

SUMMARY
Single-cell screens enable high-throughput functional assessment of enhancers in their endogenous genomic context. However, the design of current studies limits their application to identifying the primary gene targets of enhancers. Here, we improve the experimental and computational parameters of single-cell enhancer screens to identify the secondary gene targets of enhancers. Our analysis of >500 putative enhancers in K562 cells reveals an interwoven enhancer-driven gene regulatory network. We find that enhancers from distinct genomic loci converge to modulate the expression of common sub-modules, including the α- and β-globin loci, by directly regulating transcription factors. Our analysis suggests that several genetic variants associated with myeloid blood cell traits alter the activity of a distal enhancer of MYB (~140 kb away), with downstream consequences on hemoglobin genes expression and cell state. These data have implications for the understanding of enhancer-associated traits and emphasize the flexibility of controlling transcriptional systems by modifying enhancer activity.
Graphical Abstract
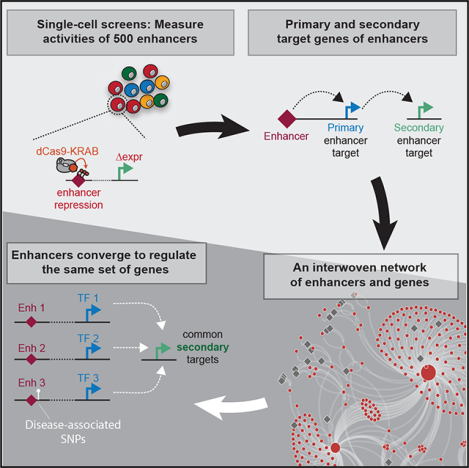
In Brief
Xie et al. apply improved strategies for single-cell screens to identify an enhancer-driven transcriptional regulatory network in K562 cells. They demonstrate that the same group of genes can be indirectly regulated by enhancers from distinct genomic loci. These data have implications for the understanding of enhancer-associated traits.
INTRODUCTION
Enhancers orchestrate gene expression programs critical to development (Levine, 2010), and genetic variants of enhancers are implicated in human disease risk (Akhtar-Zaidi et al., 2012; Franco et al., 2018; Harismendy et al., 2011). An ongoing goal has been to assign genome-wide association study (GWAS)-linked enhancers with the genes mediating disease risk. Current efforts have focused largely on identifying the direct target genes of enhancers, using methods including computational correlation of epigenetic modifications (Andersson et al., 2014; Shen et al., 2012), three-dimensional chromatin looping (Fang et al., 2016; Mumbach et al., 2016), and high-throughput perturbation of enhancers (Gasperini et al., 2019; Xie et al., 2017). However, these approaches rely on the critical assumption that the direct target gene of an enhancer plays the central role in disease risk. In contrast, evidence suggests that indirect targets can also play important roles. For example, a risk-associated enhancer for persistent fetal hemoglobin alters the expression of the transcriptional repressor BCL11A (Bauer et al., 2013; Canver et al., 2015; Uda et al., 2008) (Figure 1A). However, it is not BLC11A per se that is directly responsible for the associated phenotype. Rather, it is de-repression of the BCL11A target gene fetal hemoglobin that elicits the phenotype (Liu et al., 2018). Here, we use the terms “direct,” “primary,” and “local” interchangeably to refer to target genes in close physical proximity to an enhancer (±2 Mb). Conversely, we use the terms “indirect,” “secondary,” and “global” interchangeably to refer to other enhancer targets.
Figure 1. Reducing False-Positive Hits in Single-Cell Enhancer Screens.

(A) Schematic of primary and secondary targets of enhancers. An enhancer directly regulates the expression of a primary target, which then regulates expression of a secondary target gene.
(B) A single-cell enhancer screen of 500 enhancers with ten sgRNAs per enhancer.
(C) Shown is the genomic neighborhood near ARL15 and a proximal enhancer targeted for dCas9-KRAB-mediated repression.
(D) Manhattan plot represents uncorrected p values of genes upon suppression of ARL15-enh. The y-axis represents the raw p values. Genes are ordered on the basis of the positions on chromosomes (x-axis). The vertical dashed line indicates the position of target regions. For visualization purposes, all genes with zero log10(p value) not shown.
(E) MA plotfrom bulk RNA-seq in which ARL15-enh is repressed. The MA plot compares ten sgRNAs with two negative control sgRNAs. Red and blue transparent dots represent significantly up- or downregulated genes, respectively. Solid red/blue dots represent hits that overlap with Mosaic-seq.
(F) Examples of significance scores(SS) from three genes. Histograms show the distribution of background p values from 10,000 random iterations. Vertical black dashed lines indicate the 99.9th percentile. Red dashed lines indicate observed p values. Top two genes have positive SS; bottom two genes are negative controls.
(G) As in (D), after SS-correction. For visualization purposes, only genes with SS greater than zero are shown.
As the above example illustrates, the assignment of GWAS-associated enhancers to causal target genes, whether they are direct or indirect, remains a challenge. Functionally assessing the primary and secondary target genes of risk-associated enhancers by direct experimental perturbation can be a viable strategy. We recently reported a high-throughput single-cell enhancer screening approach called Mosaic-seq (Xie et al., 2017), which uses CRISPR/dCas9-KRABto perturb enhancers and assess their endogenous functions. However, because of the small number of single guide RNAs (sgRNAs) used in current applications of single-cell enhancer screens (Gasperini et al., 2019; Xie et al., 2017), they are limited to the identification of direct enhancer target genes.
