

Abstract
The expression and function of long noncoding RNAs (lncRNAs) in the development of hypoxic pulmonary hypertension (HPH), especially in the proliferation of pulmonary artery smooth muscle cells (PASMCs), are largely unknown. Herein, we examined the expression and role of lncRNA-maternally expressed gene 3 (lncRNA-MEG3) in HPH. lncRNA-MEG3 was significantly increased and primarily localized in the cytoplasm of hypoxic PASMCs. lncRNA-MEG3 knockdown by lung-specific delivery of small interfering RNAs (siRNAs) significantly inhibited the development of HPH in vivo. Silencing of lncRNA-MEG3 by siRNAs and gapmers attenuated proliferation and cell-cycle progression in both PASMCs from idiopathic pulmonary arterial hypertension (iPAH) patients (iPAH-PASMCs) and hypoxia-exposed PASMCs in vitro. Mechanistically, we found that lncRNA-MEG3 interacts with and leads to the degradation of microRNA-328-3p (miR-328-3p), leading to upregulation of insulin-like growth factor 1 receptor (IGF1R). Additionally, higher expression of lncRNA-MEG3 and IGF1R and lower expression of miR-328-3p were observed in iPAH-PASMCs and relevant HPH models. These data provide insights into the contribution of lncRNA-MEG3 to HPH. Upregulation of lncRNA-MEG3 sequesters cytoplasmic miR-328-3p, eventually leading to expression of IGF1R, revealing a regulatory mechanism by lncRNAs in hypoxia-induced PASMC proliferation.
Keywords: noncoding RNA, maternally expressed gene 3, hypoxic pulmonary hypertension, proliferation, microRNA-328, insulin-like growth factor 1 receptor
Graphical Abstract
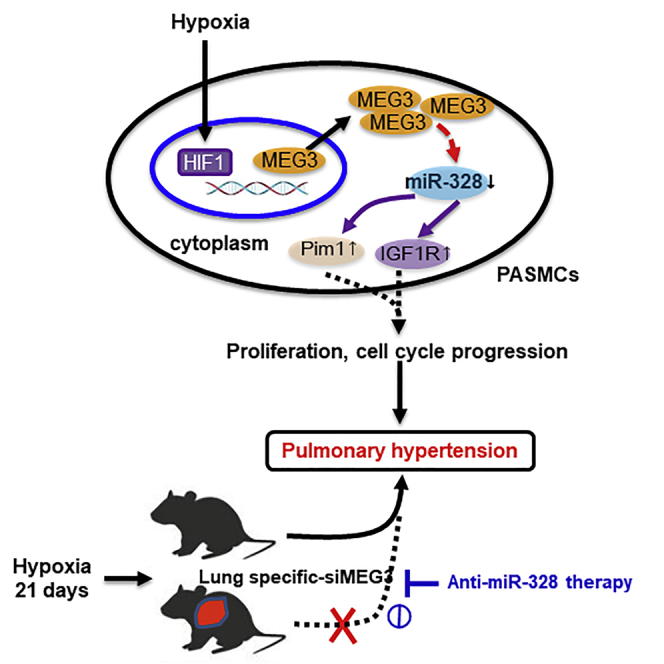
Increasing evidence indicates that lncRNAs are implicated in pulmonary hypertension. Xing et al. demonstrate that lncRNA-MEG3 contributes to PASMCs proliferation and cell-cycle progression through interacting with and facilitating miR-328-3p degradation. Lung-specific lncRNA-MEG3 knockdown rescues hypoxia-induced pulmonary hypertension, which is prevented by anti-miR-328-3p therapy.
Introduction
Pulmonary hypertension (PH) is a severe disease resulting in right heart failure and even death.1 Hypoxia is a common cause of PH in multiple diseases, such as chronic obstructive pulmonary disease, sleep apnea, and high-altitude pulmonary vascular disease, which primarily includes hypoxic pulmonary hypertension (HPH).2, 3 HPH is characterized by elevated pulmonary arterial pressure and hypoxia-induced pulmonary arterial remodeling (HPVR). Although many factors have been implicated in the susceptibility to HPVR, it is well-accepted that HPVR is primarily caused by excessive proliferation of pulmonary artery smooth muscle cells (PASMCs).4 However, the precise mechanism regulating the pathological process of HPVR is largely unknown.2
The roles of noncoding RNAs in HPH have been evaluated in many studies. MicroRNAs (miRNAs) are short noncoding RNAs associated with PH onset, progression, and treatment responsiveness.5 Previous studies from our lab and others have shown that microRNA-328-3p (miR-328-3p) is critical in the vascular remodeling and development of HPH through targeting the expression of insulin-like growth factor 1 receptor (IGF1R), L-type calcium channel-alpha 1C,6 and PIM1.7 However, specific regulation of miR-328-3p expression in PASMCs is not understood. Long noncoding RNAs (lncRNAs) are a class of noncoding RNAs longer than 200 nucleotides that function in diverse biological processes.8, 9, 10, 11, 12 Importantly, lncRNAs interact with miRNAs to modulate cell differentiation, proliferation, and death.13 In particular, the “competing endogenous RNA (ceRNA) hypothesis” has been suggested, which states that many lncRNAs act as ceRNAs by sequestrating target miRNAs; abundant lncRNAs harboring similar miRNA target sequences can sequester cytoplasmic miRNAs, indirectly regulating miRNA-targeted genes.14, 15, 16 lncRNAs have been shown to maintain cellular homeostasis and enable adaptive survival in hypoxia-associated cancer processes.8, 17, 18, 19 However, the specific roles of lncRNAs in HPH remain poorly understood.
One lncRNA involved in the response to hypoxia is maternally expressed gene 3 (MEG3), which is increased during exposure to hypoxic conditions in vascular endothelial cells.20 Elevated expression of lncRNA-MEG3 has also been detected in developing otocyst cells and differentiated neural cells.21 Numerous studies have demonstrated that lncRNA-MEG3 is a tumor suppressor.22 lncRNA-MEG3 expression is lost in multiple cancer cell lines of various tissue origins, and its overexpression inhibits tumor cell proliferation and cell-cycle progression.23, 24, 25 These controversial observations suggest a role for lncRNA-MEG3 in cellular homeostasis and adaptive survival under hypoxic stress conditions, depending on the tissue and/or cell type. Specifically, using a computational prediction approach, we predicted that lncRNA-MEG3 harbors a miR-328-3p target sequence (nucleotides [nt] 2071–2094). Thus, lncRNA-MEG may be involved in the development of HPH by interacting with miR-328-3p.
