

Abstract
Objectives
To determine the mechanisms of ubiquitination in postmenopausal osteoporosis and investigate the ubiquitinated spectrum of novel targets between healthy postmenopausal women and postmenopausal osteoporosis patients, we performed ubiquitylome analysis of the whole blood of postmenopausal women and postmenopausal osteoporosis patients.
Methods
To obtain a more comprehensive understanding of the postmenopausal osteoporosis mechanism, we performed a quantitative assessment of the ubiquitylome in whole blood from seven healthy postmenopausal women and seven postmenopausal osteoporosis patients using high‐performance liquid chromatography fractionation, affinity enrichment, and liquid chromatography coupled to tandem mass spectrometry (LC‐MS/MS). To examine the ubiquitylome data, we performed enrichment analysis using an ubiquitylated amino acid motif, Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway.
Results
Altogether, 133 ubiquitinated sites and 102 proteins were quantified. A difference of more than 1.2 times is considered significant upregulation and less than 0.83 significant downregulation; 32 ubiquitinated sites on 25 proteins were upregulated and 101 ubiquitinated sites on 77 proteins were downregulated. These quantified proteins, both with differently ubiquitinated sites, participated in various cellular processes, such as cellular processes, biological regulation processes, response to stimulus processes, single‐organism and metabolic processes. Ubiquitin conjugating enzyme activity and ubiquitin‐like protein conjugating enzyme activity were the most highly enriched in molecular function of upregulated sites with corresponding proteins, but they were not enriched in downregulated in sites with corresponding proteins. The KEGG pathways analysis of quantified proteins with differentiated ubiquitinated sites found 13 kinds of molecular interactions and functional pathways, such as glyoxylate and decarboxylate metabolism, dopaminergic synapse, ubiquitin‐mediated proteolysis, salivary secretion, coagulation and complement cascades, Parkinson's disease, and hippo signaling pathway. In addition, hsa04120 ubiquitin‐mediated proteolysis was the most highly enriched in proteins with upregulated sites, hsa04610 complement and coagulation cascades was the most highly enriched in proteins with downregulated ubiquitinated sites, and hsa04114 Oocyte meiosis was the most highly enriched among all differential proteins.
Conclusion
Our study expands the understanding of the spectrum of novel targets that are differentially ubiquitinated in whole blood from healthy postmenopausal women and postmenopausal osteoporosis patients. The findings will contribute toward our understanding of the underlying proteostasis pathways in postmenopausal osteoporosis and the potential identification of diagnostic biomarkers in whole blood.
Keywords: Postmenopausal osteoporosis, Proteome, Ubiquitylome
Introduction
The metabolism of bone cells is strongly regulated by estrogens and, therefore, postmenopausal osteoporosis is the most typical form of osteoporosis, which is characterized by low bone mass and microstructure damage of the bone tissue, leading to increased bone fragility and the risk of fracture1. Osteoporosis causes huge economic losses worldwide. Approximately 10 million men and women in the USA have osteoporosis2, 75 million people in Europe, North America and Japan are affected by it3, while the prevalence of osteoporosis in central south Chinese postmenopausal women is approximately 39.4%4.
Although the role of estrogen on bone metabolism has been documented, the mechanism of postmenopausal osteoporosis remains unclear and diagnostic strategies for postmenopausal osteoporosis are lacking.
Over the past century, many types of histone post‐translational modifications (PTM) have been identified, including lysine acetylation, arginine and lysine methylation, phosphorylation, proline isomerization, ubiquitination (Ub), ADP ribosylation, arginine citrullination, SUMOylation, carbonylation, and biotinylation5, 6. It is known that the bone remodeling cycle is a balanced process that depends on the interaction, differentiation, and functions of the mesenchymal osteoblastic lineage and the hematopoietic osteoplastic lineage to maintain homeostasis of bone mass. Many histone post‐translational modifications are involved in bone remodeling cycle regulation.
Works such as Hein et al.7 and Li et al.8 have established that advanced glycation end products (AGE) could biphasically modulate bone resorption in osteoclast‐like cells and modify proteins in osteoporotic bone. A phosphoproteome of Milani et al.9 revealed that phosphorylation is extremely important during osteoblast adhesion. In addition, a phosphoproteome study by Marumoto et al.10 further reveals that hedgehog signaling has a critical role in osteoblast morphological transitions. However, the review of Bradley et al.11 demonstrates that histone deacetylases have emerged as crucial regulators of both intramembranous and endochondral bone formation.
In summary, their results indirectly demonstrate the feasibility of revealing the mechanism of bone remodeling through the histone post‐translational modifications. The research of Zhang et al.12 provides further relevant evidence. Zhang et al. examined the dynamics of distinct histone modifications during osteogenesis, including the dynamics of: H3K9/K14 and H4K12 acetylation; H3K4 mono‐, di‐ and tri‐methylation; H3K9 di‐methylation and H3K27 tri‐methylation in osteogenic genes, runt‐related transcription factor 2 (Runx2), osterix (Osx), alkaline phosphatase, and bone sialoprotein and osteocalcin during C3H10T1/2 osteogenesis.
However, the most important post‐translational modifications are ubiquitin modifications in bone metabolism. The ubiquitin‐proteasome system is one of the major quality control pathways responsible for cellular homeostasis13, 14, 15.
Ubiquitination is a posttranslational modification of proteins that controls almost every cellular metabolic pathway through a variety of combinations of linkages, either as a single moiety or as polymers16. These cellular metabolic pathways include, but are not limited to, transcription, cell signaling17, endocytic trafficking, DNA damage and repair18, and cell‐cycle control19. The ubiquitin system in humans consists of 2 E1, 35 E2, more than 600 E3 ubiquitin ligases, and hundreds of deubiquitylases20.
For more than a decade, investigators have shown that ubiquitin‐proteasome‐mediated protein degradation is critical in regulating the balance between bone formation and bone resorption21.
In addition, several studies have found that many ubiquitinases play an important role in bone metabolism, such as poly‐ubiquitination‐mediated RUNX2 degradation, which is an important cause of bone loss22, 23. Surveys such as that conducted by Zhou et al.24 have shown that RNF185 negatively regulates osteogenesis through the degradation of Dvl2 and the downregulation of the canonical Wnt signaling pathway. Many studies indicate that the muscle‐specific RING‐finger1 (MuRF1), a muscle‐specific ubiquitin ligas, is involved in osteoblastic bone formation and osteoclastic bone resorption25, 26, 27. Xu et al. 28 established that SMURF2 is an important regulator of the critical communication between osteoblasts and osteoclasts; the bone mass phenotype in Smurf2‐deficient and Smurf1‐deficient mice is opposite, indicating that SMURF2 has a non‐overlapping and opposite function to SMURF1. Jin et al.29 found that Bre promoted the Mdm2‐mediated p53 ubiquitination and degradation by physically interacting with p53, and that Bre has a novel function in osteoblast differentiation through modulating the stability of p53.
In contrast, quantitative analysis of ubiquitylomes has proven to be a valuable tool for elucidating targets and mechanisms of the ubiquitin signaling systems13, 14, 15, as well as gaining insights into many diseases30, such as neuroblastoma31, cancer32, and Alzheimer's disease33.
To date, although the importance of ubiquitin modifications in bone metabolism has been reported, few studies have investigated ubiquitylomes in postmenopausal osteoporosis.
