients/group). Values are mean ± SEM. *P < 0.05 vs. control. See also Table S1.
4. Discussion
In investigating mechanisms causing cachexia in mice with CKD or acute diabetes, we have determined that endogenous SIRPα expression is of primary importance for several reasons: (i) SIRPα in mice with CKD is highly expressed in skeletal muscles and adipocytes, leading to impaired insulin signalling mediated by SIRPα. (ii) We identified that PKA activation promotes SIRPα expression and, hence, can be a mediator of the development of cachexia and pathologic browning of WAT. These responses resulted in decreased intracellular insulin signalling including a reduction in pAkt. (iii) We find that mice with global suppression of SIRPα exhibited improved insulin signalling and promotion of physiologic browning of WAT via a PKA‐independent pathway. In mice with CKD, the distinguishing feature of physiologic browning is suppression of SIRPα plus improved insulin sensitivity resulting in up‐regulation of pAkt in WAT as well as improvements in GTT and ITT. (iv) Notably, SIRPα is highly expressed in acute diabetes, and it is also associated with pathologic browning of WAT. (v) We find that SIRPα elimination extends the survival of mice with CKD. (vi) Skeletal muscle‐specific or adipose‐specific KO of SIRPα in mice with CKD leads to improved insulin signalling in both muscles and adipose tissues. Only mice with muscle‐specific SIRPα KO exhibited suppression of serum insulin and circulating levels of SIRPα. These responses lead to minimized loss of adipose tissue but no loss of skeletal muscle despite the presence of CKD (Figure 9). Potentially, our findings suggest that SIRPα exhibits an important, myokine‐like response mediating cachexia in mice with CKD while raising the possibility of crosstalk between adipose tissues and skeletal muscles.
Figure 9.
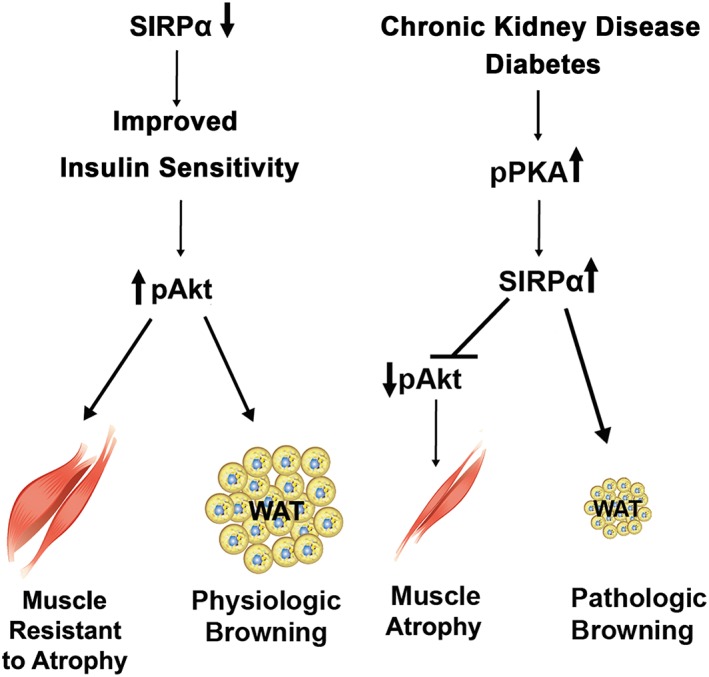
Summary diagram. Suppression of SIRPα improves insulin sensitivity leading to skeletal muscle that is resistant to atrophy plus physiologic browning of white adipose tissue (WAT). Chronic kidney disease (CKD) and acute diabetes stimulates PKA activity, an up‐regulation of SIRPα that inhibits insulin signalling causing muscle wasting and pathologic browning of WAT.