In the present study, we examined the expression of lncRNA-MEG3 in pulmonary arteries (PAs) in a hypoxia-induced PH animal model and in PASMCs. Upon hypoxia, upregulation of lncRNA-MEG3 binding to and sequestering miR-328-3p occurred, leading to the expression of insulin-like growth factor 1 receptor (IGF1R) and excessive proliferation of PASMCs. Accordingly, lncRNA-MEG3 is highly expressed in PASMCs of idiopathic pulmonary arterial hypertension (iPAH) patients, suggesting lncRNA-MEG3 as a new therapeutic target for pharmaceutical intervention of PH.
Results
Hypoxia Increases lncRNA-MEG3 Expression
To determine the expression patterns of lncRNA-MEG3 in the development of HPH, we exposed male mice to hypoxia (10% fractional inspired oxygen [FiO2]) for 21 days. Right ventricular systolic pressure (RVSP) and right ventricular to left ventricular + spatial weight ratio (RV/LV+S), two indirect indicators of pulmonary hypertension (Figures S1A and S1B), were significantly higher in the hypoxic group than in normal controls. Moreover, H&E staining (Figure S1C) demonstrated that the morphology of pulmonary vascular remodeling, the thickness of vessel wall to vessel diameter ratio, was significantly higher in hypoxia models. Therefore, the murine model of chronic HPH was validated in our study.
Next, we assessed lncRNA-MEG3 expression in isolated pulmonary arteries (PAs) from HPH mice using real-time PCR. The results indicated that lncRNA-MEG3 was potently induced by hypoxia in a time-dependent manner (Figure 1A). Fluorescence in situ hybridization (FISH) analysis showing increased expression of lncRNA-MEG3 (red) and colocalization with alpha-smooth muscle actin (αSMA) (green) in remodeling distal pulmonary arteries from the 21-day hypoxia mouse model (Figure 1B), indicating that lncRNA-MEG3 was primarily localized in smooth muscle layer. Moreover, lncRNA-MEG3 (red) was upregulated by hypoxia in a time-dependent manner, with significant induction at 4 days and maximal induction at 8–12 days. Increased expression of αSMA was also observed, while probes targeting the SUC2 gene, which served as a negative control (NC), and nonprobe controls (PBS) did not generate any signals (Figures S2A and S2B). We also detected lncRNA-MEG3 expression in other tissues (heart, lung, spleen, liver, and kidney), as well as in isolated aorta and carotid arteries. Results showed that, with the exception of PAs, lncRNA-MEG3 was increased compared to normoxia, and all other tissues displayed decreased or unchanged expression (Figures S2C and S2D). Next, we further examined lncRNA-MEG3 expression in PASMCs from iPAH patients (iPAH-PASMCs). Clinical information of the iPAH patients is shown in Table S1. The use of these iPAH-PASMCs followed recommendations by Bonnet et al.26 Real-time PCR consistently showed that the expression of lncRNA-MEG3 was higher in iPAH-PASMCs than in control PASMCs (Figure 1C, left panel). To further confirm the effect of hypoxia on lncRNA-MEG3 expression, we exposed two cell lines—human PASMCs (hPASMCs) (Figure 1C, middle panel) and mouse PASMCs (mPASMCs) (Figure 1C, right panel)—to 3% FiO2 to induce hypoxia and confirmed via real-time PCR that hypoxia augmented lncRNA-MEG3 expression in both cell lines in a time-dependent manner.
Figure 1.

Effects of Hypoxia on lncRNA-MEG3 Expression
(A) Mice were exposed to hypoxia (10% FiO2) for indicated times, and lncRNA-MEG3 expression was detected by real-time PCR in isolated PAs (n = 6). (B) FISH analysis of lncRNA-MEG3 (red) and colocalization with αSMA (green) in PAs from 21-day hypoxia-exposed mice. (C) Expression of lncRNA-MEG3 was detected in PASMCs of iPAH patients and in PASMCs from human and mouse (n = 6). (D) FISH showing the distribution lncRNA-MEG3 (red, CY3 staining) in the nucleus (blue, stained with 4,6-diamidino-2-phenylindole) and cytoplasm. (E) Quantification of FISH fluorescence signals of lncRNA-MEG3 in the nuclear and cytoplasmic fractions. U6 and 18S RNA were used as controls for localization of the nucleus and cytoplasm, respectively. The graph represents the means ± SEM from >50 cells from three independent experiments. (F) Isolation of cytoplasmic and nuclear fractions followed by real-time PCR (n = 4). Data represent means ± SEM from indicated independent experiments. Student’s t test (for two means) or one-way ANOVA followed by Dunnett’s test (for >2 means). *p < 0.05, **p < 0.01.
To examine the subcellular localization of lncRNA-MEG3, we evaluated lncRNA-MEG3 expression in cytoplasmic versus nuclear fractions using lncRNA FISH probes (red) in cultured mPASMCs. As a control, probes targeting U6 or 18S RNA were used as markers of the nucleus or cytoplasm, respectively. lncRNA-MEG3 was detected in both the cytoplasm and nucleus of PASMCs under normoxia conditions. After cells were exposed to hypoxia (3% FiO2) for 24 h, lncRNA-MEG3 was still highly enriched in the cytoplasm but was decreased in the nucleus of mPASMCs (Figure 1D). lncRNA-MEG3 distribution in the cytoplasm increased from 69.46% ± 2.47% to 86.5% ± 2.55% yet decreased from 30.54% ± 2.47% to 13.5% ± 2.55% in the nucleus (Figure 1E). Localization of U6 and 18S RNA were not significantly changed by hypoxic conditions (Figure 1E). Moreover, by isolating cytoplasmic and nuclear RNA for real-time PCR, we confirmed that lncRNA-MEG3 expression was primarily increased in the cytoplasm but decreased in the nucleus (Figure 1F). These results indicate that lncRNA-MEG3 translocates from the nucleus to the cytoplasm during hypoxia exposure.
Because hypoxia-inducible factor (HIF) transcription factors play an important role in the hypoxia response, we examined whether lncRNA-MEG3 expression is regulated by HIF. HIF isoforms were knocked down by siRNA targeting HIF1α, HIF1β, HIF2α, and HIF2β or were overexpressed by plasmids (represent images shown in Figure S3A). Chromatin immunoprecipitation experiments were performed, and we found that HIF1α, HIF1β, and HIF2α were able to bind to the promoter region of lncRNA-MEG3 (Figure S3B). We found that increased expression of lncRNA-MEG3 in response to hypoxia was markedly attenuated by siHIF1α and siHIF1β but not by siHIF2α or siHIF2β (Figure S3C). As a positive control, cobalt chloride (CoCl2, a known activator of HIF) evoked significant expression of lncRNA-MEG3, which was attenuated by siHIF1α and alleviated by siHIF1β (Figure S3D). Consistently, lncRNA-MEG3 was potently induced by HIF1α overexpression and moderately upregulated by HIF1β overexpression (Figure S3E). We also examined the role of histone acetylation in regulating lncRNA-MEG3, and results showed that hypoxia-induced lncRNA-MEG3 expression was prevented by C646 (histone acetyltransferase inhibitor) (Figure S3F). Altogether, our results revealed that lncRNA-MEG3 expression is induced by hypoxia in a HIF1-dependent manner (primarily by HIF1α).
