

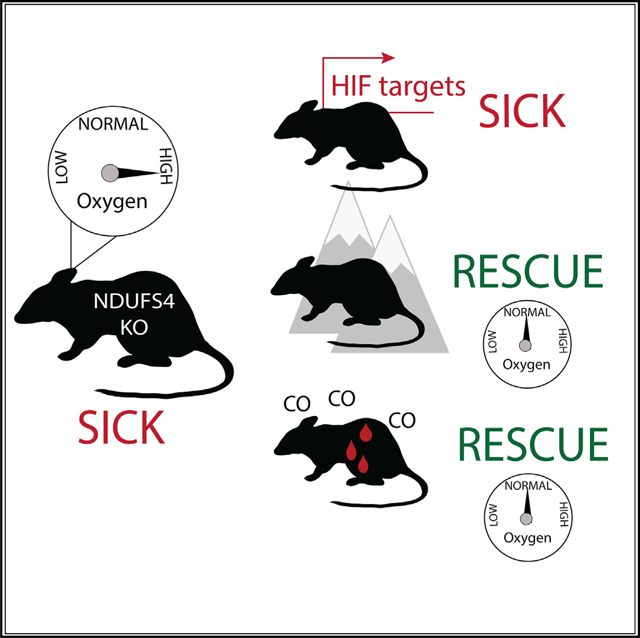
SUMMARY
Leigh syndrome is a devastating mitochondrial disease for which there are no proven therapies. We previously showed that breathing chronic, continuous hypoxia can prevent and even reverse neurological disease in the Ndufs4 knockout (KO) mouse model of complex I (CI) deficiency and Leigh syndrome. Here, we show that genetic activation of the hypoxia-inducible factor transcriptional program via any of four different strategies is insufficient to rescue disease. Rather, we observe an age-dependent decline in whole-body oxygen consumption. These mice exhibit brain tissue hyperoxia, which is normalized by hypoxic breathing. Alternative experimental strategies to reduce oxygen delivery, including breathing carbon monoxide (600 ppm in air) or severe anemia, can reverse neurological disease. Therefore, unused oxygen is the most likely culprit in the pathology of this disease. While pharmacologic activation of the hypoxia response is unlikely to alleviate disease in vivo, interventions that safely normalize brain tissue hyperoxia may hold therapeutic potential.
In Brief
Leigh syndrome is a severe mitochondrial disorder. Here, Jain et al. show, in a mouse model of the disease, that excess oxygen in the brain is a likely cause of tissue damage and that distinct interventions that reduce oxygen delivery to the brain can prevent and even reverse the neurological disease, likely by normalizing tissue hyperoxia.
Graphical Abstract
INTRODUCTION
Mitochondrial disorders comprise a large collection of individually rare, inborn errors of metabolism, with an estimated incidence of at least 1 in 4,300 births (Gorman et al., 2015). The most severe forms of disease occur because of mutations in structural components or assembly factors of the electron transport chain (ETC) and ATP synthase (CV). Of these disorders, Leigh syndrome is the most common pediatric manifestation of mitochondrial diseases (Lake et al., 2015). Mutations in over 75 distinct genes can underlie this syndrome, with patients first showing symptoms at any stage from birth to childhood (Lake et al., 2015). Patients often develop seizures, failure-to-thrive, ataxia, developmental delay, and clinical regression, ultimately leading to death, typically within the first few years of life. Diagnosis of Leigh syndrome is based on the clinical presentation and radiographic findings of bilaterally symmetric lesions on T2-weighted brain MRI. While significant advances have been made in defining their genetic basis, there are no generic treatments with proven efficacy in these patients. Recently, we demonstrated that chronic, continuous normobaric exposure to hypoxia (110025 FiO2) can prevent and even reverse the neurodegenerative brain disease in the Ndufs4 mouse model of Leigh syndrome, not only extending lifespan, but also normalizing body weight, body temperature, disease biomarkers, and improving behavioral performance (Ferrari et al., 2017; Jain et al., 2016).
The initial motivation for testing hypoxia as a therapy arose from our cell culture experiments, where we found that artificially activating the vHL-PHD-HIF hypoxia transcriptional program was sufficient to rescue cell proliferation defects caused by ETC inhibition (Jain et al., 2016). However, the in vivo therapeutic benefit was achieved by exposing Ndufs4 KO mice with a partial complex I deficiency to breathing chronic, normobaric 11% O2. In subsequent work, we showed that neither intermittent hypoxia nor more moderate hypoxia (17% O2) could rescue the neurodegenerative disease (Ferrari et al., 2017). Moreover, breathing 55% O2, which is harmless in wild-type mice, was extremely toxic for Ndufs4 KO mice, leading to death within days, suggesting the possibility of HIF-dependent and independent mechanisms.
The aim of this study was to explore the sufficiency of the hypoxia transcriptional response in the hypoxic rescue of disease, as well as the role of brain oxygen levels in pathogenesis. We report that genetic activation of the canonical vHL-PHD-HIF hypoxia response is insufficient to prevent disease in the Ndufs4 KO mouse. Rather, we find that oxygen consumption is impaired and brain oxygen tissue levels increase in KO mice, likely resulting in increased oxygen toxicity. We show that chronic hypoxic breathing normalizes brain PO2 in Ndufs4 KO mice, and that other diverse interventions that reduce brain oxygen levels are effective at preventing neurological disease in this mouse model.
RESULTS
Activation of the Hypoxia Response Pathway Is Not Sufficient to Rescue Disease
We had originally performed a genome-wide CRISPR screen and identified activation of the hypoxia transcriptional response as being protective in cell culture models of mitochondrial disease. However, we never tested whether activation of this pathway is sufficient to rescue the Ndufs4 KO mouse model. A major component of the cellular and organismal hypoxia response is mediated by the hypoxia-inducible transcription factors (HIFs). In well-oxygenated environments, the prolyl hydroxylase enzymes (PHDs) use molecular oxygen as a substrate and hydroxylate the HIF transcription factors (Wang et al., 1995; Majmundar et al., 2010; Ivan et al., 2001). It is this hydroxylated form that is recognized by the E3-ligase, von Hippel Lindau (vHL) factor and targeted for proteasomal degradation. There are three known PHD enzymes involved in oxygen sensing, PHD1–3. PHD2 inhibition is typically associated with HIF activation. However, PHD1 and PHD3 have also been reported to have redundant roles in signaling to the HIF transcriptional program. There are also two isoforms of HIF–HIF1α and HIF2α. While these transcription factors have partially overlapping roles, HIF1α is thought to primarily regulate cellular hypoxia adaptations (e.g., increasing glycolysis, modifications of the tricarboxylic acid cycle [TCA]) (Majmundar et al., 2010). HIF2α is more relevant for whole-body physiological adaptations such as erythropoiesis and angiogenesis. In mouse models, genetic disruption of the PHD enzymes or vHL activate the hypoxia transcriptional program and have been previously utilized to explore the therapeutic potential of targeting this pathway in a range of conditions, including stroke and hindlimb ischemia (Quaegebeur et al., 2016). Here, we used the same genetic models to assess whether activating the HIF transcriptional program is sufficient to rescue neurological disease in the Ndufs4 KO mouse model.
