

Summary
We previously assessed the kinetics of T cell turnover in vivo by labeling cells with 2H‐H2O over 42 days in individuals with type 1 diabetes (T1D) and demonstrated an increased turnover of CD4 memory T cells. We have now tested T cell turnover in individuals at risk for T1D using a 3–4‐day labeling protocol with 2H‐glucose. We studied 30 relatives with T1D with and without autoantibodies, and 10 healthy controls. Peripheral blood mononuclear cells (PBMC) were flow‐sorted into T cell subsets of interest; 2H‐DNA enrichment was measured by mass spectrometry and in‐vivo turnover was calculated as maximum fractional enrichment of deuterated adenosine (F max). Among CD4+ cells, F max was highest in regulatory T cells (Treg), followed by effector and central memory T cells and lowest in naive cells. Similarly, CD8+ central and effector memory T cells had a higher turnover than CD8+ terminally differentiated effector memory T cells (TEMRA) and CD8+‐naive T cells. Relatives as a group showed significantly increased Treg turnover by F max compared to controls (1·733 ± 0·6784% versus 1·062 ± 0·3787%, P = 0·004), suggesting pre‐existing immune dysfunction within families with T1D. However, there was no significant difference in F max between groups according to autoantibody or glucose tolerance status. Repeat testing in 20 subjects 1 year later demonstrated relatively higher within‐subject compared to between‐subject variability for the measurement of F max in various T cell subsets. The short labeling protocol with 2H‐glucose should be applied in the context of a clinical trial in which the therapy is expected to have large effects on T cell turnover.
Keywords: Autoantibodies, CD4 T cells, CD8 T cells, Deuterium labeled glucose (heavy glucose), Isotope labeling, Regulatory T cells, T Cell kinetics, Type 1 diabetes
We tested T cell turnover in individuals at risk for T1D using a 3–4‐day labeling protocol with 2H‐glucose. We found that relatives as a group showed significantly increased Treg turnover by F max compared to controls, suggesting pre‐existing immune dysfunction within families with T1D.
Introduction
T cells are thought to play a key role in the pathogenesis of type 1 diabetes (T1D) 1, 2. Despite mounting evidence that T cells probably behave abnormally in individuals with T1D, consistent patterns have not been reported in terms of the number, phenotype or frequency of T cells and their association with disease outcomes 3, 4. One explanation for this is that the frequency of each T cell type is impacted by the rate of T cell turnover which, in itself, can differ between individuals or between groups with differing disease status. Using in‐vivo metabolic labeling of lymphocyte DNA with the heavy isotope of deuterated water over 42 days, as first described by Neese et al. 5, we previously reported that CD4 memory T cells from T1D patients, despite being present at a normal number, show evidence of increased proliferation and disappearance rates compared to control subjects, indicating elevated turnover 6.
In the current study, we evaluated in‐vivo T cell kinetics in individuals at risk for T1D. The Type 1 Diabetes TrialNet Pathway to Prevention study has identified groups of relatives of those with T1D with markedly different risk for disease. The 5‐year risk for those who repeatedly test negative for all antibodies is almost negligible. Those repeatedly found to have just one antibody, normal glucose tolerance and a diabetes prevention trial‐type 1 (DPT) risk score for T1D < 6·5 7 have a 5‐year risk of < 3%. Once two antibodies are confirmed in the presence of normal glucose tolerance the 5‐year risk is ~35%, while the 5‐year risk in those with multiple antibodies and abnormal glucose tolerance exceeds 80%. Importantly, the risk for developing T1D in those with two antibodies does not appear to level off over time. It is generally accepted that almost all these individuals will eventually develop T1D8; however, we cannot yet predict when an individual will progress to clinical disease. We therefore aimed to determine if increased turnover of CD4 memory T cells and/or other subsets of T cells is a phenotype that precedes disease onset, and if there is any association with autoantibody status. We tested this hypothesis using heavy glucose in lieu of heavy water and a shorter protocol, as previously described 9, which could be feasibly implemented in the context of future clinical trials.
Research design and methods
Subjects
Subjects were recruited under an approved ancillary study to the Type 1 Diabetes TrialNet TN01 Pathway to Prevention Study. After TrialNet Ancillary Studies Committee and Benaroya Research Institute (BRI) Institutional Review Board approval, 30 subjects (10 subjects in each group) were recruited from TrialNet Pathway to Prevention including those with (1) no antibodies (very low risk for T1D), (2) only one antibody (low risk for T1D), (3) two or more antibodies (significant risk for T1D) and (4) healthy controls (n = 10) from the general population without family history of T1D. All at‐risk subjects were first‐degree relatives of those with T1D.
