

Summary
The past two decades have seen an explosion in research that aims to understand how the dynamic interplay with the gut microbiota impacts host health and disease, establishing a role for the gut microbiota in a plethora of pathologies. Understanding how health‐promoting microbiota are established and how beneficial host–microbiota interactions are maintained is of immense biomedical importance. Despite the enormous progress that has been made, our knowledge of the specific microbiota members that mediate these effects and the mechanisms underlying these interactions is rudimentary. The dearth of information regarding the nature of advantageous host–microbiota interactions, and the factors that cause these relationships to go awry, has hampered our ability to realize the therapeutic potential of the microbiota. Here we discuss key issues that limit current knowledge and describe a path forwards to improving our understanding of the contributions of the microbiota to host health.
Keywords: microbiota, immune homeostasis, mucosal immunology, microbiome, tolerance
The gut microbiota is a critical regulator of host immune function, and profoundly impacts host health and disease. However, our understanding of the specific microbiota members that mediate these effects, and the mechanisms that underlie these interactions, is rudimentary, in large part due to technical difficulties associated with study of the microbiota. Strategies that help overcome these challenges, allied to the development of model systems that more accurately reflect host–microbiota interactions in humans, will advance our understanding of how this dialogue shapes host health and will pave the way for the development of microbiota‐based therapeutics.
Abbreviations
- ATP
adenosine triphosphate
- CD
Crohn's disease
- CRISPR
clustered regularly interspaced short palindromic repeats
- IBD
inflammatory bowel disease
- IL
interleukin
- RAG
recombination activating gene
- SFB
segmented filamentous bacteria
The gut microbiota as a regulator of host health
Born devoid of microbes, our intestines gradually become densely colonized by a complex collection of bacteria, eukaryotes, archaea and viruses that are collectively referred to as the gut microbiota. Rather than being mere passengers, the gut microbiota profoundly shapes many facets of host physiology, including aiding in the acquisition and provision of nutrients, educating the immune system, and providing resistance to invading pathogens.1, 2 Although interest in this complex ecosystem is often touted as new, the microbiota has proved a source of fascination for scientists for well over a century. Indeed, immunological interest in the role of the gut microbiota can be traced to Elie Metchnikoff, who was obsessed with how the gut microbiota could be detrimental to health, and how this could be remedied through administration of health‐promoting bacteria (probiotics), in the early twentieth century.3 Although study of the microbiota long‐remained a fringe area of research, a number of intrepid investigators continued to provide key insights into the contributions of our gut microbes to host health, laying the groundwork for modern microbiota research.4, 5, 6, 7, 8 The field was reinvigorated with the advent of new and improved approaches for characterizing the composition of the microbiota and provocative findings that suggested the microbiota could modulate susceptibility to an ever‐increasing variety of diseases.
The development of next‐generation sequencing technologies, and analytic approaches accessible to non‐experts, provided researchers with the opportunity to interrogate the structure of complex microbial communities at an unprecedented scale, advancing earlier efforts to understand the microbial and viral ecology of the intestinal tract.6, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 These approaches allowed researchers to define the microbiota composition from large numbers of individuals across time and geographical boundaries, at different developmental stages, and in response to a variety of perturbations.20, 21, 22 These culture‐independent approaches, which continue to grow in scale and resolution, revealed that the microbiota reaches a relatively stable and functionally mature state by 3 years of age22 and exhibits large interpersonal variation,14, 23 with each individual carrying a largely unique set of strains that persist for extended periods of time.21, 24 Gut microbial community composition is modified by many environmental factors, such as geographical location,21, 22 host diet25 and administration of antibiotics26 and other medicines.27 Despite inter‐individual differences in community composition, a core set of genes shared across individuals is encoded by the microbiota, suggesting that distinct community compositions can provide similar functional outputs.13, 14 Strikingly, it was demonstrated that, relative to healthy controls, differences in microbial community structure (often termed dysbiosis) could be observed in individuals suffering from a variety of disorders including obesity,28, 29 inflammatory bowel disease (IBD),30, 31, 32 cardiovascular disease,33 malnutrition,34 and type 2 diabetes,35 many of which had not previously been linked to the activity of microbes. Furthermore, transplantation of fecal microbiota from human donors with such diseases to recipient germ‐free/gnotobiotic mice was shown to transmit features associated with the disease. This includes obesity,29 malnutrition,34, 36 autoimmunity,37, 38 food allergy39 and IBD,40, 41 providing proof of principle of a causal role for the microbiota in elicitation or exacerbation of disease. Hence, a picture emerged where the unique constellation of microbial strains within an individual represented a critical factor that, in conjunction with host genetics and environmental influences, could influence disease susceptibility. The finding that alterations in microbiota composition were not merely correlated with disease, but could play a causal role in the development of a number of distinct host pathologies captured the imagination of scientists and the public alike, placing the microbiota at the forefront of biomedical research. Furthermore, experimental studies demonstrating that ‘microbiota repair', through administration of complete fecal communities or select microbial taxa, could alleviate disease in both humans and mice,42, 43 highlighted the therapeutic potential of next‐generation probiotics. This has led to a flurry of work from scientists in disparate fields to provide insight into the microbes responsible for modulating host health, and has allowed microbiota researchers to adopt a framework akin to Koch's Postulates44 for microbiota‐driven phenotypes.
Immune modulation by the gut microbiota
The gut microbiota profoundly impacts the phenotype and function of the host immune system, the details of which have been reviewed expertly elsewhere.2, 45, 46, 47 Germ‐free mice present with numerous immune defects, including impaired T‐cell differentiation and accumulation in the intestine,48, 49, 50 decreased production/secretion of immunoglobulin A (IgA),8, 51 reduced anti‐microbial peptide production,52 and elevated levels of systemic IgE.53 Importantly, many of these deficits can be reversed through administration of a fecal microbiota from murine and human donors, or their derived bacterial strains, as well as by fungi or viruses, reinforcing the potential contribution of non‐bacterial members of the community.48, 49, 54, 55, 56, 57, 58 Although not the first description of immunomodulation by the microbiota, the discovery that different frequencies of T helper type 17 (Th17) cells were present in the small intestine of C57BL/6 mouse substrains obtained from two different commercial vendors, and that this was attributable to the presence of a single microbe, segmented filamentous bacteria (SFB),49, 55, 56 prompted intensive efforts to identify additional microbes that could control the development of various arms of the immune system. Understanding the molecular basis of such microbiota–host interactions could provide a basis to develop novel agents that shape immune function in a targeted fashion to benefit host health. However, the massive complexity of the gut microbiota poses a daunting challenge to researchers, and identifying effector strains (microbial taxa that shape phenotypes of interest) remains a major goal for the field. Several different strategies have been used to overcome these difficulties and provide detailed insight into immune–microbiota interactions.
