

Abstract
Ecotropic virus integration site 1 (EVI1), whose overexpression characterizes a particularly aggressive subtype of acute myeloid leukemia (AML), enhanced anti-leukemic activities of all-trans retinoic acid (atRA) in cell lines and patient samples. However, the drivers of leukemia formation, therapy resistance, and relapse are leukemic stem cells (LSCs), whose properties were hardly reflected in these experimental setups. The present study was designed to address the effects of, and interactions between, EVI1 and retinoids in AML LSCs. We report that Evi1 reduced the maturation of leukemic cells and promoted the abundance, quiescence, and activity of LSCs in an MLL-AF9-driven mouse model of AML. atRA further augmented these effects in an Evi1 dependent manner. EVI1 also strongly enhanced atRA regulated gene transcription in LSC enriched cells. One of their jointly regulated targets, Notch4, was an important mediator of their effects on leukemic stemness. In vitro exposure of leukemic cells to a pan-RAR antagonist caused effects opposite to those of atRA. In vivo antagonist treatment delayed leukemogenesis and reduced LSC abundance, quiescence, and activity in Evi1high AML. Key results were confirmed in human myeloid cell lines retaining some stem cell characteristics as well as in primary human AML samples. In summary, our study is the first to report the importance of EVI1 for key properties of AML LSCs. Furthermore, it shows that atRA enhances, and a pan-RAR antagonist counteracts, the effects of EVI1 on AML stemness, thus raising the possibility of using RAR antagonists in the therapy of EVI1high AML.
Subject terms: Cancer stem cells, Haematological cancer
Introduction
Acute myeloid leukemia (AML) is organized in a hierarchical manner, i.e., the bulk of the leukemic cell mass is derived from mostly quiescent leukemic stem cells (LSCs), which are the source of disease emergence, therapy resistance, and relapse1. AML is caused by specific cytogenetic aberrations, point mutations, and epigenetic and transcriptional changes2,3, which occur in hematopoietic stem cells (HSCs) or progenitor cells, causing their transformation into LSCs1,4,5. One such change is overexpression of EVI1, which is present in around 10% of patients and associated with a dismal prognosis6–8. EVI1 is located in chromosome band 3q26 and codes for a zinc finger transcription factor9,10. In normal hematopoiesis, EVI1 is highly expressed in immature cells, but rapidly down-regulated during differentiation11,12; accordingly, it promoted the abundance, quiescence, and activity of murine HSCs11,13. Experimental expression or knock-down of Evi1 in mouse models of AML promoted or delayed myeloid leukemogenesis, respectively12–15, and activation of EVI1 through vector integration caused AML in a gene therapy trial for chronic granulomatous disease16. The most prominent causes of EVI1 overexpression in human AML are juxtaposition to a strong enhancer, or transcriptional induction by leukemia-associated fusion proteins. Specifically, EVI1 is brought under control of the GATA2 enhancer in cases with inv(3)(q21q26) or t(3;3)(q21;q26)17,18, and up-regulated through direct promoter binding by lysine methyltransferase 2A (KMT2A; more commonly known as mixed lineage leukemia, MLL) fusion proteins, which result from 11q23 rearrangements12,19. MLL fusion proteins transformed both murine HSCs and progenitor cells, but enhanced Evi1 expression only in the former. This suggested that the presence or absence of EVI1 overexpression, each observed in about half of the patients, reflects the cell type in which the transforming event occurred also in human MLL rearranged AML4,7,12,19,20.
All-trans retinoic acid (atRA) acts through nuclear receptor transcription factors composed of a retinoic acid receptor (RAR) and a retinoid X receptor (RXR) subunit, and promotes both normal granulocytic differentiation and the abundance, quiescence, and activity of HSCs21–24. Furthermore, atRA is highly effective as a therapeutic agent in acute promyelocytic leukemia (APL), a subtype of AML characterized by fusion proteins involving RARα. The most frequent of these, PML-RARα, does not respond to physiological doses of atRA, yet myeloid differentiation is restored by pharmacological levels of this agent25. In contrast, even though atRA also promoted the differentiation of non-APL AML blasts, clinical trials have failed to reveal any clear therapeutic benefit in these patients26–28. Certain molecularly or genetically defined subgroups of non-APL AML were suggested to gain a survival advantage from atRA, but no consistent picture has yet emerged26,27,29,30.
atRA regulated expression of EVI1 both in cell lines and in primary AML cells30,31. Conversely, EVI1 acted as a modulator of transcriptional responses to atRA, and augmented anti-leukemic activities of atRA in human myeloid cell lines and primary AML cells29,30. However, since AML is a stem cell-driven disease, it is important to understand the impact of potential therapeutics on LSCs. Relatively little is known about the effects of atRA on, and even less about the role of EVI1 in, AML LSCs. Here, we report that EVI1 promoted essential properties of LSCs, and atRA enhanced its effects. Furthermore, EVI1 strongly augmented atRA regulated gene transcription in LSC enriched cells, and one of their joint targets, Notch4, was a relevant mediator of their effects on leukemic stemness. Conversely, a pan-RAR antagonist reduced AML stemness and delayed leukemogenesis, raising the possibility of using RAR antagonists in the therapy of EVI1high AML.
Methods
Ethics statement
Animal experiments were approved by the Animal Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science, and Research (GZ66.009/0308-WF/V/3b/2015). Federation of European Laboratory Animal Science Associations guidelines to minimize animal distress and suffering were followed. Experiments with primary AML samples were approved by the Ethics Committee of the Medical University of Vienna (EK 1394/2019) and conducted in accordance with the declaration of Helsinki.
Generation of MA9-driven murine AML with high or low expression of Evi1
HSC enriched Lin− Sca-1+ c-Kit+ (LSK) cells and common myeloid progenitors (CMPs; Lin− Sca-1− c-Kit+ CD34+ CD16/CD32low cells; Supplementary Fig. S1A) were isolated from bone marrow (BM) of 6–8 week old C57BL/6 mice (Department of Laboratory Animal Science & Genetics, Himberg, Austria), transduced with pMSCV_MA9_IRES_Venus, and transplanted into sub-lethally irradiated congenic recipient mice4,32. Venus+ BM or spleen cells from terminally ill mice were considered leukemic cells (LCs), and are referred to as LCLSK_MA9 and LCCMP_MA9, respectively. To knock down Evi1 in LCLSK_MA9, they were transduced with lentiviral vectors (pLKO.1_puro_CMV_TagRFP) containing shEvi1_41, shEvi1_43, shEvi1_44 or control shRNA SHC012 (shCtrl; Sigma-Aldrich). Venus+ RFP+ cells were used for transplantation. Venus+ RFP+ cells from BM or spleen of terminally ill recipient mice were designated LCLSK_MA9_shEvi1 and LCLSK_MA9_shCtrl, respectively.
Ex vivo culture and flow cytometric analyses of cells from leukemic mice
BM cells from leukemic mice were cultured in IMDM medium containing 10% FBS, 1% l-Glutamine (all from Thermo Fisher Scientific), 50 ng/ml mSCF, 10 ng/ml mIL-3, 10 ng/ml mTPO, 10 ng/ml mFlt3L (all from Peprotech), and 10 ng/ml mIL-6 (Biolegend). For treatment, cells were seeded at a density of 2 × 105 per ml and incubated with 1 µM atRA (Sigma-Aldrich), 1 µM pan-RAR antagonist AGN193109 (Tocris), 5 µM γ-secretase inhibitor DAPT (Stem Cell Technologies), or the corresponding amounts of DMSO (Sigma-Aldrich) for 72 h, unless indicated otherwise. By gating on fluorescence marker positive cells, all analyses were restricted to LCs. LC differentiation and the proportion of LSC enriched cells (LSCe; Venus+ or Venus+ RFP+, Lin− Sca1− c-Kit+ CD34+ CD16/CD32hi cells4,5) were determined by flow cytometric analysis of BM cells stained with the respective antibodies (Supplementary Table S1).