A prominent feature of our results is the development of pathologic browning of adipose tissue. In WT mice with CKD, browning was associated with increased energy expenditure and thermogenesis (Figure S3A–D). But when SIRPα is suppressed in mice with CKD, insulin signalling improves, and there were no significant increases in oxygen consumption, the RER, or thermogenesis. The absence of SIRPα improved insulin signalling and also prevented losses of body and muscle weight as well as adipocyte tissue mass. Interestingly, expression of UCP1 during physiologic browning was not associated with thermogenesis. We speculate that the mechanism for this response reflects suppressed activation of PKA (Figure 4A–C).
Recently, a novel mechanism was described to explain the development of cachexia in mice with CKD or cancer.16, 20 In these models, the anti‐catabolic mechanism consists of interrupting the PTH receptor, preventing losses of muscle and adipose tissues. But when PKA was stimulated by PTH or the sympathetic nervous system was activated, cachexia developed. The authors noted that a high level of circulating PTH triggered pathologic browning, which stimulated loss of muscle proteins. Our findings differ: SIRPα Mt mice exhibited improved insulin signalling and WAT browning and experienced no loss of muscle mass. Specifically, we found that SIRPα elimination stimulated UCP1 expression and WAT browning by a mechanism that was independent of PTH or PKA activation. Also, global KO or muscle and adipose‐specific KO of SIRPα exhibited improved insulin sensitivity (e.g. improved GTT plus increased pAkt expression in WAT and muscle), despite the presence of CKD. Finally, in mice with either CKD or acute diabetes, cachexia was mediated by PKA stimulation of SIRPα, promoting impaired insulin signalling.
Regarding the shortcomings of our studies, we recognize that SIRPα is a membrane‐bound protein. Therefore, we hypothesize that inter‐organ communication between skeletal muscles and adipose tissues is due to circulating SIRPα, but it is unclear if it is behaving as a myokine or related to macrophage circulation. For example, Willingham et al. suggests that macrophages express SIRPα protein.21 To examine circulating factors or the role of macrophage expression of SIRPα is beyond the scope of these experiments but requires further investigation.
Browning of WAT can be promoted by myostatin inhibition in mice fed a high fat diet.22 Moreover, inhibition of myostatin in mice with CKD promotes insulin sensitivity and increases muscle mass.5 In muscles of SIRPα Mt mice with CKD, myostatin and IL‐6 production were suppressed in skeletal muscles (Figure 1G). Additionally, Kong et al. found that increased expression of IRF4 in brown adipocytes participates in suppressing myostatin and increasing exercise capacity in skeletal muscles.23 This is relevant because global KO of SIRPα suppresses myostatin expression while improving muscle protein losses in CKD.
Bostrom et al., reported that PGC1α overexpression stimulates a myokine, irisin, which promoted the conversion of white to brown adipocytes.24 As WAT in SIRPα Mt mice with CKD developed increased grip strength plus up‐regulation of UCP1 and PGC1α, we propose that our results suggest the development of an ‘exercise‐like' phenotype (Figures 1D and 3G–J) despite lacking significant changes in physical activity (Figure S3E). Peng et al. recently reported that irisin released from skeletal muscle interacts with injured kidney tubule cells by improving ATP production and energy metabolism. The result is more rapid recovery of injured kidney tubular cells. These findings suggest that besides muscle and adipocytes, metabolic responses may change in other organs.25
In summary, SIRPα is a novel regulator of lost muscle protein and adipose tissues in two models of cachexia, CKD and acute diabetes. We have found evidence for increased expression of SIRPα and UCP1 in adipose tissues plus increased SIRPα in skeletal muscles and serum samples of patients with advanced CKD. These findings could lead to potential therapeutic targets for combating cachexia in conditions of CKD and type 1 diabetes while promoting survival in patients suffering from these catabolic illnesses.
Disclaimer
The contents of this publication do not express the views of the US Department of Veterans Affairs or the United States Government.
Author contributions
Conceptualization: SST. Investigation: JW, JD, and SST. Methodology: JW, JD, and SST. Validation: JW and SST. Visualization: JW, JD, and SST. Resources: SST, KH, DV, ZH, and GG. Supervision: SST. Writing, reviewing, and editing: SST and WEM. Funding acquisition: SST.
Conflict of Interest
None declared.