Strategies to identify effector strains
An optimal strategy to identify effector strains should have the following key features; (i) require no prior knowledge of the effector strain; (ii) not be focused solely on highly abundant community members; (iii) be agnostic to currently known functions of different taxonomic classifications; (iv) provide strain‐level resolution; and (v) be capable of identifying complex relationships where the effects of a particular microbe could be modified by other microbes.59, 60
The primary strategy for identifying effector microbes involves approaches that compare community composition (membership and relative abundance) in a community that modifies immune function with one that does not. This has been employed successfully in the identification of SFB as a potent stimulator of the intestinal immune system.56 However, it has many limitations. First, strains whose abundance correlates with a phenotype of interest may not play causal roles in that phenotype.61 Second, comparing the structure of distinct microbial communities that imprint different host phenotypes may lead to the generation of a large list of taxa that are differentially represented and therefore, potentially mediating the phenotypic difference, making the identification of target agents a difficult task. An elegant solution to this problem uses co‐housing of mice that have been colonized with different microbiota that are associated with distinct phenotypes.62, 63 As co‐housing of different mice leads to incomplete exchange of gut microbiota, this approach generates ‘sublines' of mice harboring distinct mixtures of the two ‘parental' microbiota. By determining shared microbiota constituents that correlate with the phenotype of interest in the sublines and parental mice, the microbes responsible can be parsed in a more efficient fashion. Third, until recently, sequencing‐based studies lacked strain‐level resolution, and used ≥97% 16S rRNA gene sequence similarity as a proxy for identifying species. Yet, inferring function based on this level of taxonomic resolution is flawed given that strain‐level variation, where strains of a given species vary in their function or the host phenotypes they imprint, is a well‐described feature of gut microbes (here we refer to strains as isolates of the same species that exhibit distinguishable genotypic or phenotypic characteristics64). For example, strains of Bacteroides fragilis that express the enterotoxin known as B. fragilis toxin can promote the development of colon cancer,65 colitis,66 and malnutrition‐associated cachexia,67 whereas B. fragilis toxin‐negative strains lack this capacity. Conversely, a non‐enterotoxigenic strain of B. fragilis is associated with the induction of regulatory T (Treg) cells and interleukin‐10 (IL‐10).68 This is not a peculiarity of B. fragilis, and profound differences linked to strain‐level variation among other gut bacteria have been documented.69 Hence, efforts to validate findings based on sequencing, which typically involves the use of a commercially available strain of the species of interest, may lead to use of strains lacking the desired activity, deeming the effort a failure. Finally, even when key effector strains/strain consortia can be identified, they may not be themselves sufficient and instead require the presence of specific additional community members for the phenotype to manifest. For example, in the ‘TRUC' model of transmissible colitis in T‐bet−/−RAG−/− mice, Klebsiella pneumoniae and Proteus mirabilis elicit the development of colitis, but only when in the presence of an endogenous microbial community.70 Similarly, while Helicobacter hepaticus induces robust typhlocolitis in IL‐10−/− mice, mono‐colonization of germ‐free IL‐10−/− mice with H. hepaticus is insufficient to promote intestinal pathology.71, 72, 73 Hence, microbes that do not vary between communities may be necessary to observe a phenotype and will be missed by sequencing‐dependent efforts.
Another commonly employed approach involves the use of anti‐microbial agents to winnow the number of potential effector strains. These have included antibiotics that target specific classes of bacteria (i.e. Gram‐positive, Gram‐negative, anaerobes) and chloroform or ethanol treatment of intestinal material to enrich for spore‐forming bacteria, which have also proved to be of major utility in identifying modifiers of the immune system.48, 49, 54, 74, 75 However, such strategies are prone to some of the same issues listed above, and can preclude the identification of some complex relationships where distinct microbes must interact for a phenotype to manifest (for example if Gram‐negative and Gram‐positive bacteria cooperate). Moreover, some of these anti‐microbial agents can also affect host physiology, which can confound interpretation of their effects.76 Mono‐colonization with large panels of microbes can also be used to determine their effects on host immune responses,77, 78 but such efforts require that the microbial taxa used are sufficient to promote the phenotype of interest, require the ability of the specific taxa used to successfully mono‐colonize mice, and will miss scenarios where microbe–microbe interactions are key to imprinting phenotypic responses.