To determine the cell cycle distribution of LSCe, BM cells were stained for LSCe surface markers, fixed and permeabilised in Cytofix/Cytoperm (BD Biosciences), stained with Ki-67 antibody (Supplementary Table S1) and DAPI (Sigma-Aldrich), and subjected to flow cytometry. The cut-off for Ki-67 positivity was determined using an isotype control antibody. Among cells in the LSCe gate, Ki-67- cells with a 2N DNA content were considered to be in G0. Flow cytometry was performed on an LSR Fortessa SORP (BD Biosciences), and data were analyzed with FlowJoX software (Treestar).
Serial replating assay
For serial replating assays, BM LCs were incubated with 1 µM atRA, 5 µM DAPT, or the corresponding amounts of DMSO for 72 h, or left untreated when obtained from antagonist treated mice. 2000 cells per condition were seeded into methyl cellulose (MethoCult GF M3434; Stem Cell Technologies). Colonies were counted after 7 days, and 2000 cells per condition were used for replating.
RNA sequencing (RNA-seq) and bioinformatics analyses
LSCeLSK_MA9_shCtrl (from three different mice) and LSCeLSK_MA9_shEvi1 (shEvi1_41, shEvi1_43, and shEvi1_44) were isolated from spleens of terminally ill mice, recovered for 24 h, and incubated with 1 µM atRA or the corresponding amount of DMSO for another 24 h. RNA isolation, library preparation, RNA-seq, and data analysis are described in Supplementary Methods. By applying a false discovery rate (FDR) of <0.05, genes regulated by EVI1 in the absence (Er_D) or presence (Er_A) of atRA, and genes regulated by atRA in the absence (Ar_shE) or presence (Ar_shC) of EVI1 were identified. Furthermore, genes were identified whose expression patterns mirrored the observed biological effects, i.e., which showed little or no regulation by atRA in shEvi1 cells, and whose regulation by EVI1 was enhanced by atRA (Er_D/Ar_shC; see Supplementary Methods for detailed definition). Heatmaps were generated based on z-score transformation of mean normalized counts across all four conditions. RNA-seq data were deposited in the Gene Expression Omnibus (GSE123255).
In vivo limiting dilution assay
BM LCLSK_MA9_shCtrl and LCLSK_MA9_shEvi1_44 were incubated with 1 µM atRA or the corresponding amount of DMSO. After 72 h, 5000, 2000, 750, 250, 125, and 25 cells were transplanted into sub-lethally irradiated 6–8 week old female C57BL/6 recipient mice (5 mice per condition). Two independent experiments were performed, and data were combined for the final analysis. Mice were sacrificed when signs of disease became evident, or at the end of the experiment (4 months after transplantation), and BM cells were collected for flow cytometric analysis. Recipient mice with less than 1% Venus+ RFP+ cells in BM were considered as non-responders. LSC frequency was calculated by applying the maximum likelihood method using ELDA software33.
In vivo treatment with the pan-RAR antagonist AGN193109 and secondary transplantation
Sub-lethally irradiated C57BL/6J mice were transplanted with 40,000 LCLSK_MA9_shCtrl. Seven days post transplantation (at which time Venus+ RFP+ cells started to appear in PB), mice were randomly divided into two groups (n = 4/group) and treated daily with either AGN193109 (1 mg/kg) or an equivalent amount of vehicle (2.55% DMSO in PBS) by intraperitoneal injection for 14 days24,34,35. Terminally ill mice were sacrificed, and the proportion, cell cycle distribution, and activity of LSCe/LSCs in BM were determined as described above. For secondary transplantations, sub-lethally irradiated C57BL/6J mice were transplanted with 20,000 BM LCs derived from terminally ill, AGN193109 or vehicle treated mice (n = 5/group).
Knock-down of EVI1 in human myeloid cell lines
Culture of HNT-34 and UCSD/AML1 cells, their transduction with shEVI1_1, shEVI1_2 (Open Biosystems), and control shRen in LT3REVIR36, as well as atRA treatment and biological assays are described in Supplementary Methods.
Primary AML samples
Cryopreserved primary AML samples were provided by the Hanusch hospital (Vienna, Austria). They were thawed in a 25 °C water bath and washed with RPMI medium containing 5 µg/ml DNAse (Sigma-Aldrich). Cells were incubated for 60 min at 37 °C and 5% CO2 with RPMI medium containing 50 µg/ml DNAse to prevent cell clumping, washed with PBS, and cultured in RPMI medium supplemented with 10% FBS, 1% glutamine, 1% Penicillin/Streptomycin, and 100 ng/ml each of SCF, IL3, and G-CSF (all from Peprotech). For treatment, cells were seeded at a density of 2 × 105/ml and incubated with 1 µM atRA, 1 µM AGN193109, or the corresponding amount of DMSO for 72 h. At the end of the incubation time, cells were harvested, stained with the respective antibodies (Supplementary Table S1), and subjected to flow cytometric analysis of CD11b expression or of the cell cycle distribution of the LSC enriched CD34+ CD38− population. Another portion of cells was transferred to methyl cellulose (MethoCult H4434, Stemcell Technologies) at a concentration of 1 × 105 cells per well of a 6-well plate. Technical duplicates were performed, and total colonies were counted after 14 days.
Statistical analyses
For each experiment, independent biological replicates were performed; their numbers are indicated in the figure legends. In the case of ex vivo experiments with murine LCs, cells for these replicates were obtained from different mice. Bar graphs depict means±SEM. Significance of differences between two independent groups was calculated using two-sided Student’s t-test; significance of differences between multiple groups was determined by two-way ANOVA followed by Bonferroni’s post-hoc test. The log-rank test was used to evaluate survival differences between groups of mice. Analyses were performed using GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA). LSC frequencies were calculated by applying the maximum likelihood method using ELDA software and statistical significance of their differences was assessed using the Chi-square test. p-values <0.05 were considered statistically significant.
Additional methods
Additional and more detailed methods are described in Supplementary Methods.
Results
Evi1high LCLSK_MA9 exhibit a higher degree of stemness than Evi1low LCCMP-MA9, and atRA further enhances LSC properties only in the former
To investigate the effects of Evi1 and atRA on AML stemness, an MA9-driven mouse model was used (Fig. 1a). In agreement with previous reports4,37, both LSK cells and CMPs transduced with pMSCV_MA9_IRES_Venus caused AML-like disease upon transplantation into sub-lethally irradiated mice, but LSK-derived disease displayed shorter disease latencies and higher white blood cell counts (WBC) (Fig. 1b; Supplementary Fig. S1B, C). Also as shown before12,19,20, Evi1 expression levels were substantially (>100-fold) higher in LCLSK_MA9 than in LCCMP_MA9 (Fig. 1c). To determine the impact of the cell of origin, and of atRA, on LC maturity, LCLSK_MA9 and LCCMP_MA9 were treated with atRA or solvent and subjected to flow cytometry for the myeloid differentiation markers Gr-1 and CD11b38. The proportion of immature (Gr-1−) among myeloid (CD11b+) cells was higher for LCLSK_MA9 than for LCCMP_MA9. Treatment with atRA for three or seven days significantly enhanced the frequency of immature cells in LCLSK_MA9, but had only a small, non-significant effect in LCCMP_MA9 (Fig. 1d; Supplementary Fig. S1D, E).