Here, we optimize the parameters of single-cell enhancer screens to enable the identification of secondary enhancer targets. By constructing enhancer-driven regulatory networks (ERNs), we show that secondary enhancer targets often converge onto a small number of genes. We use these networks to link genetic variants to myeloid blood cell traits.
RESULTS
A Dataset to Benchmark Experimental Parameters of Single-Cell Enhancer Screens
We performed Mosaic-seq to test the activities of 518 enhancers in K562 cells. To enable downstream analysis to benchmark the experimental parameters of single-cell enhancer screens, we created a high-quality dataset in which each enhancer was perturbed by a large number of sgRNAs (10 per enhancer) and in many cells (median 1,276 cells/enhancer) sequenced to high depth (average 7,785 unique molecular identifiers [UMIs]/cell) (Figure 1B). We targeted 10 sgRNAs in a ±200 bp region around each enhancer, for a total of 5,170 sgRNAs, including positive and negative controls (Table S1). We performed single-cell RNA sequencing (scRNA-seq) of 106,670 cells and identified sgRNAs expressed in each cell (Table S2). Overall, we sequenced a median of 1,276 perturbed cells per enhancer (Figure S1A).
Reducing False-Positive Hits in Single-Cell Enhancer Screens
Previous single-cell enhancer screens focused on identifying primary target genes in close physical proximity to perturbed enhancers. By reducing the search space for target genes, this approach reduces the problem of multiple hypothesis testing. However, we observed that simply applying these statistical procedures to test for differential expression across all genes genome-wide and over all perturbed enhancers yielded poor results. For example, our single-cell screen targeted a putative enhancer 16 kb upstream of the ARL15 gene (denoted ARL15-enh) (Figure 1C). We performed differential expression analysis on cells receiving sgRNAs targeting ARL15-enh compared with other cells and observed that ARL15 as well as a neighboring long noncoding RNA (IncRNA), RP11–461C13.1, were both repressed upon ARL15-enh repression (Figure 1D). In addition, this basic analysis suggested that many genes across the genome could also be differentially expressed upon ARL15-enh repression. To test these single-cell results, we repeated the experiments in bulk by generating cells lines for each of the 10 sgRNAs targeting ARL15-enh. Traditional bulk RNA sequencing (RNA-seq) confirmed repression of ARL15 and RP11–461C13.1, but most other hits were not recovered (Figure 1E), suggesting that successfully identifying secondary enhancer targets will likely require different computational strategies.
We noticed that differential expression analysis on cells derived from 10 randomly selected sgRNAs could yield significant p values (Figure 1F), perhaps because of the stochasticity of single-cell RNA-seq measurements and the relatively small number of cells sequenced per sgRNA. Some genes were more prone to this phenomenon, which motivated the need for gene-specific correction. Therefore, we developed a simulation-based strategy to model the significance of each gene individually and correct for false discovery (Figure 1F). We denote these adjusted p values as “significance scores” (SS). Applying this procedure to ARL15-enh, we observe a significant reduction in false positives while maintaining true positives (Figures 1G and S1E–S1G). Applied to the full dataset, we identified 94 enhancers in which recruitment of dCas9-KRAB caused a significant change of expression of a direct gene target (Figures S1B–S1D; Table S3). Overall, we identified 127 enhancer-gene pairs (false discovery rate [FDR] ~0.03). In agreement with previous observations (Gasperini et al., 2019; Xie et al., 2017), several independent lines of evidence support these enhancer hits: (1) hits are highly enriched for known markers of active enhancers including H3K27ac, p300, and RNA polymerase II (Figures S1H and S1I); (2) we observe that 74% of hits regulate one local gene (Figure S1J) and 84% regulate the closest expressed gene; and (3) most hits regulate genes less than 400 kb away (Figure S1K).
Increasing the Number of sgRNAs Reduces False-Positive Hits in Single-Cell Enhancer Screens
Next, we optimize the experimental parameters for robust identification of enhancer targets (Figure S2). One key parameter of single-cell enhancer screens is the number of sgRNAs used to perturb each enhancer. Early single-cell enhancer screens used only two or three sgRNAs per enhancer. However, as some sgRNAs have off-target or non-target activity, approaches using fewer sgRNAs could dilute signal or lead to false positives (Figures 2A and 2B). For example, if only two sgRNAs are designed per enhancer, and one has off-target activity, then determining correct target genes can be difficult. However, if ten sgRNAs are designed per enhancer, statistical signal from more frequent on-target sgRNAs can be expected to exceed that of off-target sgRNAs.
Figure 2. Increasing the Number of sgRNAs Reduces False-Positive Hits in Single-Cell Enhancer Screens.

(A) Improving signal-to-noise with more sgRNAs in single-cell screens. Increasing the number of sgRNAs targeting the enhancer increases the number of sequenced on-target cells relative to off-target cells.
(B) Overview of the computational simulation strategy. For each enhancer, we randomly discarded one to nine sgRNAs and replaced them with an equal number of random sgRNAstargeting other enhancers. Each replacement was repeated 100times, and significance scores(SS) were calculated for the top local hit of the enhancer.
(C) Scatterplot summarizing simulation results for 94 enhancers with local hits. Colors indicate the number of on-target sgRNAs required to identify the gene.
(D) Changes of SS for three example enhancers. Solid dots represent the median of 100 iterations. Transparent dots represent all iterations. Dashed boxes indicate parameters allowing known target genes to be identified (SS > 0).
See also Figure S2.