More recently, researchers have developed and implemented newer, less biased approaches to address these limitations. The most fruitful of these methods involves assessing the coating of gut microbes with host‐derived IgA, the dominant antibody class in the gut lumen, to identify immunogenic microbes within a complex community (i.e. microbes coated with IgA). In this strategy, microbes from intestinal contents that are coated by host IgA are detected through flow cytometry and IgA‐positive and IgA‐negative fractions purified by fluorescence‐activated cell sorting are subjected to 16S rRNA gene sequencing for identification, or transferred to recipient gnotobiotic mice to determine their ability to transmit a phenotype36, 41, 79, 80 (Fig.1). Using this approach to study microbiota from individuals with IBD, it has been demonstrated that bacteria enriched in IgA coating support development of more severe dextran sodium sulfate‐induced colitis than non‐targeted bacteria, identifying putative candidates that modulate the susceptibility to IBD.41 A similar strategy identified a strain of Escherichia coli that was enriched for IgA targeting in microbiota from a subset of patients with Crohn's disease with spondyloarthritis and that could elicit potent activation of mucosal CD4+ T cells.81 IgA targeting was also able to identify pathogenic Enterobacteriaceae from the microbiota of a child suffering from Kwashiorkor (a form of severe acute edematous malnutrition) that could induce a high degree of mortality upon transplantation to gnotobiotic mice consuming a nutrient‐deficient diet. In addition, IgA‐targeted microbes from a healthy microbiota donor could in turn prevent this mortality phenotype, so pathogenic and protective microbes can be enriched for targeting by IgA and identified using this strategy.36 This suggested that IgA targeting generally reflects microbes with immune modulatory potential, rather than pathogenic strains per se. However, a recent systematic assessment of the specificities of intestinal IgA has revealed a large degree of polyreactivity, suggesting that binding by IgA may not actually be due to B‐cell stimulation by a given IgA‐coated target,82 but rather the expression of an epitope targeted by polyreactive IgA. Furthermore, this strategy captures only those microbes that have been bound by IgA, and microbes that evade antibody responses, or which shape immune responses without eliciting an immune response against themselves (for example, a microbe that enhances the fitness of a strongly immunogenic strain could alter immune responses in the gut without being recognized by the immune system) will not be detected using this approach.
Figure 1.
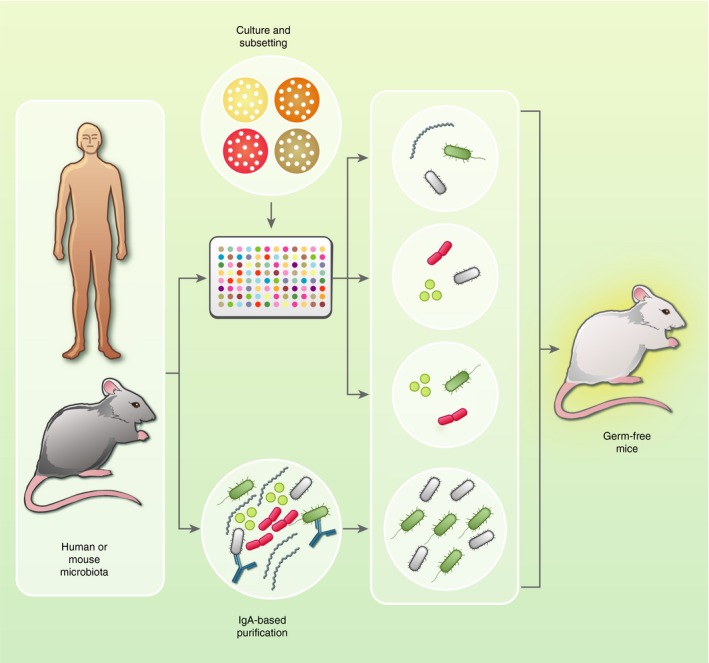
Identifying immunomodulatory microbes within the gut microbiota: As depicted above, newer strategies employ intensive culturing and microbiota subsetting, and/or IgA‐based identification of immunomodulatory microbes, followed by selective reconstitution of germ‐free mice, to map phenotypes to defined microbiota components. These efforts should provide key insights into the prominent modifiers of host immune phenotypes and help to develop next‐generation probiotics that beneficially manipulate immune function.
One of us (PPA) recently developed a strategy, termed combinatorial gnotobiotics, that provides a more systematic assessment of the microbes that shape host phenotypes of interest.83 This approach (described in ref.59) involves isolation of microbial strains from a community of interest in clonally arrayed culture libraries,84 and administration of randomly selected microbial consortia of differing but known composition to gnotobiotic mice, to assess how individual microbes, either alone or together with other community members, modulate phenotypes of interest (Fig. 1). This strategy has the advantage of providing strain‐level resolution, requires no prior knowledge of the strain, is agnostic as to whether a host response targets the microbe, and by using subsets containing different numbers of microbes has the ability to identify higher‐order interactions where two or more strains cooperate or compete to elicit a response. This strategy effectively identified complex interactions that regulated the cecal metabolite pool, and identified greater redundancy in colonic Treg cell accumulation than had been previously appreciated.83 In addition to requiring a larger number of gnotobiotic mice than the other approaches discussed, a primary challenge of this strategy is the need to capture agents in pure culture, which would have missed the important contributions of SFB to mucosal immunity because of the difficulties associated with its cultivation.85 Although much maligned, recent years have witnessed a rebirth of culture‐based approaches to interrogate gut microbiota composition and function.74, 84, 86, 87 Resistance to culture‐based strategies can be explained in part by the notion that much of the microbiota was ‘unculturable'.87 A resurgence in efforts to culture these microbes, led by environmental microbiologists, has seen dramatic improvements in the proportion of the gut microbiota that can be isolated. In a landmark study, 212 different culturing conditions, involving perturbations to media composition, growth temperature and pH among other variables, were tested leading to the isolation of 340 bacterial species, many of which had not been previously found in the human gut.86 Importantly, just 20 different conditions were sufficient to isolate a majority of the strains, and it has been demonstrated by other groups that a great deal of the diversity within a given community can even be captured using a single rich medium,84, 88 enhancing the tractability of isolation efforts. As improvements continue to be made in culturability, aided by metagenomics‐based identification of potential growth factors, isolation of strains of interest will likely be less problematic, thereby advancing studies that begin with a culture‐based step.