Fig. 1. Differential effects of atRA on maturation and stemness of Evi1high LCLSK_MA9 and Evi1low LCCMP_MA9.

a Schematic of experimental design. BM, bone marrow. b Kaplan–Meier plot of mice transplanted with MA9 transduced LSK cells and CMPs (300,000 cells/mouse). n = 3/group; **p < 0.01; log-rank test. c Relative Evi1 mRNA levels in BM LCLSK_MA9 and LCCMP_MA9 (left panel) and the corresponding LSCe (right panel). qRT-PCR; n = 3; **p < 0.01; t-test. d–g BM cells (d–f) or BM LCs (Venus+ cells; panel g) from terminally ill mice were treated with 1 µM atRA or solvent for 3 days. n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant; ANOVA followed by Bonferroni's post-hoc test. d Myeloid differentiation. e Proportions of LSCe among LCs. f Proportions of quiescent LSCe (LSCe in G0). g Colony formation in methyl cellulose, presented as percent of solvent-treated LCLSK_MA9 in each round of plating.
In MA9-driven murine AML, LSCs are strongly enriched in a cell population carrying the immuno-phenotype of granulocyte macrophage progenitors4,5. We therefore refer to fluorescence marker positive cells with this immuno-phenotype as LSCe (LSCeLSK_MA9, LSCeCMP_MA9). Like in bulk LCs, the expression of Evi1 was substantially higher in LSCeLSK_MA9 than in LSCeCMP_MA9 (Fig. 1c). In agreement with their derivation from a stem cell enriched population, LCLSK_MA9 contained a higher proportion of LSCe than LCCMP_MA9. Also, atRA increased LSCe abundance among LCLSK_MA9, but had a smaller, non-significant effect in LCCMP_MA9 (Fig. 1e; Supplementary Fig. S1F, G). Moreover, a higher proportion of LSCeLSK_MA9 than of LSCeCMP_MA9 accumulated in the G0 phase of the cell cycle, and atRA further enhanced the proportion of quiescent LSCeLSK_MA9, but not LSCeCMP_MA9 (Fig. 1f; Supplementary Fig. S1H). In a serial replating assay, which reflects stem cell activity, LCLSK_MA9 displayed higher colony formation potential than LCCMP_MA9, and atRA increased clonogenic activity of LCLSK_MA9, but not LCCMP_MA9 (Fig. 1g). In summary, our data demonstrate that atRA prevented LC maturation, and promoted LSC properties more efficiently in Evi1high LCLSK_MA9 than in Evi1low LCCMP_MA9.
Evi1 increases stemness, and facilitates further augmentation of stemness by atRA, in MA9-driven murine AML
The previous experiment employed different cells of origin (LSK cells vs. CMPs) to generate Evi1high and Evi1low LCs, mimicking the situation in human MA9-driven AML. However, the resulting LCs differ from each other by the expression not only of Evi1, but of multiple genes37. To test whether the above described differences between LCLSK_MA9 and LCCMP_MA9 and the LSCe/LSCs therein were at least partially due to differences in Evi1 expression, a knock-down approach was used. LCLSK_MA9 were transduced with lentiviral vectors containing shRNAs against Evi1 (validated by immunoblotting, Supplementary Fig. S2A) or shCtrl, and transplanted into recipient mice (Fig. 2a). Consistent with earlier reports12,13, knock-down of Evi1 significantly delayed leukemogenesis, and furthermore decreased WBC and the percentage of LCs in spleen of terminally ill mice (Fig. 2b; Supplementary Fig. S2B, C). qRT-PCR confirmed strongly decreased Evi1 mRNA expression in LCLSK_MA9_shEvi1 vs. LCLSK_MA9_shCtrl and the corresponding LSCe (Fig. 2c). While atRA increased the proportion of immature cells among LCLSK_MA9_shCtrl, down-regulation of Evi1 strongly reduced the proportion of immature cells both in the absence and presence of atRA (Fig. 2d, Supplementary Fig. S2D). Evi1 knock-down also decreased the proportion of LSCe among LCs, the fraction of LSCe in G0, and the activity of LSCs. Moreover, these parameters were augmented by atRA in LCLSK_MA9_shCtrl, but not LCLSK_MA9_shEvi1 (Fig. 2e–g; Supplementary Fig. S2E-G). Thus, experimental manipulation of Evi1 expression reproduced the effects of the cell of origin (LSK cells vs. CMPs) on key LC/LSCe/LSC properties in MA9-driven murine AML, indicating that Evi1 represents a central determinant of the characteristics of transformed HSCs. Furthermore, Evi1 unleashed the ability of atRA to promote immaturity and stemness of AML.
Fig. 2. Evi1 prevents LC maturation and promotes LSC properties, and its effects are enhanced by atRA.

a Schematic of experimental design. FC, flow cytometry. b Kaplan–Meier plot of mice transplanted with shCtrl or shEvi1-transduced LCLSK_MA9 (250,000 cells/mouse). n = 4/group; *p < 0.05; log-rank test. c Relative Evi1 mRNA levels in BM LCLSK_MA9_shCtrl and LCLSK_MA9_shEvi1 (left panel) and the corresponding LSCe (right panel). qRT-PCR; n = 3; ***p < 0.001; t-test. d–g BM cells (d–f) or BM LCs (Venus+ RFP+ cells; panel g) from terminally ill mice were treated with 1 µM atRA or solvent for 3 days. n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant; ANOVA followed by Bonferroni's post-hoc test. d Myeloid differentiation. e Proportions of LSCe among LCs. f Proportions of quiescent LSCe (LSCe in G0). g Colony formation in methyl cellulose, presented as percent of solvent-treated LCLSK_MA9_shCtrl in each round of plating.
Evi1 and atRA augment LSC activity and mutually enhance their effects in an in vivo limiting dilution assay
The in vivo limiting dilution assay represents the gold standard for measuring LSC activity, and was used to further confirm the effects of Evi1 and atRA on LSCs (Fig. 3a). Knock-down of Evi1 significantly decreased LSC frequency compared to control (Fig. 3b). Furthermore, atRA significantly enhanced LSC frequency among LCLSK_MA9_shCtrl. The effect of atRA in LCLSK_MA9_shEvi1 was substantially smaller and not significant (Fig. 3b). Taken together, these data demonstrate that Evi1 is a critical positive regulator of LSC function, and its expression augments atRA promoted stemness in MA9-driven murine AML.
Fig. 3. Evi1 and atRA cooperate to augment LSC activity in an in vivo limiting dilution assay.

a Schematic of experimental design. BM, bone marrow; FC, flow cytometry. b In vivo limiting dilution assay to determine the frequency of functional LSCs. Upper: table showing the numbers of responders (defined by the presence of ≥1% Venus+ RFP+ cells in BM at the time of sacrifice) and the total numbers of evaluable recipients for each cell dose. Data from two independent experiments were combined, and LSC frequencies were calculated by applying the maximum likelihood method using ELDA software. Lower: Plot showing the logarithmized fractions of non-responding recipients of different numbers of LCs. Statistical significance was assessed using the Chi-square test.