Here, we used ubiquitylomes analysis of the whole blood of postmenopausal women and postmenopausal osteoporosis patients, to discover the mechanism of ubiquitination in postmenopausal osteoporosis and investigate the ubiquitinated spectrum of novel targets between healthy postmenopausal women and postmenopausal osteoporosis patients. This expanded our understanding of the spectrum of novel targets that are differentially ubiquitinated in whole blood from healthy postmenopausal women and postmenopausal osteoporosis patients. Overall, the study highlights the utility of liquid chromatography coupled to tandem mass spectrometry (LC‐MS/MS) in performing comprehensive mapping of the human blood ubiquitylome changes in postmenopausal osteoporosis, which provides insight into underlying proteostasis pathways and targets that may have potential as novel diagnostic biomarkers in blood.
Materials and Methods
Human Subjects
This study included seven consecutive, unrelated, late postmenopausal women with osteoporosis and seven healthy postmenopausal women who visited the Second Affiliated Hospital of Zhejiang Chinese Medicine University. All subjects were Chinese Han females. Postmenopausal status was defined as no menses for at least 1 year after their last menses. Bone mineral density (BMD) of the lumbar spine (L1–L4) and femoral neck was measured by dual‐energy X‐ray absorptiometry using a QDR‐4500w instrument (Hologic, USA) at the Second Affiliated Hospital of Zhejiang Chinese Medicine University. The average age of the osteoporosis patients was 66.1 ± 3.4 years, with a range of 54–81 years. Seven age‐matched postmenopausal healthy volunteers (average age 68.9 ± 2.5 years, with a range of 58–79 years) were also recruited in Hangzhou.
Diagnostic Criteria
The diagnosis of osteoporosis was based on the criteria recommended by the World Health Organization34, which included that the bone density (g/cm2) of the lumbar vertebra normal position and femoral neck was surveyed using dual‐energy X‐ray absorptiometry, compared with a normal adult of the same gender and race; T ≤ −2.5 could be diagnosed as osteoporosis, where T = (the standard deviation of measured value − peak bone mass)/normal adult bone density.
Inclusion Criteria
Patients were included in the study based on the following criteria: (i) they conformed to the osteoporosis diagnostic criteria; (ii) they were postmenopausal women; and (iii) they were between 50 and 89 years old.
Exclusion Criteria
Patients were excluded from the study based on the following criteria, which are derived from a previous study35: (i) all individuals with disorders known to cause abnormalities in the metabolism of bone or calcium, such as diabetes, Cushing's syndrome, functional change of the thyroid or parathyroid, osteomalacia, rheumatoid arthritis, multiple myeloma, bone tumor, osteoarthrosis, Paget's disease, and osteogenesis imperfecta; (ii) the individuals that also had severe primary cardiac diseases, or diseases of the cerebral vessels or hematopoietic system; (iii) the individuals that also had severe liver function or renal insufficiencies; (iv) the individuals who had taken drugs within the past 6 months that affect bone metabolism, such as estrogen, steroid hormones, calcitonin, parathyroid hormones, bisphosphonates, fluoride, vitamin D, anticonvulsant drugs, and diuretics; (v) the individuals who had a medical history of mental illness; and (vi) the individuals who had Alzheimer's disease.
Ethical Review
The study protocol was approved by the local Ethics Committee of the Second Affiliated Hospital of Zhejiang Chinese Medicine University and informed consent was obtained from all subjects.
Trypsin Enzymatic Hydrolysis
We added 8 mol urea to the sample to adjust the volume, then added dithiothreitol to a final concentration of 5 mmol, reducing at 56 °C for 30 min. Iodoacetamide was then added to a final concentration of 11 mmol and incubated for 15 min at room temperature in the dark. Finally, the urea concentration of the sample was diluted to less than 2 mol. Trypsin was added at a mass ratio of 1:50 (pancreatin: protein) and digested overnight at 37 °C. Trypsin was added at a mass ratio of 1:100 (pancreatin: protein) and continued to digest for 4 h.
High‐Performance Liquid Chromatography Fractionation
The peptides were fractionated by high pH reverse phase high‐performance liquid chromatography and the column was Agilent 300 Extend C18 (5 μm particle size, 4.6 mm id, 250 mm long). Finally, the fractional gradient of the peptide was 8%–32% acetonitrile, pH 6.0, allowing 60 min time to separate 60 components, and then the peptides were combined into four components, and the combined components were vacuum freeze‐dried for subsequent operations.
Affinity Enrichment
The peptide was dissolved in IP buffer solution (100 mmol NaCl, 1 mmol EDTA, 50 mmol Tris‐HCl, 0.5% NP‐40, pH 8.0), and the supernatant was transferred to the pre‐washed ubiquitinated resin (resin number) PTM‐1104, from Hangzhou Jingjie Biotechnology, PTM Bio, placed on a rotary shaker at 4°C, gently shaken, and incubated overnight. Then, the resin was washed four times with IP buffer solution and twice with deionized water. Finally, the resin‐bound peptide was 0.1% trifluoroacetic acid eluate, eluted three times in total, and the eluate was collected and vacuum‐dried and drained. The salt was removed according to the C18 ZipTips instructions, vacuum‐dried, and drained for liquid‐mass spectrometry analysis.
LC‐MS/MS Analysis
The tryptic peptides were dissolved in phase A in an aqueous solution of 0.1% formic acid and 2% acetonitrile; buffer B was an aqueous solution of 0.1% formic acid and 90% acetonitrile. Liquid phase gradient setting: 0–26 min, 5%–22% B; 26–34 min, 22%–35% B; 34–37 min, 35%–80% B; 37–40 min, 80% B, flow rate maintenance at 350 nL/min. The peptides were separated using an ultra‐high‐performance liquid phase system and injected into the NSI ion source for ionization, and then analyzed by Orbitrap FusionTM (Thermo) mass spectrometry. The ion source voltage was 2.0 kV, and the peptide precursor and its secondary fragments were detected and analyzed by high‐resolution Orbitrap. The primary mass spectrometer scan range was 350–1550 m/z and the scan resolution was set to 60 000; the secondary mass spectrometry scan range was 100 m/z and the Orbitrap scan resolution was 15 000. After the first‐stage scanning, the first 20 peptides with the highest signals were selected to enter the HCD collision cell and 35% of the fragmentation energy was used for secondary mass spectrometry. Finally, the automatic gain control (AGC) was set to 5E4, the signal threshold to 5000 ions/s, the maximum injection time to 200 ms, and the dynamic exclusion time of the tandem mass spectrometry to 15 s to avoid the parent ion. Then the scan was repeated.
Database Search
Secondary mass spectral data was retrieved using Maxquant (v1.5.2.8). Search parameter settings: The database used was SwissProthuman (20 203 sequences), the anti‐library was added to calculate the false positive rate (FDR) caused by random matching, and a common pollution database was added to the database to eliminate the effect of contaminated proteins on the results. The enzyme digestion mode was Trypsin/P; the number of missed sites was 2; the minimum length of the peptide was seven amino acid residues; the maximum number of peptides was five; First search range was set to 5 ppm for precursor ions. Main search range set to 5 ppm and 0.02 Da for fragment ions. The mass error tolerance was 20 and 5 ppm. Finally, the fixed modification was cysteine alkylation, Author: the variable modification was methionine oxidation, protein N‐terminal acetylation, and lysine ubiquitination. The FDR for protein identification and PSM identification was 1%.
Bioinformatics Analysis
The Gene Ontology (GO) annotation proteome was derived from the UniProt‐GOA database (http://www.ebi.ac.uk/GOA/) and the InterProScan software36, 37. First, identified protein ID was converted to UniProt ID and then mapped to GO IDs by protein ID. If some identified proteins were not annotated by the UniProt‐GOA database, the InterPro Scan software would be used to annotate proteins’ GO functional classification based on the protein sequence alignment method. Finally, proteins are classified by GO annotation based on three categories: biological process, cellular component, and molecular function.