Clearly, no single approach encompasses all the discovery features required for a truly unbiased and fully systematic approach, nor are any of these strategies suited to all questions. Instead, leveraging the advantages of these different approaches may provide an optimal way towards increasing our understanding of gut microbiota regulation of immune function. As a proof‐of‐principle of the utility of combining these strategies, studies merging the power of sequencing of IgA‐bound bacteria with isolation in culture and phenotypic assessment in gnotobiotic mice, have already been employed to identify potential harmful and beneficial bacterial taxa.36, 41
Of mice and men
An important facet of gut microbiota research using gnotobiotic mice involves the decision about whether to study microbes derived from humans or those obtained from mice (Fig.2). The rationale for studying human‐derived microbes is self‐evident, in that it appears more translationally relevant. Microbial communities from humans with various diseases of interest can be assessed for their capacity to transmit disease susceptibility and microbes implicated in the modulation of human health can be identified.[29, 34, 36, 37, 38, 39, 40, 41, 42] This approach has led to the successful identification of the gut microbiota as a critical regulator of a variety of diseases, and defined specific human‐derived microbial taxa that can shape the host immune system.54, 83 However, a number of caveats exist that should be considered. First, many human‐associated microbial taxa poorly colonize mice, if at all, and the relative abundance of those taxa that do successfully colonize may poorly reflect that observed in the donor community.25, 89 The study of a complex human microbiota in gnotobiotic mice may therefore significantly overstate or underestimate the contributions of particular taxa to phenotypes of interest. Second, human‐derived fecal microbiota communities may be less well adapted to the mouse intestine. When mice harboring a human‐derived or mouse‐derived fecal microbiota were co‐housed to facilitate exchange of their respective microbiota, many strains from the murine‐derived community were effectively able to invade the human‐derived gut microbiota and stably establish themselves, while the members of the human‐derived community were largely unable to establish themselves within the murine community.90 These data suggest that although human‐derived microbiota can form a stable community within a gnotobiotic mouse, they lack fitness in this environment relative to the murine community. Finally, host specificity in host–microbe interactions is a long‐appreciated phenomenon (particularly in microbial pathogenesis)91 and individual microbial taxa may have evolved to stimulate or evade the immune system of the host in which they normally reside. Isolates of SFB from mice and rats have shown exquisite specificity in the induction of intestinal Th17 responses, with rat‐derived SFB priming Th17 responses in the rat but not the mouse, and mouse‐derived SFB priming Th17 responses in the mouse but not the rat.92 Moreover, some human‐derived fecal microbiota were demonstrated to be poor stimulators of immune system maturation in selected lymphoid and non‐lymphoid sites in gnotobiotic mice, providing reduced resistance to pathogenic intestinal infection when compared with the recipients of murine fecal microbiota.55, 89 We posit that much may be learned about human immune interactions with their endogenous microbiota by comparatively studying other microbial communities in their natural habitat, i.e. the study of murine‐derived microbiota in the mouse intestine may better delineate shared paradigms of host–microbiota interactions than the study of human‐derived microbiota in gnotobiotic mice. This is not to suggest precluding the study of human‐derived microbes in mice, from which we have already learned much,25, 34, 36, 40, 41, 42, 50, 54, 83 but instead calls for judicious interpretation of data using such microbes. Indeed, study of the molecular basis by which human‐derived microbiota adapt and evolve during colonization and long‐term residence of the mouse intestine, in addition to delineating differences in the shaping of immune responses, represents a powerful strategy to uncover key facets of host–microbiota interplay. Complimentary approaches to the discovery process are also necessary given the mouse‐intensive nature of most current strategies and the fact that important human–microbiota interactions may not be modeled in a murine system. For example, primary intestinal organoids represent a particularly useful tool to study human epithelium–microbe interactions for several reasons, including: they can be generated directly from humans (with or without disease‐associated genetic polymorphisms); they can be genetically modified using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas technology; they recapitulate important epithelial cell lineages and physical features of the intestinal tract; and they can be seeded with microbes of interest.93 Another promising approach is to identify factors like metabolites that can impact a response of interest and work to identify the microbes that are responsible for their production.94, 95 As knowledge of the key mechanisms of host–microbiota interactions are revealed, our accuracy in predicting immunomodulatory activity based on the genomic content of a strain or community should markedly improve.
Figure 2.
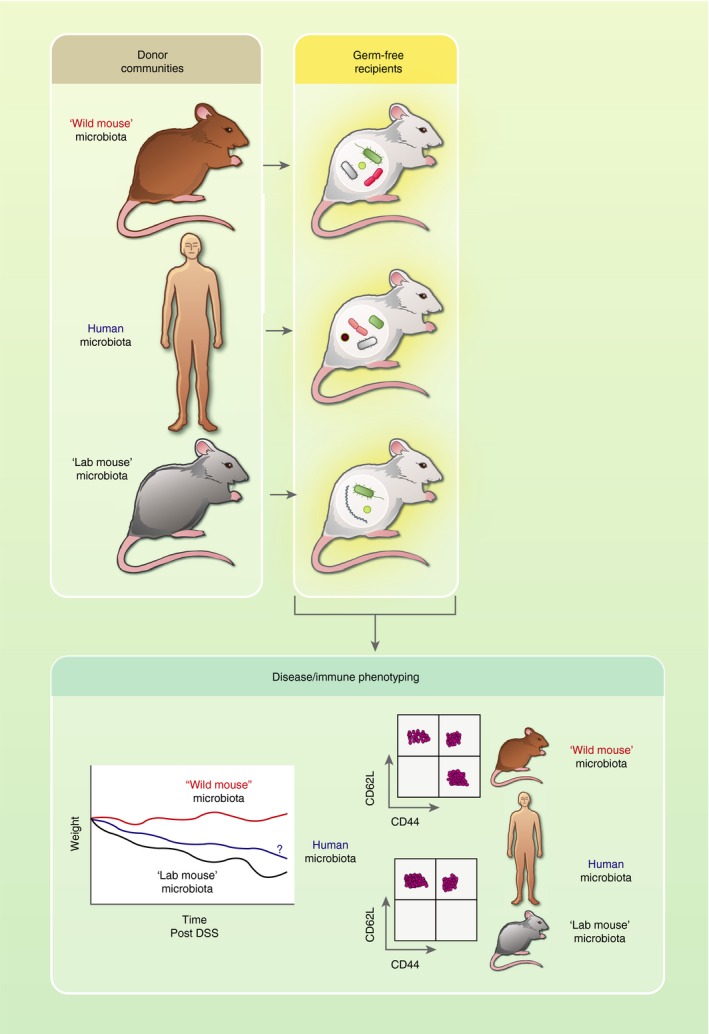
Optimizing microbial communities to model microbiota–host interactions: The best source of gut microbiota to allow faithful modeling in mice of human host–microbiota interactions remains an important question. Recent work suggests that human‐derived microbiota do not fully recapitulate the immune phenotype seen using murine‐derived communities. In addition, the gut microbiota harbored by ‘wild' (feral) mice may imprint a mature immune phenotype more comparable to that of adult humans than to the immature immune system found in common laboratory mice, priming development of more activated CD8+ T cells and providing increased resistance to weight loss induced by treatment with azoxymethane plus dextran sodium sulfate. Identification of the mechanisms that underlie these phenomena will facilitate the development of microbial consortia that allow more accurate representation of human physiology in laboratory mice.