EVI1 regulates a large number of genes, and enhances atRA dependent gene regulation, in LSCe
Both EVI1 and atRA exert their biological effects mainly through the regulation of gene transcription9,10. To explore the molecular basis of their impact on LSCs, RNA-seq was performed on atRA or solvent treated LSCeLSK_MA9_shCtrl and LSCeLSK_MA9_shEvi1. 1315 and 1469 genes were significantly (FDR < 0.05) regulated by EVI1 in the absence and presence of atRA, respectively; 936 of these (71 and 64%, respectively) responded to EVI1 in either condition (Fig. 4a, Supplementary Table S2A, B). Biological pathways enriched among these genes are listed in Supplementary Table S2C. Reaffirmingly, a transcription factor analysis (Metacore) showed that the lists of genes regulated by EVI1 in LSCe were enriched for previously reported targets of EVI1 (MECOM), as well as for targets of transcription factors known to be regulated by, or to functionally interact with, EVI1 (e.g., GATA-2, c-Fos, c-Jun, RUNX1, SMAD3, and PU.1;9,15,39–41 Supplementary Table S2D, E). Furthermore, gene set enrichment analysis (GSEA)42 revealed that gene expression profiles associated with stemness and/or poor outcome in AML, as well as EVI1-dependent profiles previously identified in other experimental systems, were enriched in our EVI1-dependent signature (albeit not always in the expected direction; Supplementary Table S2F).
Fig. 4. EVI1 augments transcriptional responses to atRA in LSCe, and Notch4 is a relevant joint target.

LSCeLSK_MA9_shCtrl and LSCeLSK_MA9_shEvi1 were treated with atRA or solvent for 24 h and subjected to RNA-sequencing. a Numbers of genes regulated by EVI1 or atRA at an FDR < 0.05. b Heatmap of genes that were minimally affected by atRA in LSCeLSK_MA9_shEvi1, but whose regulation by EVI1 was enhanced by atRA in LSCeLSK_MA9_shCtrl. c Pathway enrichment analysis of genes shown in b. d–f BM cells from mice terminally ill after transplantation with MA9 transduced LSK cells were further transduced with shCtrl, shNotch4_1, or shNotch4_2. Venus+ RFP+ cells were treated with 1 µM atRA or an equivalent amount of solvent for 3 days. n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant; ANOVA followed by Bonferroni's post-hoc test. d Proportions of LSCe among LCs. e Proportions of quiescent LSCe (LSCe in G0). f Colony formation in methyl cellulose, presented as percent of solvent-treated, shCtrl transduced LCLSK_MA9 in each round of plating.
atRA altered the expression of 811 genes in LSCeLSK_MA9_shCtrl, but of only 343 genes in LSCeLSK_MA9_shEvi1; 287 of these genes (35 and 84%, respectively) responded in both cell types (Fig. 4a, Supplementary Table S3A, B). These numbers indicated that gene regulation by atRA in LSCe was strongly dependent on EVI1, because EVI1 down-regulation reduced the number of genes affected by atRA by more than half. Pathways enriched among atRA regulated genes are listed in Supplementary Table S3C, and GSEA results in Supplementary Table S3F. The transcription factor analysis further illustrated the strong impact of EVI1 on gene regulation by atRA: the list of genes regulated by atRA in LSCeLSK_MA9_shCtrl, but not the corresponding list from LSCeLSK_MA9_shEvi1, was enriched for reported targets of PU.1, several C/EBP family members, c-Fos, SMAD3, and GATA-2. Remarkably, even RARα, RARβ, and RXRγ targets were enriched only among genes regulated by atRA in the presence, but not the absence, of EVI1 (Supplementary Table S3D, E).
Finally, we searched for genes with a transcriptional pattern reflecting the biological response pattern to EVI1 and atRA. Genes that were only minimally affected by atRA in LSCeLSK_MA9_shEvi1, but that were regulated by EVI1 in a manner that was enhanced by atRA, were defined as described in the Supplementary Methods. 33 genes conformed to this definition; 24 of these were induced and 9 repressed by EVI1 (Fig. 4b; Supplementary Table S4A). Despite this small number of genes, several pathways were significantly enriched among them (Fig. 4c; Supplementary Table S4B).
Collectively, these data show that Evi1 and atRA interacted not only to regulate key biological properties of LSCe/LSCs, but also with respect to the regulation of gene transcription.
Notch4 is a downstream mediator of the effects of Evi1 and atRA on leukemic stemness
Among the genes whose up-regulation by EVI1 was enhanced by atRA, Notch4 represented a particularly interesting candidate to mediate at least some of their biological effects on LSCs: firstly, Notch4 responded strongly to both EVI1 and atRA (Fig. 4b; Supplementary Table S4A), secondly, NOTCH4 expression was higher in AML compared to normal BM and to normal hematopoietic stem and progenitor cells (Supplementary Fig. S3A–C), thirdly, NOTCH4 has previously been implicated in tumor aggressiveness43–47 (see Discussion for details), and fourth, Evi1 acted through Notch to promote HSC emergence during zebrafish embryogenesis48. Joint regulation of Notch4 by EVI1 and atRA was confirmed by qRT-PCR (Supplementary Fig. S3D). The γ-secretase inhibitor DAPT reduced LSCe/LSC abundance, quiescence, and activity in an Evi1-dependent manner, and diminished the effects of atRA on leukemic stemness (Supplementary Fig. S3E–I). To confirm that the effects of DAPT were specific to Notch4 rather than other γ-secretase targets, bone marrow cells from terminally ill LSK_MA9 recipient mice were transduced with two different shRNAs against Notch4 (shNotch4_1, shNotch4_2, validated by qRT-PCR and flow cytometry, Supplementary Fig. S3J, K) or with shCtrl. Knock-down of Notch4 reduced the abundance, quiescence, and activity of LSCe/LSCs, and counteracted the effects of atRA on these parameters (Fig. 4d–f). Together, these data establish Notch4 as a relevant downstream mediator of the effects of Evi1 and atRA on LSCs.
A pan-RAR antagonist decreases LSCe abundance and quiescence in an Evi1-dependent manner
To determine whether the above described effects of Evi1 on LC immaturity and leukemic stemness may have been augmented by trace amounts of atRA present in the cell culture media49, LCLSK_MA9, LCCMP_MA9, LCLSK_MA9_shCtrl, and LCLSK_MA9_shEvi1 were treated with the pan-RAR antagonist AGN193109 or solvent for three days. AGN193109 had no significant effect on LC maturity (Fig. 5a). Regarding LSCe properties, AGN193109 decreased the abundance and quiescence of LSCeLSK_MA9 and LSCeLSK_MA9_shCtrl, but not of LSCeCMP_MA9 or LSCeLSK_MA9_shEvi1 (Fig. 5b, c). This demonstrates that the impact of atRA on LSCe was not only dependent on Evi1, but conversely, the effects of Evi1 were partially dependent on activated retinoic acid receptors.
Fig. 5. The pan-RAR antagonist AGN193109 decreases LSCe abundance and quiescence in an Evi1-dependent manner.

BM cells from terminally ill recipients of MA9-transduced LSK cells or CMPs, or of shRNA transduced LCLSK_MA9 were treated with 1 µM AGN193109 or solvent for 3 days. n = 3; *p < 0.05; **p < 0.01; ns, not significant; ANOVA followed by Bonferroni's post-hoc test. a Myeloid differentiation. b Proportions of LSCe among LCs. c Proportions of quiescent LSCe (LSCe in G0).
