

Abstract
The 18 kDa translocator protein (TSPO) is a widely used target for microglial PET imaging radioligands, but its expression in post‐mortem normal and diseased human brain is not well described. We aimed at characterizing the TSPO expression in human control (CTRL) and Alzheimer's disease (AD) brains. Specifically, we sought to: (1) define the cell type(s) expressing TSPO; (2) compare tspo mRNA and TSPO levels between AD and CTRL brains; (3) correlate TSPO levels with quantitative neuropathological measures of reactive glia and AD neuropathological changes; and (4) investigate the effects of the TSPO rs6971 SNP on tspo mRNA and TSPO levels, glial responses and AD neuropathological changes. We performed quantitative immunohistochemistry and Western blot in post‐mortem brain samples from CTRL and AD subjects, as well as analysis of publicly available mouse and human brain RNA‐Seq datasets. We found that: (1) TSPO is expressed not just in microglia, but also in astrocytes, endothelial cells and vascular smooth muscle cells; (2) there is substantial overlap of tspo mRNA and TSPO levels between AD and CTRL subjects and in TSPO levels between temporal neocortex and white matter in both groups; (3) TSPO cortical burden does not correlate with the burden of activated microglia or reactive astrocytes, Aβ plaques or neurofibrillary tangles, or the cortical thickness; (4) the TSPO rs6971 SNP does not significantly impact tspo mRNA or TSPO levels, the magnitude of glial responses, the cortical thickness, or the burden of AD neuropathological changes. These results could inform ongoing efforts toward the development of reactive glia‐specific PET radioligands.
Keywords: Alzheimer's disease, amyloid plaques, astrocytes, microglia, neurofibrillary tangles, neuroinflammation, peripheral benzodiazepine receptor, positron emission tomography (PET), rs6971 SNP, 18 kDa translocator protein (TSPO)
Abbreviations
- Aβ
amyloid β peptide
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- ANOVA
analysis of variance
- BA
Brodmann area
- CD31
cluster differentiation 31
- CD68
cluster differentiation 68
- CTRL
control
- DLB
dementia with Lewy bodies
- FPKM
fragments per kilobase of transcript per million mapped reads
- GFAP
glial fibrillary acidic protein
- HABs
high‐affinity binders
- LABs
low‐affinity binders
- MABs
medium/mixed affinity binders
- MADRC
Massachusetts Alzheimer's Disease Research Center
- mRNA
messenger ribonucleic acid
- MS
multiple sclerosis
- NA
numerical aperture
- NDS
normal donkey serum
- NFTs
neurofibrillary tangles
- NP
neuritic plaque
- PAGE
polyacrylamide gel electrophoresis
- PBR
peripheral benzodiazepine receptor
- PD
Parkinson's disease
- PET
positron emission tomography
- PHF1
paired helical filament 1
- PMI
post‐mortem interval
- RIN
RNA integrity number
- ROI
region of interest
- SMA
smooth muscle actin
- SDS
sodium dodecyl sulfate
- SNP
single nucleotide polymorphism
- TBI
traumatic brain injury
- TBS
Tris‐buffered saline
- TSPO
18 kDa translocator protein
Introduction
Besides amyloid plaques and neurofibrillary tangles, glial responses in the form of activated microglia and reactive astrocytes surrounding dense‐core amyloid plaques are part of the neuropathological landscape of the Alzheimer's disease (AD) brain 38, 39, 40, 41, 42. Accumulating evidence from neuropathology, genetics and experimental studies in mouse models has implicated microglial dysfunction in AD pathophysiology, so that interest in microglia and the role of neuroinflammation in AD is gaining momentum 37. The development of PET imaging radioligands to depict the microglial activation in vivo in human brain diseases is one of the ongoing efforts in microglia research 18, 50. The 18 kDa translocator protein (TSPO), formerly known as peripheral benzodiazepine receptor (PBR), has been proposed as a target for PET radioligands of microglial activation in a number of neurological conditions including acute brain injuries such as stroke and traumatic brain injury (TBI), autoimmune diseases such as multiple sclerosis (MS), and neurodegenerative diseases such as AD, amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD) and dementia with Lewy bodies (DLB). TSPO is a transmembrane protein located in the outer mitochondrial membrane and highly expressed in lungs, kidney, spleen and steroid‐producing organs such as adrenal glands and gonads, whereas its levels in the brain are relatively low. Its putative functions include neurosteroid synthesis and cholesterol transport to the mitochondria, whereas other potential functions remain under investigation 46.
Besides a qualitative study describing the expression of TSPO in activated microglial cells and astrocytes in post‐mortem brain specimens of patients with MS, TBI, HIV dementia and AD 8, little is known about the expression of TSPO in the post‐mortem brain in health and disease. Here we focused on characterizing the expression of TSPO in the healthy control (CTRL) and AD brain and attempted to answer the following five questions: (1) which cell type(s) express TSPO in the brain? (2) is tspo mRNA level increased in the AD brain? (3) is TSPO (protein) level increased in the AD brain? (4) do TSPO levels correlate with quantitative neuropathological measures of glial reaction and AD neuropathological changes? and (5) does TSPO rs6971 SNP have any impact on tspo mRNA and TSPO levels, glial responses or AD neuropathological changes?
Material and Methods
Nomenclature
In this paper, we use the following nomenclature: tspo for the mRNA, TSPO for the protein and TSPO for the gene. For clarity purposes, we will specify “tspo mRNA” when referring to the transcript, but will not specify “TSPO protein” when referring to the protein due to redundancy, as the acronym “TSPO” already includes the word “protein”.
Human post‐mortem brain specimens
8‐μm‐thick formalin‐fixed paraffin‐embedded sections from the temporal pole (Brodmann Area 38) and frozen cerebellar, temporal and frontal association cortex samples were obtained from the Massachusetts Alzheimer's Disease Research Center (MADRC) brain bank. AD subjects met clinical and neuropathological criteria for the diagnosis of AD, whereas control individuals did not meet pathological diagnostic criteria for any neurodegenerative disease. Subjects or their next of kin provided a written informed consent for the brain autopsy and this study was approved under the MADRC Brain Bank Institutional Review Board. The temporal association cortex was selected because it is a region of early amyloid plaque and NFT deposition, with prominent glial responses 38, 40, 41, 42. Conversely, the cerebellum was selected because it is relatively spared from AD neuropathological changes and glial responses 39 and has been often used as a reference region in TSPO‐based PET imaging studies in AD [reviewed in 20]. The frontal association cortex was chosen as another AD‐affected area to confirm some of the findings in the temporal cortex.
Brain protein extraction
Protein extracts from human brain samples were obtained by homogenization in 2% sodium dodecyl sulfate (SDS) buffer followed by ultracentrifugation to obtain the SDS‐soluble fraction. Briefly, small pieces of temporal and frontal cortex, and cerebellum (0.3‐0.4 g) were dissected, weighted and homogenized in 5 v/w of 2% SDS in Tris‐buffered saline (TBS) with protease inhibitors (Complete Mini Protease Inhibitor Cocktail from Sigma‐Aldrich, 04693124001‐Roche) using 5 mL of glass Dounce homogenizers with Teflon pestles inserted in a rotor and 25 strokes at low speed. Homogenates were incubated at 37ºC for 30 minutes and then ultracentrifuged in a Beckman ultracentrifuge using a 70.1Ti rotor at 43,000 rpm (≈100,000 g) at 20ºC for 30 minutes. The resulting supernatants were used as SDS‐soluble extracts, snap frozen in liquid nitrogen and stored at −80ºC until use for Western blot.
SDS‐PAGE and Western blot
The protein concentration of SDS‐soluble extracts was determined with the Pierce BCA Protein Assay Kit (Thermo Scientific, 23225) in a Perkin Elmer Wallac 1420 Victor Microplate Reader following manufacturer's instructions. NuPAGE LDS Sample Buffer (4×) (Thermo Scientific, NP007) and NuPAGE Reducing Agent (10×) (Thermo Scientific, NP009) were added to protein extracts, before boiling them at 95ºC for 5 minutes for protein denaturalization. 5 μg of protein per temporal cortex or cerebellar sample and 20 μg per frontal cortex sample were loaded into 17‐well 1.0‐mm‐thick NuPAGE 12% Bis‐Tris gels (Thermo Scientific, NP0343BOX). Precision Plus Dual Color Protein Standards (5 μL) (Bio‐Rad, 1610374) or SeeBlue Plus2 Pre‐stained Protein Standard (Thermo Scientific, LC5925) were used as molecular weight ladders. SDS‐PAGE was run in NuPAGE M.E.S. SDS running buffer (NP00202) at 120 V for 2 h in an XCell SureLockTM Mini‐Cell (Thermo Scientific, EI0001). Gels were transferred to nitrocellulose membranes (0.45 μm pore diameter, Thermo Scientific, 88018) using a wet method in an XCell IITM Blot Module (Thermo Scientific, EI9051) at 200 mA for 3 h. Membranes were blocked with Odyssey Blocking Buffer (TBS) (LI‐COR Biosciences, P/N 927‐50000) for 1 h at room temperature (RT), followed by incubation with primary antibodies in the Odyssey Blocking Buffer (TBS) overnight at 4ºC. Primary antibodies were rabbit anti‐PBR monoclonal antibody for TSPO (clone EPR5384, Abcam, ab109497, 1:1000) and chicken anti‐GAPDH polyclonal antibody (Millipore, AB2302, 1:5000) as housekeeping for loading control. On the next day, the membranes were washed with TBS‐Tween20 (2 × 10 minutes) and incubated with the secondary antibodies in the Odyssey Blocking Buffer (TBS) for 2 h at RT. Secondary antibodies were donkey anti‐rabbit IgG (H+L) IRDye 680RD (LI‐COR Biosciences, P/N‐925‐68073, 1:5000) and donkey anti‐chicken IgG (H+L) IRDye 800CW (LI‐COR Biosciences, P/N‐925‐32214, 1:5000). Finally, the membranes were washed in TBS‐Tween20 and TBS and scanned in the 2‐channel Odyssey Near‐Infrared Imaging System (LI‐COR Biosciences). Bands were quantified using the FIJI gel tool. The TSPO band was normalized to the housekeeping GAPDH band.