Using the elegant approaches discussed above, the identity of specific microbes with potent immunomodulatory activity has been elucidated, especially those that shape the intestinal T‐cell compartment. A variety of bacterial strains from distinct phyla, operating individually or as part of multi‐strain consortia, prime the accumulation of peripherally derived colonic Treg cells and the production of the immune suppressive cytokine IL‐10. These include Clostridia from clusters IV, XIVa and XVIII (Firmicutes),48, 54 various Bacteroides species (Bacteroidetes)68, 78, 83, 96 or Helicobacter species (Proteobacteria),97, 98, 99 Bifidobacterium bifidum (Actinobacteria),100 and the Altered Schaedler Flora (multiple phyla).101 An isolate of Bifidobacterium adolescentis (an Actinobacteria) obtained from humans that could induce Th17 cells was also identified, confirming that this was not unique to SFB.102 A cocktail of human‐derived strains from multiple phyla promote the development of interferon‐γ‐secreting CD8+ T cells50 and intraepithelial lymphocytes are also potently regulated by gut microbes, with a murine‐derived strain of Lactobacillus reuteri shown to elicit intraepithelial CD4+CD8αα+ T‐cell generation,75 while SFB is known to increase T‐cell receptor αβ‐positive intraepithelial lymphocytes.51 Mechanistically, microbial metabolites have been shown to mediate many of these effects. Short‐chain fatty acids, products of microbial fermentation in the gut, promote Treg cell accumulation,103, 104, 105 in addition to regulating many other immune effector functions.106, 107, 108 Microbiota‐derived ATP can stimulate Th17 development,109 and tryptophan breakdown products like indole‐3‐lactic acid promote the development of intraepithelial CD4+CD8αα+ T cells through stimulation of the aryl hydrocarbon receptor.75 Bacterially derived polysaccharides such as polysaccharide A from B. fragilis, 68 β‐glucan/galactan polysaccharides from Bifidobacterium bifidum 100 or crude carbohydrate preparations from H. hepaticus 110 can prime Treg or IL‐10 production. Intriguingly, atypical antigen acquisition pathways111 have also been shown to be important for SFB‐induced Th17 development. Collectively, these studies demonstrate that the intestinal immune system can be activated by a diverse array of microbial taxa through a variety of distinct mechanisms, highlighting a large degree of redundancy in the processes which help to maintain these mutualistic relationships. Interestingly, some of the microbial taxa that impart beneficial effects on the immune system have also been implicated in the pathogenesis of inflammatory disease models, and so rather than being inherently beneficial, their positive effects are dependent on host factors and the context of the interaction.61, 112, 113, 114, 115 Additionally, an important time‐window where colonization must take place has also been established for particular phenotypes, emphasizing the importance not just of exposure to select microbes, but of encounter at the appropriate developmental stage.116, 117, 118 Given the shorter lifespan of mice relative to humans, it will be critical to determine how such developmental windows translate between species, and the consequences for interventions that aim to repair poorly developed or damaged microbiota for therapy.
Developing a consistent approach to assessing microbiota effects
The identification of select microbial taxa that modulate various arms of the intestinal immune system that have variable presence in mouse facilities49, 75, 99, 119 points to an unwanted source of variability in mouse models, highlighting the need for more defined microbiota to facilitate comparison of data across different laboratories. Although co‐housing of different groups of mice has been reported to be able to transfer microbiota‐dependent phenotypes in many instances, as noted above, the exchange of microbiota between adult mice is incomplete. In the absence of gnotobiotic facilities, current best practice for minimizing microbiota variables recommends using heterozygous or wild‐type littermate controls in studies of genetically modified mice, as well as standardizing housing conditions and diets across experimental groups.120 In addition, pedigrees can be tracked, to enable retrospective analysis of ‘cage effects' that may have arisen as the result of distinct parental microbiota. However, these processes will not homogenize microbiota across breeding cohorts in different specific‐pathogen‐free (SPF) facilities, meaning that phenotypes that can be influenced by microbiota may not appear readily reproducible. Indeed, for the purposes of knowledge discovery, it could be argued that global homogeneity is undesirable, as such discrepancies provide a basis for identifying microbiota constituents that regulate phenotypes.56, 75 As an example, one can consider the conflicting reports on the incidence of spontaneous intestinal inflammation in Tlr5 −/− mice121, 122 or dextran sodium sulfate‐induced colitis susceptibility in Nlrp6 −/− mice,123, 124, 125 highlighting the contextual expression of these phenotypes depending on environmental factors.
In addition to variability in microbiota, another caveat of using laboratory mice to model the human immune system was highlighted by the finding that the immune system of adult laboratory mice housed under SPF conditions is phenotypically representative of a newborn's immature immune system, in contrast to the mature immune system found in adult mice living in the wild or in pet stores.126 The adult‐like immune phenotype of wild or pet‐store mice has been linked to infectious agents,127 but a gut microbiota from wild mice, devoid of known mouse pathogens, also beneficially modified the response of gnotobiotic mice to infectious or chemical insult, relative to a microbiota derived from SPF laboratory mice.128 Hence, although the SPF mouse has been of enormous value in removing confounding variables like infections, a reassessment of the optimal microbiota composition for laboratory mice appears to be in order (Fig. 2).