In vivo treatment with pan-RAR antagonist delays leukemogenesis and reduces stemness of Evi1high AML
Since atRA enhanced, and the pan-RAR antagonist AGN193109 decreased, key LSCe/LSC properties in an Evi1 dependent manner ex vivo, we next asked whether AGN193109 would inhibit leukemia formation and stemness in vivo. Mice were transplanted with LCLSK_MA9_shCtrl and treated with AGN193109 or vehicle for two weeks (Fig. 6a). Notably, AGN193109 significantly improved overall survival compared to control (Fig. 6b). Even though it had no effects on the proportion of LCs in BM, WBC, or spleen weight of terminally ill mice (Fig. 6c; Supplementary Fig. S4A, B), it decreased the percentage of LCs in spleen (Fig. 6c; Supplementary Fig. S4C) and promoted myeloid differentiation (Fig. 6d; Supplementary Fig. S4D). Furthermore, in vivo exposure to AGN193109 decreased LSCe abundance and quiescence (Fig. 6e, f; Supplementary Fig. S4E, F), and caused down-regulation of Notch4 (Supplementary Fig. S4G). In a serial replating assay with in vivo treated cells, AGN193109 increased progenitor activity (first plating), but decreased LSC activity (replatings; Fig. 6g). Furthermore, re-transplantation experiments showed that AGN193109 treatment of primary recipients delayed leukemogenesis (Fig. 6h) and decreased spleen weight (Supplementary Fig. S4H) in secondary recipients. Taken together, in vivo treatment with AGN193109 delayed the emergence of Evi1high AML, and this was associated with enhanced myeloid differentiation and decreased stemness.
Fig. 6. The pan-RAR antagonist AGN193109 delays leukemogenesis and decreases stemness in MA9-driven, Evi1high murine AML.

a Schematic of experimental design. b Kaplan–Meier plot of mice transplanted with LCLSK_MA9_shCtrl (40,000 cells/mouse) and treated with AGN193109 (1 mg/kg) or vehicle (2.55% DMSO in PBS) by daily intraperitoneal (i.p.) injection for 14 days. n = 4/group; **p < 0.01; log-rank test. c–f Flow cytometric analysis of spleen (c) and BM cells (c–f) derived from terminally ill, AGN193109 or vehicle treated mice. LCs were defined as Venus+ RFP+ cells, but similar results were obtained if the analyses were not restricted to shCtrl expressing cells, but included all leukemic (Venus+) cells. n = 4; *p < 0.05; **p < 0.01; ns, not significant; t-test. c Percentages of LCs among BM and spleen cells. d Myeloid differentiation. e Proportions of LSCe among LCs. f Proportions of quiescent LSCe (LSCe in G0). g Colony formation in methyl cellulose by LCs, presented as percent of colonies from vehicle-treated mice in each round of plating. n = 3; *p < 0.05; ***p < 0.001; ANOVA followed by Bonferroni's post-hoc test. h Kaplan–Meier plot of mice transplanted with BM LCs derived from terminally ill, AGN193109 or vehicle treated mice (20,000 cells/mouse). n = 5/group; **p < 0.01; log-rank test. 2nd BMT, secondary bone marrow transplantation.
EVI1 promotes quiescence in human AML cell lines with stem cell characteristics, and atRA enhances its effect
To determine whether the results obtained with the MA9 mouse model could be confirmed in human cells, the AML derived cell lines HNT-34 and UCSD/AML1 were used. Both cell lines express high levels of the stem cell gene EVI1, and are positive for the stem and progenitor cell associated surface marker CD34. They were transduced with lentiviral vectors expressing shRNAs targeting EVI1 or a control shRNA (shRen) in a doxycycline-inducible manner. Immunoblot analysis confirmed the down-regulation of EVI1 in HNT-34_shEVI1 and UCSD/AML1_shEVI1 cells (Fig. 7a, b; Supplementary Fig. S5A, B). Knock-down of EVI1 reduced CD34 expression in UCSD/AML1, but not HNT-34 cells (Supplementary Fig. S5C, D), probably reflecting differences between the two cell lines regarding the contributions of other regulators to the expression of CD34. In both cell lines, EVI1 knock-down slightly increased mean fluorescence intensity of the myeloid differentiation marker CD11b, and re-sensitized cells to atRA-induced differentiation (Fig. 7c, d; Supplementary Fig. S5E, F). Accordingly, it decreased, and enhanced the atRA-induced reduction, in viability (measured via the proxy, metabolic activity; Fig. 7e, f; Supplementary Fig. S5G, H). Thus, like in primary murine cells, EVI1 acted to prevent myeloid maturation in these cell lines. In contrast, atRA revealed its previously described contradictory effects21–24 in the two systems, preventing and promoting differentiation in primary mouse cells vs. human cell lines, respectively. Similar to the mouse model, EVI1 and atRA interacted to promote quiescence of HNT-34 and UCSD/AML1 cells: down-regulation of EVI1 decreased the proportion of cells in G0, and reduced the atRA-induced increase in cell quiescence (Fig. 7g, h; Supplementary Fig. S5I, J). In summary, like in primary mouse cells, the combined effects of EVI1 and atRA in these human AML cell lines, which retain some stem cell characteristics, were the prevention of differentiation and the promotion of quiescence.
Fig. 7. EVI1 and atRA cooperatively promote quiescence in human myeloid cell lines with stem cell characteristics.

a, b Immunoblot analyses of HNT-34 (a) and UCSD/AML1 (b) derivative cell lines. Cells were treated with doxycycline for 2 days to induce shRNAs. Left panels, representative experiments. Right panels, quantification; n = 3; **p < 0.01; ***p < 0.001; t-test. (c–h) HNT-34 and UCSD/AML1 derivative cell lines were treated with doxycycline for 2 days; 1 µM atRA or solvent was added for another 3 days. n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant; ANOVA followed by Bonferroni's post-hoc test. c, d Myeloid differentiation (mean fluorescence intensity (MFI) of CD11b). HNT-34 (c), UCSD/AML1 (d). e, f Relative viability (measured using metabolic activity as a proxy). HNT-34 (e), UCSD/AML1 (f). g, h Proportion of quiescent cells (cells in G0). HNT-34 (g), UCSD/AML1 (h). For all panels, control experiments performed in the absence of doxycycline are shown in Supplementary Fig. S5.
atRA increases, and AGN193109 decreases, stemness in primary human AML samples in a manner related to the expression of EVI1
Finally, we tested the effects of atRA and AGN193109 on cell differentiation, LSCe quiescence, and stem/progenitor activity of primary human AML samples. Samples from six AML patients (four EVI1high and two EVI1low as determined by qRT-PCR, Supplementary Fig. S6A) were included in the study; their clinical characteristics are summarized in Supplementary Table S5. Effects of a three day treatment with atRA or AGN193109 on CD11b expression of these primary cells were small and did not show a clearly EVI1 related pattern (Supplementary Fig. S6B). In contrast, atRA enhanced the fraction of LSC enriched cells (CD34+ CD38- cells, LSCe) in G0 in 3/4 EVI1high samples, but had the opposite or no effect in the two EVI1low samples (Fig. 8a, Supplementary Fig. S6C). Antagonist treatment did not yield a clear pattern in this assay. However, atRA increased, and AGN193109 decreased, clonogenic capacity in 3/4 EVI1high samples, while in EVI1low samples, atRA slightly decreased colony formation, and AGN193109 had only minimal effects (Fig. 8b). In summary, even though AGN193109 effects in these experiments were relatively small, probably reflecting low atRA concentrations in the media, our data show that atRA promotes, and the pan-RAR antagonist counteracts, stem/progenitor properties in a manner related to the expression of EVI1 also in primary AML samples.
Fig. 8. Effects of retinoids on stem cell/progenitor properties of primary AML samples with variable EVI1 expression.