lncRNA-MEG3 Knockdown Prevents Increased Proliferation and Reduced Apoptosis Induced by Hypoxia
To determine whether lncRNA-MEG3 is required for increasing levels of proliferation and reducing levels of apoptosis in response to hypoxia, we inhibited pulmonary vessel lncRNA-MEG3 using a lung-specific delivery system (R8 peptide conjugated PEG2000-lipid [R8-Lip] that modified siRNA targets to MEG3 [R8-Lip-siMEG3]) in vivo. R8-Lip-siMEG3 was injected into the tail vein under hypoxic conditions on days 1, 4, 7, and 14, and siRNA targeted to MEG3 without any modification (siMEG3) and R8-Lip were used as controls. R8-Lip targeting to the pulmonary smooth muscle layer was confirmed by double staining of labeled siMEG3 with αSMA but was not observed between labeled siMEG3 with CD31 (Figure S4A). The cytotoxicity of R8-Lip-siMEG3 was evaluated by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay (Figure S4B), which revealed that R8-Lip-siMEG3 was not cytotoxic to PASMCs over 24 h. R8-Lip-siMEG3 significantly and efficiently suppressed lncRNA-MEG3 expression (red), which was confirmed by tissue FISH (Figures 2A and 2B), while siMEG3 and R8-Lip did not affect lncRNA-MEG3 expression under hypoxic conditions. FISH probes targeting αSMA mRNA (green) were used as positive controls, while probes targeting the SUC2 gene served as NCs and nonprobe control (PBS), which did not generate any signals. The inhibitory effect of R8-Lip-siMEG3 on lncRNA-MEG3 expression under hypoxic conditions was further confirmed by real-time PCR (Figure 2C) in isolated PAs from the HPH mouse model. R8-Lip-siMEG3 inhibited the elevation of proliferating cell nuclear antigen (PCNA) expression (Figure 2D). We observed decreased caspase-3 and caspase-9 expression in hypoxic mice as well, and in vivo silencing of MEG3 using R8-Lip-siMEG3 attenuated this decreasing expression. However, neither siMEG3 without modification nor R8-Lip control prevented the decreased apoptosis under hypoxia conditions (Figure 2E). These findings confirm that increased lncRNA-MEG3 leads to hypoxia-induced PASMCs proliferation and reduces apoptosis in vivo.
Figure 2.

Pulmonary-Specific lncRNA-MEG3 Knockdown Prevented Hypoxia-Induced Pulmonary Hypertension
(A) Efficiency of R8-liposome-loaded siRNA targeting to lncRNA-MEG3 (R8-Lip-siMEG3) on knockdown of lncRNA-MEG3 expression was confirmed by FISH. FISH probes targeting αSMA mRNA were used as positive controls, probes targeting to SUC2 gene were used as NCs, and nonprobe PBS was also used as a control. (B) Bar graph indicating lncRNA-MEG3-positive staining by FISH. (C) Real-time PCR revealed lncRNA-MEG3 in isolated PAs. (D) PCNA expression in PAs detected by western blot. (E) Caspase-3 and caspase-9 expression was detected in isolated PAs. Data represent means ± SEM from six independent experiments. One-way ANOVA followed by Dunnett’s test was used. *p < 0.05, **p < 0.01.
Next, we further explored the function of lncRNA-MEG3 by knocking down its expression with siMEG3 or gapmers in cultured mPASMCs. Two different siRNAs (siMEG-936 and siMEG3-1417) and gapmers targeting two different sequences were designed to knock down lncRNA-MEG3 expression. All siRNAs and gapmers efficiently inhibited lncRNA-MEG3 expression in a dose-dependent manner (Figure S5A). Because no difference was observed in the knockdown of lncRNA-MEG3 expression between the two siRNAs, siMEG-936 was used for further in vivo and in vitro studies. siMEG3 treatment reduced lncRNA-MEG3 levels compared to scramble control RNAs (NC). Hypoxia increased cell viability in PASMCs, and silencing of lncRNA-MEG3 attenuated the effects of hypoxia compared to NCs (Figure 3A, left panel). Indeed, in the analysis of PASMC proliferation, hypoxia markedly enhanced bromodeoxyuridine (BrdU) incorporation (Figure 3A, right panel), PCNA expression (Figure 3B), and Ki67 staining (Figure 3C). In the presence of siMEG3, these effects were reduced nearly to baseline levels.
Figure 3.

MEG3 Is Involved in Hypoxia-Induced PASMC Proliferation, Cell-Cycle Progression, and Cellular Migration
(A) Cell viability determined by MTT (left panel) and BrdU incorporation (right panel) of PASMCs transfected with siRNA targeting lncRNA-MEG3 (siMEG3) or NC nucleotide, followed by exposure to hypoxia for 24 h. (B) PCNA expression determined by western blot, and (C) Ki67 staining performed by immunofluorescence. (D) Flow cytometry analyzed cell-cycle progression. (E) Cyclins and CDKs and (F) caspase-3 and caspase-9 expression was detected by western blot. (G) Cellular migration experiment. PCNA represents proliferating cell nuclear antigen. CDK represents cyclin-dependent kinases. Data represent means ± SEM from at least six independent experiments. One-way ANOVA followed by Dunnett’s test was used. *p < 0.05, **p < 0.01.
Next, we investigated the function of lncRNA-MEG3 in cell-cycle progression. As expected, hypoxia promoted S+G2/M phase, whereas lncRNA-MEG3 knockdown attenuated the effects of hypoxia (Figure 3D). Furthermore, knockdown of lncRNA-MEG3 prevented the increased expression of cyclin A, B, and D (Figure 3E) and coordinated the activation of important cell-cycle proteins, including cyclin-dependent kinases (CDK1, CDK2, and CDK4) (Figure 3E), which were induced by hypoxia. Moreover, we assessed the role of lncRNA-MEG3 in apoptosis. As indicated by the expression of caspase-3 and caspase-9 (Figure 3F), lncRNA-MEG3 knockdown increased cellular apoptosis under hypoxia conditions. Furthermore, knockdown of lncRNA-MEG3 by siRNA inhibited hypoxia-induced cell migration (Figure 3G). Gapmers of lncRNA-MEGs displayed similar effects in preventing cell proliferation, cell-cycle progression, and cellular migration induced by hypoxia (Figures S5B–S5F).