Supporting information
Table S1. Characteristics of Patients with CKD and Age Matched Healthy Controls. (Related to Fig. 8) (A) Patient characteristics defined from skeletal muscle and adipose tissue biopsies (n = 7‐9 patients/group). Values are a mean±SEM. *p < 0.05 vs. Control. (B) Serum was obtained from patients with CKD vs. healthy controls and their known characteristics are defined. (n = 21‐29 patients/group).
Figure S1. Suppression of SIRPα Improves Intracellular Insulin Signaling in Skeletal Muscles of Mice with CKD. (Related to Fig. 1). (A) Gastrocnemius (Gas) lysates were immunoblotted for SIRPα, GLUT4, pAkt (ser 473), and GAPDH. (n = 4‐6 mice/group).
Figure S2. Blocking SIRPα in Mice with CKD Improves Insulin Intracellular Signaling and Ectopic Lipids. (Related to Fig. 2). (A) iWAT lysates were immunoblotted for SIRPα, pAkt (ser 473), Akt, and GAPDH. (B) Lipid content in liver was measured. (A‐B: n = 4‐6 mice/group). Values are expressed as mean±SEM; *p < 0.05, Sham vs. CKD and #p < 0.05, WT vs. SIRPα Mt mice.
Figure S3. The Absence of SIRPα Did Not Generate Heat or Expend Energy Despite the Presence of CKD. (Related to Fig. 3). (A‐E) VO2, CO2, Respiratory Exchange Ratio, Heat, and Physical Activity were monitored by indirect calorimetry. (F) eWAT and (G) iWAT lysates were immunoblotted for UCP1, PPARγ and GAPDH and the relative band densities to GAPDH are shown. (A‐G: n = 4‐6 mice/group). Values are expressed as mean±SEM; *p < 0.05, Sham vs. CKD and #p < 0.05, WT vs. SIRPα Mt mice.
Figure S4. Stimulation of PKA Downregulates Intracellular Insulin Signaling. (Related to Fig. 5).
(A) White adipocytes were treated with or without norepinephrine (NE) for 6 h and lysates were immunoblotted for pAkt, Akt, and GAPDH. (B) Primary cultures of iWAT were treated with or without insulin 1 nM for 10 minutes or the PI3K inhibitor (LY294002) 50 μM, norepinephrine (NE) 100 nM, H89 50 μM for 30 min and lysates were immunoblotted for phosphorylated protein kinase A (pPKA (RRXS/T)) and GAPDH. (C) Primary cultures of iWAT from WT or SIRPα Mt mice were treated with or without a PI3K inhibitor at 50 μM for 6 h and lysates were immunoblotted for pPKA (RRXS/T) and GAPDH. (D) 3T3‐L1 adipocytes were transfected with a SIRPα plasmid stimulating PKA expression and compared to results from a plasmid that expresses green fluorescent protein (GFP). Lysates were immunoblotted for pPKA (RRXS/T) and GAPDH (n = 3 independent experiments).
Figure S5. Blocking SIRPα Prevents Acute Diabetes‐Induced Cachexia. (A) Mice were treated with streptozotocin (STZ) to create insulinopenia. (B) Hyperglycemic streptozotocin (STZ)‐treated mice were studied. (C) Organ weights (Wt) were normalized to tibia length (TL). (D) Grip strength in Newtons (N) was determined. (E) mRNA levels of autophagy genes in gastrocnemius skeletal muscle were measured using RT‐ qPCR analysis. (F) eWAT and (G) iWAT (H) Gastrocnemius (Gas) lysates were immunoblotted (top panel) for SIRPα, pAkt (ser 473), Akt, UCP1 and GAPDH. The relative band densities to GAPDH or pAkt to Akt are shown (bottom panel). (I) eWAT and (J) iWAT lysates were immunoblotted (left panel) phosphorylated protein kinase A (pPKA (RRXS/T)), pHSL, pCREB, and GAPDH. The relative band densities to GAPDH were determined (right panel; A‐J: n = 4‐6 mice/group). Values are a mean SEM; *p < 0.05, Sham vs. STZ, #p < 0.05, WT vs. SIRPα Mt.