Importantly, this anti‐TSPO antibody is directed against the amino acid residues 150 to C‐terminal in TSPO and has been previously validated in tspo null mice 46. We confirmed the specificity of the antibody for TSPO by Western blot with recombinant TSPO peptide and tissues (brain, lungs and adrenal glands) from tspo null mice and floxed littermates kindly provided by Dr. Vimal Selvaraj (Cornell University, Ithaca, NY) (Figure S1).
Immunohistochemistry
TSPO immunohistochemistry was performed with the rabbit anti‐PBR monoclonal antibody from Abcam (clone EPR5384, ab109497) at a concentration of 1:100. Briefly, paraffin was cleared with xylenes and sections were rehydrated with decreasing concentrations of ethanol before an antigen retrieval step consisting of microwaving them in boiling citrate buffer 0.01 M at pH 6.0 with 0.05% Tween20 for 20 minutes at 95ºC. After cooling, sections were washed with TBS, blocked with 10% normal donkey serum (NDS) in TBS for 1 h at RT and incubated overnight at 4ºC with the above primary antibody in 5% NDS. On the next day, sections were washed thoroughly with TBS and incubated in secondary antibody solution consisting of donkey anti‐rabbit Cy3‐conjugated antibody (Jackson ImmunoResearch, 23225, 1:200) in 5% NDS in TBS for 2 h at RT, washed in TBS, covered with Fluoromount‐G mounting media with DAPI (Southern Biotech, 0100‐01) and the coverslips sealed with nail polish. Selected sections were subjected to double fluorescent immunohistochemistry with the above rabbit anti‐PBR monoclonal primary antibody and either a goat anti‐IBA1 polyclonal antibody (Abcam, ab107159, 1:100), a mouse anti‐cluster differentiation 68 (CD68) monoclonal antibody (clone KP‐1, Agilent Dako, M0814, 1:50), a mouse anti‐glial fibrillary acidic protein (GFAP) monoclonal antibody (clone G‐A‐5, Sigma, G3893, 1:1000), a mouse anti‐cluster differentiation 31 (CD31, also known as PECAM‐1) monoclonal antibody (clone 89C2, Cell Signaling Technology, 3528, 1:100), a mouse anti‐smooth muscle actin (SMA) monoclonal antibody (clone 1A4, Agilent Dako, M0851, 1:500), or a mouse anti‐HuC/HuD monoclonal antibody (clone 15A7.1, Millipore, MABN153, 1:100). For these primary antibodies, the species‐appropriate secondary antibody conjugated with AlexaFluor488 was obtained from Thermo Scientific. Nearly adjacent sections were stained for TSPO with the peroxidase‐DAB method using the Vectastain Universal Elite ABC HRP kit (Vector Labs, PK‐6200). Briefly, after clearance with xylenes and before rehydration with decreasing concentrations of ethanol, sections were treated with a solution of 0.3% hydrogen peroxide in methanol for 20 minutes to block the endogenous peroxidase activity. After antigen retrieval as above, sections were incubated with horse serum for 1 h at RT followed by overnight incubation at 4ºC with the same anti‐TSPO antibody at a concentration of 1:1000. On the next day, the sections were washed with TBS (2 × 10 minutes), incubated with the horse anti‐rabbit/mouse universal biotinylated secondary for 2 h at RT, washed again with TBS (2 × 10 minutes), incubated with the avidin–biotin complex (ABC) for 30 minutes for signal amplification, washed with TBS (5 minutes) and, finally, incubated with 3,3′‐diaminobenzidine (DAB) as a peroxidase substrate (Vector Labs., SK‐4100) for 1 minute to develop the TSPO brown signal. Sections were counterstained with Mayer's hematoxylin (Sigma, MHS32) and coverslipped with Permount mounting media (Fisher Scientific, SP15‐500). Similarly, immunohistochemistry for Aβ (mouse monoclonal anti‐Aβ antibody, clone 10D5, Elan‐Lilly, 1:50), GFAP (rabbit anti‐GFAP polyclonal antibody, Sigma, G9269, 1:1000), CD68 (mouse anti‐CD68 monoclonal antibody, clone KP‐1, Dako, M0814, 1:100) and phospho‐tau (mouse monoclonal anti‐TaupSer396/404 antibody, clone PHF1, Dr. Peter Davies' generous gift, 1:200) was performed with the DAB‐peroxidase method as described above and elsewhere 41. Negative control sections without primary antibodies were run in parallel and showed no staining (not shown).
Confocal microscopy
After double fluorescent immunohistochemistry, sections were imaged either on a Zeiss 510 Meta confocal laser scanning inverted microscope, which is equipped with a tunable argon (488 nm) laser, a helium‐neon (543 nm) laser, a femtosecond‐pulsed Chameleon Ti:Sapphire laser (Coherent Inc., Santa Clara, CA) for two‐photon excitation and a 63× water‐immersion objective (NA 1.4), or on an Olympus FV3000 confocal laser scanning inverted microscope, which is equipped with 405, 488, 561 and 640 nm lasers (Coherent OBIS, Santa Clara, CA) and a 60× oil‐immersion objective (NA 1.30).
Quantitative neuropathological methods
The TSPO burden in cortex and white matter was measured as the area occupied by TSPO‐immunoreactivity (TSPO‐ir burden). Briefly, TSPO‐immunostained sections were placed on the motorized stage of an Olympus BX51 epifluorescence microscope (Olympus, Tokyo, Japan) controlled by the stereology software CAST (Olympus, Tokyo, Japan). The cortex and the subjacent white matter were outlined in the DAPI channel under the 4× objective using the drawing tool of the software. Next, each of these regions of interest (ROIs) were randomly and systematically sampled under the 20× objective with a sampling fraction of 5% for the cortex and 10% for the white matter, and the resulting fields were photographed at the same exposure time of 300 ms. Images were then opened with the public domain software FIJI and converted to 8‐bit images; immunoreactive particles were highlighted with the “Threshold” tool and quantified using the “Analyze Particles” tool to obtain the area fraction (%) or TSPO‐ir burden. Measures were obtained by a single researcher (YG), who was blind to diagnosis. To account for possible batch differences in staining intensity, the TSPO‐ir burden from AD cases was normalized to the average TSPO‐ir burden of the CTRL cases within the same batch.
Stereology‐based quantification of GFAP+ reactive astrocytes, CD68+ activated microglia and PHF1+ NFTs, and amyloid burden and cortical thickness measurements were performed as described before 41. Briefly, GFAP+ astrocytes, CD68+ microglial cells and PHF1+ NFTs (both intra and extracellular) were counted using the optical disector probe of the BIOQUANT NOVA PRIME, version 6.90.10 (MBSR, Nashville, TN), which is coupled with the motorized stage of an upright Leica DMRB microscope (Leica, Wetzlar, Germany). The objective/disector size used was 40×/50 × 50, 100×/20 × 20 and 40×/150 × 150 μm respectively, and the endpoint was prespecified as either 100 profiles or 1000 optical disectors. Densities were obtained by dividing the number of profiles observed by the total area of disectors analyzed and then corrected by cortical atrophy to obtain a total number of profiles in a 1‐cm‐long full‐width strip of cortex. Amyloid burden was measured as the percentage of total cortical surface stained by the N‐terminal‐specific anti‐Aβ mouse monoclonal antibody 10D5 (Elan‐Lilly) in a 1‐cm‐long full‐width strip of cortex, using the optical threshold tool of the same software. Cortical thickness was measured as the average of 20 measurements in random sites distributed throughout the cortical ribbon, using the xy measure tool of the CAST software.