We crossed the Ndufs4 KO strain with mice lacking Phd1, Nestin-Phd2, Phd3, or the Chuvash mouse harboring a point mutation in Vhl (Vhlch/ch) (Quaegebeur et al., 2016; Hickey et al., 2007). We asked whether the hypoxia response was indeed activated in the different genetic models of the hypoxia transcriptional program. The Phd2 strain consistently showed elevation of canonical HIF targets in the cerebellum, where disease pathology eventually appears (Figure 1C). For example, Epo expression was increased 100× in this strain. Moreover, Ldha and Vegfa transcripts were similarly elevated in Phd1, Phd2, and Vhl mice. Phd3 mice did not show a canonical HIF response in the cerebellum. These findings are consistent with previous reports that Phd2 and vHL are more direct mediators of the HIF response. Furthermore, HIF2α activation results in elevated hematocrit levels. In our hands, only Nestin-Phd2 mice had an elevated hematocrit (>60%, Figure 1D).
Figure 1.

Genetic Activation of the Hypoxia Response Is Insufficient to Prevent Leigh Disease
(A) Kaplan-Meier curves for double KOs relative to single Ndufs4−/− KO mice. Log-rank p values and sample size within insert. Single KOs are homozygous-null for Ndufs4 locus and WT or Het for other genetic locus of given cross.
(B) MRI of vestibular nucleus (red arrow for Phd1, Phd2 and Phd3 crosses) of late-stage disease mice for each genotype. In Vhl cross, red arrow corresponds to lateral lesions (dentate nucleus) and midline lesion (central lobule, cerebellum).
(C) qPCR for canonical HIF targets (mean ± SD, n = 4 per group). Mice are homozygous for given genotype and WT or Het for Ndufs4 genotype.
(D) Hematocrit of control or Phd and Vhl genetic models (WT or Het for Ndufs4) (mean ± SEM, Ctrl [n=3], Vhl [n=6], Phd1 [n=3], Phd2 [n=4], Phd3 [n=5]). One-tailed unpaired t test p value shown for genetic crosses relative to control mice. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA, with Dunnett’s test for multiple comparison).
Notably, none of these genetic crosses were sufficient to prevent disease. Overall survival of double KOs was equivalent or even worse than Ndufs4−/− mice (Het or WT for Phd/Vhl genes) (Figure 1A). Specifically, Ndufs4 KO mice that were lacking Phd1 had a median survival of 45 days versus control Ndufs4 KO mice with a median survival of 57 days (matched for genetic background). Even more strikingly, Ndufs4−/−; Nestin-Phd2−/− had a median survival of 29 days versus matched Ndufs4 KO controls which had a median survival of 64 days. Ndufs4−/−; Phd3−/− survived for a median of 69 days versus matched Ndufs4−/− with median survival of 61 days. Finally, Ndufs4−/−; Vhlch/ch lived to 48 days versus 69 days for matched Ndufs4−/− mice. Notably, loss of Phd2 or Vhl robustly led to HIF activation, and these same strains were particularly sensitive to complex I deficiency. Moreover, all four genetic crosses resulted in double KOs that developed lesions in the vestibular nucleus (and additional cerebellar lesions in the Vhl cross) as imaged by T2-weighted MRI (Figure 1B). These genetic crosses indicate that artificially activating the HIF response is insufficient to rescue disease. Thus, small molecule inhibitors of the PHD enzymes or vHL enzyme are predicted to be ineffective in vivo, and in fact, could be toxic, in the context of a whole organism with complex I deficiency and Leigh syndrome.
Impaired Whole-Body Oxygen Consumption and Brain Hyperoxia in Ndufs4 KO Mice
Mitochondria consume 90% of the oxygen in the body (Rich, 2003). Thus, it is possible that genetic defects in the electron transport chain will have a secondary impact on tissue oxygenation. In fact, venous hyperoxia has been documented in patients with mitochondrial disease and has also been proposed to be a diagnostic test in patients with mitochondrial myopathy (Taivassalo et al., 2002). We monitored whole body and cerebral oxygenation in WT and Ndufs4 KO animals. We continuously measured whole body oxygen consumption in WT and KO animals, as a function of age. At ~37 days of age, whole body oxygen consumption (normalized for total body weight) was not significantly different between KO and WT animals. However, in awake mice this parameter decreased in KO animals as a function of age, reaching nearly half the value of WT animals at ~45 days of age (Figure 2A). Thus, it appears that the decline in whole body oxygen consumption correlates with the overt disease phenotype until death at ~60 days of age. This difference is observed during both active and inactive periods of the murine day (Figure 2B).
Figure 2.

Whole-Body and Brain Oxygenation of KO and WT Mice
(A and B) Whole-body oxygen consumption (mean SEM), normalized for body weight, during (A) active hours or (B) inactive hours. Data smoothened over 70 collection points (~5.5 h). Values normalized to body weight at each collection point (n = 6 per group). Unpaired t test p value shown for each genotype comparison at each day.
(C) Tissue PO2 (mean ± SEM) measured stereotaxically in hypothalamus of WT and KO mice as a function of age using a Clark electrode (young = 36–41 days, old 50–54 days). n = 6 for WT, n = 5 for young KO and n = 5 for old KO. Dotted line represents PO2 of WT mice breathing room air. Unpaired t test p value shown for comparison of KOs to WT breathing room air.
(D) Tissue PO2 (mean ± SD) measurement using an optical PO2 probe stereotaxically placed near vestibular nuclei of WT and KO mice breathing varying oxygen tensions (n = 4–6 per group). Dotted line represents PO2 of WT mice breathing room air.
(E) Hydrogen peroxide released during complex I-mediated respiration by isolated mitochondria from WT and KO brains as a function of dissolved oxygen.