Acknowledgements
We acknowledge the support by the VA Career Development Award IK2BX002492 to S.S.T. from the US Department of Veterans Affairs, Biomedical Laboratory Research and Development Program and Michael E. DeBakey VA Medical Center for Translational Research on Inflammatory Diseases (CTRID) and in part from the National Institutes of Health 2R01DK037175‐32 to W.E.M. We acknowledge the generous support of Dr. and Mrs. Harold Selzman. We also acknowledge Yifu Fang for his technical assistance. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia,Sarcopenia and Muscle: update 2017.26
Wu J., Dong J., Verzola D., Hruska K., Garibotto G., Hu Z., Mitch W. E., and Thomas S. S. (2019) Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk, Journal of Cachexia, Sarcopenia and Muscle, 10, 1210–1227. 10.1002/jcsm.12459.
References
- 1. Huang CX, Tighiouart H, Beddhu S, Cheung AK, Dwyer JT, Eknoyan G, et al. Both low muscle mass and low fat are associated with higher all‐cause mortality in hemodialysis patients. Kidney Int 2010;77:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas SS, Zhang L, Mitch WE. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int 2015;88:1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meuwese CL, Snaedal S, Halbesma N, Stenvinkel P, Dekker FW, Qureshi AR, et al. Trimestral variations of C‐reactive protein, interleukin‐6 and tumour necrosis factor‐alpha are similarly associated with survival in haemodialysis patients. Nephrol Dial Transplant 2011;26:1313–1318. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marshall S, Podlecki DA, Olefsky JM. Low pH accelerates dissociation of receptor‐bound insulin. Endocrinology 1983;113:37–42. [DOI] [PubMed] [Google Scholar]
- 7. de Brito‐Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009;20:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest 2009;119:3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol 2014;10:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fliser D, Pacini G, Engelleiter R, Kautzky‐Willer A, Prager R, Franek E, et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int 1998;53:1343–1347. [DOI] [PubMed] [Google Scholar]
- 11. Cecchin F, Ittoop O, Sinha MK, Caro JF. Insulin resistance in uremia: insulin receptor kinase activity in liver and muscle from chronic uremic rats. Am J Physiol 1988;254:E394–E401. [DOI] [PubMed] [Google Scholar]
- 12. Thomas SS, Dong Y, Zhang L, Mitch WE. Signal regulatory protein‐alpha interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int 2013;84:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997;386:181–186. [DOI] [PubMed] [Google Scholar]
- 14. Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, et al. A novel membrane glycoprotein, SHPS‐1, that binds the SH2‐domain‐containing protein tyrosine phosphatase SHP‐2 in response to mitogens and cell adhesion. Mol Cell Biol 1996;16:6887–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 2011;474:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 2016;23:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavi‐Moshayoff V, Wasserman G, Meir T, Silver J, Naveh‐Many T. PTH increases FGF23 gene expression and mediates the high‐FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 2010;299:F882–F889. [DOI] [PubMed] [Google Scholar]
- 18. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, et al. The systemic nature of CKD. Nat Rev Nephrol 2017;13:344–358. [DOI] [PubMed] [Google Scholar]
- 19. Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, et al. Muscle wasting in insulinopenic rats results from activation of the ATP‐dependent, ubiquitin–proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest 1996;98:1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47‐signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 2012;109:6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin‐mediated cross talk between muscle and adipose tissues. Int J Obes (Lond) 2016;40:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong X, Yao T, Zhou P, Kazak L, Tenen D, Lyubetskaya A, et al. Brown adipose tissue controls skeletal muscle function via the secretion of myostatin. Cell Metab 2018;28:631–643.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1‐alpha‐dependent myokine that drives brown‐fat‐like development of white fat and thermogenesis. Nature 2012;481:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peng H, Wang Q, Lou T, Qin J, Jung S, Shetty V, et al. Myokine mediated muscle‐kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun 2017;8:1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of Patients with CKD and Age Matched Healthy Controls. (Related to Fig. 8) (A) Patient characteristics defined from skeletal muscle and adipose tissue biopsies (n = 7‐9 patients/group). Values are a mean±SEM. *p < 0.05 vs. Control. (B) Serum was obtained from patients with CKD vs. healthy controls and their known characteristics are defined. (n = 21‐29 patients/group).