Pulse DNA labeling with oral 2H‐glucose and DNA enrichment monitoring
Subjects were admitted to the Clinical Research Center for oral 2H‐glucose administration over 10 h. Subjects received a total of 1·0 g/kg of heavy glucose mixed with 240 ml of water to be consumed at 30‐min intervals. During the labeling period, fingerstick (capillary) blood was sampled periodically to assess glucose enrichment.
In order to measure the enrichment of 2H in the DNA of proliferating lymphocytes, blood was drawn on days 3 and 4 after the initial 2H‐glucose consumption visit.
In‐vivo T cell turnover measurement
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized fresh blood samples by Ficoll gradient centrifugation, surface‐stained and flow‐sorted into CD4‐ and CD8‐naive, effector memory and central memory T cells; CD4 regulatory T (Treg) cells; and CD8 CD45RA terminally differentiated effector memory T cells (TEMRA) (Fig. 1). Antibodies used for sorting were purchased from BD (Franklin Lakes, NJ, USA) and eBioscience (San Diego, CA, USA). DNA was immediately isolated from sorted populations after proteinase K digestion using DNEasy minicolumns (Qiagen Sciences, Valencia, CA, USA). 2H enrichment in DNA was measured by mass spectrometry using an Agilent model 5973/6890 GC (Agilent Technologies/Quantum Analytics, Foster City, CA, USA) and in‐vivo T cell turnover was calculated as fractional enrichment of deuterated adenosine (F max) as previously reported 6, 9, 10. F represents the fraction of newly synthesized DNA strands or, equivalently, the fraction of newly divided cells and was calculated by the following method. First, the 2H‐adenosine enrichment of sorted cells is normalized to observed plasma 2H‐glucose, or precursor enrichment (b) using the formula b = trapezoidal area under the curve (AUC)/pulse time and then the percentage of 2H adenosine (F) is calculated using the formula F = % 2H adenosine/precursor enrichment (b). The maximum of F max between days 3 and 4 was used to represent T cell turnover rate.
Figure 1.
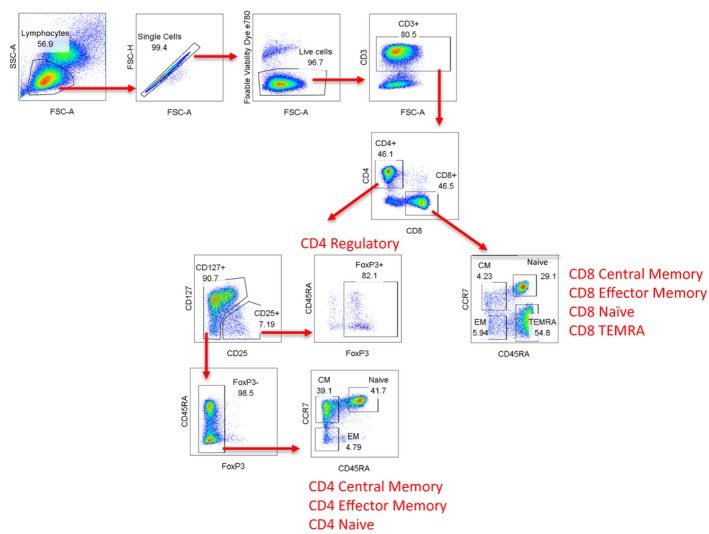
Overview of sorting strategies for T cell subsets.
Statistical analyses
The group mean of F max in T cell subsets among the four studied groups was compared using the Kruskal–Wallis test. The group mean of F max between relatives and normal controls was compared using the Mann–Whitney U‐test. The data were presented as mean ± standard deviation (s.d.). Differences were considered significant at P < 0·05. The effect size for the Mann–Whitney U‐test was calculated by dividing the Mann–Whitney Z‐value by the square root of the sample size. Intraclass correlation coefficients (ICC) were used to measure within‐ and between‐subject variability for repeat testing. The statistical analyses and graphical displays were performed using sas version 9.4, jmp Pro version 14 and GraphPad Prism statistical software.