It is well recognized that iPAH-PASMCs exhibit a “cancer-like” hyperproliferative and apoptosis-resistant phenotype when cultured in vitro. We examined the role of lncRNA-MEG3 knockdown in cultured iPAH-PASMCs. We observed reduced cell viability, PCNA expression, and cyclin expression and increased expression of caspases, as well as resistance to migration after MEG3 knockdown in iPAH-PASMCs (Figure S6). These data suggest that lncRNA-MEG3 knockdown prevents hyperproliferation and reduced apoptosis phenotype of iPAH-PASMCs.
To confirm the function of lncRNA-MEG3 in hypoxia-induced proliferation and cell-cycle progression in PASMCs, we overexpressed one segment of lncRNA-MEG3 (mouse transcript varies 1, nt 1563–3207, 1644 bp, Figure 5A) using a lentiviral vector. As shown in Figure 4, overexpression of lncRNA-MEG3 increased cell proliferation (as measured by cell viability, BrdU incorporation, PCNA expression, and Ki67 staining), cell-cycle progression (as measured by flow cytometry, cyclins, and CDKs expression), and cell migration in the absence of hypoxia. Taken together, these results suggest that lncRNA-MEG3 is involved in hypoxia-induced PASMC proliferation, cell-cycle progression, cell migration, and reduced apoptosis in PASMCs.
Figure 5.

lncRNA-MEG3 Binds to and Decreases the Expression of miR-328-3p
(A) Diagram showing the genomic structure of the relative oligos, miR-328-3p binding sequences, mutation sequence used for luciferase experiments, PCR product for MST, and overexpression segment (left panel). miR-328-3p expression detected by real-time PCR in mPASMCs transiently transfected with 30 nM NC siRNA or siRNA against MEG3 (siMEG3), followed by hypoxia for 24 h (right panel). (B) Detection of miR-328-3p expression in PAs, lung, and right ventricular from hypoxia PH mouse. (C) Detection of lncRNA-MEG3 (left panel) and miR-328-3p (right panel) expression in PAs from SUGEN-hypoxia mouse models. (D) Expression of miR-328-3p was detected in iPAH-PASMCs (n = 3). (E) mPASMCs were transiently transfected with the indicated agents, followed by transient transfection with 25 ng of psiCHECK plasmid harboring the lncRNA-MEG3 binding region (nt 2071–2094) (left panel) or mutated sequence (right panel). Luciferase activity was assessed 36 h after transfection and normalized to the activity of Renilla luciferase. (F) MST analysis indicated that miR-328-3p mimics interaction with lncRNA-MEG3 (2030–2193, 163-nt PCR product), IGF1R, and NC nucleotide. (G) RNA immunoprecipitation by biotin-labeled MEG3 segment (nt 2030–2193) was subjected to pulldown, following real time-PCR to detect miR-328-3p expression. Data represent means ± SEM from six (except where indicated) independent experiments. Student’s t test (for two means) or one-way ANOVA followed by Dunnett’s test (for >2 means). *p < 0.05, **p < 0.01.
Figure 4.

Overexpression of MEG3 Induces PASMC Proliferation, Cell-Cycle Progression, and Cellular Migration
(A) One segment of lncRNA-MEG3 (mouse transcript varies 1, nt 1563–3207, 1644 bp) was amplified by real-time PCR, and then inserted into lentiviral vectors. Expression of lncRNA-MEG3 in mPASMCs after transfection with different doses of plasmid. (B) Cell viability determined by MTT. (C) BrdU incorporation. (D) PCNA expression detected by western blot. (E) Cell-cycle progression detected by flow cytometry. (F) Cyclin expression and (G) CDK expression detected by western blot. (F) Cellular migration. Data represent means ± SEM from four independent experiments. One-way ANOVA followed by Dunnett’s test was used. *p < 0.05, **p < 0.01.
Association of lncRNA-MEG3 with miRNA-328-3p
To understand the molecular mechanism by which lncRNA-MEG3 regulates hypoxia-induced PASMC proliferation and cell-cycle progression, we used computational approaches to assess the binding propensity of lncRNA-miRNAs. This analysis identified seven potential lncRNA-MEG3-interacting miRNAs, including miR-21, miR-30c, miR-138, miR-145, miR-328-3p, miR-1906-1, and miR-770 (Figure 5A, left panel; Figure S7A). To confirm the relationship between these miRNAs and lncRNA-MEG3, we assessed the expression of these miRNAs under hypoxic conditions with and without lncRNA-MEG3 knockdown, observing that expression of miR-30c and miR-328-3p were inhibited, whereas those of miR-138, miR-145, and miR-21 were increased (Figure 5A; Figure S7B) by hypoxia. However, among these miRNAs, we focused on miR-328-3p because a previous study indicated that miR-328-3p may play an important role in HPH.6 Results indicated that miR-328-3p is regulated by lncRNA-MEG3, as knockdown of lncRNA-MEG3 reversed the downregulation of miR-328-3p under hypoxic conditions (Figure 5B; Figure S7C). Furthermore, real-time PCR revealed that miR-328-3p was decreased in PAs under hypoxic conditions in a time-dependent manner (Figure S7C), and lung-specific knockdown of lncRNA-MEG3 by R8-Lip-siMEG3 rescued this downregulation, while siMEG3 without modification or R8-Lip control showed no effects (Figure 5C). To determine whether the relationship between lncRNA-MEG3 and miR-328-3p is applicable in other models of PH, we used SUGEN-hypoxia mouse models and iPAH-PASMCs. Results revealed that lncRNA-MEG3 was increased but miR-328-3p was decreased in PAs from SUGEN-hypoxia mouse models (Figure 5D) and in iPAH-PASMCs (Figure 5D). Using a computational approach, we found that nt 2071–2094 of lncRNA-MEG3 may directly bind to miR-328-3p (Figure 5A). To confirm this hypothesis, we cloned the putative miR-328-3p target-binding sequence into a luciferase construct, and results showed that miR-328-3p repressed and AMO-328 (inhibitor nucleotide of miR-328-3p) increased luciferase activity (Figure 5E, left panel). Mutation in the putative miR-328-3p binding site (marked in bold in Figure 5A) decreased the response to miR-328-3p (Figure 5E, right panel). In addition, we used MicroScale thermophoresis (MST) to analyze the interaction between lncRNA-MEG3 and miR-328-3p. The results indicated that miR-328-3p mimics binding with lncRNA-MEG3 (nt 2030–2193, Figure 5A) with a Kd of 7.46 ± 0.166 μM. As controls, a nonbinding mutant miR-328-3p (Kd of 3940 ± 46 μM) and an IGF1R mRNA sequence were used (Kd of 10.24 ± 0.298 μM) (Figure 5F). Furthermore, RNA immunoprecipitation using biotin labeled MEG3 followed by biotin pulldown and miR-328-3p expression was determined by real-time PCR. miR-328-3p expression was detected in the biotin group and input group, but not in the immunoglobulin G (IgG) control group, indicating direct binding between MEG3 and miR-328-3p (Figure 5G, left panel). Moreover, we found binding of MEG3 with miR-328-3p was increased in iPAH-PASMCs (Figure 5G, right panel). These results indicate that lncRNA-MEG3 directly binds to miR-328-3p in a sequence-specific manner and upregulation of lncRNA-MEG3 decreases the expression of miR-328-3p in vivo and in vitro under hypoxia conditions.