Crucially, SPF conditions are currently defined by the absence of specific pathogenic microbes rather than being defined by the microbes that are present. Furthermore, the precise criteria used to classify colonies as SPF varies among vendors and facilities, with many potent immunomodulatory microbial taxa, such as SFB and Helicobacter spp., not routinely included in the list of proscribed microbes. New microbial cocktails containing known immunomodulatory microbes as noted above, in addition to non‐pathogens found in wild mice that shape host physiology (yet to be described), could be developed and used similarly to the Altered Schaedler Flora129 to provide the consistency desired by researchers, while improving the translatability of mouse systems. However, this approach poses many challenges. For example, developing a consensus as to which microbes should be used in such a cocktail and implementing strategies that allow for maintenance of these microbes without the invasion of true pathogens or other immunomodulatory microbes, is far from simple. Generation and characterization of purified stable consortia, potentially composed of bacteria, fungi or viruses that provide a more homogeneous microbiota, driving a consistent level of immune maturation that more accurately reflects the immune‐activation levels in adult humans, is an essential first step. It is very likely that there will not be a single microbiota type that is relevant for all questions, and the most appropriate community will critically depend on the human population to be modeled, as well as the environmental challenges under investigation. However, the community should be fully characterized, should have minimal variance over time and should reflect the human population to be studied as closely as possible. These consortia could then be used to regularly seed breeder colonies to help ensure their sustained presence within a colony, but the large‐scale feasibility of such an approach and the precise protocols required to ensure consistent microbiota composition remain to be fully determined. Furthermore, this approach would require careful monitoring, maintenance of high levels of hygiene and husbandry and strict control of diet. The expansion of gnotobiotic facilities will likely expedite the generation of new ‘minimal essential' stable consortia, with parallel characterization of the level of immune maturation in the murine host. In the meantime, in addition to the breeding and husbandry strategies outlined above, investigators should consider reporting the composition of the microbiota of mice used in their studies wherever possible. Much in the same way that sentinel mice are used to declare a colony as SPF, assessment of the microbiota of sentinels using 16S rRNA gene sequencing or shotgun metagenomic sequencing could be reported in published manuscripts, addressing an important variable that could lead to issues associated with reproducibility of data. Of course, because of constraints in resources and infrastructure, this reporting is unlikely to be feasible in all cases, but as sequencing costs reduce and bioinformatic training becomes more routine, reporting of microbiota composition should become a standard practice in many institutes. In addition, making such data available should also facilitate in silico approaches by other investigators to identify potential members, or interacting cohorts of microbes, that regulate immune maturation or experimental phenotypes. Together, such efforts will iteratively improve our understanding of how microbiota shape host immunity and disease phenotypes.
Conclusion
We are at an exciting time in microbiota research, with an increasing number of diseases being connected to the dynamic functions of the microbiota. The next steps, including the development of novel probiotic strains to treat disease, will require detailed understanding of the most beneficial microbes for host health, the context in which they operate, and molecular‐level details underpinning these effects. Indeed, with the strategies described here, allied to efforts to characterize the mechanisms of action of the microbiota and microbiota‐derived products in health and disease, we are well on our way.
Disclosures
The authors have no competing interests or conflicts of interest to declare.
Acknowledgements
We thank Anagha Kadam, Morgan Engelhart and Naseer Sangwan for helpful comments. We apologize to the many researchers whose outstanding work we did not cite because of space constraints. KJM is supported by a Wellcome Trust Investigator Award (102972). We dedicate this review to our late friend and colleague, Dr. Mark Asquith, who helped shape our thinking on microbiota–host interactions and many other topics.
Contributor Information
Philip P. Ahern, Email: ahernp@ccf.org.
Kevin J. Maloy, Email: Kevin.Maloy@glasgow.ac.uk.
References
- 1. Hooper LV, Midtvedt T, Gordon JI. How host–microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 2002; 22:283–307. [DOI] [PubMed] [Google Scholar]
- 2. Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol 2007; 19:59–69. [DOI] [PubMed] [Google Scholar]
- 3. Mackowiak PA. Recycling metchnikoff: probiotics, the intestinal microbiome and the quest for long life. Front Public Health 2013; 1:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orcutt RP, Gianni FJ, Judge RJ. Development of an Altered Schaedler Flora for Nci gnotobiotic rodents. Microecol Ther 1987; 17:59. [Google Scholar]
- 5. Reyniers JA, Sacksteder MR. The use of germfree animals and techniques in the search for unknown etiological agents. Ann N Y Acad Sci 1958; 73:344–56. [DOI] [PubMed] [Google Scholar]
- 6. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 1977; 31:107–33. [DOI] [PubMed] [Google Scholar]
- 7. Schaedler RW, Dubs R, Costello R. Association of Germfree mice with bacteria isolated from normal mice. J Exp Med 1965; 122:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun 1999; 67:1992–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 2010; 7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al Diversity of the human intestinal microbial flora. Science 2005; 308:1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error‐correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 2008; 5:235–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D et al Direct sequencing of the human microbiome readily reveals community differences. Genome Biol 2010; 11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al A core gut microbiome in obese and lean twins. Nature 2009; 457:480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P et al Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol 2003; 185:6220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD et al The human gut virome: inter‐individual variation and dynamic response to diet. Genome Res 2011; 21:1616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC et al The gut mycobiome of the human microbiome project healthy cohort. Microbiome 2017; 5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F et al Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010; 466:334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scupham AJ, Presley LL, Wei B, Bent E, Griffith N, McPherson M et al Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microbiol 2006; 72:793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dominguez‐Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N et al Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 2010; 107:11971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F et al Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019; 176:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez‐Bello MG, Contreras M et al Human gut microbiome viewed across age and geography. Nature 2012; 486:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A et al Genomic variation landscape of the human gut microbiome. Nature 2013; 493:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al The long‐term stability of the human gut microbiota. Science 2013; 341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 2008; 6:e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S et al Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015; 528:262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005; 102:11070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444:1027–31. [DOI] [PubMed] [Google Scholar]