(F) Median survival of KO mice as a function of inhaled oxygen, using data from this paper and from previously published studies (Ferrari et al., 2017; Jain et al., 2016).
We reasoned that such overt defects in oxygen consumption might affect brain tissue PO2. We used two different types of probes for measuring the partial pressure of oxygen in the brain–an amperometric Clark electrode, and an optical probe whose phosphorescence is sensitive to oxygen levels (see STAR Methods). Using a Clarke electrode, we stereotaxically monitored cerebral PO2 in the hypothalamus (as a representative brain region that was easily accessible) of anesthetized KO and WT mice as a function of age (Figure 2C). In 30-day-old KO mice, the partial pressure of oxygen (PO2) in brain tissue was significantly higher than in WT mice (51 versus 31 mmHg, p = 0.003). Remarkably, KO mice showed an age-dependent increase in brain tissue PO2, reaching 64 mmHg at 40–50 days of age (p < 0.001 versus WT mice and p = 0.04 versus 30-day-old KO mice). Once again, age-dependent changes in brain tissue PO2 appear to correlate with the disease severity in KO mice. We then asked how breathing various oxygen concentrations translated into changes in brain PO2. We monitored brain PO2 in the vestibular nuclei of anesthetized KO and WT mice while FiO2 was increased from 11% to 100%. These measurements were made using an optical probe. For any given FiO2, brain PO2 was significantly higher in late-stage KO mice versus agematched WT mice. Indeed, 11% FiO2 in KO mice resulted in an equivalent brain PO2 as 21% FiO2 in WT mice (Figure 2D).
We determined the dose-response relationship between FiO2 and survival of Ndufs4 KO mice. Combining previously published data (Ferrari et al., 2017) with newly generated survival curves, we show a remarkable drop-off in survival at 11%–13% FiO2 (Figure 2F). Survival between 17% and 55% FiO2 also shows a clear worsening in the disease state. Therefore, it is likely that localized and age-dependent changes in brain PO2 result in tissue hyperoxia, which directly causes brain tissue damage.
Previous studies have demonstrated a strong dependence of isolated mitochondrial superoxide production on local O2 concentrations (Kussmaul and Hirst, 2006; Stepanova et al., 2019). We asked whether this relationship varied between KO and WT tissue. We collected forebrain and midbrain tissue from ~60-day-old mice from each genotype, isolated mitochondria, and assessed hydrogen peroxide production as a function of local oxygen tension. We modulated oxygen concentration in the medium with respiring mitochondria while simultaneously measuring the resulting H2O2 production. Using malate and pyruvate as a substrate combination for complex I, we find that this relationship is comparable in KO and WT mice (Figure 2E). Therefore, for a given oxygen tension, mitochondria from both genotypes produce an equivalent amount of H2O2. In principle, the increased brain PO2 observed in KO mice is predicted to provide a higher rate of ROS production at a fixed FiO2.
Collectively, these findings suggest that impaired complex I activity in aged KO mice results in relative tissue hyperoxia, presumably due to a mismatch between oxygen delivery and its utilization. Future work is needed to understand how this elevated oxygen pressure results in the observed neuroinflammatory signature and neuronal cell death.
Chronic Hypoxia Normalizes Brain Po2 in Ndufs4 KO Mice
In the context of acute hypoxia exposure, we observed a decrease in brain PO2 in both WT and KO mice (Figure 2C). We asked whether a similar effect would be observed in mice chronically exposed to 11% oxygen. During chronic hypoxia a compensatory increase in Hb concentration and ventilation may preserve oxygen delivery to the brain with no change in brain PO2. We exposed both WT and KO mice to 11% O2 and we measured brain PO2 after three weeks. All hypoxic mice had a lower brain PO2 as compared to mice breathing 21% O2 with the same genotype (17 mmHg versus 30 mmHg, p = 0.0007 in WT and 29 versus 64 mmHg, p < 0.0001 in KO, Figure 2D). As we observed in the acute setting, chronic 11% O2 exposure in KO mice resulted in a brain PO2 similar to WT mice breathing air. These results show that chronic hypoxic breathing normalizes the brain PO2 of Ndusf4 KO mice despite an increase in the circulating Hb level (Jain et al., 2016).
CO Decreases Oxygen Content, Normalizes Brain Tissue PO2, and Rescues Brain Disease
If brain PO2 is a key parameter dictating neurological disease progression in the mouse model of Leigh syndrome, other interventions that lower brain PO2 should similarly rescue neurodegenerative disease. Hypoxia breathing reduces arterial oxygen content by lowering both arterial oxygen saturation and the partial pressure of arterial oxygen (PaO2). A well-known mode of altering tissue oxygenation is exposure to carbon monoxide (Blumenthal, 2001). Carbon monoxide binds to hemoglobin with a 200-fold increased affinity relative to oxygen, outcompeting the binding pocket for oxygen. In doing so, CO displaces oxygen from hemoglobin, forming carboxyhemoglobin (COHb) and lowering arterial oxygen saturation (O2Hb). CO also increases the affinity of hemoglobin for oxygen, reducing the off-loading of bound oxygen to the tissues. We decided to test whether breathing a high but sublethal concentration of CO in 21% O2 could rescue neurological disease in Ndufs4 KO mice.
Exposure to chronic, continuous 600 ppm CO in air resulted in ~40% arterial COHb and ~60% arterial O2Hb (Figure 3C). Of note, breathing 11% O2 results in a similar arterial O2Hb saturation of 55%–60%. After three weeks of CO treatment hematocrit showed a marked increase from 48% to 79% in WT and from 53% to 76% in KO mice, suggesting this dose was sufficient to cause renal hypoxia and erythropoietin production (Figures 3A and 3B). CO treatment was started in KO mice at a late-stage of brain disease, after body weight loss had already occurred and MRI lesions were apparent. Upon commencing 600 ppm CO treatment, body weight gradually regained, similar to the trend observed during chronic inhaled hypoxia treatment in previous experiments (Figure 3D) (Ferrari et al., 2017). Moreover, survival was substantially prolonged in KO mice with CO treatment, with a median survival of ~150 days (Figure 3E). We additionally performed MRI in late-stage mice before and after treatment with CO. Remarkably, CO treatment for 2 to 3 weeks completely reversed lesions in the vestibular nucleus (Figure 3F). Since chronic 11% O2 breathing normalized the relative brain hyperoxia observed in KO mice breathing room air, we asked whether CO treatment similarly acted by reducing brain tissue PO2. Both WT and KO mice chronically breathing CO at 600 ppm in air had a lower brain PO2 as compared to WT and KO mice breathing air (19 versus 30 mmHg, p = 0.0008 in WT and 36 versus 64 mmHg, p = 0.001 in KO, Figure 3G), based on measurements using an optical probe. The results show that chronic administration of CO at 600 ppm reduces brain PO2, improves survival, and reverses brain disease in Ndufs4 KO mice.