Lastly, nearly adjacent sections immunostained for TSPO, CD68 and GFAP with the DAB‐peroxidase method were scanned in a VS120 Olympus slide scanner under the 40× objective. A full width of 1‐ to 2‐cm‐long strip of the cortical ribbon was outlined on the resulting images using the polygon ROI tool of the “Count and Measure” module of the cellSens image analysis software (Olympus, Tokyo, Japan). The TSPO, CD68 and GFAP‐ir burdens were measured as the immunoreactive area fraction within these ROIs using the manual HSV threshold tool of the software. After pilot studies, saturation was set constant for all sections at 90 for TSPO, 70 for CD68 and 130 for GFAP. Given the punctate pattern of their staining, no lower limit of pixels was set for TSPO and CD68, whereas a lower limit of 30 pixels was set for GFAP to avoid saturation in some sections.
TSPO rs6971 SNP genotyping
Genomic DNA was extracted from cerebellar frozen samples using the PureLink Genomic DNA Extraction Mini Kit (Thermo Scientific, K182002) following manufacturer's instructions. The DNA concentration was measured in a DS‐11 spectrophotometer (DeNovix Inc). TaqMan genotyping assay for the rs6971 SNP in TSPO was purchased from Thermo Scientific (Assay ID C__2512465_20). For the TaqMan assay, DNA samples were diluted to a concentration of 1.8 ng/μL. The polymerase chain reaction (PCR) mix consisted of 1.25 μL of 20× TaqMan SNP genotyping assay, 12.50 μL of 2× TaqMan Fast Universal PCR Master Mix, no AmpErase UNG (Thermo Scientific, 4352042) and 11.25 μL of DNA sample (20 ng), for a total volume of 25 μL per well. DNA samples were run in duplicate. TaqMan assay was run in a Bio‐Rad CFX96 Touch Real‐Time PCR Detection System with the following program: 95ºC × 10 minutes (ramp 1ºC/s), 95ºC × 15 s and 60ºC × 1 minute, for 45 cycles. Principal component analysis of Vic vs. Fam fluorescence (corresponding to base A vs. G) allowed the discrimination between the AA, AG and GG genotypes.
Analysis of publicly available RNA‐Seq datasets
The cell types expressing tspo mRNA were ascertained in the mouse and human RNA‐Seq datasets made publicly available by the Barres 1, 53, 54 and Betsholtz 2, 16, 48 labs. For the analyses of tspo mRNA levels across CERAD scores of neuritic plaques, Braak NFT stages and TSPO rs6971 SNP genotypes, the transcriptome data from the Religious Orders Study and Memory and Aging Project (ROSMAP) cohort 28 was obtained from the Accelerating Medicines Partnership‐Alzheimer's Disease (AMP‐AD) Knowledge Portal 3, 17. Briefly, this is a bulk RNA‐Seq study performed on the dorsolateral prefrontal cortex (BA9) of several hundred subjects using the Illumina HiSeq platform with 101‐bp paired‐end reads for a target coverage of 50 million reads (except for the first 12 samples, which achieved a coverage of 150 million reads). Only cases with Agilent Bioanalyzer RNA integrity number (RIN) >5 and RNA quantity threshold of 5 μg were included 28. Processed RNA‐Seq datasets (Fragments Per Kilobase of transcript per Million mapped reads, FPKM) were downloaded and further analysis was conducted as described in Bihlmeyer et al. 5. Gene expression level is expressed in the log2 scale.
Statistical analyses
Normality of datasets was evaluated with D'Agostino‐Pearson omnibus test. Unpaired t test or Mann–Whitney U test was used for pairwise comparisons of normally and nonnormally distributed independent datasets (ie, TSPO levels by Western blot in AD vs. CTRL), respectively. Comparisons of paired data were performed with a paired t test or Wilcoxon matched‐pairs signed‐rank test for normally and nonnormally distributed datasets (ie, TSPO levels in temporal vs. cerebellar cortex in AD or CTRL groups), respectively. For comparisons among three or more independent groups, one‐way ANOVA with Tukey's multiple comparison test was run if datasets were normally distributed and Kruskal–Wallis ANOVA with Dunn's multiple comparison test if one or more datasets were not normally distributed. TSPO rs6971 genotype distribution (AA vs. AG vs. GG) and allele distribution (A vs. G) were compared between AD and CTRL groups with a Chi‐square and a Fisher's exact test, respectively. For some analyses, a two‐way ANOVA with diagnosis (CTRL vs. AD) and either region (repeated measures two‐way ANOVA for cortex vs. white matter or temporal cortex vs. cerebellum) or TSPO rs6971 genotype (ordinary two‐way ANOVA for AG vs. GG) as factors was performed. Correlations between TSPO‐ir burden and post‐mortem interval (PMI), the burden of amyloid plaques, PHF1+ NFTs, GFAP+ astrocytes, and CD68+ microglia and cortical thickness were performed with Pearson's or Spearman's rank correlation test for normally and nonnormally distributed variables, respectively. All statistical analyses and graphs were performed with GraphPad Prism version 8.0 (GraphPad Inc., La Jolla, CA). Error bars in scatter dot plots represent median and interquartile range.
Results
TSPO is expressed in microglial cells, astrocytes, endothelial cells, and vascular smooth muscle cells
To identify the cell types expressing TSPO in the human brain, we used confocal microscopy on formalin‐fixed paraffin‐embedded sections subjected to double fluorescent immunohistochemistry. TSPO and IBA1 coimmunostaining demonstrated a punctate staining of TSPO in the processes of IBA1+ microglia. The pattern of staining was clearly distinct from the diffuse cytosolic pattern of the calcium‐binding protein IBA1 and suggested a preferential localization of microglial mitochondria along the microglial cell processes rather than within the somas (Figure 1A‐D). This punctate pattern of TSPO staining in microglial processes resembled that of the phagocytic lysosomal marker CD68 but, as expected for markers of two different organelles, there was little colocalization between them (Figure 1E‐H).
Figure 1.
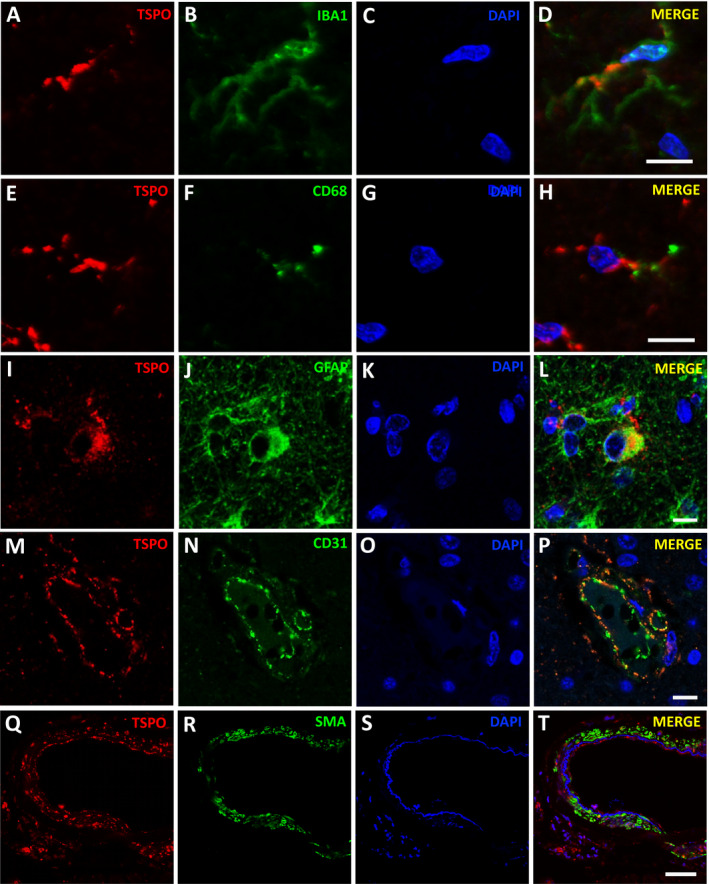
TSPO expression in microglia, astrocytes, endothelial cells, and vascular smooth muscle cells. Confocal microscopy images showing TSPO expression in IBA1+ (A‐D) and CD68+ (E‐H) microglia, GFAP+ astrocytes (I‐L), CD31+ endothelial cells of a capillary vessel (M‐P), and SMA+ vascular smooth muscle cells of a leptomeningeal artery (Q‐T). Note the punctate pattern of TSPO immunostaining along microglial processes (D, H), but its limited colocalization with CD68+ puncta depicting microglial lysosomes (H). Note also the limited colocalization of TSPO+ puncta with smooth muscle actin (SMA) filaments in vascular smooth muscle cells (T).
In addition to microglial cells, TSPO expression was also localized to astrocytes depicted with GFAP (Figure 1I‐L), endothelial cells costained for CD31 (Figure 1M‐P), and vascular smooth muscle cells costained for smooth muscle actin (SMA) (Figure 1Q‐T). In astrocytes, TSPO appeared to be mainly expressed in the cell soma and proximal branches, but not in more distal finer processes. The staining in endothelial and vascular smooth muscle cells was particularly conspicuous and present in leptomeningeal, cortical and white matter vessels of all sizes including the capillary endothelial cells. Sections costained for TSPO and HuC/D as a neuronal marker confirmed no expression of TSPO in neurons (not shown). Of note, negative control sections were devoid of microglial, astrocytic and vascular signal (not shown) and the specificity of the anti‐TSPO antibody was validated here (Figure S1) and elsewhere 46 in tissues from tspo null mice.