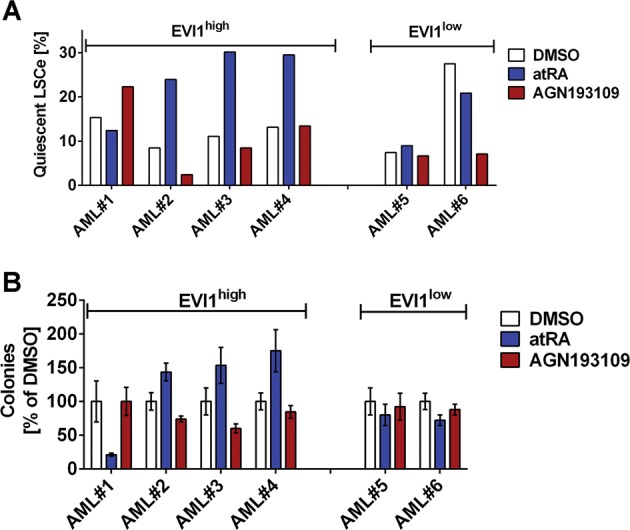
Primary AML samples were treated with 1 µM atRA, 1 µM AGN193109, or corresponding amounts of solvent (DMSO) for 3 days and subjected to flow cytometric analysis or transferred to methyl cellulose. a Proportion of quiescent LSCe (LSCe (CD34+ CD38- cells) in G0). b Colony formation in methyl cellulose. Mean +/- SD from duplicate measurements, presented as percent of DMSO-treated cells.
Discussion
EVI1 overexpression is associated with a particularly poor prognosis in AML6–8. Although its roles in leukemogenesis, normal HSCs, and chronic myeloid leukemia LSCs have been studied extensively11–15,50, its impact on AML LSCs, which are the key drivers of this disease1, has not been investigated so far. Here, we establish a previously unreported role of Evi1 in augmenting key LSC properties in an MA9-driven mouse model of AML, and show that atRA further promoted AML stemness in an EVI1-dependent manner. Indeed, in the MA9 model, the activity of atRA was abolished in Evi1low LSC/LSCeCMP_MA9 or LSC/LSCeLSK_MA9_shEvi1. In the human AML cell lines, analogous effects of, and interactions between, EVI1 and atRA were observed, but these were only reduced by the EVI1 knock-down, likely due to its incompleteness. Results were further confirmed in primary AML samples with high and low EVI1 expression, with the exception of AML #1, which expressed high levels of EVI1, but did not respond to atRA in the expected manner. Since AML is a genetically and molecularly heterogeneous disease, this may be due to the presence of additional lesions that may be able to counteract the effects of EVI1 (see below).
In addition to their roles in LSCs, EVI1 and atRA interacted with respect to LC differentiation: in the mouse model, they cooperated to maintain cells in a more immature state; in the human cell lines, EVI1 prevented atRA-induced differentiation. Thus, in both models, atRA-treated cells that expressed EVI1 were less mature than those not expressing this gene. No strong effect of atRA on myeloid differentiation was observed in primary AML samples. This is in contrast to a previous report30, which may be due to differences in incubation time, atRA dose, and/or additional genetic and molecular lesions present in the primary samples.
Several EVI1/MECOM mRNA and protein variants have been described51. The most abundant and best characterized of these are (i) the originally described 1051 amino acid variant usually referred to as EVI1, (ii) splice variant EVI1Δ324, which lacks part of the first of two zinc finger domains present in EVI1, and (iii) alternative promoter variant MDS1/EVI1, which has an N-terminal extension containing a PR-domain as compared to EVI151. EVI1Δ324 is often co-expressed with EVI1, but appears to be of subordinate functional importance52,53. This is confirmed by the fact that it is not targeted by shEVI1_2, which nevertheless caused the same phenotypes as shEVI1_1 in our experiments. All other shRNAs used in this study target all three EVI1 mRNA variants. Both EVI1 and MDS1/EVI1 are expressed in the MA9 model, but, as is usually the case in cells with 3q26 rearrangements17,18, only EVI1 is expressed in UCSD/AML1 and HNT-34 cells. This may indicate that the PR-domain lacking variant EVI1 causes the increased stemness and decreased differentiation observed in our model systems. This would also be in line with a widespread assumption that EVI1 is the oncogenic protein variant, while MDS1/EVI1 may act as its antagonist54,55. However, roles for Mds1/Evi1 in HSCs and in MA9-driven murine AML have also been reported56,57. Therefore, future experiments will have to resolve the question which EVI1 protein variant(s) mediate(s) the phenotypes described in the present report.
The situation is even more complex for the retinoic acid receptor, which is composed of an RAR and an RXR subunit, each of which is encoded by three paralogous genes that additionally are subject to alternative splicing58,59. In normal hematopoiesis and an AML1-ETO driven mouse model of AML, atRA promoted the maintenance of stem cells and the differentiation of more mature myeloid cells through the actions of Rarg and Rara, respectively23,38. In an acute promyelocytic leukemia cell line, the splice variants RARA1 and RARA2 had differentiation promoting and inhibiting functions, respectively59. By analogy, RARG may mediate the effects of atRA on stemness, and RARA those on differentiation, in our models. However, further experiments are required to directly address this issue.
RNA-seq on atRA or solvent treated LSCeLSK_MA9_shCtrl and LSCeLSK_MA9_shEvi1 revealed a pivotal role of EVI1 also in augmenting transcriptional effects of atRA in LSCe. Among the genes that were jointly regulated by EVI1 and atRA, Notch4 appeared as a particularly promising candidate to mediate their biological effects in LSCs. NOTCH4 belongs to a family of transmembrane receptor proteins. NOTCH4 up-regulation correlated to metastasis formation in melanoma60 and colorectal cancer44, and its experimental manipulation in cancer cell lines revealed a role in promoting metastasis-related properties60–62. NOTCH4 was also up-regulated in high-risk B-ALL patients, and its inhibition sensitized B-ALL cells to chemotherapeutic drugs both in vitro and in vivo47. Experimental expression of Notch4 inhibited myeloid differentiation and promoted HSC expansion in mice63. Furthermore, genetic or pharmacologic inhibition of NOTCH4 synergized with FLT3 inhibition to more effectively eliminate FLT3-ITD+ AML cells46. Our data extend these previous findings and suggest an important role of Notch4 in promoting AML stemness.
Although a number of clinical trials have investigated the therapeutic potential of atRA in non-APL AML26–28,64, only few reports have addressed the impact of atRA on AML LSCs. atRA decreased LSC frequency in Nup98-HoxD13/FLT3-ITD-driven murine AML65, but had the opposite effect in murine BM cells expressing the AML1-ETO fusion protein38, suggesting that the effects of atRA on LSCs are strongly influenced by the identity of the respective genetic driver lesions. Supporting this notion, our study revealed that atRA promoted leukemic stemness in the MA9 mouse model, in human AML cell lines, and in primary AML samples in an EVI1 dependent manner. These data establish EVI1, a gene whose expression reflects the immaturity of the originally transformed cell at least in MLL-rearranged AML, as an important determinant of LSC responses to atRA.