To estimate the number of functional sgRNAs needed to confidently identify enhancer targets, we performed computational simulations. For a given enhancer targeted by ten sgRNAs, we simulated loss of signal by randomly replacing one to nine sgRNAs with non-targeting sgRNAs. We performed this analysis for each of the 94 enhancers with local hits, over 100 iterations per sgRNA replacement (Figure 2C). As expected, we find that SS are higher with more on-target sgRNAs, suggesting that multiple sgRNAs support the same hit (Figure 2D). In addition, while the number of sgRNAs required to identify a hit varies depending on the target, all hits require at least three sgRNAs. Thus, with our experimental parameters, at least 30% of sequenced cells must support a gene target to confidently call a hit. Conversely, this analysis also suggests that if >30% of sequenced cells support an off-target, then the off-target could be falsely called a hit. As it is unlikely for multiple sgRNAs to share off-targets, to reduce the possibility of false positives, we recommend designing at least four sgRNAs for each targeted region. This guideline to increase specificity effectively dilutes the signal from each sgRNA to less than 30%, such that true hits must be supported by multiple sgRNAs.
Identifying Secondary Target Genes of Enhancers
When an enhancer’s primary target is a DNA binding transcription factor (TF) as defined by Gene Ontology, we define the enhancer’s secondary targets as the target genes of the TF. We focused on these annotated TFs because they are well-studied regulators. Previous single-cell enhancer screens have focused on identifying primary target genes of enhancers. Our unique experimental approach to target each enhancer with ten sgRNAs, combined with new analysis procedures, increases the specificity of hits and enables the confident identification of secondary enhancer targets.
For each enhancer with a primary gene target, we performed genome-wide identification of secondary gene targets (Figures 3A and 3B; Figures S3A–S3D; Table S3). As expected, we observe that the strongest hits for most enhancers correspond to primary gene targets, with few secondary targets (Figure S3E). In contrast, we find that enhancers regulating TFs as primary targets can also have many secondary targets across the genome (Figures 3A and 3B; Figures S3A–S3D). For example, an enhancer ~140 kb downstream of MYB has 191 secondary gene targets, in addition to primary targets MYB and AHI1. Importantly, many of the secondary targets for this enhancer are the known downstream targets of MYB, such as KCNH2, ALAS2, and the hemoglobin genes (Lorenzo et al., 2011).
Figure 3. Global Analysis Reveals Enhancer-Driven Regulatory Networks (ERNs).

(A) Genome Browser snapshots illustrate the local epigenetic features of several targeted enhancers near LMO2, NFE2, and MYB/AHI1.
(B) Similar to Figure 1G. In addition, the dot size represents fold change.
(C) An enhancer-driven regulatory network (ERN) consists of an enhancer’s primary (direct) targets and its secondary (indirect) targets.
(D) An example ERN.
(E) ERN sub-network highlighting PTRF, CD24, and VIM as common secondary targets of enhancers across the genome.
Several lines of evidence support our identification of secondary enhancer targets. First, enhancers from the same locus share significantly more global targets than enhancers from other loci (Figure S3E), which is consistent with the notion that they regulate the same primary target. Second, the results are consistent across multiple biological replicates. Third, secondary hits are consistent with those obtained from bulk RNA-seq validation experiments (Figure S1G; Table S4). However, consistent with previous publications, the false-negative rate of single-cell screens is high (> 80%), particularly for lowly expressed genes (Dixit et al., 2016; Kharchenko et al., 2014; Xie et al., 2017). As scRNA-seq technology matures to enable higher quality data with increased sensitivity and reduced cost, we expect detection rates to improve. Together, our data demonstrate that both primary and secondary target genes can be specifically identified in single-cell enhancer screens.
An ERN Converges onto Common Gene Targets
Traditional gene regulatory networks consist of linkages between TFs and the genes they regulate. Our analysis identifying each enhancer’s primary and secondary gene targets motivated us to construct an ERN. In an ERN, nodes represent enhancers and genes. Edges connect (1) enhancers with their primary gene targets (TFs) and (2) these TFs with their direct targets (which are secondary enhancer targets) (Figure 3C). Importantly, each edge in an ERN is supported by direct experimental perturbation through the single-cell enhancer screen.
Constructing an ERN on all 94 enhancers with primary/secondary targets, we identified 37 connected subnetworks. More than half of the enhancers belong to one subnetwork, which consists of 410 nodes (49 enhancers and 361 genes) and 853 edges (76 primary and 777 secondary edges) (Figure 3D). Within this subnetwork, 10 enhancers regulate the expression of several TFs: MYB, CBFA2T3 (also known as ETO2), LMO2, and NFE2. Each of these TFs is the primary target of at least one distinct enhancer.
Strikingly, we observe that the secondary enhancer targets in this subnetwork converge onto a common set of significantly overlapping genes (Figures 3E and 4A; Table S3; STAR Methods). Notably, these include members of the α/β-globin gene clusters, whose expression is tightly regulated in the hemoglobin-producing K562 cell line. By examining the ERN, we observe several mechanisms through which enhancers indirectly regulate globin expression. For example, enhancers of NFE2 and LMO2 act as activators of hemoglobin genes. This is consistent with the known roles of NFE2 and LMO2 as components of the GATA1/TAL1 complex that binds locus control regions to regulate hemoglobin genes (Goardon et al., 2006; Moi et al., 1994; Song et al., 2010) (Figure 3D). In contrast, enhancers of MYB and CBFA2T3 function as suppressors of hemoglobin expression. This is consistent with observations that c-Myb suppresses hemoglobin gene expression indirectly, possibly via KLF1 activation of BCL11A (Bauer and Orkin, 2011; Bianchi et al., 2010).
Figure 4. Linking Distal MYB Enhancers to Blood-Associated Traits.