Figure 3.

Breathing Carbon Monoxide Reduces Brain Hyperoxia and Reverses Disease in Ndufs4 KO Mice
(A) Hemoglobin and (B) Hematocrit (mean ± SEM) of WT and KO mice exposed to air or ~600 ppm carbon monoxide in air for ~3 weeks. Unpaired t test p value shown within inserts.
(C) Percentage of carboxyhemoglobin (mean ± SEM) of WT mice breathing 11% FiO2 versus 600 ppm CO.
(D) Body weight trajectory of mice exposed to preventative (gray) hypoxia therapy, late-stage hypoxia therapy (black), or late-stage carbon monoxide therapy (red). (n = 8 for KO prevention in gray, n = 8 for KO disease reversal with hypoxia breathing in black, and n = 6 for KO disease reversal with CO breathing). Note: hypoxia body weight curves taken from historical data previously reported in (Ferrari et al., 2017) and adapted for this figure.
(E) Survival of mice breathing room air (historical data from (Ferrari et al., 2017) versus 600 ppm CO in room air. Log-rank p value shown within insert. n = 12 for CO survival in red, n = 14 for mice breathing room air in black.
(F) Reversal of T2-weighted MRI lesions in mice exposed to 600 ppm CO starting a late stage of disease. Pre-treatment scans on left, post-treatment of same mice after 2 to 3 weeks of CO on right. Three rows are three individual mice.
(G) Brain (vestibular nucleus) PO2 of WT and KO (mean ± SD) mice breathing 21% FiO2, 11% FiO2, or 600 ppm CO in 21% O2. (data for WT taken from same data as Figure 2).
(H) T2-weighted MRI scan sections through late-stage KO mouse breathing room air versus lesions observed in KO mouse chronically exposed to 600 ppm CO.From left to right in 3H, lesions correspond to top 1 (vestibular nucleus), bottom 2 (red nucleus), bottom 4 (caudoputamen) and top 5 (olfactory bulb). Typical disease lesions in vestibular nucleus shown with red arrow. New lesions in CO-treated KO mice shown with orange arrow.
Of course, CO presents a significant health hazard and high doses can result in death or permanent neurological dysfunction (Blumenthal, 2001). We asked whether KO mice that were exposed to chronic low-dose CO were able to chronically maintain healthy brain tissue, both in the regions affected by CI deficiency and more generally. We scanned KO mice that had been exposed to CO for 2 to 3 months. While the Leigh’s disease lesions were not present, these mice did develop hyperintense lesions apparent in the caudoputamen region of the brain, presumably due to chronic CO treatment (Figure 3H). Lesions that are present in humans that have suffered from CO poisoning can be found in different anatomical structures, such as the globus pallidus. However, this pattern is not completely stereotyped and can vary between individuals or states of poisoning (Jeon et al., 2018; O’Donnell et al., 2000). Although inhaled CO is unlikely to be utilized clinically as a therapeutic strategy given its remarkably narrow therapeutic index, these data (Figure 3) provide valuable proof of concept that reducing brain oxygen delivery alleviates the progression of Leigh disease in this mouse model.
Severe Anemia Can Reverse Disease Progression in Ndufs4 KO Mice
The ability of CO treatment to rescue disease serves as a proof of concept that additional therapeutic regimens which affect oxygen delivery might also be protective. In theory, affecting several physiological parameters (arterial Hb saturation, Hb content, cardiac output, oxygen consumption, etc.) should also lower brain PO2 and rescue disease (Figure 4A). We explored an additional mode of decreasing oxygen delivery– extreme anemia achieved by phlebotomy in combination with dietary iron deficiency.
Figure 4.

Severe Anemia Decreases Brain Oxygenation and Rescues Neurological Disease in the Ndufs4 KO Mouse
(A) Schematic of variables affecting tissue oxygenation. Theoretical therapeutic interventions shown in red, physiological variables represented in blue or black.
(B) Gradual decrease in hemoglobin following serial phlebotomy every 2 to 3 days for ~20 days, in combination with an Fe-deficient diet (mean ± SEM, n = 7).
(C) Brain PO2 (vestibular nuclei) of WT and KO mice that are untreated or made anemic using phlebotomy, in combination with an Fe-deficient diet (WT data same as Figure 2).
(D) Survival of untreated (Ferrari et al., 2017) or anemic Ndufs4 KO mice.
(E) MRI of anemic Ndufs4 KO mice at 80 days or 170 days of age. Note: Mice scanned in left and right panels are distinct. Rows correspond to different mice to show reproducibility. Red arrow depicts lesions in vestibular nuclei.
One simple strategy to decrease oxygen delivery is to limit the number of circulating red blood cells. In humans, chronic anemia causes weakness, lethargy, and reversible hair loss (Lopez et al., 2016). However, phlebotomy remains the first line therapy for a small number of diseases, including certain forms of polycythemia and hemochromatosis. We therefore phlebotomized our KO mice. Mice were bled ~200–400 microliters by tail vein, 5 to 6 times with 2 to 3 days in between bleeding sessions to allow blood volume to recover. Mice were also placed on an Fe-deficient diet to prevent the regeneration of RBCs. Using this therapeutic regimen, we were able to cause extreme anemia in KO mice, reaching an average Hb of 2 to 3 g/dL (Figure 4B). While this is an extreme level of anemia, mice appeared relatively normal and active even at these low levels of Hb. To determine the effects of anemia on brain PO2, we once again used the optical probe to measure cerebral oxygenation in WT and KO mice that were untreated or subjected to our therapeutic anemia protocol. Similar to CO treatment and inhaled hypoxia, anemia reduced the brain hyperoxia observed in Ndufs4 KO mice, down to WT levels (Figure 4C). This anemia protocol extended the life of KO mice from a median of 55 days [4] to a median of ~130 days (Figure 4D). Moreover, MRI lesions were not apparent at 50–70 days of age when they normally appeared in untreated mice. However, brain lesions did become apparent closer to death at ~150 days (Figure 4E). The ability of this relatively simple procedure—severe anemia—to rescue disease, further establishes proof of concept that brain tissue oxygenation is a key parameter in the pathogenesis of neurological disease in Leigh syndrome.