Previously, EVI1 and atRA were found to cooperate to enhance anti-leukemic activities in AML samples and cell lines29,30, while our study, focusing on LSCs, indicated the opposite, resulting in diverging assumptions about the possible utility of retinoids in the therapy of EVI1high AML. In fact, patients with EVI1high AML were not reported to specifically benefit from atRA in any of the pertinent clinical trials26–28,64. Our observation that in vivo treatment with the pan-RAR antagonist AGN193109 delayed leukemogenesis and reduced stemness in an Evi1high, MA9-driven AML model even raises the possibility that some subgroups of AML may benefit from RAR antagonists. Albeit corresponding effects of AGN193109 in primary AML cells were small, this may be due to low concentrations of atRA in the in vitro setting precluding strong antagonist effects. This assumption is supported by the finding that in the mouse model, AGN193109 effects were also more pronounced in vivo than ex vivo (Figs. 5 and 6). RAR antagonists are being explored as treatments for diverse ailments, including malignancies and hematopoietic diseases66,67, and did not cause any serious toxicities in corresponding mouse models66–68. Future studies will have to identify the most suitable antagonist (possibly RAR isoform specific23) for the treatment of AML. Furthermore, the extent of the therapeutic window given the role of atRA in normal HSCs23,24, the identity of additional AML subgroups potentially benefitting from such a therapy, and the timing of retinoid application in the context of combination therapy will have to be addressed.
In summary, we demonstrate that EVI1 promoted key LSC properties in AML. Furthermore, atRA and EVI1 cooperated to enhance AML stemness and to regulate gene transcription in LSCe. Their biological effects on LSCs were at least partly mediated by Notch4. Conversely, a pan-RAR antagonist delayed leukemogenesis and reduced stemness of EVI1high AML, suggesting potential novel treatment options for this aggressive AML subtype.
Supplementary information
Acknowledgements
This work was funded by the Austrian Science Fund (FWF), project no P28256-B28 to R.W. The FWF did not play any role in the design of the study, the writing of the manuscript, or its submission to “Cell Death & Disease". The authors gratefully acknowledge Christoph Bock and Michael Schuster from the Biomedical Sequencing Facility at the Center for Molecular Medicine, Vienna, Austria, for advice on and assistance with RNA-sequencing. Johannes Schmöllerl and Petra Aigner, Ludwig Boltzmann Institute for Cancer Research, Vienna, Austria, provided important practical advice regarding retroviral transduction of primary murine cells. Andreas Spittler, Core Facility Flow Cytometry, Medical University of Vienna, Vienna, Austria, performed cell sorts and gave invaluable advice for flow cytometry applications. Dr. Takuro Nakamura, Cancer Institute of JFCR, Tokyo, Japan, kindly provided pMSCV_Flag-Evi1_IRES_GFP. The authors wish to thank Dr. Georg Krupitza, Clinic of Pathology, and Dipl.-Ing. Alexander Grandits, Clinic of Medicine I, Medical University of Vienna, for critically reading the manuscript.
Author contributions
C.H.N., D.S., J.Z., P.B.S., A.H.K., L.E.P., F.G. and R.W. designed experiments. C.H.N., K.B., A.S. and A.H. performed experiments. C.H.N., K.B., D.S., F.G. and R.W. interpreted experimental data. H.H. performed bioinformatics analyses of RNA-seq data. E.K. provided patient AML samples. C.H.N., H.H. and R.W. wrote the manuscript. R.W. designed and supervised the study. All authors critically read the manuscript and approved of the final version, as well as its submission to “Cell Death & Disease".
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by T. Kaufmann
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41419-019-2172-2).
References
- 1.Zagozdzon R, Golab J. Cancer stem cells in haematological malignancies. Contemp. Oncol. (Pozn.) 2015;19:A1–A6. doi: 10.5114/wo.2014.47127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grimwade D, Ivey A, Huntly B. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. doi: 10.1182/blood-2015-07-604496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krivtsov A, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krivtsov A, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 6.Haas K, et al. Expression and prognostic significance of different mRNA 5'-end variants of the oncogene EVI1 in 266 patients with de novo AML: EVI1 and MDS1/EVI1 overexpression both predict short remission duration. Genes Chromosomes Cancer. 2008;47:288–298. doi: 10.1002/gcc.20532. [DOI] [PubMed] [Google Scholar]
- 7.Groschel S, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J. Clin. Oncol. 2010;28:2101–2107. doi: 10.1200/JCO.2009.26.0646. [DOI] [PubMed] [Google Scholar]
- 8.Groschel S, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J. Clin. Oncol. 2013;31:95–103. doi: 10.1200/JCO.2011.41.5505. [DOI] [PubMed] [Google Scholar]
- 9.Bard-Chapeau E, et al. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc. Natl. Acad. Sci. USA. 2012;109:2168–2173. doi: 10.1073/pnas.1119229109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass C, et al. Global identification of EVI1 target genes in acute myeloid leukemia. PLoS ONE. 2013;8:e67134. doi: 10.1371/journal.pone.0067134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataoka K, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J. Exp. Med. 2011;208:2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bindels E, et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood. 2012;119:5838–5849. doi: 10.1182/blood-2011-11-393827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goyama S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe-Okochi N, et al. The shortest isoform of C/EBPbeta, liver inhibitory protein (LIP), collaborates with Evi1 to induce AML in a mouse BMT model. Blood. 2013;121:4142–4155. doi: 10.1182/blood-2011-07-368654. [DOI] [PubMed] [Google Scholar]
- 15.Ayoub E, et al. EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat. Commun. 2018;9:4239. doi: 10.1038/s41467-018-06208-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stein S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 17.Groschel S, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Yamazaki H, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21; q26) by activating EVI1 expression. Cancer Cell. 2014;25:415–427. doi: 10.1016/j.ccr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arai S, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–6314. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 20.Stavropoulou V, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30:43–58. doi: 10.1016/j.ccell.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Purton L, Bernstein I, Collins S. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin(-)c-kit(+)Sca-1(+)) while enhancing the terminal maturation of committed granulocyte monocyte progenitors. Blood. 1999;94:483–495. doi: 10.1182/blood.V94.2.483. [DOI] [PubMed] [Google Scholar]
- 22.Purton L, Bernstein I, Collins S. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood. 2000;95:470–477. doi: 10.1182/blood.V95.2.470. [DOI] [PubMed] [Google Scholar]
- 23.Purton L, et al. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 2006;203:1283–1293. doi: 10.1084/jem.20052105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabezas-Wallscheid N, et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. 2017;169:807–823 e819. doi: 10.1016/j.cell.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Testa U, Lo-Coco F. Targeting of leukemia-initiating cells in acute promyelocytic leukemia. Stem Cell Investig. 2015;2:8. doi: 10.3978/j.issn.2306-9759.2015.04.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burnett A, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood. 2010;115:948–956. doi: 10.1182/blood-2009-08-236588. [DOI] [PubMed] [Google Scholar]