(A) ERN sub-network highlighting hemoglobin genes as common secondary targets of enhancers across the genome.
(B) MYB-enh-3 exhibits DNase I hypersensitivity in CD34+ common myeloid progenitor cells. SNPs and associated traits are highlighted.
(C) Gene set enrichment analysis (GSEA) of the DEGs upon MYB-enh-3 knockdown. Genes upregulated are positively correlated with hematopoietic differentiation genes.
(D) Heatmap indicates the expression pattern of genes that are identified in (C). Several hematopoietic differentiation genes are indicated.
See also Figure S4.
Highlighting the complexity of the system, we observe that the same enhancers can serve opposing roles in the secondary regulation of different genes. For example, another group of common secondary enhancer targets consists of CD24, VIM, and PTRF, which are positively regulated by enhancers of MYB and CBFA2T3 but negatively affected by enhancers of LMO2 (Figure 3E).
Together, these results indicate that enhancer-driven networks converge onto common gene targets, and they can be used to propose hypotheses of enhancer activity on secondary gene targets.
Genetic Variants of a MYB Enhancer Influence Blood Cell State through Secondary Targets
To highlight one potential application of ERNs, we focus on enhancers of MYB (Figure 4A). GWAS studies have identified variants in the MYB locus that are associated with blood cell differentiation (Figure 4B). As some of these variants are located near enhancers within the gene body of AHI1, previous studies have suggested that these variants affect AHI1 expression (Astle et al., 2016; Kichaev et al., 2019; Sud et al., 2017). However, as our direct perturbation data indicate that these enhancers regulate MYB, we instead hypothesize that these genetic variants alter MYB expression, with secondary consequences on blood cell state.
Focusing on MYB-enh-3, we find several lines of evidence supporting this hypothesis. First, to determine the cellular context in which MYB-enh-3 is active, we examined published epigenetic data (Kundaje et al., 2015). Among normal human blood cell types, we find that MYB-enh-3 exhibits DNase I hypersensitivity specifically in CD34+ common myeloid progenitor cells (Figures 4B and S4D). Consistently, this region is highly conserved among all vertebrates (Figure S4A). In mice, this region exhibits DNase I hypersensitivity in liver-derived myeloid progenitor cells (Figure S4B). These data suggest that MYB-enh-3 is active in the context of myeloid progenitor cells.
Second, to assess whether MYB is a primary gene target of MYB-enh-3 in this cellular context, we examined whether DNase I hypersensitivity at MYB-enh-3 correlates with MYB activity. Indeed, we find that myeloid progenitor cells, which have the most open chromatin at MYB-enh-3, also exhibit the greatest expression of MYB among blood lineages (Figures S4C and S4D). Additionally, in the context of myelogenous K562 cells, three-dimensional chromatin conformation analysis indicates that MYB-enh-3 loops to the MYB gene (Figure S4E). Together, these data support our direct perturbation experiments that MYB-enh-3 regulates MYB in myeloid cells.
Third, we assessed if the secondary gene targets of MYB-enh-3 are consistent with the phenotypes associated with these variants such as red cell distribution width and mean corpuscular hemoglobin. Consistently, the ERN suggests that MYB-enh-3 regulates erythrocytic gene clusters including hemoglobin genes and glycophorins (GYPA, GYPB, and GYPC) as secondary targets through a MYB-dependent mechanism (Figure 4A).
More generally, the phenotypes associated with these variants reflect changes in myeloid cell state. Indeed, it has been previously reported that repression of MYB is necessary for the final stages of erythroid maturation (Weiss et al., 1994), and mouse studies have shown that knockout of MYB leads to impaired proliferation and accelerated differentiation of blood cell lineages (Lieu and Reddy, 2009). Consistently, we observe that direct repression of MYB-enh-3 in K562 cells decreases cell proliferation (Figure S1A), and perturbed cells adopt an altered cellular state in which hematopoietic differentiation genes are coordinately activated (Figures 4C and 4D).
Taken together, ERN analysis suggests that genetic variants near MYB-enh-3 may contribute to blood cell traits by reducing the activity of MYB-enh-3, with consequent reduction in the primary target gene MYB and changes in the expression of secondary targets, with impacts on cell state. Additional experiments will be required to test this hypothesis.
DISCUSSION
Secondary enhancer targets can contribute to disease risk, and identifying these targets can illuminate disease-associated molecular mechanisms (Bauer and Orkin, 2011). Although single-cell enhancer screens can reliably identify primary targets (Gasperini et al., 2019; Xie et al., 2017), current studies lack the statistical power to confidently identify secondary targets. Here, we find that increasing the number of sgRNAs in enhancer perturbations can remedy this problem. As it is unlikely that multiple sgRNAs support the same off-targets, diluting signal across many sgRNAs amplifies on-target signal and decrease off-target signal.
Even though we targeted enhancers throughout the genome without a priori knowledge about their potential primary targets, we observed that many enhancers often share the same set of secondary target genes. We show that this is mediated through the enhancers’ primary TF targets. Thus, enhancers and their primary/secondary gene targets form a highly connected regulatory network. One implication of this network structure is that there are many ways in which genetic variants of enhancers can indirectly contribute to disease through secondary target genes. One notable example is the GWAS-associated enhancer for BCL11A, which indirectly controls the expression of fetal hemoglobin (Bauer et al., 2013; Canver et al., 2015; Liu et al., 2018; Uda et al., 2008). One caveat to our approach is that, if our analysis identifies multiple primary targets as TFs, we cannot distinguish which of these TFs regulate secondary targets. Similarly, our approach cannot distinguish secondary from tertiary or higher order interactions.