lncRNA-MEG3 Functions through Its Association with miR-328-3p
To investigate the interplay of lncRNA-MEG3 and miR-328-3p on hypoxia-induced cell proliferation and cell-cycle progression, we used miR-328-3p mimics or an inhibitor (AMO-328) to examine the role of lncRNA-MEG3 knockdown on hypoxia-induced biological processes. Both increasing the expression of endogenous miR-328-3p by knockdown of lncRNA-MEG3 and applying exogenous miR-328-3p mimics reversed the increased cell viability (Figure 6A, left panel), BrdU incorporation (Figure 6B, right panel), PCNA expression (Figure 6B), and Ki67 staining (Figure 6C), as well as accelerated the cell cycle into the S+G2/M phase (Figure 6D), and increased the expression of cyclins A, B, and D (Figure 6D) and CDK1, CDK2, and CDK4 (Figure 6D) induced by hypoxia. Application of AMO-328 (which disrupts the binding of miR-328-3p with its targets) reversed the effects of lncRNA-MEG3 knockdown and miR-328-3p mimics, whereas application of miR-328-3p mimics during lncRNA-MEG3 knockdown had no additional effects. Our previous study showed that miR-328-3p is involved in HPH by regulating the expression of IGF1R.6 Computational approaches indicated the binding propensity of miR-328-3p with IGF1R mRNA. We also observed that IGF1R mRNA and protein (Figure 6E) expression was upregulated in iPAH-PASMCs. Next, we detected IGF1R expression in mPASMCs. As shown in Figure 6F, the knockdown of lncRNA-MEG3 by siMEG3, application of miR-328-3p mimics, or siMEG3 plus miR-328-3p mimics inhibited the increased expression of IGF1R mRNA and protein levels in response to hypoxic conditions. Disruption of miR-328-3p binding with AMO-328 reversed the inhibitory effects of siMEG3.
Figure 6.

lncRNA-MEG3/miR-328-3p Regulates IGF1R Expression
(A) Mouse PASMCs were transiently transfected with indicated agents, followed by exposure to hypoxia for 24 h. Cell viability determined by MTT (left panel) and BrdU incorporation (right panel). (B) PCNA expression examined by immunoblotting. (C) Ki67 staining performed by immunofluorescence. (D) Cell-cycle progression detected by flow cytometry (left panel), cyclin expression (middle panel), CDK expression (right panel) detected by western blot. (E) Binding propensity between miR-328-3p and IGF1R mRNA (top panel). Real-time PCR (lower left panel) and western blot (lower right panel) revealed expression of IGF1R in iPAH-PASMCs of patients (n = 3). (F) AMO-328 (miR-328-3p-specific 2-O-methyl antisense inhibitory oligoribonucleotide) reversed the effects of lncRNA-MEG3 knockdown on IGF1R expression in cultured mPASMCs. Student’s t test (for two means) or one-way ANOVA followed by Dunnett’s test (for >2 means). *p < 0.05, **p < 0.01.
Moreover, we examined whether anti-miR-328-3p therapy in vivo could reverse the pulmonary vascular remodeling effects of lncRNA-MEG3 knockdown by injecting AMO-328. The data showed that lung-specific lncRNA-MEG3 knockdown by R8-Lip-siMEG3 prevented increases in Ki67 expression (Figure 7A) and decreased RVSP (Figure 7B), RV/LV+S (Figure 7C), cardiac output, mean pulmonary artery pressure (mPAP), and total pulmonary vascular resistance (PVR) (Figure 7D). Fibrosis and increasing percentage of medial wall thickness to the external diameter accessed by Masson trichrome staining (Figure 7E) was induced by hypoxia, whereas siMEG3 and R8-Lip did not show significant inhibitory effects. Furthermore, upregulation of IGF1R (Figure 7F; Figure S8A) and PIM1 (Figure S9) in PAs from HPH mouse models were prevented by R8-Lip-siMEG3, which was rescued by AMO-328. Right ventricular function was examined in our in vivo study, and hypoxia induced IG1R mRNA and protein expression (Figure S8B), cardiomyocyte hypertrophy (Figure S8C), collagen deposition (Figure S8D), and fibrosis (Figure S8E), reduced apoptosis (Figure S8F), and increased microvessel density (Figure S8G), which was expressed as a proportion of CD31+ cells measured in the whole section, as suggested by Potus et al.27 All of these effects were significantly attenuated by lung-specific MEG3 knockdown, and anti-miR-328-3p therapy by AMO-328 significantly reversed the effects of R8-Lip-siMEG3.
Figure 7.

lncRNA-MEG3/miR-328-3p Regulates Hypoxia-Induced Pulmonary Artery Hypertension
(A) Immunohistochemistry of Ki67 expression. (B) RVSP was measured by right heart catheter. (C) RV/LV+S. (D) Cardiac output (CO, left panel) and mean pulmonary artery pressure (mPAP, middle panel) measured by ultrasound, total pulmonary vascular resistance, which was calculated as the ratio of mean pulmonary arterial pressure to cardiac output (right panel). (E) Masson trichrome staining. (F) IGF1R expression in isolated PAs was detected by real-time PCR and western blot. For histology section data, a minimum of 10 resistance PAs (<100 μm external diameter) were analyzed per animal. Data represent means ± SEM from six (except where indicated) independent experiments. Student’s t test (for two means) or one-way ANOVA followed by Dunnett’s test (for >2 means). *p < 0.05, **p < 0.01.
To assess the beneficial effect of R8-Lip-siMEG3 once PH was established, we exposed mice to hypoxia for 2 weeks to establish PAH, and R8-Lip-siMEG3 was subsequently injected through the tail vein for another 3 weeks. R8-Lip and siMEG3 without modification served as controls. To confirm that lung-specific MEG3 knockdown was dependent on miR-328-3p, we injected anti-miR-328-3p therapy with AMO-328 in the R8-Lip-siMEG3 group. We found that hypoxia-induced PH was reversed by R8-Lip-siMEG3. Moreover, anti-miR-328-3p therapy completely prevented the increase in muscularization of PAs as detected by Masson trichrome staining (Figure S10A) and by measurement of RVSP (Figure S10B), RV/LV+S (Figure S10C), cardiac output (Figure S10D), mPAP (Figure S10E), and total PVR (Figure S10F). These data indicate that lung-specific delivery of R8-Lip-siMEG3 was able to reverse hypoxia-induced PH.