We used the Kyoto Encyclopedia of Genes and Genomes (KEGG) database for protein pathway analysis38. First, we used the KEGG online service tool KAAS to annotate proteins’ KEGG database description. Then, we mapped the annotation result on the KEGG pathway database using the KEGG online service tool KEGG mapper. The protein complex analysis was conducted using the CORUM protein complex database. We used the InterPro domain database for protein domain annotation and Motif‐x for protein analysis.
Results
Overview of Global Ubiquitylome upon Postmenopausal Osteoporosis
Altogether, 133 ubiquitinated sites and 102 proteins were quantified. The analysis of ubiquitylomes the whole blood in seven healthy postmenopausal women and seven postmenopausal osteoporosis patients revealed that 32 ubiquitinated sites on 25 proteins were upregulated and 101 ubiquitinated sites on 77 proteins were downregulated. The difference is more than 1.2 times as significant upregulation and less than 0.83 as a significant downregulation (P < 0.05, Table 1). Here, there are multiple ubiquitination sites on the same protein, some of which are rising and some are different.
Table 1.
The differentially expressed ubiquitinated sites and proteins in ubiquitylome of postmenopausal osteoporosis patients and healthy postmenopausal women
| Protein accession | Ratio | Regulated type | Protein description | Gene name | Score |
|---|---|---|---|---|---|
| P02656 | 0.212 | Down | Apolipoprotein C‐III | APOC3 | 161.24 |
| O43765 | 0.564 | Down | Small glutamine‐rich tetratricopeptide repeat‐containing protein alpha | SGTA | 131.66 |
| P00918 | 0.608 | Down | Carbonic anhydrase 2 | CA2 | 125.73 |
| Q04323 | 0.26 | Down | UBX domain‐containing protein 1 | UBXN1 | 88.708 |
| Q9P107 | 13.569 | Up | GEM‐interacting protein | GMIP | 50.314 |
| P02675 | 0.373 | Down | Fibrinogen beta chain | FGB | 117.84 |
| P02675 | 0.192 | Down | Fibrinogen beta chain | FGB | 132.81 |
| P00736 | 0.115 | Down | Complement C1r subcomponent | C1R | 70.165 |
| P21980 | 0.595 | Down | Protein‐glutamine gamma‐glutamyltransferase 2 | TGM2 | 102.53 |
| Q9BQE3 | 0.276 | Down | Tubulin alpha‐1C chain | TUBA1C | 137.18 |
| Q9BQE3 | 0.546 | Down | Tubulin alpha‐1C chain | TUBA1C | 115.54 |
| Q9BQE3 | 0.611 | Down | Tubulin alpha‐1C chain | TUBA1C | 70.783 |
| Q9BQE3 | 0.416 | Down | Tubulin alpha‐1C chain | TUBA1C | 95.618 |
| Q9BQE3 | 0.122 | Down | Tubulin alpha‐1C chain | TUBA1C | 119.39 |
| P10599 | 0.336 | Down | Thioredoxin | TXN | 92.19 |
| P02774 | 0.121 | Down | Vitamin D‐binding protein | GC | 50.786 |
| P15153 | 0.09 | Down | Ras‐related C3 botulinum toxin substrate 2 | RAC2 | 50.108 |
| P02647 | 0.483 | Down | Apolipoprotein A‐I | APOA1 | 247.38 |
| P28066 | 0.23 | Down | Proteasome subunit alpha type‐5 | PSMA5 | 161.68 |
| P32119 | 26.6 | Up | Peroxiredoxin‐2 | PRDX2 | 92.039 |
| P32119 | 0.696 | Down | Peroxiredoxin‐2 | PRDX2 | 145.46 |
| Q13228 | 5.732 | Up | Selenium‐binding protein 1 | SELENBP1 | 141.07 |
| P63104 | 0.564 | Down | 14‐3‐3 protein zeta/delta | YWHAZ | 199.8 |
| P16157 | 0.589 | Down | Ankyrin‐1 | ANK1 | 150.11 |
| P16157 | 0.525 | Down | Ankyrin‐1 | ANK1 | 220.63 |
| P16157 | 0.184 | Down | Ankyrin‐1 | ANK1 | 154.72 |
| P16157 | 0.384 | Down | Ankyrin‐1 | ANK1 | 271.08 |
| P16157 | 2.199 | Up | Ankyrin‐1 | ANK1 | 122.28 |
| P16157 | 1.482 | Up | Ankyrin‐1 | ANK1 | 239.87 |
| P02649 | 0.183 | Down | Apolipoprotein E | APOE | 160.18 |
| Q14687 | 0.598 | Down | Genetic suppressor element 1 | GSE1 | 116.54 |
| P20618 | 0.416 | Down | Proteasome subunit beta type‐1 | PSMB1 | 132.13 |
| P02671 | 0.805 | Down | Fibrinogen alpha chain | FGA | 81.017 |
| P02671 | 0.554 | Down | Fibrinogen alpha chain | FGA | 71.153 |
| P04040 | 2.279 | Up | Catalase | CAT | 109.83 |
| P00441 | 0.442 | Down | Superoxide dismutase [Cu‐Zn] | SOD1 | 80.96 |
| Q9C0C9 | 1.597 | Up | (E3‐independent) E2 ubiquitin‐conjugating enzyme | UBE2O | 112.26 |
| Q9C0C9 | 4.967 | Up | (E3‐independent) E2 ubiquitin‐conjugating enzyme | UBE2O | 68.536 |
| Q9C0C9 | 0.581 | Down | (E3‐independent) E2 ubiquitin‐conjugating enzyme | UBE2O | 97.597 |
| Q9C0C9 | 4.099 | Up | (E3‐independent) E2 ubiquitin‐conjugating enzyme | UBE2O | 88.021 |
| Q9C0C9 | 0.809 | Down | (E3‐independent) E2 ubiquitin‐conjugating enzyme | UBE2O | 124.21 |
| P62258 | 0.227 | Down | 14–3‐3 protein epsilon | YWHAE | 76.847 |
| P25789 | 0.473 | Down | Proteasome subunit alpha type‐4 | PSMA4 | 105.2 |
| P25789 | 0.349 | Down | Proteasome subunit alpha type‐4 | PSMA4 | 127.56 |
| P27348 | 0.36 | Down | 14‐3‐3 protein theta | YWHAQ | 120.46 |
| P22314 | 95.567 | Up | Ubiquitin‐like modifier‐activating enzyme 1 | UBA1 | 168.62 |
| P22314 | 0.166 | Down | Ubiquitin‐like modifier‐activating enzyme 1 | UBA1 | 118.32 |
| P28070 | 0.489 | Down | Proteasome subunit beta type‐4 | PSMB4 | 205.62 |
| P23634 | 0.405 | Down | Plasma membrane calcium‐transporting ATPase 4 | ATP2B4 | 85.355 |
| P0C0L4 | 0.43 | Down | Complement C4‐A | C4A | 109.71 |
| P62158 | 12.941 | Up | Calmodulin | CALM1 | 62.099 |
| P30153 | 1.362 | Up | Serine/threonine‐protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | PPP2R1A | 120.03 |
| Q96BN8 | 0.071 | Down | Ubiquitin thioesterase otulin | OTULIN | 109.07 |
| P11171 | 0.457 | Down | Protein 4.1 | EPB41 | 184.24 |
| P11171 | 2.742 | Up | Protein 4.1 | EPB41 | 63.691 |
| P11171 | 0.416 | Down | Protein 4.1 | EPB41 | 125.97 |
| P11171 | 0.266 | Down | Protein 4.1 | EPB41 | 54.772 |
| P67775 | 0.352 | Down | Serine/threonine‐protein phosphatase 2A catalytic subunit alpha isoform | PPP2CA | 78.149 |
| P02042 | 1.625 | Up | Hemoglobin subunit delta | HBD | 120.23 |
| P68036 | 1.742 | Up | Ubiquitin‐conjugating enzyme E2 L3 | UBE2L3 | 90.793 |
| P16452 | 1.313 | Up | Erythrocyte membrane protein band 4.2 | EPB42 | 360.48 |
| P16452 | 0.72 | Down | Erythrocyte membrane protein band 4.2 | EPB42 | 189.43 |
| P02652 | 0.223 | Down | Apolipoprotein A‐II | APOA2 | 89.123 |
| P02652 | 0.568 | Down | Apolipoprotein A‐II | APOA2 | 124.12 |
| P02765 | 0.573 | Down | Alpha‐2‐HS‐glycoprotein | AHSG | 134.99 |