Results
Study subject characteristics
Demographic and clinical attributes of the study groups are summarized in Table 1. Participants were age‐, sex‐ and BMI‐matched among the groups, and were Caucasian non‐Hispanic. There were seven subjects with impaired glucose tolerance (IGT), including one in the single autoantibody‐positive group and six in the two or more autoantibody‐positive group. The islet autoantibody profiles and HLA risk alleles for T1D in each group are listed in Table 1.
Table 1.
Demographics of the subjects in each group
| Normal control (n = 10) | Relatives without antibody (n = 10) | Relatives with one antibody (n = 10) | Relatives with two or more antibodies (n = 10) | |
|---|---|---|---|---|
| Age (mean ± s.d.), years | 29·6 ± 7·8 | 30·3 ± 7·3 | 29·1 ± 8·6 | 30·5 ± 8·7 |
| BMI (mean ± s.d.) | 28·7 ± 5·4 | 28·9 ± 8·3 | 24·7 ± 5·5 | 26·7 ± 3·8 |
| Sex (male/female) | 5/5 | 4/6 | 5/5 | 5/5 |
| Race/ethnicity | ||||
| Non‐Hispanic white | 10/10 (100) | 10/10 (100) | 10/10 (100) | 10/10 (100) |
| OGTT | ||||
| IGT | 0/10 (0) | 0/10 (0) | 1/10 (10) | 6/10 (60) |
| Islet autoantibodies | ||||
| GAD65 | 0 | 0 | 10/10 (100) | 8/10 (80) |
| ICA | 0 | 0 | 0 | 6/10( 60) |
| ICA512 | 0 | 0 | 0 | 7/10 (70) |
| mIAA | 0 | 0 | 0 | 1/10 (10) |
| ZnT8 | 0 | 0 | 0 | 6/10 (60) |
| HLA | ||||
| DR3 or DR4; not DQA1*0102 or DQB1*0602 | 4/10 (40) | 10/10 (100)* | 9/10 (90) | 9/10 (90) |
Values are mean ± s.d. or n (%).
P < 0·05 versus normal controls.
BMI = body mass index; s.d. = standard deviation; OGTT = oral glucose tolerance test; GAD65 = glutamate decarboxylase; ICA = islet cell antibodies; mIAA = microinsulin autoantibody; ZnT8 = zinc transporter 8; HLA = human leukocyte antigen.
T cell kinetics in individuals at risk for T1D based on antibody status
We evaluated the kinetics of peripheral blood CD4+ and CD8+ T cell subset turnover in the three groups of subjects at varying risk for T1D and in healthy controls using in‐vivo 2H‐glucose labeling and mass spectrometry. Specifically, we determined the in‐vivo turnover of flow‐sorted CD4+ central memory T cells, CD4+ effector memory T cells, CD4+ Treg and CD4+‐naive T cells; CD8+ central memory T cells, CD8+ effector memory T cells, CD8+ TEMRA (CD45RA+ effector memory T cells) and CD8+‐naive T cells. The fractional enrichment with deuterium (F max, representing turnover) was the highest in CD4+ Treg, followed by CD4+ effector and memory T cells, and the lowest in naive CD4+ T cells in all subjects (Fig. 2). CD8+ memory T cells had higher turnover than CD8+‐naive T cells and TEMRA. Thus, these results were consistent with our previous findings using the longer clinical protocol and labeling with heavy water 6.
Figure 2.
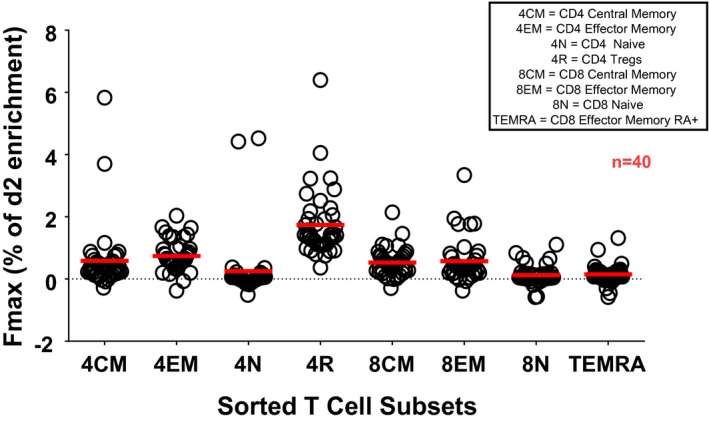
Enrichment of deuterated adenosine in various T cell subsets in all participants.