Together, these results demonstrate that lncRNA-MEG3 binds to and sequesters miR-328-3p, eventually increasing downstream target gene IGF1R expression to regulate cell proliferation, cell-cycle progression, cellular migration, and apoptosis during HPH development.
Discussion
In the present study, we identified lncRNA-MEG3 as a key regulator of HPH that interacts with miR-328-3p. We showed that lncRNA-MEG3 was upregulated in response to hypoxia and was involved in hypoxia-induced PASMC proliferation and cell-cycle progression. Mechanistically, lncRNA-MEG3 directly binds to miR-328-3p using nt 2071–2094. Upregulation of lncRNA-MEG3 sequesters miR-328-3p, leading to increased expression of IGF1R and regulating the development of HPH. This finding implicates lncRNAs in HPH and suggests that modulation of lncRNA-MEG3 activity and its downstream targets, such as miR-328-3p, is a novel therapeutic approach for treating this fatal disease.
The most important finding of our study was that lncRNA-MEG3, upregulated by hypoxia, is involved in hypoxia-induced PASMC proliferation. Although the majority of previous studies showed that overexpression of lncRNA-MEG3 suppresses cell proliferation in multiple cancer cell lines,23, 24, 25 our data showed that upregulation of lncRNA-MEG3 is involved in hypoxia-induced PASMC proliferation. Knockdown of lncRNA-MEG3 expression reversed hypoxia-induced RVSP and the RV/LV+S weight ratio. We also showed that silencing lncRNA-MEG3 attenuated hypoxia-induced PASMC proliferation in PAs, which was confirmed by PCNA expression and Ki67 staining. Through knockdown of lncRNA-MEG3 in cultured PASMCs, we further showed that hypoxia-induced PASMC proliferation and cell-cycle progression occurred in a lncRNA-MEG3-dependent manner. Finally, lncRNA-MEG3 was increased in iPAH-PASMCs and in SUGEN-hypoxia models. Together, these results indicate that lncRNA-MEG3 plays an important role in the development of pulmonary hypertension.
Previous studies have indicated that downregulation of lncRNA-MEG3 is correlated with hypoxic microenvironment-related cancer metastasis or recurrence,22, 23, 24, 25 depending on the tissue- and/or cell type.28 Specifically, lncRNA-MEG3 was downregulated in human PASMCs under hypoxic conditions.29, 30 One possible reason for these distinct results compared with our observations could be due to the different genetic background of PASMCs and culture system (i.e., cultured medium) used in these experiments. Another possibility could be the different transcript variants of MEG3 detected by specifically designed primers, because there are 15 transcript variants (1–10, 12–16) of Homo sapiens MEG3. We using primers designed based on the conserved region of all the 15 transcript variants in human PASMCs. Northern blotting can be used to clarify the specific transcript variants of MEG3 underlying these opposing results. Furthermore, using an HPH mouse model and in vitro cultured cells, we observed that lncRNA-MEG3 expression was specifically increased by hypoxia in PASMCs, while downregulation of lncRNA-MEG3 expression in all other tissues was detected, even in lung tissue (Figure S2). We think this could be due to the tissue- and/or cell-specific expression pattern of MEG3. For example, even in endothelial cells, MEG3 was significantly lower in arterial compared to venous or microvascular endothelial cells. Similarly, another lncRNA, MALAT1, is highly expressed in the lung microvasculature compared to the cardiac microvasculature. Our results differed from reports of other diseases, suggesting that the expression of lncRNA-MEG3 induced by hypoxia is specifically elevated in PASMCs and that upregulation of lncRNA-MEG3 contributes to PASMC proliferation and cell-cycle progression. In addition to tissue-specific distribution, our study also showed that lncRNA-MEG3 was primarily localized to the cytoplasm in PASMCs. These results are consistent with those of a previous study showing that lncRNA-MEG3 retains its cytoplasmic subcellular localization in otocyst cells undergoing proliferation.21 These results suggest that the effect of lncRNA-MEG3 on PASMC proliferation is not related to the cellular localization of lncRNA-MEG3 in pulmonary hypertension but rather is dependent on the expression of lncRNA-MEG3. Based on our experimental results, lncRNA-MEG3 was only increased by hypoxia in PASMCs, and this alternative mechanism of PASMC proliferation by lncRNA-MEG3 greatly differs from those in other diseases, even in hypoxic microenvironment-related diseases. The specific effect of lncRNA-MEG3 on PASMCs may depend on lung vascular responses to hypoxia. Further studies are needed to evaluate these alternative effects among PASMCs and other tumor cell lines.
We observed an increase in lncRNA-MEG3 expression in hypoxic PAs and PASMCs. The precise factors regulating lncRNA-MEG3 expression in response to hypoxia remain unclear. A previous study indicated that the regulation of lncRNA-MEG3 by miR-29a was methylation dependent.23 Cyclic AMP stimulates MEG3 gene expression in cells through a cyclic AMP-response element site in the promoter region,31 suggesting that an alternative regulatory pathway functions in PASMCs during hypoxia. By analyzing the promoter of lncRNA-MEG3, we found there are multiple binding sites for transcript factors, such as NFAT2, STAT3, and HIF. Our data indicated that lncRNA-MEG3 was induced by hypoxia in a HIF1-dependent manner (primarily HIF1α) (Figure S3). The role of HIF in oxygen-dependent regulation of lncRNA-MEG3 in vivo remains to be elucidated. LncRNA-MEG3 expression was only induced in PAs but not in other organs, which may be a lung-system-specific response to hypoxia. For example, the pulmonary artery contracts while systemic vasculature dilates in response to hypoxia. Indeed, whether other transcription factors and epigenetic regulatory factors, such as DNA methylation or histone acetylation, are involved in manipulating lncRNA-MEG3 expression during hypoxic conditions in PASMCs requires further analysis. Moreover, lncRNA-MEG3 is a sexually dimorphic gene, showing significant female bias with expression 1.36-fold higher expression in the female cortex and hippocampus than in males.32 Whether the response of lncRNA-MEG3 to hypoxia varies between females and males, and such sex-dependent expression in HPH, cannot be excluded and therefore will be pursued in future studies.
Previous studies by our lab and others have demonstrated that miR-328-3p is downregulated in PAs from experimental animals under hypoxic conditions and in PAH patients and overexpressing miR-328 remarkably attenuated hypoxia-increased RVSP and PA wall thickness.6, 7 These results suggest a critical role for miR-328 in the development of pulmonary hypertension. The mechanism of miR-328 might be through regulating the expression of IGF1R, L-type calcium channel-alpha 1C, and PIM1. We assessed the expression of IGF1R both in in vivo and in vitro. The increased expression of IGF1R in pulmonary vessels and right ventricular induced by hypoxia was inhibited by lncRNA-MEG3 knockdown, and anti-miR-328 therapy restored IGF1R expression. Moreover, alteration of IGF1R, cardiomyocyte hypertrophy, collagen deposition, fibrosis, reduced apoptosis, and increasing microvessel density (Figure S8) occurred simultaneously. These results, combined with others,33 indicate the important role of IGF1R in pulmonary remodeling and right ventricular failure in HPH. PIM1 expression was also detected in pulmonary vessels, and results suggested regulatory effects of lncRNA-MEG3/miR-328-3p on PIM1 expression. Because PIM1 is critical for vascular remodeling,34, 35 we consider that lncRNA-MEG3/miR-328-3p may modulate multiple targets during the development of HPH.