Figure S1. Suppression of SIRPα Improves Intracellular Insulin Signaling in Skeletal Muscles of Mice with CKD. (Related to Fig. 1). (A) Gastrocnemius (Gas) lysates were immunoblotted for SIRPα, GLUT4, pAkt (ser 473), and GAPDH. (n = 4‐6 mice/group).
Figure S2. Blocking SIRPα in Mice with CKD Improves Insulin Intracellular Signaling and Ectopic Lipids. (Related to Fig. 2). (A) iWAT lysates were immunoblotted for SIRPα, pAkt (ser 473), Akt, and GAPDH. (B) Lipid content in liver was measured. (A‐B: n = 4‐6 mice/group). Values are expressed as mean±SEM; *p < 0.05, Sham vs. CKD and #p < 0.05, WT vs. SIRPα Mt mice.
Figure S3. The Absence of SIRPα Did Not Generate Heat or Expend Energy Despite the Presence of CKD. (Related to Fig. 3). (A‐E) VO2, CO2, Respiratory Exchange Ratio, Heat, and Physical Activity were monitored by indirect calorimetry. (F) eWAT and (G) iWAT lysates were immunoblotted for UCP1, PPARγ and GAPDH and the relative band densities to GAPDH are shown. (A‐G: n = 4‐6 mice/group). Values are expressed as mean±SEM; *p < 0.05, Sham vs. CKD and #p < 0.05, WT vs. SIRPα Mt mice.
Figure S4. Stimulation of PKA Downregulates Intracellular Insulin Signaling. (Related to Fig. 5).
(A) White adipocytes were treated with or without norepinephrine (NE) for 6 h and lysates were immunoblotted for pAkt, Akt, and GAPDH. (B) Primary cultures of iWAT were treated with or without insulin 1 nM for 10 minutes or the PI3K inhibitor (LY294002) 50 μM, norepinephrine (NE) 100 nM, H89 50 μM for 30 min and lysates were immunoblotted for phosphorylated protein kinase A (pPKA (RRXS/T)) and GAPDH. (C) Primary cultures of iWAT from WT or SIRPα Mt mice were treated with or without a PI3K inhibitor at 50 μM for 6 h and lysates were immunoblotted for pPKA (RRXS/T) and GAPDH. (D) 3T3‐L1 adipocytes were transfected with a SIRPα plasmid stimulating PKA expression and compared to results from a plasmid that expresses green fluorescent protein (GFP). Lysates were immunoblotted for pPKA (RRXS/T) and GAPDH (n = 3 independent experiments).
Figure S5. Blocking SIRPα Prevents Acute Diabetes‐Induced Cachexia. (A) Mice were treated with streptozotocin (STZ) to create insulinopenia. (B) Hyperglycemic streptozotocin (STZ)‐treated mice were studied. (C) Organ weights (Wt) were normalized to tibia length (TL). (D) Grip strength in Newtons (N) was determined. (E) mRNA levels of autophagy genes in gastrocnemius skeletal muscle were measured using RT‐ qPCR analysis. (F) eWAT and (G) iWAT (H) Gastrocnemius (Gas) lysates were immunoblotted (top panel) for SIRPα, pAkt (ser 473), Akt, UCP1 and GAPDH. The relative band densities to GAPDH or pAkt to Akt are shown (bottom panel). (I) eWAT and (J) iWAT lysates were immunoblotted (left panel) phosphorylated protein kinase A (pPKA (RRXS/T)), pHSL, pCREB, and GAPDH. The relative band densities to GAPDH were determined (right panel; A‐J: n = 4‐6 mice/group). Values are a mean SEM; *p < 0.05, Sham vs. STZ, #p < 0.05, WT vs. SIRPα Mt.