DISCUSSION
Chronic, continuous 11% O2 breathing results in a striking rescue of brain disease in the Ndufs4 KO mouse model of Leigh syndrome (Jain et al., 2016; Ferrari et al., 2017). Our initial studies were motivated by a CRISPR/Cas9 screen that spotlighted the hypoxia response and suggested that shifting metabolism from oxidative phosphorylation toward glycolysis can alleviate growth defects in cultured cells. In cell culture, forced activation of this pathway is sufficient to buffer the growth defects in cells with impaired electron transport chains (Jain et al., 2016; Li et al., 2018). However, subsequent clues suggested that this pathway may be insufficient for the hypoxic rescue in vivo of the Ndufs4 KO mouse. For example, intermittent hypoxia and more moderate hypoxia were able to trigger HIF-dependent hematocrit elevation but did not result in even an intermediate rescue (Ferrari et al., 2017). In our current work, we show that genetically activating the hypoxia transcriptional program by decreasing or eliminating activity of PHD1/2/3 or vHL is insufficient to rescue brain disease, even though canonical HIF targets are activated. While our work strongly argues against sufficiency of HIF activation for the rescue of Leigh syndrome, we have not tested the necessity of this pathway for rescue by hypoxia.
Not only were the genetic crosses we performed unable to rescue brain disease, but in some cases disease progression was hastened. At a systemic level, hypoxia adaptations mediated by HIF include several homeostatic mechanisms to increase oxygen delivery and dampen oxygen consumption. Thus, if these pathways are activated in normoxic conditions, they may paradoxically increase the amount of local brain tissue PO2 (the key culprit identified in the current study) thereby exacerbating disease. One caveat is that the PHD enzymes are also involved in oxygen sensing more broadly. For example, PHD1 inhibition is protective during ischemia stroke (Quaegebeur et al., 2016). In this case, the proposed mechanism is shunting of glucose toward the pentose phosphate pathway and increased NADPH production for ROS-scavenging enzymes. Several other HIF-independent, PHD-dependent adaptations have been proposed including an increase in flux through gluconeogenesis. If PHD enzymes have redundant roles in HIF-independent hypoxia adaptations, it is possible that distinct hypoxia adaptations contribute to disease rescue. While our studies demonstrate that genetic activation of the hypoxia response is not sufficient to rescue Leigh syndrome, we cannot exclude the possibility that the vHL-PHD-HIF program is necessary for some aspects of hypoxia’s in vivo rescue of disease trajectory.
Mitochondria are widely regarded as the energy-producing organelles of the cell and the textbook explanation for mitochondrial disease is centred on defective ATP production. However, in most tissues mitochondria are also the primary oxygen-consumers, allowing intracellular oxygen tensions to remain low and preventing the toxic oxidation of intracellular biomolecules. In fact, humans with mitochondrial disease are known to exhibit venous hyperoxia and impaired oxygen extraction which is most apparent during exercise (Taivassalo et al., 2002), and this venous hyperoxia has even been proposed as a biomarker for the disease. We show that with advanced disease, CI deficiency results in a decline in whole body oxygen consumption. This is associated with elevated brain tissue PO2, which is normalized by chronic hypoxic exposure. A recent study showed that the defect in CI-mediated electron transport occurs in a regional and temporal manner in the Ndufs4 KO mouse. Kayser et al. showed that CI activity is relatively well-preserved in young mice but declines with age (Kayser et al., 2016). It is tempting to speculate that a vicious cycle could lead to the end pathology: an initial defect in mitochondrial CI could lead to local hyperoxia that exacerbates mitochondrial dysfunction, creating a feed-forward loop of ETC damage. Since whole body oxygen consumption declines with age, it is likely that other tissues are also experiencing relative hyperoxia in KO mice.
The current study proposes that in the face of mitochondrial dysfunction, unused oxygen can accumulate and this can be directly toxic. Oxygen toxicity is traditionally associated with the damaging effects of superoxide radicals or hydrogen peroxide. In principle, the generation of such reactive oxygen species should be higher in the brains of Ndufs4 KO mice, as both NADH and O2 can contribute to the generation of toxic reactive oxygen species (Kussmaul and Hirst, 2006; Hirst et al., 2008). Complex I deficiency leads to a direct increase in NADH/NAD+ ratio and as we have shown in the current paper, will lead to increased brain O2 tensions in vivo. Moreover, an impaired ETC with structural defects may cause increased one-electron leak to molecular oxygen. However, we have shown that isolated mitochondria from KO and WT mice produced similar amounts of H2O2 at a given oxygen tension, when respiring on malate and pyruvate. If ROS are the culprit species, KO mice may be producing greater amounts of ROS simply because more oxygen is available as a substrate. An alternative hypothesis for oxygen toxicity is that the increased oxygen levels may be directly oxidizing and damaging biomolecules, e.g., those containing Fe-S clusters or iron centers, without the need to invoke the formation of toxic reactive oxygen species, as we have recently shown for the Fe-S cluster biosynthetic machinery itself (Ast et al., 2019). Consistent with this perhaps unconventional hypothesis for oxygen toxicity, various antioxidant strategies have been tested in mitochondrial diseases with little or no proven benefit. It is possible that hypoxia is more effective than antioxidants, since it prevents oxygen toxicity at its root base. Further work is needed to determine the nature of dioxygen toxicity in the in vivo setting.
A key finding from the current work is that very different strategies that reduce brain oxygenation are protective in the Ndufs4 KO mouse model of Leigh syndrome. Our original work (Ferrari et al., 2017; Jain et al., 2016) utilized hypoxic gas mixtures to create normobaric inhaled hypoxia. Here, we have reported the use of two different strategies to reduce oxygen delivery to also alleviate disease in this mouse model. First, we have demonstrated that lowering brain tissue PO2 by using CO to decrease arterial oxygen content (without altering arterial PO2), is sufficient to rescue disease (Figure 3). Second, we have used severe anemia (through a mix of an iron deficient diet and phlebotomy) to reduce oxygen delivery to the brain (Figure 4). Both of these interventions are severe, but serve as important proof of concept that decreasing arterial oxygen content (rather than arterial PaO2) or overall oxygen delivery should rescue disease by lowering tissue PO2. Although low-dose CO (250 ppm) is known to have anti-inflammatory effects and is in phase 1 clinical trials for idiopathic pulmonary fibrosis (Rosas et al., 2018) in humans, its therapeutic index is too narrow and hence unlikely to serve as a candidate therapeutic in humans. Similarly, although phlebotomy is used as a therapy for other disorders, including hemochromatosis, its extension to mitochondrial disorders will be challenging, as anemia is known to be a risk factor for worse prognosis in certain inherited mitochondrial diseases, including POLG deficiency (Hikmat et al., 2017). While hypoxia breathing, inhaled CO, and severe anemia are each individually highly pleiotropic and could be having a beneficial effect on the Ndufs4 KO mice by distinct mechanisms, the most parsimonious explanation is that they are all alleviating Leigh syndrome by directly reducing brain oxygen tensions.