| P40925 | 4.17 | Up | Malate dehydrogenase, cytoplasmic | MDH1 | 95.531 |
| P54727 | 0.629 | Down | UV excision repair protein RAD23 homolog B | RAD23B | 167.89 |
| P54252 | 0.464 | Down | Ataxin‐3 | ATXN3 | 72.315 |
| P05198 | 0.564 | Down | Eukaryotic translation initiation factor 2 subunit 1 | EIF2S1 | 103.02 |
| Q04917 | 0.154 | Down | 14–3‐3 protein eta | YWHAH | 54.259 |
| P00491 | 1.549 | Up | Purine nucleoside phosphorylase | PNP | 140.96 |
| Q13875 | 2.37 | Up | Myelin‐associated oligodendrocyte basic protein | MOBP | 73.233 |
| Q13875 | 2.37 | Up | Myelin‐associated oligodendrocyte basic protein | MOBP | 73.233 |
| P01857 | 0.489 | Down | Ig gamma‐1 chain C region | IGHG1 | 156.7 |
| P27169 | 0.147 | Down | Serum paraoxonase/arylesterase 1 | PON1 | 55.064 |
| O15554 | 1.412 | Up | Intermediate conductance calcium‐activated potassium channel protein 4 | KCNN4 | 47.039 |
| P05154 | 0.405 | Down | Plasma serine protease inhibitor | SERPINA5 | 119.68 |
| Q5T4S7 | 0.751 | Down | E3 ubiquitin‐protein ligase UBR4 | UBR4 | 98.175 |
| Q5T4S7 | 0.263 | Down | E3 ubiquitin‐protein ligase UBR4 | UBR4 | 75.764 |
| P25786 | 0.528 | Down | Proteasome subunit alpha type‐1 | PSMA1 | 169.89 |
| P49720 | 0.326 | Down | Proteasome subunit beta type‐3 | PSMB3 | 190.26 |
| Q58WW2 | 0.464 | Down | DDB1‐ and CUL4‐associated factor 6 | DCAF6 | 121.61 |
| Q58WW2 | 0.327 | Down | DDB1‐ and CUL4‐associated factor 6 | DCAF6 | 147.4 |
| P02549 | 1.491 | Up | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 156.67 |
| P02549 | 4.173 | Up | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 124.08 |
| P02549 | 0.779 | Down | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 155.66 |
| P02549 | 0.624 | Down | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 156.36 |
| P02549 | 0.687 | Down | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 228.19 |
| P02549 | 0.301 | Down | Spectrin alpha chain, erythrocytic 1 | SPTA1 | 158.29 |
| P61088 | 2.626 | Up | Ubiquitin‐conjugating enzyme E2 N | UBE2N | 190.41 |
| P54725 | 0.532 | Down | UV excision repair protein RAD23 homolog A | RAD23A | 168.59 |
| Q00610 | 0.208 | Down | Clathrin heavy chain 1 | CLTC | 65.231 |
| P29144 | 0.572 | Down | Tripeptidyl‐peptidase 2 | TPP2 | 104.18 |
| P51811 | 0.666 | Down | Membrane transport protein XK | XK | 138.89 |
| Q9BSL1 | 9.707 | Up | Ubiquitin‐associated domain‐containing protein 1 | UBAC1 | 180.88 |
| P04075 | 0.53 | Down | Fructose‐bisphosphate aldolase A | ALDOA | 139.47 |
| P02743 | 0.338 | Down | Serum amyloid P‐component | APCS | 121.99 |
| Q16531 | 10.591 | Up | DNA damage‐binding protein 1 | DDB1 | 123.63 |
| Q5VW32 | 0.264 | Down | BRO1 domain‐containing protein BROX | BROX | 57.532 |
| P30043 | 0.511 | Down | Flavin reductase (NADPH) | BLVRB | 187.78 |
| P62987 | 0.595 | Down | Ubiquitin‐60S ribosomal protein L40 | UBA52 | 136.96 |
| P45974 | 0.742 | Down | Ubiquitin carboxyl‐terminal hydrolase 5 | USP5 | 173.72 |
| P02655 | 0.238 | Down | Apolipoprotein C‐II | APOC2 | 80.585 |
| O14818 | 0.484 | Down | Proteasome subunit alpha type‐7 | PSMA7 | 56.916 |
| Q15843 | 9.91 | Up | NEDD8 | NEDD8 | 206.18 |
| Q15843 | 3.159 | Up | NEDD8 | NEDD8 | 207.82 |
| Q15843 | 13.049 | Up | NEDD8 | NEDD8 | 84.365 |
| P13716 | 0.402 | Down | Delta‐aminolevulinic acid dehydratase | ALAD | 53.453 |
| Q9NR09 | 0.242 | Down | Baculoviral IAP repeat‐containing protein 6 | BIRC6 | 45.357 |
| P0CG05 | 0.383 | Down | Ig lambda‐2 chain C regions | IGLC2 | 131.82 |
| Q96GG9 | 0.645 | Down | DCN1‐like protein 1 | DCUN1D1 | 120.76 |
| P55072 | 0.18 | Down | Transitional endoplasmic reticulum ATPase | VCP | 45.883 |
| P55072 | 0.622 | Down | Transitional endoplasmic reticulum ATPase | VCP | 94.114 |
| P01009 | 0.13 | Down | Alpha‐1‐antitrypsin | SERPINA1 | 71.451 |
| P02679 | 0.305 | Down | Fibrinogen gamma chain | FGG | 123.86 |
| Q93034 | 2.055 | Up | Cullin‐5 | CUL5 | 211.64 |
| P07738 | 0.593 | Down | Bisphosphoglycerate mutase | BPGM | 134.44 |
| P23526 | 13.226 | Up | Adenosylhomocysteinase | AHCY | 49.188 |
| P04406 | 0.66 | Down | Glyceraldehyde‐3‐phosphate dehydrogenase | GAPDH | 92.439 |
| P27105 | 0.51 | Down | Erythrocyte band 7 integral membrane protein | STOM | 134.73 |
| P00915 | 0.322 | Down | Carbonic anhydrase 1 | CA1 | 65.428 |
| P00915 | 0.705 | Down | Carbonic anhydrase 1 | CA1 | 147.17 |
| P00915 | 0.116 | Down | Carbonic anhydrase 1 | CA1 | 110.34 |
| P68371 | 0.547 | Down | Tubulin beta‐4B chain | TUBB4B | 167.61 |
| P28289 | 0.655 | Down | Tropomodulin‐1 | TMOD1 | 95.822 |
| P69905 | 0.717 | Down | Hemoglobin subunit alpha | HBA1 | 199.81 |
| P69905 | 3.223 | Up | Hemoglobin subunit alpha | HBA1 | 168.55 |
| P11277 | 0.748 | Down | Spectrin beta chain, erythrocytic | SPTB | 108.63 |
| P11277 | 0.47 | Down | Spectrin beta chain, erythrocytic | SPTB | 142.89 |
| P11166 | 0.387 | Down | Solute carrier family 2, facilitated glucose transporter member 1 | SLC2A1 | 136.57 |
| Q8IZP2 | 0.627 | Down | Putative protein FAM10A4 | ST13P4 | 127.36 |
| Q99436 | 0.099 | Down | Proteasome subunit beta type‐7 | PSMB7 | 103.88 |
| Q99436 | 0.17 | Down | Proteasome subunit beta type‐7 | PSMB7 | 103.88 |
Gene Ontology of Differentially Quantified Proteins
To investigate the features of the whole blood differentially expressed proteins upon postmenopausal osteoporosis patients and healthy postmenopausal women, classification of GO annotation was performed. GO is an important bioinformatics analysis method and tool for expressing various properties of genes and gene products. It is divided into three broad categories: biological process, cellular component, and molecular function. In quantified proteins with upregulated ubiquitinated sites, cellular process (14%), single‐organism process (14%), biological regulation process (12%), response to stimulus process (12%), and metabolic process (12%) were leading categories when biological processes were evaluated (Fig. 1A); cell (24%) and organelle (22%)‐related proteins stood out in the cellular component analysis (Fig. 1B); molecular function analysis showed that the top two functions were binding (42%) and catalytic activity (23%, Fig. 1C).