Intrigued by these observations, we sought to determine whether tspo mRNA has been found in cell types other than microglia. We entered tspo into the search box of publicly available RNA‐Seq databases from isolated mouse and human brain cell subpopulations 1, 2, 16, 48, 53, 54. We confirmed that tspo mRNA is detectable not only in microglial cells but also present in high levels in endothelial and smooth muscle cells isolated from the mouse brain (Figure S2A,C), and in microglia, endothelial cells and astrocytes purified from the normal human brain (Figure S2B).
Quantification of brain tspo mRNA levels reveals a substantial overlap between AD and healthy control subjects
Next, we asked whether the transcription of TSPO is upregulated in AD. To this end, we interrogated the public domain RNA‐Seq dataset from the ROSMAP 28. This is a population‐based clinic‐pathological longitudinal cohort study that recently conducted a bulk RNA‐Seq experiment in the dorsolateral prefrontal cortex (BA9) of 638 participants. We plotted the tspo mRNA levels as a function of Braak NFT stage (Figure 2A) and CERAD score of neuritic plaques (Figure 2B). Surprisingly, no significant differences in tspo mRNA levels were found across the different stages of AD neuropathological changes (one‐way ANOVA, P > 0.05).
Figure 2.
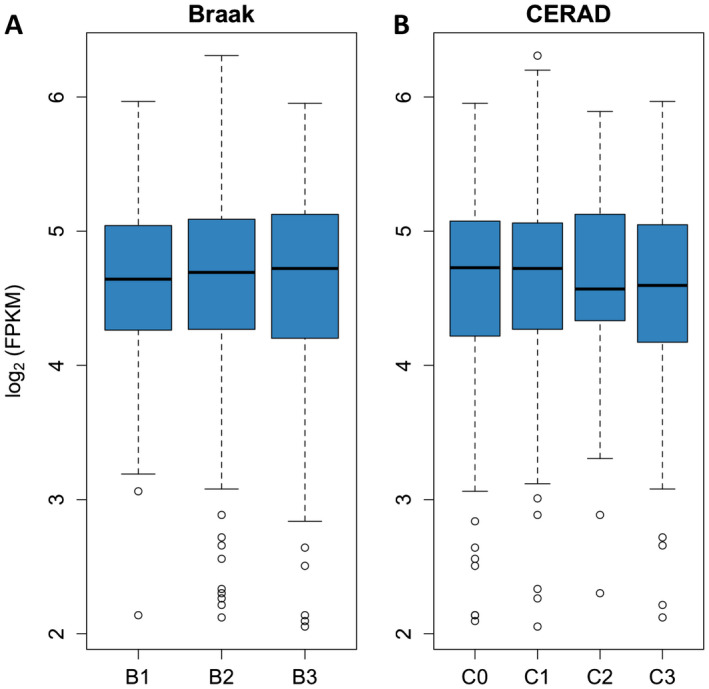
tspo mRNA levels do not differ across different stages of AD neuropathological change. tspo mRNA levels from the dorsolateral prefrontal cortex (BA9) were obtained from the publicly available Religious Orders Study and Memory Aging Project RNA‐Seq dataset (n = 638, see methods) and plotted against the severity of AD neuropathological changes. Neither Braak NFT stages (A) nor CERAD neuritic plaque scores (B) impact tspo mRNA levels (one‐way ANOVA, P > 0.05). Braak NFT stages: B1 = Braak 0/I/II (n = 112); B2 = Braak III/IV (n = 386), and B3 = Braak V/VI (n = 140). CERAD neuritic plaque scores: C0 = none (n = 165), C1 = sparse (n = 67), C2 = moderate (n = 219), and C3 = frequent (n = 187).
Quantification of brain TSPO levels reveals a substantial overlap between AD and healthy control subjects
We next asked whether the levels of TSPO are elevated in the AD brain vs. healthy CTRL brains. To this end, we quantified the levels of TSPO with both quantitative immunohistochemical methods and Western blot in a sample of AD and CTRL cases. Post‐mortem interval (PMI) did not significantly impact TSPO levels measured by either method (data not shown).
Quantification of the TSPO‐ir burden, measured as area fraction of TSPO immunoreactivity in randomly selected 20× fields comprising 5% of the cortical ribbon and 10% of the underlying white matter of the temporal lobe (BA38), is shown in Figure 3A,B. AD subjects had a marginally significant higher TSPO‐ir burden in the cortex and a statistically significant higher TSPO‐ir burden in the white matter compared to CTRL subjects (cortex: Mann–Whitney U = 175.5, P = 0.0516; white matter: Mann–Whitney U = 161.5, P = 0.0235). In contrast, no significant difference was observed in TSPO‐ir burden between cortex and white matter in either group (CTRL: P = 0.3558; AD: P = 0.5183, Wilcoxon matched‐pairs signed‐rank test). The repeated measures two‐way ANOVA with region (cortex vs. white matter) and diagnosis (CTRL vs. AD) as cofactors revealed that diagnosis only represented ≈7% of the variance in TSPO immunoreactivity [F (1,44) = 4.035, P = 0.0507], whereas the variance accounted for by the region and the interaction between region and diagnosis was negligible (≈0.1%) and nonsignificant. Indeed, while GFAP+ astrocytes and IBA1+ or CD68+ microglia are known to cluster within and around dense‐core (Thioflavin‐S‐positive) amyloid plaques 38, 41, 42, this was less evident for TSPO immunoreactivity (Figure S3).
Figure 3.
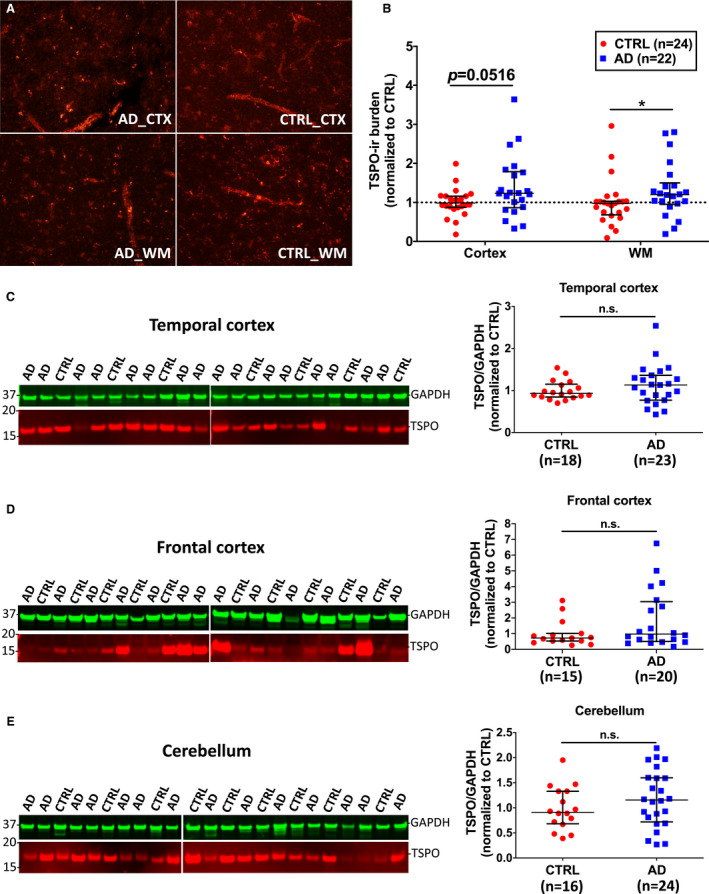
Substantial overlap of TSPO levels between AD and CTRL brains. (A) Representative images of TSPO fluorescent immunohistochemistry in the cortex (CTX) and white matter (WM) from the temporal pole of an AD and a control (CTRL) individuals. (B) Quantification of TSPO‐immunoreactive (TSPO‐ir) burden in 24 CTRL and 22 AD subjects rendered marginally significant (Mann–Whitney U = 175.5, P = 0.0516) higher levels in the cortex and statistically significant higher levels in the white matter (Mann–Whitney U = 161.5, P = 0.0235) in the AD group. (C‐E) Representative Western blots of TSPO and GAPDH (housekeeping for loading control) and scatter dot plots showing the quantification in temporal (C), frontal (D) and cerebellar (E) cortex of AD and CTRL subjects. No significant differences were observed between both groups in either region (temporal cortex: Mann–Whitney U 168.5, P = 0.3180; frontal cortex: Mann–Whitney U 112, P = 0.2112; cerebellum: unpaired t test, t = 1.151, df = 38, P = 0.2568).