We anticipate that other approaches, such as small molecule HIF inhibitors, now in trials for renal cell cancer (Martínez-Sáez et al., 2017), may be more practical to limit an adaptive increase in RBC production, or orally available oxy-Hb left-shifters, which are now in clinical trials for sickle cell anemia Oksenberg et al., 2016, are worthy of future exploration. Further understanding of the precise mechanisms by which elevated tissue oxygenation contributes to end-organ pathology in mitochondrial disease may lead to additional novel and practical combination therapies that are needed to treat Leigh syndrome.
Limitations of Study
(1) Our study demonstrates that HIF activation is not sufficient to rescue complex I deficiency in mouse models. However, it is possible that HIF is still necessary. Future studies will be required to determine whether HIF is necessary for the inhaled hypoxia rescue mechanism in vivo. (2) We show increased oxygen levels in the brains of the Ndufs4 KO mouse model. While this suggests oxygen toxicity, we have not directly measured toxic reactive oxygen species in vivo. We have also not determined the exact nature of the damage (e.g., which enzymes and biomolecules are failing). (3) Our current work suggests that therapeutic strategies which decrease oxygen delivery might be protective in this mouse model. However, future studies are needed to optimize such a therapeutic strategy and investigate any long-term negative side effects.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vamsi K. Mootha (vamsi@hms.harvard.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal work in manuscript was approved by the Subcommittee on Research Animal Care and the Institutional Animal Care and Use Committee of Massachusetts General Hospital. Ndufs4−/− were obtained from Palmiter laboratory (University of Washington). Mice lacking Phd1 (Aragoné s et al., 2008), nestin-Phd2 (Mazzone et al., 2009) and Phd3 (Bishop et al., 2008) were provided by the Carmeliet laboratory (KU Leuven). VhlLch/ch mice were provided by Josef Prchal (University of Utah). Genetic crosses were performed by crossing Ndus4+/+ mice with remaining strains. Throughout this study, Ndufs4+/+ and Ndufs4+/− mice were indiscriminately used as healthy controls, since there is no apparent phenotypical difference between these genotypes. When possible, controls from the same genetic cross were used, in order to make comparisons between reasonably similar genetic backgrounds. Pups were genotyped and weaned from mothers at 25–30 d of age. Cages and water were changed weekly. Humane euthanasia criteria were used throughout the study. Natural death or 20% body weight loss were used to generate survival curves. Animals were housed in either conventional or barrier housing. Males and females were used throughout the study. The NDUFS4, PHD1 and PHD3 KO strain are C57/BL6. The nestin-PHD2 strain is mixed background (C57/BL6 and 129S6).
METHOD DETAILS
Experiments were conducted in experimental and technical replicates (sample size described throughout figures). Mice were randomized within each genotype. Careful consideration was used to ensure groups were balanced for gender, age and starting weight. When possible, samples were blinded. For example, survival curves, body weight measurements, brain PO2 measurements, etc. were all made blinded. Sample sizes and power calculations were based on data from our previous studies using the Ndufs4 KO mouse model (Ferrari et al., 2017; Jain et al., 2016).
Respirometry
Whole body oxygen consumption was monitored using the Sable system (Promethion Cages) continuously from ~30 d to ~45 d of age. During the experiment, mice were continuously provided food/water and exposed to a day/night cycle of 12h/12h. A smoothening window of 3h was used for each timepoint during the analysis. Oxygen consumption values were normalized to body weight.
H2O2 Assay
A high-resolution respirometer (O2k, Oroboros Instruments GmbH) equipped with home-built two-channel fluorescence optical module was used to monitor simultaneously oxygen concentration and H2O2 production as described in detail in [14]. The module was connected to the amperometric port of the respirometer and the fluorescence was recorded using the Datlab software. Mitochondrial H2O2 release was measured as the fluorescence of resorufin formed from Amplex UltraRed in the presence of horseradish peroxidase (excitation 525nm, emission filter 580 nm cut off). The fluorescence signal was calibrated into an H2O2 concentration by addition of 90nM of H2O2 to the chamber at the end of each experiment. To assess H2O2 release at different oxygen level, humidified gaseous pre-purified argon was purged into the 1ml gas headspace of respirometer chamber at a flow rate of 10–60ml/min to rapidly decrease oxygen concentration in the measuring assay.
Intact brain mitochondria (0.1–0.5 mg/ml) were added to assay media containing 125 mM KCl, 14 mM NaCl, 0.2 mM EGTA, 4 mM KH2PO4, 2 mM MgCl2, 20 mM HEPES-Tris (pH 7.4), supplemented with 2 mg/ml BSA, 10 mM Amplex UltraRed, 4 U/ml horseradish peroxidase, and 40 U/ml Cu/Zn superoxide dismutase at 37° C. Combination of 2 mM malate and 5 mM pyruvate was used to reduce matrix NADH for respiration mediated by CI. Pyruvate is oxidized by pyruvate dehydrogenase generating NADH and acetyl-CoA which reacts with matrix oxaloacetate to shift the malate dehydrogenase reaction towards oxidation of malate and generation of another NADH molecule.