Figure 1.
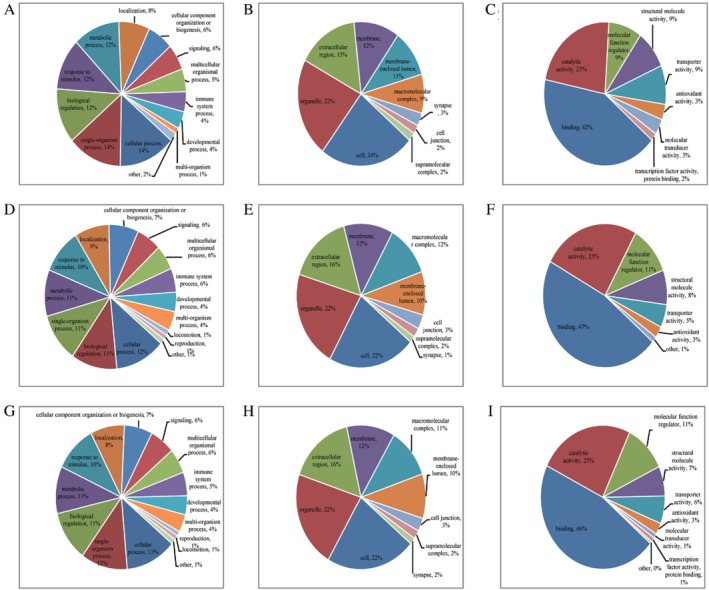
Gene Ontology annotations of quantified proteins with differential sites: (A) biological process annotation of quantified proteins with upregulated ubiquitinated sites; (B) cellular component annotation of quantified proteins with upregulated ubiquitinated sites; (C) molecular function annotation of quantified proteins with upregulated ubiquitinated sites; (D) biological process annotation of quantified proteins with downregulation ubiquitinated sites; (E) cellular component annotation of quantified proteins with downregulation ubiquitinated sites; (F) molecular function annotation of quantified proteins with downregulation ubiquitinated sites; (G) biological process annotation of all quantified proteins; (H) cellular component annotation of all quantified proteins; and (I) molecular function annotation of all quantified proteins.
As for quantified proteins with downregulation ubiquitinated sites, cellular process (12%), biological regulation process (11%), single‐organism process (11%), metabolic process (11%), response to stimulus process (10%) and localization process (9%) were important in biological processes (Fig. 1D); cell (22%) and organelle (22%) related proteins still stood in leadership in the cellular component analysis (Fig. 1E); the top two functions of molecular function were binding (47%) and catalytic activity (25%, Fig. 1F).
To explore the cellular functions of differentially regulated proteins in whole blood from healthy postmenopausal women and postmenopausal osteoporosis patients, functional enrichment was conducted in the GO pathway. In the GO functional clustering analysis of upregulated sites with corresponding proteins, ubiquitin conjugating enzyme activity and ubiquitin‐like protein conjugating enzyme activity were equally highly enriched in molecular function, spectrin‐associated cytoskeleton was the most highly enriched in cellular component, and purine‐containing compound catabolic process was the most enriched in biological process (Fig. 2A). However, the threonine‐type peptidase activity and threonine‐type endopeptidase activity were equally highly enriched in molecular function; blood microparticles were the most highly enriched in cellular component; and negative regulation of multicellular organismal process was the most enriched in biological process in the GO functional clustering analysis of downregulated ubiquitinated sites with corresponding protein (Fig. 2B). Simultaneously, in the GO functional clustering analysis of all differentially expressed proteins, structural molecule activity was the most highly enriched in molecular function; extracellular space was the most highly enriched in cellular component, followed by blood microparticles; regulation of cell adhesion was the most highly enriched in biological process (Fig. 2C).
Figure 2.
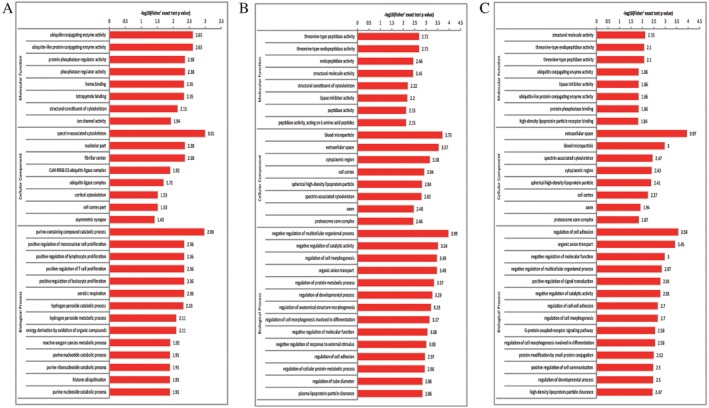
Gene Ontology enrichment results for upregulated ubiquitinated sites with corresponding proteins (A), downregulated ubiquitinated sites with corresponding proteins (B), and all differentially expressed proteins (C), the horizontal axis value is a negative logarithmic transformation of significant P values (P < 0.05).
KEGG Pathway Analysis of Differentially Quantified Proteins
KEGG is an information network that links known intermolecular interactions (http://www.kegg.jp/ or http://www.genome.jp/kegg/), as well as an encyclopedia of genes and genomes. The KEGG pathway mainly includes: metabolism, genetic information processing, environmental information processing, cellular processes, human diseases, drug development, and the like39.
The KEGG pathways of quantified proteins with upregulation ubiquitinated sites included glyoxylate and decarboxylate metabolism (Fig. 3A), ubiquitin mediated proteolysis (Fig. 3B), dopaminergic synapse (Fig. 3C), salivary secretion (Fig. 3D) and Parkinson's disease (Fig. 3E). However, the KEGG pathways of quantified proteins with downregulation ubiquitinated sites were coagulation and complement cascades (Fig. 4A), nitrogen metabolism (Fig. 4B), hippo signaling pathway (Fig. 4C), and the PPAR signaling pathway (Fig. 4D). The KEGG pathways of all quantified proteins were nucleotide excision repair (Fig. 5A), adrenergic signaling in cardiomyocytes (Fig. 5B), hepatitis C (Fig. 5C), hippo signaling pathway (Fig. 5D), coagulation and complement cascades (Fig. 5E), and oocyte meiosis (Fig. 5F).