The interaction between lncRNAs and miRNAs has been shown to play important roles in diverse biological processes. With respect to the clinical utility of miRNAs in the diagnostic and therapeutic aspects of PAH, the objective of the present study was to provide greater insight into the pervasive roles of lncRNAs in the regulation of miR-328-3p expression. Notably, we showed that lncRNA-MEG3 uses nt 2071–2094 to bind miR-328-3p, leading to downregulation of miR-328-3p. This was confirmed by the following findings: first, hypoxia inhibited miR-328-3p expression, which was attenuated by lncRNA-MEG3 knockdown; second, the luciferase reporter assay indicated that lncRNA-MEG3 binds to miR-328-3p in a sequence-specific manner; third, MST results confirmed the interaction of lncRNA-MEG3 with miR-328-3p; fourth, knockdown of lncRNA-MEG3 inhibited the expression of IGF1R and PIM1 (miR-328-3p-regulated genes); and finally, increased lncRNA-MEG3, decreased miR-328-3p, and increased IGF1R were observed in iPAH-PASMCs. Although we excluded four other miRNAs displaying high binding propensity with lncRNA-MEG3, miR-1906-1 and miR-770 were also regulated by lncRNA-MEG3, and it would be interesting to determine whether miR-1906-1, miR-770, and/or other miRNAs or proteins plays important roles in the proliferation and migration of PASMCs. Specifically, why such an interaction would lead to degradation of miR-328-3p is still unknown. One possible reason may be cytoplasmic retention or degradation by RNase H. lncRNA-MEG3 is known to affect the p53 and transforming growth factor β pathways,36 so it is important to determine whether this association is involved in PASMC proliferation under hypoxic conditions. MEG3−/− mice would help to confirm the role of MEG3 in the development of HPH.
The delivery system of R8-modified liposomes has shown potential as a pulmonary drug delivery system for PAH treatment.37 We modified this lung-specific delivery system using siRNA-loaded liposomes (R8-Lip-siMEG3) and found that the biodistribution of R8-Lip in the lungs reached high levels within 1 h, which were maintained for 24 h. Thus, we repeated this application of R8-Lip-siMEG3 during hypoxia treatment. Real-time PCR and FISH confirmed the knockdown efficiency of this approach, whereas nonmodified siRNA and R8-Lip showed no significant effects (Figures 2A and 2B). Moreover, RVSP and RV/LV+S indirectly indicated the efficiency of R8-Lip-siMEG3 in preventing increased pulmonary artery pressure (Figures 2C and 2D). Therefore, using R8-Lip-siMEG3 for knockdown of lung lncRNA-MEG3 expression may provide a foundation for designing new mechanism-based therapies for HPH.
In summary, the present study revealed the involvement of lncRNA-MEG3 in HPH, which functions as a molecular sponge for miR-328-3p. Through additional studies of how lncRNAs function in various diseases,38 lncRNAs may become new pharmaceutical targets and treatment targets for hypoxia-induced pulmonary hypertension.
Materials and Methods
Hemodynamic assessment, histological and morphometric analyses, cell culture, RNA isolation and real-time PCR, protein preparation and western blot analysis, immunofluorescence assay, cell-cycle progression analysis, cell viability assay, and BrdU assay are described in the Supplemental Information.
Animal Model of Pulmonary Hypertension and Tissue Preparation
Adult male C57BL/6 mice (mean weight 30 g) were obtained from the Experimental Animal Center of Harbin Medical University, which is accredited by the Institutional Animal Care and Use Committee. This animal study was approved by the ethics review board of Harbin Medical University ([2012]-006). Study design attempted to follow the guideline for preclinical study of pulmonary hypertension by Provencher et al.,39 including but not limited to two PH models, invasive hemodynamic assessment, image-based data, randomization, and blinded assessment of standardized outcomes. Mice were randomized for exposure for the indicated times to normal and hypoxic environments with FiO2 levels of 0.21 and 0.12, respectively. After the indicated hypoxia exposure period, mice were anesthetized with chloral hydrate (40 mg/kg, intraperitoneally). RVSP and RV/LV+S, two indices of pulmonary artery systolic pressure, were measured to confirm the success of producing the HPH model. Organs were subsequently excised for additional experiments. For SUGEN models, animals received Su5416 (20 mg/kg subcutaneously, dissolved in DMSO; Sigma-Aldrich, St. Louis, MO, USA) immediately followed by hypoxia exposure. After 3 weeks of hypoxia, animals were returned to room air for another 4 weeks.
Fluorescence In Situ Hybridization
To confirm the cellular localization of lncRNA-MEG3 in PASMCs, we performed FISH. The lncRNA-MEG3 FISH Probe Mix was designed and synthesized by RuiBo Biology (#C10910, Guanzhou, China). Experiments were performed according to the manufacturer’s protocol. Briefly, excised organs were embedded in paraffin, and 4-μm cryosections were deparaffinized. Following rehydration in phosphate-buffered saline and acetylation, slides were hybridized at 23°C for 3 h and then incubated with 200 ng custom-designed digoxigenin-labeled lncRNA-MEG3 probes (probe sequences: 5′-GGTCCCTCTCTGGCAACTGTTCATTCATTTGATGC-3′, 5′-CTCAGGCCTGTCGCG TCTTCCTGTGCCATTTGCTG-3′, 5′-TTTACAAATGGACTCTTGTTCCGTCGTTTCCC ACC-3′) in 200 μL hybridization buffer at 60°C overnight. Following sequential washing in 5 × saline sodium citrate (SSC), 0.2 × SSC (20 × SSC contains 3 M NaCl, 0.3 M Na-citrate, pH 7.0), and Tris-buffered saline (TBS), slides were blocked using 10% fetal calf serum in TBS for 2 h followed by incubation with cy3-labeled anti-digoxigenin antibodies (3.75 U/mL; Roche, Basel, Switzerland) in TBS at 4°C overnight. After washing with TBS, bound antibodies were detected by microscopy. Probes targeting αSMA mRNA (5′-CCTTGAAGTACCCGATCGAACATGGCATCA-3′; 5′-CTGACTACCTCAT GAAGATCCTGACTGAGC-3′; 5′-GGAATCTGCTGGCATCCATGAAACCACC TA-3′) were used as positive controls. NC probes were custom designed using the yeast SUC2 gene, which does not express in human and mice tissue (5′-AGCTATTCTCTTGATG GTGGTTACACTTTTACTGA -3′; 5′-ACCCTGTTTTAGCTGCCAACTCCACTCAATTC AGA-3′; 5′-GATCATCCATGTCTTTGGTCCGCAAGTTTTCTTTG-3′), and a no-probe control using PBS with hybridization buffer. Then, samples were treated the same as the lncRNA-MEG3 FISH probes.