Our data underscore the complexity of understanding functional consequences of enhancer-related traits. Direct perturbation of GWAS-associated enhancers holds potential to identify other molecular mechanisms through which secondary enhancer targets contribute to human disease.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be addressed to and will be fulfilled by Lead Contact Gary Hon (Gary.Hon@utsouthwestern.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
Human K562 cells (Female) were cultured in IMDM with 10% FBS and pen/strep at 37°C and 5% CO2, and HEK293T cells (Female) were cultured in DMEM with 10% and pen/strep. Both cells were purchased from ATCC and are tested monthly to ensure they are mycoplasma free.
METHOD DETAILS
Selection of Candidate Regions and sgRNAs
Since scRNA-Seq has lower sensitivity than bulk RNA-Seq, changes of expression for lowly expressed genes are less likely to be detected in Mosaic-seq. Therefore, targeting enhancers that are close to highly expressed genes will increase the chances of detecting hits. Thus, we selected all the topological associated domains (TADs) which have at least one gene with CPM > 50 in K562 cells. We identified 1021 candidate TADs from a total list of 2290, and only focused on enhancers located within these candidate TADs. The putative enhancers targeted in this study are defined by DNase-seq peaks which are at least 2kb away from any annotated TSS and that also harbor H3K4me1 signal (CPM > 1, which is commonly used as a marker for poised or active enhancers).
We used a mixed strategy to select candidate enhancers. About half of the enhancers were unbiasedly chosen from DNase I hypersensitive sites on chromosome 18 and the remaining half were selected on the basis of strong p300 enrichment, a well-known marker of enhancer activity (Visel et al., 2009; Xie et al., 2017). We filtered our target regions based on two different criteria. First, we selected all putative enhancers on chromosome 18 that are within candidate TADs. Second, we biased toward strong enhancers. As p300 was the best predictor for enhancer activity from our small scale Mosaic-seq experiment (Xie et al., 2017), we selected enhancers with strong p300 enrichment. We also manually added control regions including promoters or enhancers identified in our previous study. In total, we targeted 267 regions on chromosome 18 and 251 regions on other chromosomes.
Plasmids and sgRNA Library Construction
We used lenti-dCas9-KRAB-blast for dCas9-KRAB expression (Addgene ID: 89567) and CROPseq-Guide-Puro plasmid for sgRNA expression (Addgene ID: 86708). For sgRNA library construction, a single-strand sgRNA oligo library containing 5166 sgRNAs (Table S1) was synthesized by Custom Array. The library was amplified by NEBNext High-Fidelity 2X PCR master mix (New England Biolabs) to make it double-stranded and then was inserted into the BsmBI digested CROPseq-Guide-Puro plasmid through Gibson Assembly (New England Biolabs). Then the circulized product was purified and electroporated to Endura ElectroCompetent cells (Lucigen) following the manufacturer’s protocol. The cells were then cultured in LB medium with 100μg/ml Ampicillin at 30°C overnight and the plasmid was extracted using the ZymoPURE plasmid maxiprep kit (Zyme Research). We also designed primers (Table S1) to amplify the spacer sequences of the sgRNA library and verified the complexity of the library by Illumina sequencing. See our previous publication for a full protocol (Xie and Hon, 2019).
Virus Packaging, Titration and Infection
The virus-related methods have been previously described (Xie and Hon, 2019). Briefly, the Ientivirus packaging plasmids MD2.G and psPAX2 (Addgene ID 12259 and 12260) were co-transfected with the carrier plasmid to HEK293T cells with linear polyethylenimine (PEI, Polysciences). Supernatant was collected 72 h after transfection and filtered with a 0.45μm filter. The virus was further purified by using Lenti-X Ientivirus concentrator (Clontech). For virus titration, we performed infection of K562 cells with serially diluted virus, and measured the survival rate of the cells after the antibiotic selection. For the single-cell enhancer screen, we performed infection of K562 cells in 24-well. We tried different multiplicity of infection (MOIs) in order to maximize the number of sgRNAs we could test per cell. We found a non-linear relationship between the MOI of virus and the actual number of sgRNA detected (Table S2). By using MOI = 2000, we were able to detect about 10 sgRNAs per cell.
Single-cell RNA-seq
All the single-cell RNA-seq libraries were prepared with the 10X Genomics Chromium 3′ V2 kit, following the standard protocol. To construct the sgRNA enrichment libraries, 20–30 ng of cDNA product was used to perform an enrichment PCR by the SP-RI primer and sgRNA enrichment primers. Then the PCR product was purified by performing a 0.8X SPRI beads cleanup. The expected size of the final library is about 500bp. See our previous publication for more details (Xie and Hon, 2019).
Quantitative Real-time PCR and Bulk RNA-seq
RNA was extracted by using TRIzol (Invitrogen), following the manufacturer’s instructions. For Real-time qPCR, the RNA was reverse transcribed using Maxima H Minus Reverse Transcriptase (Thermo Fisher) and then the relative expression of genes was quantified by qPCR, using the expression of ACTB as reference. For bulk RNA-seq, we followed the protocol from LM-seq (Hou et al., 2015). At least two biological duplicates were performed for each on-target / control sgRNA.