Brain protein SDS‐extracts from the cerebellum, frontal and temporal neocortex of AD and healthy control subjects were subjected to SDS‐PAGE followed by immunoblot for TSPO with the same commercially available rabbit monoclonal antibody. Quantification of TSPO band with GAPDH as housekeeping for loading control revealed no statistically significant differences between CTRL and AD subjects in either temporal neocortex (Figure 3C, Mann–Whitney U 168.5, P = 0.3180), frontal neocortex (Figure 3D, Mann–Whitney U 112, P = 0.2112), or cerebellum (Figure 3E, unpaired t test, t = 1.151, df = 38, P = 0.2568). A comparison between temporal and cerebellar TSPO levels from the same CTRL and AD cases is shown in Figure S4. With some exceptions, TSPO levels were generally higher in temporal neocortex than in cerebellum in both CTRL and AD groups. However, these regional differences did not reach statistical significance in either group (CTRL: P = 0.1250 and AD: P = 0.1310, Wilcoxon matched‐pairs signed‐rank test). Repeated measures two‐way ANOVA with diagnosis and region as cofactors revealed a modest effect of region (10.86% of variance, P = 0.1045), whereas diagnosis and the interaction between region and diagnosis were not significant and only explained <1% of the variance in TSPO levels.
TSPO levels do not correlate with the burden of AD neuropathological changes or the magnitude of glial responses
Recent PET imaging studies have reported a positive correlation between TSPO radioligand uptake and both amyloid and tau PET radioligand uptake 10, 32. Thus, we inquired whether post‐mortem TSPO levels correlate with the burden of AD neuropathological changes (10D5+ amyloid plaques and PHF1+ NFTs) or with the magnitude of glial responses—reactive (GFAP+) astrocytes and activated (CD68+) microglia—within the AD temporal neocortex. We observed no significant correlation between the TSPO‐ir burden and cortical thickness, amyloid plaque burden or stereology‐based counts of PHF1+ NFTs obtained from nearly adjacent formalin‐fixed paraffin‐embedded sections (Figure 4A‐C). Similarly, no significant correlation was observed between the TSPO‐ir burden and either the GFAP‐ir or the CD68‐ir burden within the same ROI of the temporal neocortex (Figure 4D,E), or stereology‐based measures of GFAP+ astrocytes and CD68+ microglia (not shown).
Figure 4.
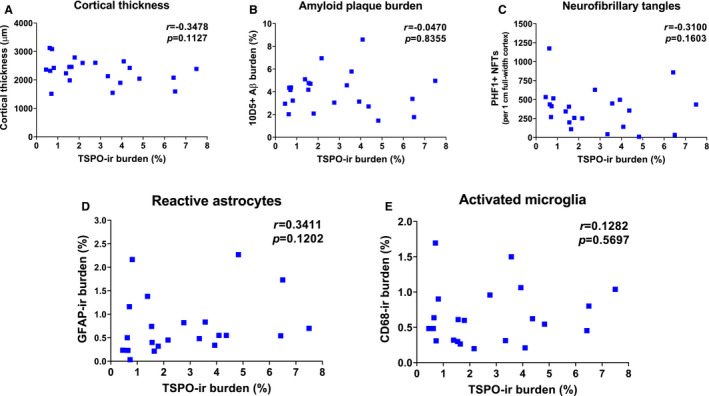
TSPO levels do not correlate with the local burden of AD neuropathological changes or the magnitude of glial responses. Correlation plots of TSPO‐ir burden and cortical thickness (A), amyloid plaque burden (B), stereological counts of PHF1+ NFTs (C), GFAP‐ir burden (reactive astrocytes) (D) and CD68‐ir burden (activated microglia) (E) in the temporal pole neocortex (BA38) of n = 22 AD subjects. Correlation coefficients and P‐values can be found in each figure of the panel.
TSPO rs6971 SNP has no impact in tspo mRNA or TSPO levels, the magnitude of glial responses or the burden of AD neuropathological changes
The rs6971 SNP in the TSPO gene predicts an alanine to threonine substitution in position 147 (Ala147Thr) of TSPO that has been associated with a variable uptake of the TSPO radioligands. In vitro studies have suggested that this is due to a variable affinity between radioligands and TSPO. Individuals homozygous for the minor allele (AA) are low‐affinity binders (LABs), heterozygous (AG) are medium affinity binders (MABs), and homozygous for the major allele (GG) are high‐affinity binders (HABs) 15, 19, 30, 52.
Here we tested the hypothesis that, rather than just having a pharmacodynamic effect on radioligand binding, TSPO rs6971 SNP could impact tspo mRNA or TSPO levels or the number of reactive glial cells. Firstly, we interrogated the ROSMAP RNA‐Seq dataset for differential tspo mRNA levels across TSPO rs6971 genotypes and observed no significant differences between LABs (n = 45), MABs (n = 213) and HABs (n = 215) (one‐way ANOVA, P > 0.05) (Figure 5). Secondly, we genotyped this SNP in 110 subjects (n = 86 AD and 24 CTRL) from the MADRC brain bank. Genotype distribution did not significantly differ between the AD group [AA: n = 7 (8.1%), AG: n = 44 (51.2%), GG: n = 35 (40.7%)] and the CTRL group [AA: n = 0 (0.0%), AG: n = 14 (58.4%), GG: n = 10 (41.7%)] (P = 0.3429; Chi‐square, df: 2.141, 2). Allele distribution did not differ between the AD group [A: n = 58 (33.7%), G: n = 114 (66.3%)] and the CTRL group [A: n = 14 (29.2%), G: n = 34 (70.8%)] (P = 0.6051, Fisher's exact test). We observed no significant differences between CTRL and AD groups in the TSPO‐ir burden (Figure 6A‐D) across TSPO rs6971 genotypes in either cortex or white matter (cortex: P = 0.2506; white matter: P = 0.2592, Kruskal–Wallis ANOVA with Dunn's post test). A two‐way ANOVA with diagnosis and rs6971 genotype as cofactors revealed a significant effect of diagnosis on cortical TSPO‐ir burden (diagnosis: 12.6% of variance, P = 0.0359), but not of rs6971 genotype (1.01% of variance, P = 0.5393) or the interaction between diagnosis and rs6971 genotype (0.03% of variance, P = 0.9109); however, none of these factors was significant in the white matter. Similarly, Western blot rendered no significant differences in TSPO levels across TSPO rs6971 genotypes in either group (Figure 6E‐G). Lastly, no differences in cortical thickness, amyloid plaque burden, or stereology‐based measures of PHF1+ NFTs, or glial responses (GFAP+ astrocytes and CD68+ microglia) were observed across genotypes in the group of 86 genotyped AD subjects (Figure 7).
Figure 5.
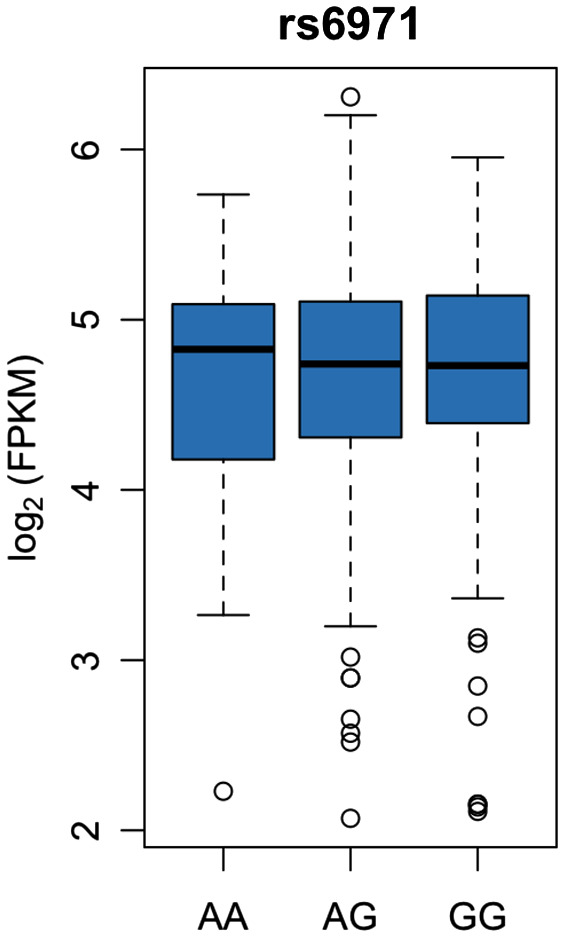
The TSPO rs6971 SNP does not impact tspo mRNA levels. tspo mRNA levels from the dorsolateral prefrontal cortex (BA9) were obtained from the publicly available Religious Orders Study and Memory Aging Project RNA‐Seq dataset (n = 473, see methods) and plotted against the TSPO rs6971 genotype. The tspo mRNA level did not differ between AA (LABs, n = 45), AG (MABs, n = 213) and GG (HABs, n = 215) subjects (one‐way ANOVA, P > 0.05).
Figure 6.