There was no significant difference in F max between at‐risk groups according to antibody or oral glucose tolerance test (OGTT) status among the subset of T cells studied (Fig. 3). Compared with controls, there was heterogeneity in the turnover of most T cell subsets among relatives as a group, especially for the Treg population. Further, there was significantly increased Treg turnover (as assessed by F max) between relatives regardless of autoantibody or OGTT status and healthy controls (1·733 ± 0·6784% versus 1·062 ± 0·3787% (mean ± s.d.), P = 0·004, effect size = 0·47), as shown in Figure 4. While there was heterogeneity in the turnover of most T cell subsets among relatives, particularly for the Treg population, in this small study no association between Treg turnover and genetic variants at IL2RA, IL2/IL21 and PTPN2 loci associated with T1D and T regulatory function was observed (data not shown).
Figure 3.
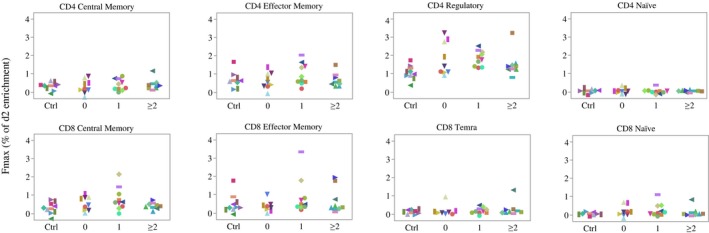
Enrichment of maximum fractional enrichment of deuterated adenosine (F max) in various T cell subsets in individuals at risk for type 1 diabetes (T1D) based on antibody status and controls. The y‐axis represents the percentage of F max. The subjects were divided into four groups, with Ctrl = normal control, 0 = relatives without autoantibody, 1 = relatives with one autoantibody and ≥ 2 = relatives with two or more autoantibodies; n = 38 (two participants, including one from the 0 antibody group and one from the two or more antibodies group, were excluded from the analysis due to poor quality of T cell sorting).
Figure 4.
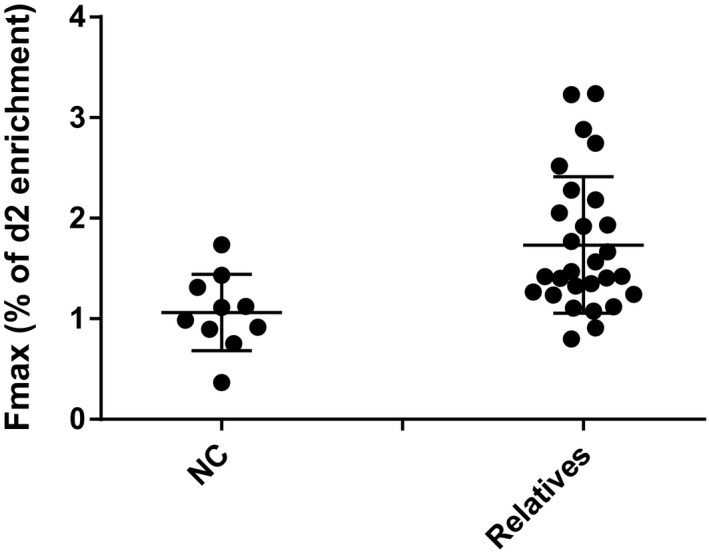
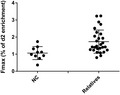
Enrichment of deuterated adenosine by maximum fractional enrichment of deuterated adenosine (F max) in CD4 regulatory T cells between controls and relatives of those with type 1 diabetes (T1D). The regulatory T cell (Treg) turnover by F max is significantly increased in relatives of those with T1D (n = 28) versus controls (n = 10) (1·733 ± 0·6784% versus 1·062 ± 0·3787% (mean ± standard deviation), P = 0·004, effect size 0·47).
Repeat measurement of T cell kinetics over 1 year
Five subjects who were available and willing to undergo additional testing 1 year later in each of the four groups were studied on a second occasion, under identical conditions with the aim of assessing variability in the measurements over time and associating results with potential changes in disease. We used ICC to measure the relative between‐ and within‐subject variability of each T cell population. Only one marker (F max of CD4 central memory T cells) had an ICC > 70%, indicating that this population was more stable within an individual over 1 year. As no subject had a change in either glucose tolerance or antibody status during the year, we were unable to evaluate relationships between T cell kinetics and disease progression.