Sequencing
All libraries were sequenced on the Illumina NextSeq 500/550 with the High Output kit (~400M reads per Flowcell). For scRNA-seq and sgRNA libraries, we used paired-end sequencing with R1–26bp, R2–58bp and idx1–8bp. For all the other libraries, we used the following configuration: R1–48bp, R2–48bp, idx1–8bp. About 1.5 billion reads were sequenced in total, which yields an average of 7785 UMIs in each cell. All the sequencing data are available on GEO:GSE129837.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Pre-processing and Mapping
scRNA-seq libraries were demultiplexed and mapped to human reference genome (hg38) using the Cellranger software (ver 2.1.0, 10X Genomics), with expected cell number of 10,000 and default parameters. Bulk RNA-seq libraries were demultiplexed with Bcl2fastq (Illumina), mapped to hg38 reference genome by Star (Dobin et al., 2013). For the gene reference, we used Gencode V28 (Derrien et al., 2012). The sgRNA enrichment libraries were processed as previously described (Xie and Hon, 2019). Briefly, all potential cell barcodes and sgRNAs were extracted from read-1 and read-2 by comparing with the white lists of cell barcodes (output of Cellranger pipeline) and sgRNAs, respectively, with the edit distance of 1. Then, the sgRNA counts were summarized for each sequenced cell, and the UMIs are calculated by using the “directional” method described in UMI-tools (Smith et al., 2017).
Assigning sgRNAs to Cells
To accurately assign sgRNAs to each single cell, we used the saturation curve method described in Drop-seq (Macosko et al., 2015). Briefly, for a given sgRNA, we calculated the cumulative distribution of the UMIs from all cells. By identifying the inflection point of the curve, we adjusted the UMIs from cells after the inflection point to be zero. The sgRNAs with the adjusted UMI count greater than 0 are considered as true sgRNAs in this cell. The analysis was performed per library and then combined for downstream analysis. The details of this method are described previously (Xie and Hon, 2019).
Hits Calling
The gene expression matrices from individual batches were first normalized by the method described previously (Xie et al., 2017), and then combined to one matrix. For each enhancer region, we combined all 10 sgRNAs together to minimize the potential off-targeting of individual sgRNAs.
We use the hypergeometric test to evaluate whether the expression of a given gene is significantly changed upon the perturbation of sgRNA. We only consider genes that are expressed in at least 2% of the cell population. Let C represent all the cells analyzed; let Eg,c represent the expression of gene g in cell c; and let Cs represent the cells with the indicated sgRNA s. Given a given gene g and an sgRNA s, the parameters for the hypergeometric test are then:
In terms of the hypergeometric test, M represents the population size, N represents the number of successful events); K represents the number of trials; and x represents the number of successful trials.
The hypergeometric p values were then calculated by using these four parameters M, N, K, and x. The p values for the downregulated genes were calculated by using the survival function of the hypergeometric test and the p values for upregulated genes were calculated by using the cumulative density function (see the manual for scipy for more details: https://docs.scipy.org/doc/scipy-0.14.0/reference/generated/scipy.stats.hypergeom.html)
In order to correct the p values in a gene specific manner, we then introduced background p values for each gene (denoted as Pbg). To calculate Pbg, we randomly chose 10 sgRNAs from our library and perform the same hypergeometric test for all genes. This step was repeated 10,000 times to get the distribution of Pbg for all genes. For local hits calling (genes within ± 2Mbp), we calculated the 99.9th percentile Pbg for each gene (Pbg99,.9) and calculated the Significance Score (SS) of each gene as:
We defined any gene with SS > 0 as a local hit for targeted region. We estimated the average false discovery rate (FDR) for the local hits calling is about 0.03 false local hit per region. For global hits calling, we focused on the 94 regions with local hits. We calculated the SS for both the genes upregulated and downregulated across the genome. To further decrease the false discovery rate in the global analysis, we tested different cutoff values for SS. We chose SS = 2.8 for the upregulated genes and SS = 1.9 for the downregulated genes, and the average FDR is less than 0.1 false global hit per region.
Down-sampling Simulations
To assess the effect of sequencing depth and cell number on identifying enhancer targets, we performed computational simulations with down-sampled reads and cells, respectively. To simplify the computational process, for all the down-sampling simulations, we focused on the 127 local region-gene pairs identified in the previous local hits calling analysis. For down-sampling of cells, we randomly sampled a specified percentage of cells and calculated the hypergeometric p values of each region-gene pairs. Using the same down-sampled matrix, the 99.9th percentile of the background p values were estimated across 1000 iterations and the SS was calculated. For each specified down sampling percentage, we repeated the whole process 100 times to get 100 SSs for each region-gene pair. The region-gene pairs with median SSs above 0 were identified as hits. The down-sampling of reads was performed in a similar manner except that only 25 iterations were used.
We observe that read depth has a strong impact on the detection of low-confidence hits, and that a median depth of ~3000 UMIs/cell is sufficient to capture most high-confidence hits (Figure S2A). In contrast, sequencing fewer cells has similar impact on both high and low confidence hits (Figure S2B), with ~900 cells per enhancer sufficient to recover ~80% of all hits. Therefore, single-cell enhancer screens that aim to access high-confidence hits can decrease sequencing depth, while screens aiming to capture both high and low confidence hits require many cells to be sequenced at high depth.