Figure 3.
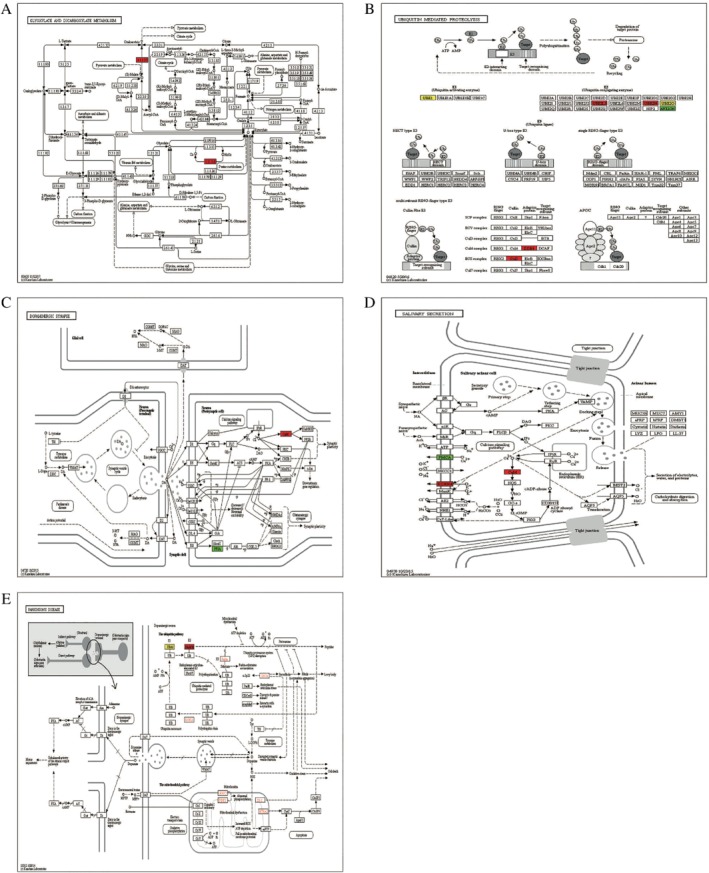
KEGG pathway of quantified proteins with upregulated ubiquitinated sites: (A) glyoxylate and decarboxylate metabolism; (B) ubiquitin mediated proteolysis; (C) dopaminergic synapse; (D) salivary secretion. (E) Parkinson's disease (red indicates the level of the protein is upregulated, bright green indicates the level of the protein is downregulated, and yellow indicates the presence of the node).
Figure 4.
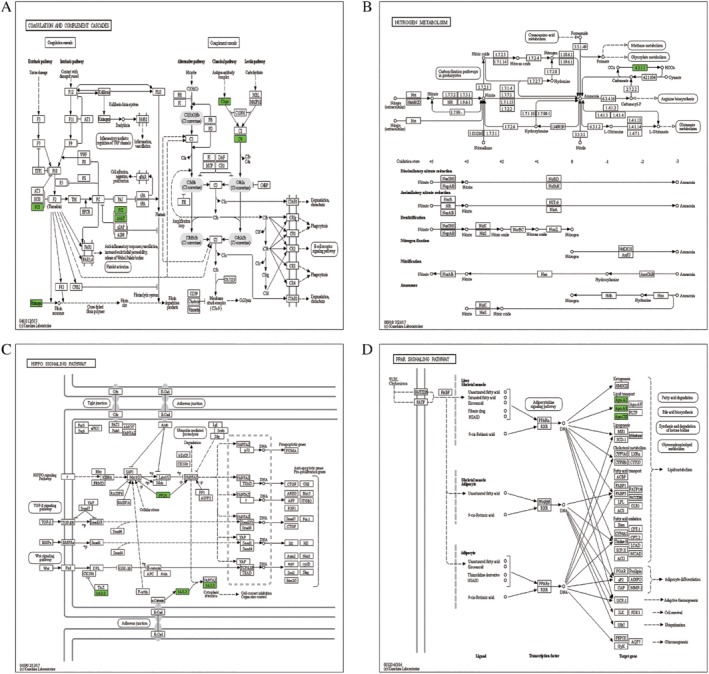
KEGG pathway of quantified proteins with downregulation ubiquitinated sites: (A) coagulation and complement cascades; (B) nitrogen metabolism (C) hippo signaling pathway (D) and the PPAR signaling pathway (green indicates the level of the protein is downregulated).
Figure 5.
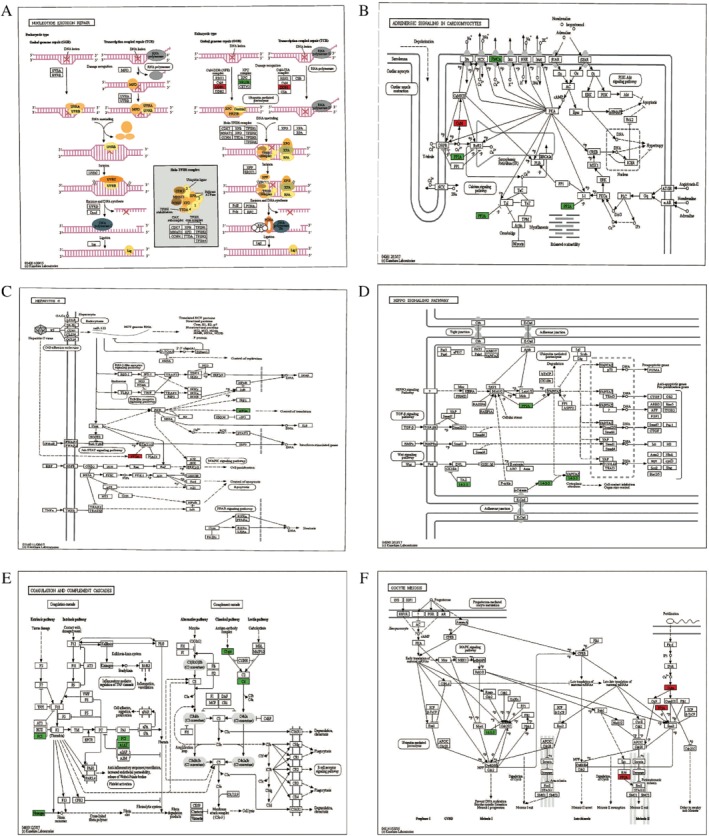
KEGG pathway of all quantified proteins: (A) nucleotide excision repair; (B) adrenergic signaling in cardiomyocytes; (C) hepatitis C; (D) hippo signaling pathway; (E) coagulation and complement cascades; and (F) oocyte meiosis (red indicates the level of the protein is upregulated, bright green indicates the level of the protein is downregulated, and yellow indicates the presence of the node).
In the KEGG functional clustering analysis, hsa04120 ubiquitin‐mediated proteolysis was the most highly enriched in upregulated sites with corresponding proteins (Fig. 6A), hsa04610 complement and coagulation cascades were the most highly enriched in downregulated ubiquitinated sites with corresponding proteins (Fig. 6B), and hsa04114 oocyte meiosis was the most highly enriched in all differentially expressed proteins (Fig. 6C).
Figure 6.