Discussion
We evaluated the kinetics of immune cell subset turnover in first‐degree relatives of individuals with T1D as well as healthy controls using heavy glucose labeling and focusing on the turnover rates of CD4 effector and central memory cells, CD4 Treg cells and CD8 effector and memory cells. We found that relatives of those with T1D as a group demonstrated significantly enhanced CD4 Treg turnover compared to healthy control individuals, suggesting pre‐existing immune dysfunction in those with a genetic predisposition for T1D. However, in this small cohort we did not detect an association of Treg turnover rate by F max with individual genetic variants in the IL2RA, IL2/IL21 and PTPN2 loci that have been associated with risk for T1D. Other studies also find phenotypical differences between unaffected relatives and controls; one report demonstrated that T1D family members have an elevated inflammatory state 11 and another found small pancreas volume in family members compared with non‐family members as controls 12.
We previously demonstrated that individuals with clinically overt T1D had increased CD4 memory T cell turnover 6; however, we did not find this in autoantibody‐positive relatives of those with T1D, suggesting that increased CD4 proliferation only occurs later in disease progression. It is important to note, however, that different protocols were used in these two studies 5, 9. To simplify the study protocol, making it more practical to use in clinical trials we modified the T cell labeling procedure, using 2H‐glucose labeling with a 3–4‐day labeling time in the current study versus using 2H‐H2O labeling with a 42‐day labeling time in the previous study. While we did not conduct a direct comparison between shorter and longer protocols, both approaches demonstrated the expected hierarchical order of greatest to least turnover of Treg cells > T memory cells > T naive cells 6.
Also, we found no relationship between T cell turnover measures and autoantibody or glucose tolerance status. Using data from repeat testing on a subset of participants, an important caveat to interpretation of these results is the generally low ICC observed for the turnover rate of most of the T cell subsets evaluated. The limited range of F max values for the T cell subsets probably contributes to the low ICC. Biomarkers with high ICC can be particularly useful to dissect disease heterogeneity. A biomarker which is stable (little technical or biological variability) within a subject when the disease is stable, but which varies between subjects with different clinical courses, could then be evaluated in relationship with clinically or biologically relevant disease measures. In this study, we were unable to demonstrate such relationships. This may be due to the low ICC for most subsets, but may also be due to limitations of our study. There were only a small number of individuals who underwent repeat testing, and the follow‐up period was too short to observe disease progression in this adult population, whose rate of progression is known to be slow. Moreover, we did not include individuals with clinical T1D in this study.
Thus, while this heavy glucose protocol is unlikely to be informative in understanding natural history, it could provide useful information in the context of clinical trials. For example, there are five immune therapies that have been demonstrated in randomized clinical trials to alter disease course in individuals with recently diagnosed T1D 13, 14, 15, 16, 17. However, as in all immune therapy, there is a range of responses among those treated with active drug. Poor responders to the B cell depletion therapy rituximab were found to have an increase in activated T cell gene transcripts 18, and those who had poor responses to abatacept had an increase in B cell gene transcripts 19. Whether or not these transcriptional changes were due to changes in proliferation could be addressed in subsequent clinical trials, with therapies expected to impact T cell turnover using the short heavy glucose labeling protocol described here.
Disclosures
There are no conflicts of interest involved in this study.
Author contributions
W. H. researched the data and wrote the manuscript. H. T. B contributed statistical support and edited the manuscript. C. S. co‐ordinated the data analysis and reviewed and edited the manuscript. K. C. performed genetic analysis and edited the manuscript. C. J. G. designed the study, reviewed and edited the manuscript. W. H. and C. J. G. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Acknowledgements
The authors thank the Diabetes Clinical Research Program staff, especially Kim Varner, for their efforts in recruitment and retention for this study. We also acknowledge Jared Odegard for early work on this study, Jim Qin and Jerill Thorpe for technical support in conduct of the flow sorting and mass spec analysis for this study, as well as Martin Salidek at the University of Washington, where the mass spec analysis was conducted. This work was supported by National Institute of Health Grant: DP3DK098217. We acknowledge the support of the Type 1 Diabetes TrialNet Study Group, which identified study participants and provided samples and follow‐up data for this study. The Type 1 Diabetes TrialNet Study Group is a clinical trials network funded by the National Institutes of Health (NIH) through the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, through co‐operative agreements U01 DK061010, U01 DK061034, U01 DK061042, U01 DK061058, U01 DK085465, U01 DK085453, U01 DK085461, U01 DK085466, U01 DK085499, U01 DK085504, U01 DK085509, U01 DK103180, U01 DK103153, U01 DK085476, U01 DK103266, U01 DK103282, U01 DK106984, U01 DK106994, U01 DK107013, U01 DK107014, UC4 DK097835, UC4 DK106993 and JDRF. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the JDRF.