Brain Tissue PO2 Measurement
Ndufs4−/− and WT mice were anesthetized with Isoflurane (induction at 2–4%, maintenance at 1–1.5%), intubated, and mechanically ventilated with a tidal volume of 8 ml/kg, a respiratory rate of 110 breaths per minute and an inspired fraction of oxygen (FiO2) of 21%. Mice were placed in a prone position and the head was stabilized using a stereotaxic frame (ASI Instruments, MI). After incision and dissection of the skin, an opening in the skull was performed using a micro-drill (MD-1200, Braintree Scientific, MA) and a PO2 probe was inserted at the desired location. The coordinates for the thalamic measurements were mediolateral [ML] = −1.00 mm, anterioposterior [AP] = −2.00 mm, and dorsoventral [DV] = −4.00 mm from the bregma, while the coordinates for the vestibular nuclei were ML = −1.25 mm, AP = −6.00 mm, and DV = −3.90 mm from the bregma. For the thalamic measurements a flexible polarographic Clarktype oxygen microprobe was used (LICOX; GMS, Kiel-Mielkendorf, Germany). For the vestibular nuclei measurements an optical PO2 probe was employed (OxyLab, Oxford Optronix, Abingdon, UK). During the brain PO2 measurement the depth of anesthesia was reduced by lowering the Isoflurane concentration to 0.5–1% to minimize the impact of anesthesia on the brain PO2. During measurements mice were ventilated with various gas mixtures according to the experimental group: 21% O2, 11% O2, 600 ppm CO balanced air. In some experiments the FiO2 was modulated over the course of the test and the brain PO2 was simultaneously measured.
Hematocrit and COHb Measurements
Eighty microliters of blood were collected by tail snip into a heparinized capillary. Hemoglobin concentration, hematocrit and the saturation of hemoglobin with oxygen (O2Hb) and carbon monoxide (COHb) were measured using a blood gas analyzer (ABL800 FLEX, Radiometer, Copenhagen, Denmark).
Chambers for Various Chronic Breathing Regimens
Mice were housed in 80-L transparent acrylic boxes. Control chambers were provided with a constant flow of medical air at 21% FiO2. A concentration of 11% or 17% oxygen was obtained by mixing medical air with nitrogen obtained from a nitrogen generator (MAG-20; Higher Peak). A concentration of 600 ppm CO was obtained by mixing medical air with CO from a cylinder containing 2% CO (20,000 ppm) (Airgas). The total gas flow through the chamber was adjusted to maintain chamber CO2 concentrations below 0.4% (CO200; Extech). Soda lime was added to the chambers as a CO2 scavenger. The oxygen concentration was measured at the outlet port of the chamber with an O2 sensor (MiniOx 1; Ohio Medical), which was calibrated using an 8.55% O2 reference tank (Airgas). In a similar way the CO concentration was measured a CO sensor (MSA Altair PRO; MSA Safety, Pittsburgh, Pa), which was calibrated using a 1000-ppm CO reference cylinder (Airgas). Oxygen levels inside the chambers were tolerated within a 0.4% offset from the target concentration by adjusting nitrogen flow as needed. Temperature was maintained at 24–26C, and humidity was maintained at 30–70%. A standard light-dark cycle of ~12h light exposure was used. Mice were housed in cages with standard bedding and given unlimited access to food and water.
Anemia
To induce anemia, mice were placed on a low iron diet. Since the time to achieve adequate levels of anemia would have been too long and KO mice would have died of their mitochondrial disease, 150–200 μL of blood were collected every other day until hemoglobin concentration reached the desired values. Within 2 weeks, hemoglobin concentrations reached approximately 3–4 g/dL and remained stable while the mice remained on a low iron diet.
MRI
MRI scans of the brain were performed as previously described (Ferrari et al., 2017), using respiratory gated T2-weighted RARE (rapid acquisition of refocused echoes) MRI images acquired on a 4.7-T small animal scanner (Pharmascan; Bruker) with the following parameters: RARE factor: 10, echo time: 60 ms, repetition time: 6000 ms, Averages: 8, 192 × 192 × 24 image matrix with a voxel size of 0.130 × 0.130 × 0.7 mm. Mice were under anesthesia (isoflurane between 0.5–1.5% in room air) during the imaging procedure. Images were generated using a DICOM reader (OsiriX, University of Geneva).
qPCR
Animals were sacrificed by CO2 asphyxiation followed by cervical dislocation. Individual cerebella were immediately harvested and snap frozen in liquid N2. The tissue was then disrupted with two 5mm stainless steel beads (Qiagen) using a Qiagen TissueLyser for 2 mins at 25 Hz. RNA was extracted with the RNeasy Lipid Tissue Mini Kit (Qiagen ), before murine leukemia virus (MLV) reverse transcription using random primers (Promega). qPCR was performed using the TaqMan technology (Life Technologies), using the following probes:
Mm00833882_m1 (EPOR)
Mm00437306_m1 (VEGFA)
Mm01612132_g1 (LDHA)
Mm00441480_m1 (SLC2A1)
Mm01202755_m1 (EPO)
Mm00437762_m1 (B2M)
All data were normalized to B2M (Mm00437762_m1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analyses were performed using GraphPad Prism 7.0 software. The two-sample Student t-test was used for two-group comparisons. Unless otherwise indicated, one-way ANOVA with Bonferroni’s correction was used for multiple comparisons. The log-rank test was performed, and HRs with 95% CIs were calculated to compare survival rates. A p value < 0.05 was considered to indicate statistical significance. Data are reported as mean ± SD.
DATA AND CODE AVAILABILITY
There are no relevant datasets or code in this study.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mannitol | Sigma | Cat # 63559 |
| Bovine Serum Albumin | Sigma | Cat # A6003 |
| Digitonin | Sigma | Cat # D141 |
| Horseradish Peroxidase | Thermo Fisher Scientific | Cat #012001 |
| EPOR TaqMan Assay | Thermo Fisher Scientific | Mm00833882_m1 |
| EPO TaqMan Assay | Thermo Fisher Scientific | Mm01202755_m1 |
| VEGF TaqMan Assay | Thermo Fisher Scientific | Mm00437306_m1 |
| LDHA TaqMan Assay | Thermo Fisher Scientific | Mm01612132_g1 |
| SLC2A1 TaqMan Assay | Thermo Fisher Scientific | Mm00441480_m1 |
| Critical Commercial Assays | ||
| TaqMan Gene Expression Master Mix | Thermo Fisher Scientific | Cat #4369016 |
| M-MLV Reverse Transcriptase | Promega | Cat # M1701 |
| Amplex UltraRed | Thermo Fisher Scientific | Cat # A36006 |
| Qiagen RNeasy Lipid Tissue Mini Kit | Qiagen | Cat # 74804 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ndufs4−/− | Richard Palmiter | N/A |
| Mouse: Phd1−/− | Peter Carmeliet | N/A |
| Mouse: Nestin-Phd2−/− | Peter Carmeliet | N/A |
| Mouse: Phd3−/− | Peter Carmeliet | N/A |
| Mouse Vhlch/ch | Josef Prchal | N/A |
Context and Significance.