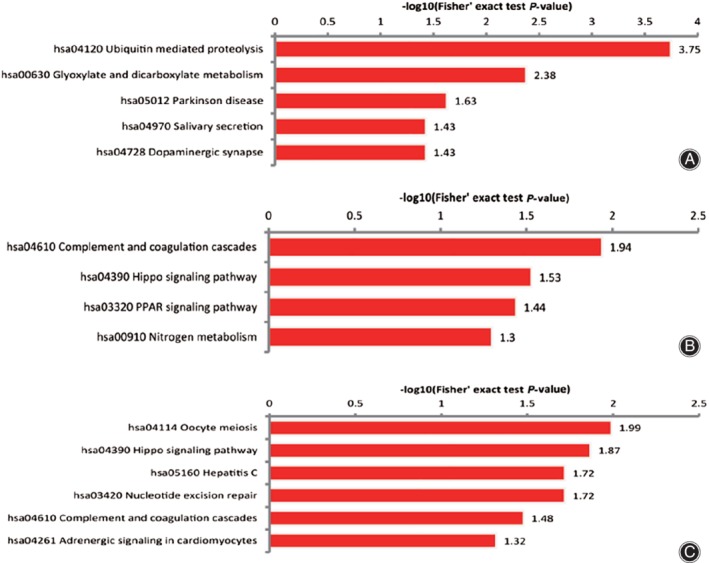
KEGG enrichment results of all quantified proteins: The horizontal axis value is a negative logarithmic transformation of significant P‐values (P < 0.05).
Discussion
Quantitative analysis of ubiquitylomes was conducted in this study. The ubiquitylomes analysis of the whole blood in seven healthy postmenopausal women and seven postmenopausal osteoporosis patients demonstrated that 32 sites on 25 proteins were upregulated and 101 sites on 77 proteins were downregulated. However, increasing the number of samples may make the results more credible.
Gene Ontology Analysis of Differentially Quantified Proteins
In our study, the GO analysis showed that cellular process, single‐organism process, biological regulation process, response to stimulus process, and metabolic process were leading biological process categories in quantified proteins both with upregulated ubiquitinated sites (Fig. 1A) and downregulation ubiquitinated sites (Fig. 1D). The top two functions of molecular function were binding and catalytic activity in quantified proteins with upregulated ubiquitinated sites (Fig. 1C) and downregulated ubiquitinated sites (Fig. 1F).
In contrast, the GO functional clustering analysis revealed that the enrichment results of the same cellular components, molecular function, and biological process vary widely between downregulated sites with corresponding proteins and upregulated sites with corresponding proteins.
Blood microparticles were the most highly enriched in cellular component of downregulated sites with corresponding proteins, and the second most highly enriched in cellular component of all differentially expressed proteins, it was not enriched in cellular component of upregulated sites with corresponding proteins. Ubiquitin conjugating enzyme activity and ubiquitin‐like protein conjugating enzyme activity were the most highly enriched in molecular function of upregulated sites with corresponding proteins, but they were not enriched in downregulated sites with corresponding proteins.
The above results suggest that there was a huge difference in cell biological activity between quantified proteins with upregulated ubiquitinated sites and downregulated ubiquitinated sites in postmenopausal osteoporosis.
KEGG Pathway Analysis of Quantified Proteins
The KEGG pathway analysis of quantified proteins with differentiated ubiquitinated sites revealed 13 kinds of molecular interactions and functional pathways. Among them, glyoxylate and decarboxylate metabolism (Fig. 3A) and dopaminergic synapse (Fig. 3C) have both found in quantified proteins with upregulation ubiquitinated sites and neither found the relationship between them and bone metabolism. On the other hand, ubiquitin‐mediated proteolysis (Fig. 3B), salivary secretion (Fig. 3D), and Parkinson's disease (Fig. 3E) were reported all related to bone metabolism. These results reflect those of Fukushima (2017)40, who found that sustained osteoclast activity is largely due to accumulation of NOTCH2 carrying a truncated C terminus that escapes FBW7‐mediated ubiquitination and degradation. Salivary secretion may contribute to postmenopausal bone health. The most important clinically relevant finding was that MPTP‐induced Parkinsonian features in mice lead to trabecular bone loss through decreased bone formation and increased bone resorption due to changes in the serum circulating factors41, and Parkinson's disease was a risk factor for osteoporosis42.
Moreover, the hippo signaling pathway (Figs 4C and 5D), coagulation, and complement cascades (Figs 4A and 5E) were enriched both in all quantified proteins and the quantified proteins with downregulated ubiquitinated sites. These results indicated they were in significant position of bone metabolism. This also accords with the KEGG functional clustering analysis in downregulated ubiquitinated sites with corresponding proteins, which showed that hsa04610 complement and coagulation cascades are the most highly enriched (Fig. 6B).
However, the hippo pathway has been considered more important in previous studies of postmenopausal osteoporosis. In multicellular organisms, the hippo pathway is an identified signaling that plays an evolutionarily conserved fundamental role in organ size control, cell proliferation, cell apoptosis and fate determination of stem cells42, 43, 44. The hippo‐signaling pathway plays an important role in the osteogenic differentiation of mouse bone marrow mesenchymal stem cells45. Importantly, the hippo‐signaling pathway plays an important role in bone metastasis from breast carcinomar45.
The other molecular interactions and functional pathways of our study may also be involved in the regulation of bone metabolism in postmenopausal osteoporosis. More research is necessary to understand their roles in postmenopausal osteoporosis. This might help us comprehend the pathogenesis of postmenopausal osteoporosis, and the quantified proteins with differential regulation ubiquitinated sites may be potential diagnostic biomarkers in whole blood.
Conclusion
The present study expands our understanding of the spectrum of novel targets that are differentially ubiquitinated in whole blood from healthy postmenopausal women and postmenopausal osteoporosis patients. Overall, these findings will contribute toward our understanding of the underlying proteostasis pathways in postmenopausal osteoporosis and the potential identification of diagnostic biomarkers in whole blood.
Supporting information
Table S1 The differentially expressed ubiquitinated sites and proteins in ubiquitylome of postmenopausal osteoporosis patients and healthy postmenopausal women.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81573754; 81873129) and the “Thirteen Five‐Year” Zhejiang Province Traditional Chinese Medicine (Integrated Chinese and Western Medicine) Key Discipline Construction Plan (No. 2017‐XK‐A16). We thank the staff of the Second Affiliated Hospital of Zhejiang University of Traditional Chinese Medicine for their support throughout these experiments.
Disclosure: The authors declare no conflict of interest.