- 30. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular‐phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104:13780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gevers D, Kugathasan S, Denson LA, Vázquez‐Baeza Y, Van Treuren W, Ren B et al The treatment‐naive microbiome in new‐onset Crohn's disease. Cell Host Microbe 2014; 15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manichanh C, Rigottier‐Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L et al Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 2006; 55:205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kelly TN, Bazzano LA, Ajami NJ, He H, Zhao J, Petrosino JF et al Gut microbiome associates with lifetime cardiovascular disease risk profile among Bogalusa heart study participants. Circ Res 2016; 119:956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J et al Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 2013; 339:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qin J et al A metagenome‐wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490:55–60. [DOI] [PubMed] [Google Scholar]
- 36. Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I et al Functional characterization of IgA‐targeted bacterial taxa from undernourished Malawian children that produce diet‐dependent enteropathy. Sci Transl Med 2015; 7:276ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z et al Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A 2017; 114:10719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA et al Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci U S A 2017; 114:10713–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feehley T, Plunkett CH, Bao R, Choi Hong SM, Culleen E, Belda‐Ferre P et al Healthy infants harbor intestinal bacteria that protect against food allergy. Nat Med 2019; 25:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R et al Microbiotas from humans with inflammatory bowel disease alter the balance of Gut Th17 and RORγt+ regulatory T cells and exacerbate colitis in mice. Immunity 2019; 50:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L et al Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014; 158:1000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O et al Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 2016; 351:aad3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM et al Duodenal infusion of donor feces for recurrent Clostridium difficile . N Engl J Med 2013; 368:407–15. [DOI] [PubMed] [Google Scholar]
- 44. Neville BA, Forster SC, Lawley TD. Commensal Koch's postulates: establishing causation in human microbiota research. Curr Opin Microbiol 2018; 42:47–52. [DOI] [PubMed] [Google Scholar]
- 45. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol 2012; 30:759–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature 2016; 535:75–84. [DOI] [PubMed] [Google Scholar]
- 47. Ost KS, Round JL. Communication between the microbiota and mammalian immunity. Annu Rev Microbiol 2018; 72:399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y et al Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB et al Specific microbiota direct the differentiation of IL‐17‐producing T‐helper cells in the mucosa of the small intestine. Cell Host Microbe 2008; 4:337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y et al A defined commensal consortium elicits CD8 T cells and anti‐cancer immunity. Nature 2019; 565:600–5. [DOI] [PubMed] [Google Scholar]
- 51. Umesaki Y, Setoyama H, Matsumoto S, Imaoka A, Itoh K. Differential roles of segmented filamentous bacteria and clostridia in development of the intestinal immune system. Infect Immun 1999; 67:3504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006; 313:1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cahenzli J, Köller Y, Wyss M, Geuking MB, McCoy KD. Intestinal microbial diversity during early‐life colonization shapes long‐term IgE levels. Cell Host Microbe 2013; 14:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H et al Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013; 500:232–6. [DOI] [PubMed] [Google Scholar]
- 55. Gaboriau‐Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C et al The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009; 31:677–89. [DOI] [PubMed] [Google Scholar]
- 56. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014; 516:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM et al Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe 2019; 25:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ahern PP, Faith JJ, Gordon JI. Mining the human gut microbiota for effector strains that shape the immune system. Immunity 2014; 40:815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rosen CE, Palm NW. Functional Classification of the Gut Microbiota: the key to cracking the microbiota composition code: functional classifications of the gut microbiota reveal previously hidden contributions of indigenous gut bacteria to human health and disease. BioEssays 2017; 39:1700032. [DOI] [PubMed] [Google Scholar]
- 61. Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL et al Commensal Bacteroides species induce colitis in host‐genotype‐specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011; 9:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Surana NK, Kasper DL. Moving beyond microbiome‐wide associations to causal microbe identification. Nature 2017; 552:244–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL et al Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dijkshoorn L, Ursing BM, Ursing JB. Strain, clone and species: comments on three basic concepts of bacteriology. J Med Microbiol 2000; 49:397–401. [DOI] [PubMed] [Google Scholar]
- 65. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR et al A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15:1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA et al Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild‐type C57BL/6 mice. Infect Immun 2009; 77:1708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wagner VE, Dey N, Guruge J, Hsiao A, Ahern PP, Semenkovich NP et al Effects of a gut pathobiont in a gnotobiotic mouse model of childhood undernutrition. Sci Transl Med 2016; 8:366ra164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T‐cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 2010; 107:12204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Arthur JC, Perez‐Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ et al Intestinal inflammation targets cancer‐inducing activity of the microbiota. Science 2012; 338:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML et al Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 2010; 8:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dieleman LA, Arends A, Tonkonogy SL, Goerres MS, Craft DW, Grenther W et al Helicobacter hepaticus does not induce or potentiate colitis in interleukin‐10‐deficient mice. Infect Immun 2000; 68:5107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nagalingam NA, Robinson CJ, Bergin IL, Eaton KA, Huffnagle GB, Young VB. The effects of intestinal microbial community structure on disease manifestation in IL‐10–/– mice infected with Helicobacter hepaticus . Microbiome 2013; 1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Whary MT, Taylor NS, Feng Y, Ge Z, Muthupalani S, Versalovic J et al Lactobacillus reuteri promotes Helicobacter hepaticus‐associated typhlocolitis in gnotobiotic B6.129P2‐IL‐10(tm1Cgn) (IL‐10–/–) mice. Immunology 2011; 133:165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD et al Culturing of 'unculturable' human microbiota reveals novel taxa and extensive sporulation. Nature 2016; 533:543–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cervantes‐Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J et al Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα + T cells. Science 2017; 357:806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang JH, Bhargava P, McCloskey D, Mao N, Palsson BO, Collins JJ. Antibiotic‐induced changes to the host metabolic environment inhibit drug efficacy and alter immune function. Cell Host Microbe 2017; 22:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Geva‐Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz‐Lopez A et al Mining the human gut microbiota for immunomodulatory organisms. Cell 2017; 168:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sefik E, Geva‐Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM et al Individual intestinal symbionts induce a distinct population of RORγ + regulatory T cells. Science 2015; 349:993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD et al Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 2015; 43:541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y et al Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014; 41:152–65. [DOI] [PubMed] [Google Scholar]