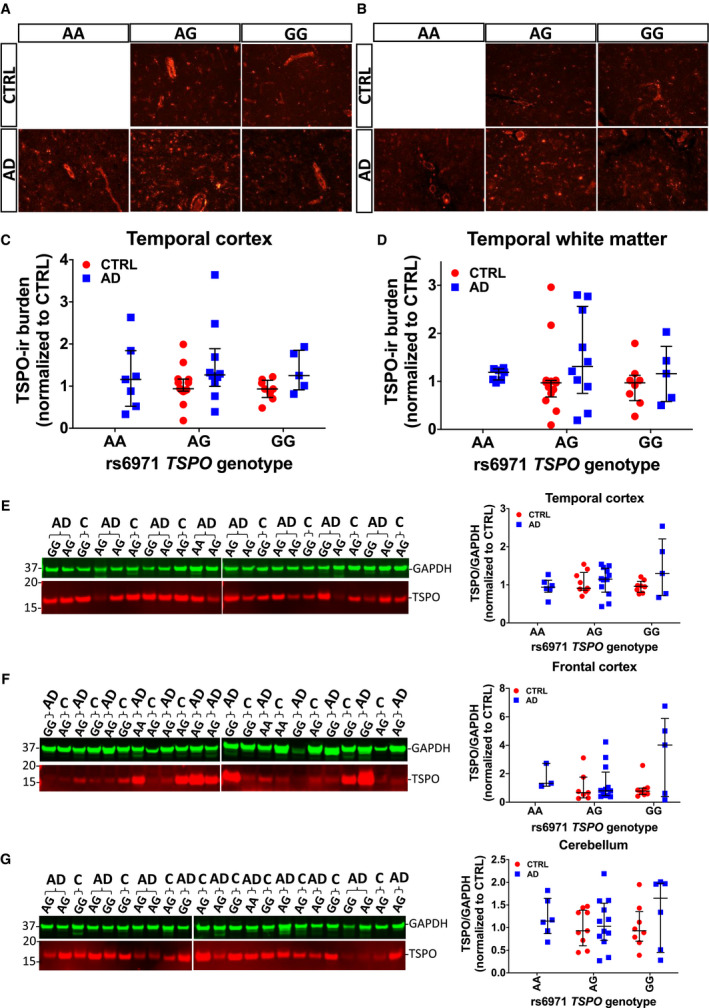
The TSPO rs6971 SNP does not impact TSPO levels. (A, B) Representative images of TSPO fluorescent immunohistochemistry in the cortex (A) and white matter (B) from the temporal pole of AD and control (CTRL) individuals split by TSPO rs6971 SNP genotype. (C, D) No significant differences in TSPO‐ir burden across genotypes were found between CTRL and AD in either region (cortex: P = 0.2506; white matter: P = 0.2592, Kruskal–Wallis ANOVA with Dunn's post test). (E‐G) Same representative Western blots of TSPO and GAPDH (housekeeping for loading control) as in Figure 3 and scatter dot plots showing the quantification in temporal (E), frontal (F) and cerebellar (G) cortex of AD and CTRL subjects, now split by TSPO rs6971 SNP genotype. No significant differences were observed between CTRL and AD groups across genotypes in either region (temporal cortex: P = 0.6850; frontal cortex: P = 0.3983; cerebellum: P = 0.7665, Kruskal–Wallis ANOVA with Dunn's post test). Note that the AA genotype was absent in the 24 CTRL cases genotyped; therefore, there is no “CTRL AA” group.
Figure 7.
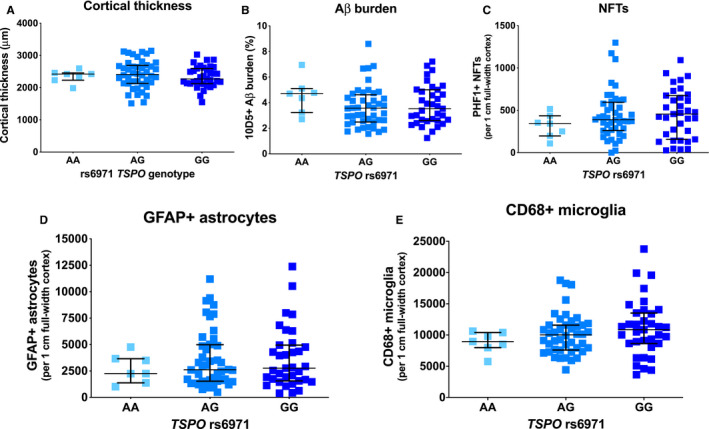
The TSPO rs6971 SNP does not impact the severity of glial responses, AD neuropathological changes or cortical atrophy. The TSPO rs6971 SNP was genotyped in n = 86 AD cases. No significant differences were observed across the three genotypes in cortical thickness (A), amyloid plaque burden (B), or stereology‐based counts of PHF1+ neurofibrillary tangles (NFTs) (C), GFAP+ astrocytes (D), or CD68+ microglia (E) in the temporal neocortex (BA38).
Discussion
Our results can be summarized as follows: (1) the expression of TSPO is not just limited to microglia, but very conspicuous also in astrocytes, and endothelial and vascular smooth muscle cells; (2) there is no significant increase in the dorsolateral prefrontal cortex tspo mRNA levels in AD compared to CTRL subjects; (3) there is a substantial overlap of TSPO levels in the temporal and frontal cortex of AD compared to CTRL subjects and in the temporal neocortex compared with the underlying white matter in both groups; (4) TSPO cortical burden does not correlate with Aβ plaque or NFT burden or with the burden of activated microglia or reactive astrocytes; and (5) the rs6971 TSPO SNP does not significantly impact tspo mRNA or TSPO levels, the magnitude of glial responses, or the burden of AD neuropathological changes.
Using double fluorescent immunohistochemistry with confocal microscopy, we have extended the previous qualitative characterization reported by others 8, 22, 25, 27, 29 and demonstrated that astrocytes and especially blood vessels––both endothelial and vascular smooth muscle cells––are major sources of TSPO expression. The interrogation of available RNA‐Seq datasets from cell populations isolated from adult mouse and human brains confirmed that these cell types have high levels of the tspo mRNA. It is noteworthy that TSPO‐based PET is often referred to as microglial PET; however, other cell types are likely contributing to the observed uptake. Nuclear emulsion autoradiography on post‐mortem brain frozen cryostat sections 26 would be needed to confirm that TSPO radioligands bind to these different cell types with similar affinity. In fact, a prominent vascular binding of TSPO radioligands has been previously acknowledged and palliated through a mathematical modeling aimed at subtracting the distribution of the radioligands in the vascular compartment 34, 35, 45, 47, 49, 51. Perhaps due to this prominent vascular TSPO expression, we found a substantial overlap of TSPO levels between AD and CTRL subjects by either quantitative immunohistochemistry or Western blot; however, TSPO‐ir burden was seemingly a more sensitive measure to detect differences than the TSPO/GAPDH ratio rendered by Western blot. Moreover, we found no significant difference in TSPO burden between cortex and underlying white matter in either group by quantitative immunohistochemistry, which would prevent an adequate grey/white matter differentiation in PET scans with TSPO radioligands. In most analyzed cases, the TSPO levels were higher in the temporal than in the cerebellar cortex, supporting the use of the latter as the reference region for radioligand uptake measurements.
The discovery that the rs6971 SNP in the TSPO gene is associated with a variable uptake of the TSPO radioligands and the subsequent concern that including low‐affinity binders (LABs) could render nonsignificant differences in radioligand uptake between diseased and CTRL subjects 36, has led researchers to restrict their PET imaging studies to medium and high‐affinity binders (MABs and HABs). It is widely accepted that this variable radioligand uptake across TSPO rs6971 genotypes is due to the differential affinity between radioligands and TSPO observed in vitro 15, 19, 30, 52. Here we show no significant difference in tspo mRNA levels across TSPO rs6971 genotypes in the population‐based ROSMAP cohort (n = 473). We also observed no significant differences in TSPO levels across TSPO rs6971 genotypes in either AD or CTRL by both quantitative immunohistochemistry and Western blot. Moreover, the TSPO rs6971 SNP did not influence the burden of AD neuropathological changes (Aβ plaques and NFTs) or the magnitude of glial responses (reactive astrocytes and activated microglia). Taken together, these data support the idea that this SNP impacts the affinity of TSPO for its ligands, rather than the abundance of TSPO or reactive glia.
TSPO functions remain to be fully understood. Although it was initially proposed to have a role in neurosteroid synthesis and cholesterol transport to the mitochondria, the tspo global null mice have normal steroid hormone levels and normal cholesterol mitochondrial levels 46, 55. While TSPO‐based PET imaging is commonly referred to as an imaging biomarker of neuroinflammation, the role of TSPO in neuroinflammation remains controversial. On one hand, TSPO agonists such as PK11195, Ro5‐4864 and etifoxine have been shown to have beneficial anti‐inflammatory and neuroprotective properties in animal models of AD 4, 7, MS 12, TBI 31, 43, stroke 23 and cerebral hemorrhage 24, among other neurological conditions that lead to glial reaction. On the other hand, selective deletion of tspo in astrocytes has been reported to prevent astrocyte and microglial activation and to reduce the levels of inflammatory cytokines, supporting a pro‐inflammatory role of TSPO in astrocytes 11. Furthermore, the contribution of the brain vascular TSPO expression to neuroinflammation remains unclear. Interestingly, TSPO PET imaging has been assayed to depict vascular inflammation in various vascular diseases such as systemic vasculitis 9, 21, 33, atherosclerosis 6, 14 and aneurysms 13. Thus, it is possible that vascular inflammation in AD and other neurodegenerative diseases contributes to the observed increased TSPO PET signal 44.