- 27.Schlenk R, et al. All-trans retinoic acid as adjunct to intensive treatment in younger adult patients with acute myeloid leukemia: results of the randomized AMLSG 07-04 study. Ann. Hematol. 2016;95:1931–1942. doi: 10.1007/s00277-016-2810-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuley-Bagheri Y, Kreuzer K, Monsef I, Lubbert M, Skoetz N. Effects of all-trans retinoic acid (ATRA) in addition to chemotherapy for adults with acute myeloid leukaemia (AML) (non-acute promyelocytic leukaemia (non-APL)) Cochrane Database Syst. Rev. 2018;8:CD011960. doi: 10.1002/14651858.CD011960.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinmetz B, et al. The oncogene EVI1 enhances transcriptional and biological responses of human myeloid cells to all-trans retinoic acid. Cell Cycle. 2014;13:2931–2943. doi: 10.4161/15384101.2014.946869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verhagen H, et al. Primary acute myeloid leukemia cells with overexpression of EVI-1 are sensitive to all-trans retinoic acid. Blood. 2016;127:458–463. doi: 10.1182/blood-2015-07-653840. [DOI] [PubMed] [Google Scholar]
- 31.Bingemann S, Konrad T, Wieser R. Zinc finger transcription factor ecotropic viral integration site 1 is induced by all-trans retinoic acid (ATRA) and acts as a dual modulator of the ATRA response. FEBS J. 2009;276:6810–6822. doi: 10.1111/j.1742-4658.2009.07398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen CH, et al. SOCS2 is part of a highly prognostic 4-gene signature in AML and promotes disease aggressiveness. Sci. Rep. 2019;9:9139. doi: 10.1038/s41598-019-45579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu, Y. & Smyth, G. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays J. Immunol. Methods347, 70–78 (2009). [DOI] [PubMed]
- 34.Cho SJ, George CL, Snyder JM, Acarregui MJ. Retinoic acid and erythropoietin maintain alveolar development in mice treated with an angiogenesis inhibitor. Am. J. Respir. Cell Mol. Biol. 2005;33:622–628. doi: 10.1165/rcmb.2005-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, et al. All-trans retinoic acid and arsenic trioxide fail to derepress the monocytic differentiation driver Irf8 in acute promyelocytic leukemia cells. Cell Death Dis. 2017;8:e2782. doi: 10.1038/cddis.2017.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fellmann C, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 37.George J, et al. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat. Commun. 2016;7:12166. doi: 10.1038/ncomms12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chee L, Hendy J, Purton L, McArthur G. ATRA and the specific RARalpha agonist, NRX195183, have opposing effects on the clonogenicity of pre-leukemic murine AML1-ETO bone marrow cells. Leukemia. 2013;27:1369–1380. doi: 10.1038/leu.2012.362. [DOI] [PubMed] [Google Scholar]
- 39.Yuasa H, et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24:1976–1987. doi: 10.1038/sj.emboj.7600679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senyuk V, et al. Repression of RUNX1 activity by EVI1: a new role of EVI1 in leukemogenesis. Cancer Res. 2007;67:5658–5666. doi: 10.1158/0008-5472.CAN-06-3962. [DOI] [PubMed] [Google Scholar]
- 41.Kurokawa M, et al. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn S, Hyeon J, Park CK. Notch1 and Notch4 are markers for poor prognosis of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2013;12:286–294. doi: 10.1016/S1499-3872(13)60046-6. [DOI] [PubMed] [Google Scholar]
- 44.Wu G, et al. NOTCH4 is a novel prognostic marker that correlates with colorectal cancer progression and prognosis. J. Cancer. 2018;9:2374–2379. doi: 10.7150/jca.26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang JW, et al. The association between Notch4 expression, and clinicopathological characteristics and clinical outcomes in patients with breast cancer. Oncol. Lett. 2018;15:8749–8755. doi: 10.3892/ol.2018.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shirley CM, et al. Genetic and pharmacologic Notch4 inhibition synergizes with FLT3 tyrosine kinase inhibition in vitro to more effectively eliminate FLT3/ITD-positive leukemia cells. Blood. 2014;124:3583. doi: 10.1182/blood.V124.21.3583.3583. [DOI] [Google Scholar]
- 47.Kamga PT, et al. Characterization of a new B-ALL cell line with constitutional defect of the Notch signaling pathway. Oncotarget. 2018;9:18341–18350. doi: 10.18632/oncotarget.24836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konantz M, et al. Evi1 regulates Notch activation to induce zebrafish hematopoietic stem cell emergence. EMBO J. 2016;35:2315–2331. doi: 10.15252/embj.201593454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohishi K, et al. Physiological concentrations of retinoic acid suppress the osteoblastic differentiation of fetal rat calvaria cells in vitro. Eur. J. Endocrinol. 1995;133:335–341. doi: 10.1530/eje.0.1330335. [DOI] [PubMed] [Google Scholar]
- 50.Sato T, et al. Evi1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene. 2014;33:5028–5038. doi: 10.1038/onc.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene. 2007;396:346–357. doi: 10.1016/j.gene.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Kilbey A, Bartholomew C. Evi-1 ZF1 DNA binding activity and a second distinct transcriptional repressor region are both required for optimal transformation of Rat1 fibroblasts. Oncogene. 1998;16:2287–2291. doi: 10.1038/sj.onc.1201732. [DOI] [PubMed] [Google Scholar]
- 53.Hoyt P, et al. The Evi1 proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech. Dev. 1997;65:55–70. doi: 10.1016/S0925-4773(97)00057-9. [DOI] [PubMed] [Google Scholar]
- 54.Soderholm J, Kobayashi H, Mathieu C, Rowley J, Nucifora G. The leukemia-associated gene MDS1/EVI1 is a new type of GATA-binding transactivator. Leukemia. 1997;11:352–358. doi: 10.1038/sj.leu.2400584. [DOI] [PubMed] [Google Scholar]
- 55.Lugthart S, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111:4329–4337. doi: 10.1182/blood-2007-10-119230. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, et al. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood. 2011;118:3853–3861. doi: 10.1182/blood-2011-02-334680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, et al. Essential role of PR-domain protein MDS1-EVI1 in MLL-AF9 leukemia. Blood. 2013;122:2888–2892. doi: 10.1182/blood-2012-08-453662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Gianni M, et al. RARa2 and PML-RAR similarities in the control of basal and retinoic acid induced myeloid maturation of acute myeloid leukemia cells. Oncotarget. 2017;8:37041–37060. doi: 10.18632/oncotarget.10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin X, et al. Notch4+cancer stem-like cells promote the metastatic and invasive ability of melanoma. Cancer Sci. 2016;107:1079–1091. doi: 10.1111/cas.12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qian CJ, et al. Notch4 inhibition reduces migration and invasion and enhances sensitivity to docetaxel by inhibiting Akt/fascin in pancreatic cancer cells. Oncol. Lett. 2016;12:3499–3505. doi: 10.3892/ol.2016.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagamatsu I, et al. NOTCH4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014;34:69–80. [PubMed] [Google Scholar]
- 63.Ye Q, Shieh JH, Morrone G, Moore MAS. Expression of constitutively active Notch4 (Int-3) modulates myeloid proliferation and differentiation and promotes expansion of hematopoietic progenitors. Leukemia. 2004;18:777–787. doi: 10.1038/sj.leu.2403291. [DOI] [PubMed] [Google Scholar]
- 64.Schlenk R, et al. Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004;18:1798–1803. doi: 10.1038/sj.leu.2403528. [DOI] [PubMed] [Google Scholar]
- 65.Ma H, et al. All-trans retinoic acid synergizes with FLT3 inhibition to eliminate FLT3/ITD+ leukemia stem cells in vitro and in vivo. Blood. 2016;127:2867–2878. doi: 10.1182/blood-2015-05-646786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toma S, Emionite L, Scaramuccia A, Ravera G, Scarabelli L. Retinoids and human breast cancer: in vivo effects of an antagonist for RAR-alpha. Cancer Lett. 2005;219:27–31. doi: 10.1016/j.canlet.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 67.Hong S, et al. Rescue of a primary myelofibrosis model by retinoid-antagonist therapy. Proc. Natl. Acad. Sci. USA. 2013;110:18820–18825. doi: 10.1073/pnas.1318974110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chung S, Wang X, Wolgemuth D. Prolonged oral administration of a pan-retinoic acid receptor antagonist inhibits spermatogenesis in mice with a rapid recovery and changes in the expression of influx and efflux transporters. Endocrinology. 2016;157:1601–1612. doi: 10.1210/en.2015-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.