- 81. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG et al Escherichia coli enriched in Crohn's disease spondyloarthritis promote TH17‐dependent inflammation. Sci Transl Med 2017; 9:eaaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M et al Wilson PC. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 2017; 358:eaan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe–host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014; 6:220ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G et al Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 2011; 108:6252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schnupf P, Gaboriau‐Routhiau V, Gros M, Friedman R, Moya‐Nilges M, Nigro G et al Growth and host interaction of mouse segmented filamentous bacteria in vitro . Nature 2015; 520:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C et al Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect 2012; 18:1185–93. [DOI] [PubMed] [Google Scholar]
- 87. Lagier JC, Dubourg G, Million M, Cadoret F, Bilen M, Fenollar F et al Culturing the human microbiota and culturomics. Nat Rev Microbiol 2018; 16:540–50. [DOI] [PubMed] [Google Scholar]
- 88. Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD et al A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 2019; 37:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB et al Gut immune maturation depends on colonization with a host‐specific microbiota. Cell 2012; 149:1578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE et al Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014; 159:253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Baumler A, Fang FC. Host specificity of bacterial pathogens. Cold Spring Harb Perspect Med 2013; 3:a010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S et al Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015; 163:367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fujii M, Clevers H, Sato T. Modeling human digestive diseases with CRISPR‐Cas9‐modified organoids. Gastroenterology 2019; 156:562–76. [DOI] [PubMed] [Google Scholar]
- 94. Koeth RA, Lam‐Galvez BR, Kirsop J, Wang Z, Levison BS, Gu X et al l‐Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest 2019; 129:373–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B et al Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wegorzewska MM, Glowacki RWP, Hsieh SA, Donermeyer DL, Hickey CA, Horvath SC et al Diet modulates colonic T cell responses by regulating the expression of a Bacteroides θιο antigen. Sci Immunol 2019; 4:eaau9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z et al Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci Immunol 2017; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kullberg MC, Jankovic D, Gorelick PL, Caspar P, Letterio JJ, Cheever AW et al Bacteria‐triggered CD4+ T regulatory cells suppress Helicobacter hepaticus‐induced colitis. J Exp Med 2002; 196:505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ et al c‐MAF‐dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018; 554:373–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Verma R, Lee C, Jeun EJ, Yi J, Kim KS, Ghosh A et al Cell surface polysaccharides of Bifidobacterium bifidum induce the generation of Foxp3+ regulatory T cells. Sci Immunol 2018; 3:eaat6975. [DOI] [PubMed] [Google Scholar]
- 101. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S et al Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011; 34:794–806. [DOI] [PubMed] [Google Scholar]
- 102. Tan TG, Sefik E, Geva‐Zatorsky N, Kua L, Naskar D, Teng F et al Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A 2016; 113:E8141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P et al Metabolites produced by commensal bacteria promote peripheral regulatory T‐cell generation. Nature 2013; 504:451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D et al Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013; 504:446–50. [DOI] [PubMed] [Google Scholar]
- 105. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly‐Y M et al The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013; 341:569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe 2016; 20:202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schulthess J, Pandey S, Capitani M, Rue‐Albrecht KC, Arnold I, Franchini F et al The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 2019; 50:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tan J, McKenzie C, Vuillermin PJ, Goverse G, Vinuesa CG, Mebius RE et al Dietary fiber and bacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep 2016; 15:2809–24. [DOI] [PubMed] [Google Scholar]
- 109. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M et al ATP drives lamina propria TH17 cell differentiation. Nature 2008; 455:808–12. [DOI] [PubMed] [Google Scholar]
- 110. Danne C, Ryzhakov G, Martínez‐López M, Ilott NE, Franchini F, Cuskin F et al A large polysaccharide produced by Helicobacter hepaticus induces an anti‐inflammatory gene signature in macrophages. Cell Host Microbe 2017; 22:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan‐Diez M, Soualhi S et al Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 2019; 363:eaat4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A et al Helicobacter hepaticus triggers colitis in specific‐pathogen‐free interleukin‐10 (IL‐10)‐deficient mice through an IL‐12‐ and γ interferon‐dependent mechanism. Infect Immun 1998; 66:5157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T‐cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2011; 108(Suppl 1):4615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T et al Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis 2007; 13:1202–11. [DOI] [PubMed] [Google Scholar]
- 115. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y et al Gut‐residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010; 32:815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Déjardin Fs. et al A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 2019; 50:e5. [DOI] [PubMed] [Google Scholar]
- 117. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk‐Hasdemir D et al Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 2014; 156:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A et al Microbial exposure during early life has persistent effects on natural killer T cell function. Science 2012; 336:489–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A et al Host–protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 2016; 167:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wullaert A, Lamkanfi M, McCoy KD. Defining the impact of host genotypes on microbiota composition requires meticulous control of experimental variables. Immunity 2018; 48:605–7. [DOI] [PubMed] [Google Scholar]
- 121. Letran SE, Lee SJ, Atif SM, Flores‐Langarica A, Uematsu S, Akira S et al TLR5‐deficient mice lack basal inflammatory and metabolic defects but exhibit impaired CD4 T cell responses to a flagellated pathogen. J Immunol 2011; 186:5406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Vijay‐Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV et al Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest 2007; 117:3909–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Elinav E, Strowig T, Kau AL, Henao‐Mejia J, Thaiss CA, Booth CJ et al NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, Platnich JM et al The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep 2017; 21:3653–61. [DOI] [PubMed] [Google Scholar]
- 125. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira‐Silva S, Yilmaz B, Martens L et al Nlrp6‐ and ASC‐dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 2017; 47:e4. [DOI] [PubMed] [Google Scholar]
- 126. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA et al Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016; 532:512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Reese TA, Bi K, Kambal A, Filali‐Mouhim A, Beura LK, Bürger MC et al Sequential infection with common pathogens promotes human‐like immune gene expression and altered vaccine response. Cell Host Microbe 2016; 19:713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K et al Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 2017; 171:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wymore Brand M, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet AM, Jergens AE et al The altered schaedler flora: continued applications of a defined murine microbial community. ILAR J 2015; 56:169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]