Conclusions
In summary, we show that TSPO is not only expressed in microglia but also in astrocytes, endothelial and vascular smooth muscle cells, and that there is a substantial overlap of tspo mRNA and TSPO levels between CTRL and AD subjects, no correlation between TSPO levels and glial responses or AD neuropathological changes and no impact of the rs6971 TSPO SNP on tspo mRNA or TSPO levels, glial responses, or AD neuropathological changes. Our findings could inform ongoing efforts toward the development of reactive glia‐specific PET radioligands 18, 50. More research is needed to understand the cell type‐specific role of TSPO in AD and other brain diseases.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Validation of anti‐TSPO antibody. Western blot for TSPO using Abcam rabbit anti‐PBR monoclonal antibody (clone EPR5384, ab109497) on recombinant TSPO (rTSPO, 1 μg), SDS protein extracts from both tspo knock out (ko) and floxed (flox) mice (5 μg, lung, adrenal gland and brain) and SDS protein extracts (5 μg) from the temporal cortex of human control (C) and AD brains. GAPDH was used as housekeeping for loading control. Note that the antibody renders a single band in the rTSPO, the floxed mouse tissues and the human brain extracts, whereas no band was detected in tspo knock out mouse tissues. Also, note that TSPO levels are much higher in mouse adrenal gland and lung than in mouse or human brain.
Figure S2. tspo mRNA is present not only in microglia but also in astrocytes, endothelial cells and vascular smooth muscle cells. (A and B) Bar graphs illustrate the levels of the tspo mRNA (in Fragments Per Kilobase of transcript per Million mapped reads or FPKM) in different cell types isolated from the mouse (A) and human (B) brains obtained from a publicly available RNA‐Seq dataset 1, 53, 54. (C) Average expression in each cell cluster in the mouse brain obtained from another publicly available RNA‐Seq dataset 2, 16, 48. Note that mouse astrocytes have very low levels of tspo mRNA, whereas mature human astrocytes express significant tspo mRNA levels. Abbreviations: PC = pericytes; vSMC = venous smooth muscle cells; aaSMC = arteriolar smooth muscle cells; aSMC = arterial smooth muscle cells; MG = microglia; FB = vascular fibroid‐like cells; OL = oligodendrocytes; EC = endothelial cells; vEC = venous endothelial cells; capiEC = capillary endothelial cells; aEC = arterial endothelial cells and AC = astrocytes.
Figure S3. TSPO immunoreactivity with respect to dense‐core (Thioflavin‐S‐positive) amyloid plaques. Confocal images of TSPO (red), thioflavin‐S (green) and DAPI (blue) in adjacent fields with (A‐D and I‐L) and without (E‐H and M‐P) dense‐core amyloid plaques in a representative AD case. Note that TSPO‐ir burden can be similar in the presence and absence of plaques. Scale bar: 50 μm.
Figure S4. TSPO levels are usually higher in temporal neocortex than in cerebellum. (A) Representative Western blot for TSPO (red) and GAPDH (housekeeping, green) in SDS extracts from temporal cortex (temp) and cerebellum (cblm) of CTRL and AD subjects. (B) Quantification of TSPO over GAPDH levels shows that, with some exceptions, TSPO levels are higher in temporal neocortex than in cerebellum; however, these regional differences did not reach statistical significance in either group (CTRL: P = 0.1250 and AD: P = 0.1310, Wilcoxon matched‐pairs signed‐rank test, n = 5 CTRL and 14 AD).
Acknowledgments
YG did most experiments; JDM did some experiments; SD did the ROSMAP RNA‐Seq analysis; BTH provided intellectual input and edited the manuscript; AS‐P conceived the idea, designed the experiments, did some experiments, and wrote the manuscript. The ROSMAP project was supported by funding from the National Institute on Aging (AG034504 and AG041232). The authors want to acknowledge patients and caregivers involved in research at the Massachusetts Alzheimer's Disease Research Center.
This work was funded by the US National Institute for Neurodegenerative Diseases and Stroke (NINDS R25NS065743 to AS‐P) and the Alzheimer's Association (AACF‐17‐524184 to AS‐P).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Anon . Available at http://www.brainrnaseq.org.
- 2. Anon . Available at http://betsholtzlab.org/VascularSingleCells/database.html.
- 3. Anon . Available at https://www.synapse.org/#!Synapse:syn2580853/wiki/493203.
- 4. Barron AM, Garcia‐Segura LM, Caruso D, Jayaraman A, Lee J‐W, Melcangi RC, Pike CJ (2013) Ligand for translocator protein reverses pathology in a mouse model of Alzheimer's disease. J Neurosci 33:8891–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bihlmeyer NA, Merrill E, Lambert Y, Srivastava GP, Clark TW, Hyman BT, Das S (2019) Novel methods for integration and visualization of genomics and genetics data in Alzheimer's disease. Alzheimers Dement 15:788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bird JLE, Izquierdo‐Garcia D, Davies JR, Rudd JHF, Probst KC, Figg N et al (2010) Evaluation of translocator protein quantification as a tool for characterising macrophage burden in human carotid atherosclerosis. Atherosclerosis 210:388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Christensen A, Pike CJ (2018) TSPO ligand PK11195 improves Alzheimer‐related outcomes in aged female 3xTg‐AD mice. Neurosci Lett 683:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cosenza‐Nashat M, Zhao M‐L, Suh H‐S, Morgan J, Natividad R, Morgello S, Lee SC (2009) Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol 35:306–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cuhlmann S, Gsell W, Van der Heiden K, Habib J, Tremoleda JL, Khalil M et al (2014) In vivo mapping of vascular inflammation using the translocator protein tracer 18F‐FEDAA1106. Mol Imaging 13. [DOI] [PubMed] [Google Scholar]
- 10. Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R et al (2018) Microglial activation correlates in vivo with both tau and amyloid in Alzheimer's disease. Brain J Neurol 141:2740–2754. [DOI] [PubMed] [Google Scholar]
- 11. Daugherty DJ, Chechneva O, Mayrhofer F, Deng W (2016) The hGFAP‐driven conditional TSPO knockout is protective in a mouse model of multiple sclerosis. Sci Rep 6:22556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daugherty DJ, Selvaraj V, Chechneva OV, Liu X‐B, Pleasure DE, Deng W (2013) A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 5:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. English SJ, Diaz JA, Shao X, Gordon D, Bevard M, Su G et al (2014) Utility of (18) F‐FDG and (11)C‐PBR28 microPET for the assessment of rat aortic aneurysm inflammation. EJNMMI Res. 4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaemperli O, Shalhoub J, Owen DRJ, Lamare F, Johansson S, Fouladi N et al (2012) Imaging intraplaque inflammation in carotid atherosclerosis with 11C‐PK11195 positron emission tomography/computed tomography. Eur Heart J. 33:1902–1910. [DOI] [PubMed] [Google Scholar]
- 15. Guo Q, Colasanti A, Owen DR, Onega M, Kamalakaran A, Bennacef I et al (2013) Quantification of the specific translocator protein signal of 18F‐PBR111 in healthy humans: a genetic polymorphism effect on in vivo binding. J Nucl Med 54:1915–1923. [DOI] [PubMed] [Google Scholar]
- 16. He L, Vanlandewijck M, Mäe MA, Andrae J, Ando K, Del Gaudio F et al (2018) Single‐cell RNA sequencing of mouse brain and lung vascular and vessel‐associated cell types. Sci Data 5:180160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hodes RJ, Buckholtz N (2016) Accelerating medicines partnership: Alzheimer's disease (AMP‐AD) knowledge portal aids Alzheimer's drug discovery through open data sharing. Expert Opin. Ther Targets 20:389–391. [DOI] [PubMed] [Google Scholar]
- 18. Horti AG, Naik R, Foss CA, Minn I, Misheneva V, Du Y et al (2019) PET imaging of microglia by targeting macrophage colony‐stimulating factor 1 receptor (CSF1R). Proc Natl Acad Sci U S A 116:1686–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL et al (2013) A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab 33:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lagarde J, Sarazin M, Bottlaender M (2018) In vivo PET imaging of neuroinflammation in Alzheimer's disease. J Neural Transm (Vienna) 125:847–867. [DOI] [PubMed] [Google Scholar]
- 21. Lamare F, Hinz R, Gaemperli O, Pugliese F, Mason JC, Spinks T et al (2011) Detection and quantification of large‐vessel inflammation with 11C‐(R)‐PK11195 PET/CT. J Nucl Med 52:33–39. [DOI] [PubMed] [Google Scholar]