References
- 1. Cano A, García‐Pérez MÁ. Postmenopausal osteoporosis Menopause: Springer, 2017; 125–140. [Google Scholar]
- 2. Wright NC, Looker AC, Saag KG, et al The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res, 2014, 29: 2520–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tu KN, Lie JD, Wan CKV, et al Osteoporosis: a review of treatment options. P T, 2018, 43: 92. [PMC free article] [PubMed] [Google Scholar]
- 4. Sheng Z, Xu K, Ou Y, et al Relationship of body composition with prevalence of osteoporosis in central south Chinese postmenopausal women. Clin Endocrinol, 2011, 74: 319–324. [DOI] [PubMed] [Google Scholar]
- 5. Hassan YI, Zempleni J. A novel, enigmatic histone modification: biotinylation of histones by holocarboxylasesynthetase. Nutr Rev, 2008, 66: 721–725. [DOI] [PubMed] [Google Scholar]
- 6. Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post‐translational modifications. Epigenetics Chromatin, 2013, 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hein G, Weiss C, Lehmann G, et al Advanced glycation end product modification of bone proteins and bone remodelling: hypothesis and preliminary immunohistochemical findings. Ann Rheum Dis, 2006, 65: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Z, Li C, Zhou Y, et al Advancedglycation end products biphasically modulate bone resorption in osteoclast‐like cells. Am J Physiol Endocrinol Metab, 2016, 310: E355–E366. [DOI] [PubMed] [Google Scholar]
- 9. Milani R, Ferreira CV, Granjeiro JM, et al Phosphoproteome reveals an atlas of protein signaling networks during osteoblast adhesion. J Cell Biochem, 2010, 109: 957–966. [DOI] [PubMed] [Google Scholar]
- 10. Marumoto A, Milani R, da Silva RA, et al Phosphoproteome analysis reveals a critical role for hedgehog signalling in osteoblast morphological transitions. Bone, 2017, 103: 55–63. [DOI] [PubMed] [Google Scholar]
- 11. Bradley EW, Carpio LR, van Wijnen AJ, McGee‐Lawrence ME, Westendorf JJ. Histone deacetylases in bone development and skeletal disorders. Physiol Rev, 2015, 95: 1359–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang YX, Sun HL, Liang H, Li K, Fan QM, Zhao QH. Dynamic and distinct histone modifications of osteogenic genes during osteogenic differentiation. J Biochem, 2015, 158: 445–457. [DOI] [PubMed] [Google Scholar]
- 13. Leestemaker Y, Ovaa H. Tools to investigate the ubiquitin proteasome system. Drug Discov Today Technol, 2017, 26: 25–31. [DOI] [PubMed] [Google Scholar]
- 14. Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem, 2017, 86: 123–128. [DOI] [PubMed] [Google Scholar]
- 15. Rose CM, Isasa M, Ordureau A, et al Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst, 2016, 3: 395–403.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Polge C, Uttenweiler‐Joseph S, Leulmi R, et al Deciphering the ubiquitin proteome: limits and advantages of high throughput global affinity purification‐mass spectrometry approaches. Int J Biochem Cell Biol, 2013, 45: 2136–2146. [DOI] [PubMed] [Google Scholar]
- 17. Kirkin V, Dikic I. Role of ubiquitin‐ and Ubl‐binding proteins in cell signaling. Curr Opin Cell Biol, 2007, 19: 199–205. [DOI] [PubMed] [Google Scholar]
- 18. Bennett EJ, Harper JW. DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol, 2008, 15: 20–22. [DOI] [PubMed] [Google Scholar]
- 19. Nakayama KI, Keiko N. Ubiquitin ligases: cell‐cycle control and cancer. Nat Rev Cancer, 2006, 6: 369–381. [DOI] [PubMed] [Google Scholar]
- 20. Heap RE, Gant MS, Lamoliatte F, Peltier J, Trost M. Mass spectrometry techniques for studying the ubiquitin system. Biochem Soc Trans, 2017, 45: 1137–1148. [DOI] [PubMed] [Google Scholar]
- 21. Vriend J, Reiter RJ. Melatonin, bone regulation and the ubiquitin‐proteasome connection: a review. Life Sci, 2016, 145: 152–160. [DOI] [PubMed] [Google Scholar]
- 22. Zhu W, He X, Hua Y, Li Q, Wang J, Gan X. The E3 ubiquitin ligase WWP2 facilitates RUNX2 protein transactivation in a mono‐ubiquitination manner during osteogenic differentiation. J Biol Chem, 2017, 292: 11178–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fei W, Huiyu Z, Yuxin D, Shiting L, Gang Z, Yinghui T. Calcitonin gene‐related peptide‐induced osteogenic differentiation of mouse bone marrow stromal cells through hippo pathway in vitro. Hua Xi Kou Qiang Yi Xue Za Zhi, 2016, 34: 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y, Shang H, Zhang C, et al The E3 ligase RNF185 negatively regulates osteogenic differentiation by targeting Dvl2 for degradation. Biochem Biophys Res Commun, 2014, 447: 431–436. [DOI] [PubMed] [Google Scholar]
- 25. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab, 2014, 307: E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kondo H, Ezura Y, Nakamoto T, et al MURF1 deficiency suppresses unloading‐induced effects on osteoblasts and osteoclasts to lead to bone loss. J Cell Biochem, 2011, 112: 3525–3530. [DOI] [PubMed] [Google Scholar]
- 27. Bettis T, Kim BJ, Hamrick MW. Impact of muscle atrophy on bone metabolism and bone strength: implications for muscle‐bone crosstalk with aging and disuse. Osteoporos Int, 2018, 29: 1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu Z, Greenblatt MB, Yan G, et al SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat Commun, 2017, 8: 14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin F, Wang Y, Wang X, et al Bre enhances osteoblastic differentiation by promoting the Mdm2‐mediated degradation of p53. Stem Cells, 2017, 35: 1760–1772. [DOI] [PubMed] [Google Scholar]
- 30. Porras‐Yakushi TR, Sonja H. Recent advances in defining the ubiquitylome. Expert Rev Proteomics, 2014, 11: 477–490. [DOI] [PubMed] [Google Scholar]
- 31. Jiang M, Hua Z, Dong Y, Liu Z, Thiele CJ, Li Z. Quantitative ubiquitylome analysis and crosstalk with proteome/acetylome analysis identified novel pathways and targets of perifosine treatment in neuroblastoma. Transl Cancer Res, 2018, 7: 1548–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theurillat JP, Udeshi ND, Errington WJ, et al Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP‐mutant prostate cancer. Science, 2014, 346: 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abreha MH, Dammer EB, Ping L, et al Quantitative analysis of the brain ubiquitylome in Alzheimer's disease. Proteomics, 2018, 18: e1800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group. Osteoporos Int, 1994, 4: 368–381. [DOI] [PubMed] [Google Scholar]
- 35. Shi XL, Li CW, Liang BC, He KH, Li XY. Weak cation magnetic separation technology and MALDI‐TOF‐MS in screening serum protein markers in primary type I osteoporosis. Genet Mol Res, 2015, 14: 15285–15294. [DOI] [PubMed] [Google Scholar]
- 36. Rédei GP . Encyclopedia of Genetics, Genomics, Proteomics, and Informatics. Dordrecht: Springer Netherlands, 2008; 752–753. [Google Scholar]
- 37. Camon E, Magrane M, Barrell D, et al The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res, 2004, 32: D262–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aoki‐Kinoshita KF, Kanehisa M. Gene annotation and pathway mapping in KEGG. Methods Mol Biol, 2007, 396: 71–91. [DOI] [PubMed] [Google Scholar]
- 39. Clemens W, Andreas DG, Andreas Z. KEGG translator: visualizing and converting the KEGG PATHWAY database to various formats. Bioinformatics, 2011, 27: 2314–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukushima H, Shimizu K, Watahiki A, et al NOTCH2 Hajdu‐Cheney mutations escape SCFFBW7‐dependent proteolysis to promote osteoporosis. Mol Cell, 2017, 68: 645–658.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ali SJ, Ellur G, Khan MT, Sharan K. Bone loss in MPTP mouse model of Parkinson's disease is triggered by decreased osteoblastogenesis and increased osteoclastogenesis. Toxicol Appl Pharmacol, 2019, 363: 154–163. [DOI] [PubMed] [Google Scholar]
- 42. Malochet‐Guinamand S, Durif F, Thomas T. Parkinson's disease: a risk factor for osteoporosis. Joint Bone Spine, 2015, 82: 406–410. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Yu W, Zhou B. Hippo signaling pathway in cardiovascular development and diseases. Hereditas, 2017, 39: 576. [DOI] [PubMed] [Google Scholar]
- 44. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell, 2005, 122: 421–434. [DOI] [PubMed] [Google Scholar]
- 45. Pan D. The hippo signaling pathway in development and cancer. Dev Cell, 2010, 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The differentially expressed ubiquitinated sites and proteins in ubiquitylome of postmenopausal osteoporosis patients and healthy postmenopausal women.