References
- 1. Herold KC, Vignali DAA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 2013; 13:243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makhlouf L, Grey ST, Dong V et al Depleting anti‐CD4 monoclonal antibody cures new‐onset diabetes, prevents recurrent autoimmune diabetes, and delays allograft rejection in nonobese diabetic mice. Transplantation 2004; 77:990–7. [DOI] [PubMed] [Google Scholar]
- 3. Peakman M, Warnock T, Vats A et al Lymphocyte subset abnormalities, autoantibodies and their relationship with HLA DR types in children with type 1 (insulin‐dependent) diabetes and their first degree relatives. Diabetologia 1994; 37:155–65. [DOI] [PubMed] [Google Scholar]
- 4. Tree TI, Roep BO, Peakman M. A mini meta‐analysis of studies on CD4+CD25+ T cells in human type 1 diabetes: report of the Immunology of Diabetes Society T Cell Workshop. Ann NY Acad Sci 2006; 1079:9–18. [DOI] [PubMed] [Google Scholar]
- 5. Neese RA, Misell LM, Turner S et al Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci USA 2002; 99:15345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bollyky JB, Long SA, Fitch M et al Evaluation of in vivo T cell kinetics: use of heavy isotope labelling in type 1 diabetes. Clin Exp Immunol 2013; 172:363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sosenko JM, Krischer JP, Palmer JP et al A risk score for type 1 diabetes derived from autoantibody‐positive participants in the diabetes prevention trial‐type 1. Diabetes Care 2008; 31:528–33. [DOI] [PubMed] [Google Scholar]
- 8. Insel RA, Dunne JL, Atkinson MA et al Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015; 38:1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Macallan DC, Fullerton CA, Neese RA, Haddock K, Park SS, Hellerstein MK. Measurement of cell proliferation by labeling of DNA with stable isotope‐labeled glucose: studies in vitro, in animals, and in humans. Proc Natl Acad Sci USA 1998; 95:708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Macallan DC, Asquith B, Irvine AJ et al Measurement and modelling of human T cell kinetics. Eur J Immunol 2003; 33:2316–26. [DOI] [PubMed] [Google Scholar]
- 11. Chen YG, Cabrera SM, Jia S et al Molecular signatures differentiate immune states in type 1 diabetic families. Diabetes 2014; 63:3960–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campbell‐Thompson ML, Filipp SL, Grajo JR et al Relative pancreas volume is reduced in first‐degree relatives of patients with type 1 diabetes. Diabetes Care 2019; 42:281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pescovitz MD, Greenbaum CJ, Krause‐Steinrauf H et al Rituximab, B‐lymphocyte depletion, and preservation of beta‐cell function. N Engl J Med 2009; 361:2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orban T, Bundy B, Becker DJ et al Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled trial. Lancet 2011; 378:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herold KC, Hagopian W, Auger JA et al Anti‐CD3 monoclonal antibody in new‐onset type 1 diabetes mellitus. N Engl J Med 2002; 346:1692–8. [DOI] [PubMed] [Google Scholar]
- 16. Rigby MR, Harris KM, Pinckney A et al Alefacept provides sustained clinical and immunological effects in new‐onset type 1 diabetes patients. J Clin Invest 2015; 125:3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haller MJ, Schatz DA, Skyler JS et al Low‐dose anti‐thymocyte globulin (ATG) preserves beta‐cell function and improves HbA1c in new‐onset type 1 diabetes. Diabetes Care 2018; 41:1917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Linsley PS, Greenbaum CJ, Rosasco M, Presnell S, Herold KC, Dufort MJ. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new‐onset type 1 diabetes. Genes Immun 2019; 20:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Linsley PS, Greenbaum CJ, Speake C, Long SA, Dufort MJ. B lymphocyte alterations accompany abatacept resistance in new‐onset type 1 diabetes. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]