- 22. Lavisse S, Guillermier M, Hérard A‐S, Petit F, Delahaye M, Van Camp N et al (2012) Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 32:10809–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H‐D, Li M, Shi E, Jin W‐N, Wood K, Gonzales R, Liu Q (2017) A translocator protein 18 kDa agonist protects against cerebral ischemia/reperfusion injury. J Neuroinflammation. 14:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M, Ren H, Sheth KN, Shi F‐D, Liu Q. (2017) A TSPO ligand attenuates brain injury after intracerebral hemorrhage. FASEB J 31:3278–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu B, Le KX, Park M‐A, Wang S, Belanger AP, Dubey S et al (2015) In vivo detection of age‐ and disease‐related increases in neuroinflammation by 18F‐GE180 TSPO microPET imaging in wild‐type and Alzheimer's transgenic mice. J Neurosci 35:15716–15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG et al (2015) Validating novel tau positron emission tomography tracer [F‐18]‐AV‐1451 (T807) on postmortem brain tissue. Ann Neurol 78:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mirzaei N, Tang SP, Ashworth S, Coello C, Plisson C, Passchier J et al (2016) In vivo imaging of microglial activation by positron emission tomography with [(11)C]PBR28 in the 5XFAD model of Alzheimer's disease. Glia 64:993–1006. [DOI] [PubMed] [Google Scholar]
- 28. Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J et al (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci 21:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nack A, Brendel M, Nedelcu J, Daerr M, Nyamoya S, Beyer C et al (2019) Expression of translocator protein and [18F]‐GE180 ligand uptake in multiple sclerosis animal models. Cells 8(2):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A et al (2012) An 18‐kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palzur E, Sharon A, Shehadeh M, Soustiel JF (2016) Investigation of the mechanisms of neuroprotection mediated by Ro5‐4864 in brain injury. Neuroscience 329:162–170. [DOI] [PubMed] [Google Scholar]
- 32. Parbo P, Ismail R, Hansen KV, Amidi A, Mårup FH, Gottrup H et al (2017) Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer's disease. Brain J Neurol 140:2002–2011. [DOI] [PubMed] [Google Scholar]
- 33. Pugliese F, Gaemperli O, Kinderlerer AR, Lamare F, Shalhoub J, Davies AH et al (2010) Imaging of vascular inflammation with [11C]‐PK11195 and positron emission tomography/computed tomography angiography. J Am Coll Cardiol 56:653–661. [DOI] [PubMed] [Google Scholar]
- 34. Rizzo G, Veronese M, Tonietto M, Bodini B, Stankoff B, Wimberley C et al (2019) Generalization of endothelial modelling of TSPO PET imaging: Considerations on tracer affinities. J Cereb Blood Flow Metab 39:874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rizzo G, Veronese M, Tonietto M, Zanotti‐Fregonara P, Turkheimer FE, Bertoldo A (2014) Kinetic modeling without accounting for the vascular component impairs the quantification of [(11)C]PBR28 brain PET data. J Cereb Blood Flow Metab 34:1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF et al (2013) Microglial activation in Alzheimer's disease: an (R)‐[11C]PK11195 positron emission tomography study. Neurobiol Aging. 34:128–136. [DOI] [PubMed] [Google Scholar]
- 37. Serrano‐Pozo A, Aldridge GM, Zhang Q (2017) Four decades of research in Alzheimer's disease (1975–2014): a bibliometric and scientometric analysis. J Alzheimers Dis 59:763–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serrano‐Pozo A, Betensky RA, Frosch MP, Hyman BT (2016) Plaque‐associated local toxicity increases over the clinical course of Alzheimer disease. Am J Pathol 186:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Serrano‐Pozo A, Gómez‐Isla T, Growdon JH, Frosch MP, Hyman BT (2013) A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 182:2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrano‐Pozo A, Mielke ML, Gómez‐Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 179:1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Serrano‐Pozo A, Muzikansky A, Gómez‐Isla T, Growdon JH, Betensky RA, Frosch MP, Hyman BT (2013) Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol 72:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon‐O'Brien E, Gauthier D, Riban V, Verleye M (2016) Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J Neuroinflammation. 13:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takeda S, Sato N, Uchio‐Yamada K, Sawada K, Kunieda T, Takeuchi D et al (2010) Diabetes‐accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 107:7036–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tomasi G, Edison P, Bertoldo A, Roncaroli F, Singh P, Gerhard A et al (2008) Novel reference region model reveals increased microglial and reduced vascular binding of 11C‐(R)‐PK11195 in patients with Alzheimer's disease. J Nucl Med 49:1249–1256. [DOI] [PubMed] [Google Scholar]
- 46. Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V (2014) Peripheral benzodiazepine receptor/translocator protein global knock‐out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem 289:27444–27454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A et al (2007) Reference and target region modeling of [11C]‐(R)‐PK11195 brain studies. J Nucl Med 48:158–167. [PubMed] [Google Scholar]
- 48. Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F et al (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480. [DOI] [PubMed] [Google Scholar]
- 49. Veronese M, Reis Marques T, Bloomfield PS, Rizzo G, Singh N, Jones D et al (2018) Kinetic modelling of [11C]PBR28 for 18 kDa translocator protein PET data: a validation study of vascular modelling in the brain using XBD173 and tissue analysis. J Cereb Blood Flow Metab 38:1227–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C, Keliher E, Zeller MWG, Wojtkiewicz GR, Aguirre AD, Buckbinder L et al (2019) An activatable PET imaging radioprobe is a dynamic reporter of myeloperoxidase activity in vivo. Proc Natl Acad Sci U S A 116:11966–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wimberley C, Lavisse S, Brulon V, Peyronneau M‐A, Leroy C, Bodini B et al (2018) Impact of endothelial 18‐kDa translocator protein on the quantification of 18F‐DPA‐714. J Nucl Med 59:307–314. [DOI] [PubMed] [Google Scholar]
- 52. Yoder KK, Nho K, Risacher SL, Kim S, Shen L, Saykin AJ (2013) Influence of TSPO genotype on 11C‐PBR28 standardized uptake values. J Nucl Med 54:1320–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S et al (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD et al (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao AH, Tu LN, Mukai C, Sirivelu MP, Pillai VV, Morohaku K et al (2016) Mitochondrial translocator protein (TSPO) function is not essential for heme biosynthesis. J Biol Chem 291:1591–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Validation of anti‐TSPO antibody. Western blot for TSPO using Abcam rabbit anti‐PBR monoclonal antibody (clone EPR5384, ab109497) on recombinant TSPO (rTSPO, 1 μg), SDS protein extracts from both tspo knock out (ko) and floxed (flox) mice (5 μg, lung, adrenal gland and brain) and SDS protein extracts (5 μg) from the temporal cortex of human control (C) and AD brains. GAPDH was used as housekeeping for loading control. Note that the antibody renders a single band in the rTSPO, the floxed mouse tissues and the human brain extracts, whereas no band was detected in tspo knock out mouse tissues. Also, note that TSPO levels are much higher in mouse adrenal gland and lung than in mouse or human brain.
Figure S2. tspo mRNA is present not only in microglia but also in astrocytes, endothelial cells and vascular smooth muscle cells. (A and B) Bar graphs illustrate the levels of the tspo mRNA (in Fragments Per Kilobase of transcript per Million mapped reads or FPKM) in different cell types isolated from the mouse (A) and human (B) brains obtained from a publicly available RNA‐Seq dataset 1, 53, 54. (C) Average expression in each cell cluster in the mouse brain obtained from another publicly available RNA‐Seq dataset 2, 16, 48. Note that mouse astrocytes have very low levels of tspo mRNA, whereas mature human astrocytes express significant tspo mRNA levels. Abbreviations: PC = pericytes; vSMC = venous smooth muscle cells; aaSMC = arteriolar smooth muscle cells; aSMC = arterial smooth muscle cells; MG = microglia; FB = vascular fibroid‐like cells; OL = oligodendrocytes; EC = endothelial cells; vEC = venous endothelial cells; capiEC = capillary endothelial cells; aEC = arterial endothelial cells and AC = astrocytes.
Figure S3. TSPO immunoreactivity with respect to dense‐core (Thioflavin‐S‐positive) amyloid plaques. Confocal images of TSPO (red), thioflavin‐S (green) and DAPI (blue) in adjacent fields with (A‐D and I‐L) and without (E‐H and M‐P) dense‐core amyloid plaques in a representative AD case. Note that TSPO‐ir burden can be similar in the presence and absence of plaques. Scale bar: 50 μm.
Figure S4. TSPO levels are usually higher in temporal neocortex than in cerebellum. (A) Representative Western blot for TSPO (red) and GAPDH (housekeeping, green) in SDS extracts from temporal cortex (temp) and cerebellum (cblm) of CTRL and AD subjects. (B) Quantification of TSPO over GAPDH levels shows that, with some exceptions, TSPO levels are higher in temporal neocortex than in cerebellum; however, these regional differences did not reach statistical significance in either group (CTRL: P = 0.1250 and AD: P = 0.1310, Wilcoxon matched‐pairs signed‐rank test, n = 5 CTRL and 14 AD).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.