Prediction of individual sgRNA Functions
With our experimental parameters, individual sgRNAs usually have few cells sequenced. Therefore, individual sgRNAs are unlikely to have the statistical power to accurately assign target genes. We reason that the efficacy of each sgRNA could be estimated by evaluating its performance when mixed with other sgRNAs targeting the same region. We speculate that functional sgRNAs with common targets will improve statistical significance, while non-functional sgRNAs that do not share common targets will decrease statistical significance. Based on this assumption, we enumerated the raw hypergeometric p values for all 1023 combinations of sgRNAs targeting each region (10 sgRNAs choose 2 minus 1). For a given sgRNA, we examined whether adding the sgRNA in combination with other sgRNAs improves the statistical significance of calling differentially expressed genes. For instance, if adding sgRNA-1 to a combination containing sgRNA-2 and −3 leads to an increase of -log10p-val greater than 0.1, this pair (from combo of sgRNA-2,3 to the combo of sgRNA-1,2,3) is denoted as ‘improved’; if the decrease of -log10p-val is greater than 0.1, it is denoted as ‘worsen’; otherwise it is denoted as ‘not changed’. Then, we counted the frequency of these three categories among all 511 pairs and used the binomial test to assess if any category was dominant. Regions with p-val < 0.05 were considered as either ‘functional” or ‘nonfunctional’, otherwise they were denoted as ‘ambiguous’.
Overall, we observe that two-thirds (66.7%) of sgRNAs exhibit on-target activity by this metric (Figure S2G). For example, our data indicate that, of the 10 sgRNAS targeting enhancers ARL15-enh, sgRNA 6 has weaker activity compared to the remaining sgRNAs (Figure S2D) (see Table S3 for denoted names of each region). This observation was confirmed by RNA-Seq experiments performed in bulk for each sgRNA (Figures S2E and S2F). Similarly, we confirmed that sgRNA-3 from MYB-enh 3 exhibits weaker activity (Figure S2D).
Mixing of sgRNAs from different loci
To test the off-targeting of sgRNAs, we performed computational simulations. For each enhancer with a primary hit, we randomly removed 1–9 sgRNAs and randomly added back the same number of sgRNAs from the pool of remaining sgRNAs. We repeated this analysis 100 times for each pairing of enhancers and number of sgRNAs removed (total of 900 simulations for each enhancer). The SS was calculated for primary target genes.
Network Analysis
A directed network was constructed by NetworkX (Hagberg et al., 2008), using both the local and global hits. Each enhancer node was first connected to its primary hit genes, and then the primary hit gene(s) was connected to the secondary hit genes. The network was then saved as a gml file and then imported to Cytoscape (Shannon et al., 2003) for visualization, using the Edge-weighted Spring Embedded layout with manual adjustment of a few overlapped nodes and edges. For visualization purpose, we deleted the nondetermining primary hits for the four major transcriptional factor regions (i.e., AHI1 for the MYB enhancers, NAT10 and TRIM44 for the LMO2 enhancers).
Identification of significantly overlapping genes in the network
We performed a binomial test to identify genes that are common secondary targets to enhancers. Given a set of enhancers E in the ERN (here 49) and a set of genes G, let M represent a matrix of dimension |E| × |G|, where Me,g = 1 if enhancer e regulates gene g and 0 otherwise. Then we define the binomial p as the fraction of 1’s in M. Next, we let the binomial n = |E|, and perform the binomial test binocdf(x, p, n) for each gene g where x is the number of times g is a target for all enhancers in E.
Analysis of the Bulk RNA-seq
After mapping and duplicates removal, featureCounts (Liao et al., 2014) was used to count the expression value of each gene. DEG analysis was performed by using edgeR (Robinson et al., 2010).
Meta-analysis with ENCODE data
All ChIP-seq and DNase-seq datasets were downloaded from ENCODE. PRO-seq, GRO-Cap (Core et al., 2014) and TT-seq (Schwalb et al., 2016) datasets are downloaded from previous publications. Read counts were summarized in a ± 1,200 bp region from the summit of DNase-seq peak by using featureCounts. The p values were calculated by the Wilcoxon rank-sum test.
GWAS Data
All the disease associated SNPs were downloaded from GWAS Catalog (Buniello et al., 2019). We displayed all the previously reported SNPs in the indicated regions.
GSEA analysis
All the GSEA analysis was performed by using GSEApy (Subramanian et al., 2005). Briefly, the read count table from featureCounts was normalized by CPM. For GSEA analysis, we use ‘signal_to_noise’ method with 1000 permutations.
DATA AND CODE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE129837. We have deposited example scripts to Github (https://github.com/russellxie/Global-analysis-K562-enhancers).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | Sigma | Cat#P8833 |
| Blasticidin | Invivogen | Cat#Ant-bl-1 |
| Critical Commercial Assays | ||
| LM-seq protocol | Hou et al., 2015 | |
| Maxima H Minus Reverse Transcriptase | Thermo Fisher | Cat#EP0753 |
| KAPA HiFi HS | KAPA | Cat#KK2502 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7572 |
| GE Healthcare Sera-Mag Magnetic SpeedBeads | Thermo Fisher | Cat#09-981-121 |
| QIAGEN MinElute PCR purification Kit | QIAGEN | Cat#28004 |
| Gibson Assembly Master Mix | NEB | Cat#E2611L |
| 10X genomics Chromium Single Cell 3′ Kit V2 | 10X Genomics | Cat#PN-120237 |
| 10X Genomics Chromium Single Cell A Chip Kit | 10X Genomics | Cat#PN-120236 |
| 10X Genomics Chromium i7 Multiplex Kit | 10X Genomics | Cat#PN-1000009 |
| Bacterial and Viral Strains | ||
| Endura ElectroCompetent Cells | Lucigen | Cat#60242–2 |
| Stellar Competent Cells | Clonetech | Cat#636766 |
| Deposited Data | ||
| Single-cell RNA-seq Data | This paper | GEO: GSE129837 |
| RNA-seq Data | This paper | GEO: GSE129837 |
| Experimental Models: Cell Lines | ||
| K562 cells | ATCC | ATCC CCL243 |
| 293T cells | ATCC | ATCC CRL-3216 |
| Recombinant DNA | ||
| Plasmid: pMD2.G | Addgene | Addgene 12259 |
| Plasmid: psPAX2 | Addgene | Addgene 12260 |
| Plasmid: lenti-dCas9-KRAB-Blast | Addgene | Addgene 89567 |
| Plasmid: CROPseq-Guide-puro | Addgene | Addgene 86708 |
| Sequence-Based Reagents | ||
| sgRNA Oligos | Table S1 | N/A |
| Custom primers for library construction | Table S1 | N/A |
| qPCR primers | Table S1 | N/A |
| Software and Algorithms | ||
| Star | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| edgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FeatureCounts | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| 10X Genomics Cellranger V2.1.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| NetworkX | https://networkx.github.io | |
| Cytoscape | https://cytoscape.org | |
| GSEApy | https://github.com/zqfang/GSEApy/blob/master/docs/introduction.rst | |
| Mosaic-seq pipeline | This Paper | https://github.com/russellxie/Global-analysis-K562-enhancers |
| Other | ||
| Illumina NextSeq 500 instrument | Illumina | N/A |
| Agilent 2200 TapeStation instrument | Agilent | N/A |
| Qubit Fluorometric Quantitation instrument | Thermo Fisher | N/A |
Highlights.