Leigh syndrome is a devastating, inherited mitochondrial disease characterized by marked neurological and metabolic deficits and early death. At present, there are no effective, approved therapies. Here, the authors show, in a mouse model, that excess, unused oxygen plays a key role in the pathology of Leigh disease. Multiple interventions that decrease oxygen delivery to the brain can normalize the elevated oxygen concentration and reverse the disease. While many pathologies are associated with reduced tissue oxygen (e.g., stroke and heart attack), this appears to be one of the first associated with excessive oxygen. These findings suggest a new mechanism of action for mitochondrial disease and may aid in the design of future therapies based on the control of oxygen delivery.
Highlights.
The Ndufs4 KO mouse model of mitochondrial Leigh syndrome exhibits brain hyperoxia
Genetically activating the hypoxia transcriptional response is not beneficial
CO treatment and anemia reverse disease by decreasing oxygen delivery
ACKNOWLEDGMENTS
We thank Alex Soukas for providing access to metabolic cages that were supported by an NIH shared instrumentation grant #1S10OD020112-01. We thank Celeste Simon and Josef Prchal for providing the Vhl Chuvash mice and Richard Palmiter for providing the Ndufs4 mice. We thank William Kaelin for useful discussions and advice on this project. The work in the A.G. lab was partially supported by MRC UK grant MR/L007339/1 and by NIH grant NS-100850. This project was supported by a fund from the Marriott Family Foundation (V.K.M.). V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
V.K.M., W.M.Z., I.H.J., and L.Z. are listed as inventors on a patent application submitted by the Massachusetts General Hospital on technology described in this paper. V.K.M. is a paid advisor to Janssen Pharmaceuticals, Raze Therapeutics, and 5AM Ventures.
REFERENCES
- Aragoné s J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, et al. (2008). Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 40, 170–180. [DOI] [PubMed] [Google Scholar]
- Ast T, Meisel JD, Patra S, Wang H, Grange RMH, Kim SH, Calvo SE, Orefice LL, Nagashima F, Ichinose F, et al. (2019). Hypoxia rescues frataxin loss by restoring iron sulfur cluster biogenesis. Cell 177, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, et al. (2008). Abnormal sympathoadrenal development and systemic hypotension in PHD3/ mice. Mol. Cell. Biol. 28, 3386–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal I (2001). Carbon monoxide poisoning. J. R. Soc. Med. 94, 270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, and Zapol WM (2017). Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. 114, E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, et al. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MM, Lam JC, Bezman NA, Rathmell WK, and Simon MC (2007). Von Hippel–Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2a signaling and splenic erythropoiesis. J. Clin. Invest. 117, 3879–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikmat O, Tzoulis C, Klingenberg C, Rasmussen M, Tallaksen CME, Brodtkorb E, Fiskerstrand T, McFarland R, Rahman S, and Bindoff LA (2017). The presence of anaemia negatively influences survival in patients with POLG disease. J. Inherit. Metab. Dis. 40, 861–866. [DOI] [PubMed] [Google Scholar]
- Hirst J, King MS, and Pryde KR (2008). The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 36, 976–980. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, and Kaelin WG Jr. (2001). HIFa targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SB, Sohn CH, Seo DW, Oh BJ, Lim KS, Kang DW, and Kim WY (2018). Acute brain lesions on magnetic resonance imaging and delayed neurological sequelae in carbon monoxide poisoning. JAMA Neurol. 75, 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Sedensky MM, and Morgan PG (2016). Region-specific defects of respiratory capacities in the Ndufs4 (KO) mouse brain. PLoS One 11, e0148219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussmaul L, and Hirst J (2006). The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake NJ, Bird MJ, Isohanni P, and Paetau A (2015). Leigh syndrome: neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 74, 482–492. [DOI] [PubMed] [Google Scholar]
- Li X, Cui XX, Chen YJ, Wu TT, Xu HX, Yin H, and Wu YC (2018). Therapeutic potential of a prolyl hydroxylase inhibitor FG-4592 for Parkinson’s diseases in vitro and in vivo: regulation of redox biology and mitochondrial function. Front. Aging Neurosci. 10, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez A, Cacoub P, Macdougall IC, and Peyrin-Biroulet L (2016). Iron deficiency anaemia. Lancet 387, 907–916. [DOI] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, and Simon MC (2010). Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Sá ez O, Gajate Borau PG, Alonso-Gordoa T, Molina-Cerrillo J, and Grande E (2017). Targeting HIF-2 a in clear cell renal cell carcinoma: a promising therapeutic strategy. Crit. Rev. Oncol. Hematol. 111, 117–123. [DOI] [PubMed] [Google Scholar]
- Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, de Almodovar CR, et al. (2009). Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Buxton PJ, Pitkin A, and Jarvis LJ (2000). The magnetic resonance imaging appearances of the brain in acute carbon monoxide poisoning. Clin. Rad. 55, 273–280. [DOI] [PubMed] [Google Scholar]
- Oksenberg D, Dufu K, Patel MP, Chuang C, Li Z, Xu Q, Silva-Garcia A, Zhou C, Hutchaleelaha A, Patskovska L, et al. (2016). GBT 440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br. J. Haematol. 175, 141–153. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A, Segura I, Schmieder R, Verdegem D, Decimo I, Bifari F, Dresselaers T, Eelen G, Ghosh D, Davidson SM, et al. (2016). Deletion or inhibition of the oxygen sensor PHD1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell Metab. 23, 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich P (2003). Chemiosmotic coupling: the cost of living. Nature 421, 583. [DOI] [PubMed] [Google Scholar]
- Rosas IO, Goldberg HJ, Collard HR, El-Chemaly S, Flaherty K, Hunninghake GM, Lasky JA, Lederer DJ, Machado R, Martinez FJ, et al. (2018). A phase ii clinical trial of low-dose inhaled carbon monoxide in idiopathic pulmonary fibrosis. Chest 153, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova A, Konrad C, Manfredi G, Springett R, Ten V, and Galkin A (2019). The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J. Neurochem. 148, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taivassalo T, Abbott A, Wyrick P, and Haller RG (2002). Venous oxygen levels during aerobic forearm exercise: an index of impaired oxidative metabolism in mitochondrial myopathy. Ann. Neurol. 51, 38–44. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, and Semenza GL (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There are no relevant datasets or code in this study.