Improving single-cell screens to identify the secondary gene targets of enhancers
Applying single-cell screens to measure the activities of >500 enhancers
Enhancers across the genome converge to regulate the expression of common modules
Linking genetic variants at enhancers to trait-associated secondary gene targets
ACKNOWLEDGMENTS
This work is supported by the Cancer Prevention Research Institute of Texas (CPRIT) (RR140023 and RP190451 to G.C.H.), the NIH (DP2GM128203 to G.C.H.), the Department of Defense (PR172060 toG.C.H.), the Welch Foundation (I-1926-20170325 to G.C.H.), the Burroughs Wellcome Fund (1019804 to G.C.H.), the Harold C. Simmons Comprehensive Cancer Center, and the Green Center for Reproductive Biology. The authors also acknowledge University of Texas (UT) Southwestern McDermott Center for providing sequencing service and BioHPC for providing high-performance computing (HPC) and storage resources.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.073.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science 336, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. (2016). The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, and Orkin SH (2011). Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr. Opin. Pediatr 23, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, et al. (2013). An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E, Zini R, Salati S, Tenedini E, Norfo R, Tagliafico E, Manfredini R, and Ferrari S (2010). c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 116, e99–e110. [DOI] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47 (D1), D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, and Lis JT (2014). Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet 46, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 26, 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, Malladi VS, Li W, Xi Y, Richardson D, Allton KL, Tanaka K, Li J, Murakami S, et al. (2018). Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 28, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. (2019). A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goardon N, Lambert JA, Rodriguez P, Nissaire P, Herblot S, Thibault P, Dumenil D, Strouboulis J, Romeo P-H, and Hoang T (2006). ETO2 coordinates cellular proliferation and differentiation during erythropoiesis. EMBO J. 25, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg A, Swart P, and Chult SD (2008). Exploring Network Structure, Dynamics, and Function Using NetworkX (Los Alamos National Laboratory).
- Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu X-D, Topol EJ, Rosenfeld MG, and Frazer KA (2011). 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 470, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Jiang P, Swanson SA, Elwell AL, Nguyen BKS, Bolin JM, Stewart R, and Thomson JA (2015). A cost-effective RNA sequencing protocol for large-scale gene expression studies. Sci. Rep 5, 9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, and Scadden DT (2014). Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G, Bhatia G, Loh P-R, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, and Price AL (2019). Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet 104, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysisof 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M (2010). Transcriptional enhancers in animal development and evolution. Curr. Biol 20, R754–R763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lieu YK, and Reddy EP (2009). Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc. Natl. Acad. Sci. USA 106, 21689–21694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173, 430–42.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo PI, Brendeford EM, Gilfillan S, Gavrilov AA, Leedsak M, Razin SV, Eskeland R, Sæther T, and Gabrielsen OS (2011). Identification of c-Myb target genes in K562 cells reveals a role for c-Myb as a master regulator. Genes Cancer 2, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M,Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, and Kan YW (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U S A 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Ga gneur J, and Cramer P (2016). TT-seq mapsthe human transient transcriptome. Science 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Shannon P,Markiel A,Ozier O,Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, and Ren B (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S-H, Kim A, Ragoczy T, Bender MA, Groudine M, and Dean A (2010). Multiple functions of Ldb1 required for beta-globin activation during erythroid differentiation. Blood 116, 2356–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud A, Thomsen H, Law PJ, Försti A, Filho MIDS, Holroyd A, Broderick P, Orlando G, Lenive O, Wright L, et al. ; PRACTICAL consortium (2017). Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat. Commun 8, 1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, et al. (2008). Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. U S A 105, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MJ, Keller G, and Orkin SH (1994). Novel insights into erythroid development revealed through in vitro differentiation of GATA-1 embryonic stem cells. Genes Dev. 8, 1184–1197. [DOI] [PubMed] [Google Scholar]
- Xie S, and Hon GC (2019). Experimental and computational approaches for single-cell enhancer perturbation assay. Methods Mol. Biol 1935, 203–221. [DOI] [PubMed] [Google Scholar]
- Xie S, Duan J, Li B, Zhou P, and Hon GC (2017). Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol. Cell 66, 285–299.e5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE129837. We have deposited example scripts to Github (https://github.com/russellxie/Global-analysis-K562-enhancers).