For in vitro cultured cell FISH, PASMCs were fixed in 4% paraformaldehyde and then incubated with lncRNA-MEG3 FISH Probe Mix overnight at 37°C. Fluorescence images were obtained using a fluorescence microscope. U6 FISH Probe Mix was used to image the nuclei, 18S FISH Probe Mix was used to localize the cytoplasm, and DAPI (4,6-diamidino-2-phenylindole) was used to label the nuclei. Images were captured by microscopy and analyzed using Image Pro Plus software.
lncRNA-MEG3 Knockdown In Vivo and In Vitro
To silence the expression of lncRNA-MEG3 in vitro, we transfected PASMCs with siRNA targets to lncRNA-MEG3 (siMEG3-936, 5′-GCGUCUUCCUGU GCCAUUUTT-3′ sense; 5′-AAAUGGCACAGGAAGACGCTT-3′ antisense), (siMEG3-1147, 5′-CC UCCUGGAUUAGGCCAAATT-3′ sense; 5′-UUUGGCCUAAUCCAGGAGG TT-3′ antisense), and nontargeted control siRNA (siNC, 5′-UUCUCCGAACGUGUC ACGUTT-3′ sense; 5′-ACGUGACACGUUCGGAGAATT-3′ antisense) using X-treme Gene siRNA Transfection Reagent (Roche). In some experiments, lncRNA-MEG3 was knocked down using gapmers (gapmer-1, 5′-TCCATTTGCCTCATAA-3′; gapmer-3, 5′-CACTCCA TCACTCATA-3′), which were designed and synthesized by Exiqon (Vedbaek, Denmark).
To knock down the expression of lncRNA-MEG3 in PAs, we loaded siRNA against lncRNA-MEG3 (siMEG3) with octaarginine (R8) conjugated PEG2000-Lipid (R8-Lip) to form a lung-specific delivery system (R8-Lip-siMEG3) as previously described with some modifications.37 Briefly, R8 and DSPE-PEG2000-Mal (molar ratio: 1.5:1) were mixed in chloroform and methanol (v/v = 2:1) at room temperature with gentle stirring for 48 h. The mixture was evaporated under a vacuum and then redissolved in chloroform after discarding the insoluble material. The supernatant (DSPE-PEG2000-R8) was evaporated again under a vacuum. Lipid compositions of the prepared liposomes were as follows: R8-modified liposomes (R8-LIP), SPC/Chol/DSPE-PEG2000/DSPE-PEG2000-R8 (molar ratio 59:33:3:5). A lipid film was produced by rotary evaporation of all lipids in chloroform. The films were left under a vacuum for 2 h. Hydration buffer was added to produce a concentration of 10 μmol/3 mL of lipid. Lipid suspensions were sonicated with a probe sonicator at 80 W for 2 min. siMEG3 and lipid ingredients, at a siRNA to lipid molar ratio of 1:40, were dissolved in chloroform to prepare siRNA-loaded liposomes. Free siRNA was removed using a Sephadex-G50 column, and pellets were subsequently collected.
To efficiently knock down the expression of lncRNA-MEG3, we repeatedly injected R8-Lip-siMEG3 into the tail vein on days 1, 4, 7, and 14 during the experiment. R8-Lip and siMEG3 plus Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) were used as controls. In some experiments, AMO-328 (miR-328-3p specific 2-O-methyl antisense inhibitory oligoribonucleotide) was loaded into the liposome together with siMEG3. The biodistribution of R8-Lip was evaluated using a labeled siRNA (si-MEG3), and fluorescence images were captured. Additionally, αSMA stains or CD31+ stains were used to confirm delivery to smooth muscle and endothelial compartments. The efficiency of MEG3 knockdown by R8-Lip-siMEG3 was confirmed using real-time PCR and FISH. To access the beneficial effect of R8-Lip-siMEG3 once PH established, we exposed mice to hypoxia for 2 weeks to establish PH and subsequently injected R8-Lip-siMEG3 (days 14, 17, 21, and 28) through the tail vein for another 3 weeks.
lncRNA-MEG3 Overexpression in PASMCs
An overexpression vector for lncRNA-MEG3 was constructed. Briefly, using Mus musculus MEG3 transcript variant 1, (GenBank: NR_003633.3) as a template, PCR was performed with 5′-TGAACCGTCAGATCGAATTCGCCACCGGGGACCTTTACAGACC-3′ and 5′-AATCCAGAGGTTGAGGATCCTTACAGGTGCACCTAATTGGATGGATA-3′ to obtain a fragment of 1644 base pairs (bp), which was the gene sequence of lncRNA-MEG3 and upstream and downstream recombination exchange arms. The pLenti-EF1a-EGFP-P2A-Puro-CMV-MCS (H145) vector (CutSmart, New England Biolabs, Ipswich, MA, USA) was digested with EcoR I-HF and BamH I-HF, and the carrier was recovered by electrophoresis after scanning for the 8.8-kb fragment. Enzyme digestion reactions contained 2 μg plasmid, 1 μL enzyme, 5 μL buffer, and double-distilled water to a total volume of 50 μl. Reactions were incubated at 37°C for 2 h. After 1% agarose gel electrophoresis, the vector was removed from the gel and recovered using a gel recovery kit. The lncRNA-MEG3 PCR product was ligated into the vector using a seamless cloning kit to obtain the target plasmid with ampicillin resistance. PASMCs were transfected with the plasmid and then harvested for RNA isolation, cell proliferation assay, cell migration assay, and flow cytometry analysis.
MicroScale Thermophoresis
Sample preparation: For the experiment, FAM-labeled single-stranded RNA (ssRNA) of lncRNA-MEG3 molecules was amplified using 5′-GTTTCTGGGCTACGGGTTTG-3′ sense; 5′-CATACTGTTGTC ACTCACCC-3′ antisense. The PCR product of ssRNA was 163 nt (2030-2193). Unlabeled single-chain miR-328-3p mimics (5′-CUGGCCCUCUCUGCCCUUCCGU-3′) or NC (5′-UUGUACUACACAAAAGUACUG-3′) was added at a 1:1 dilution. For measurement, samples were filled into standard capillaries. Measurements were performed on a NanoTemper Monolith NT.115 instrument. Analysis was performed at 22% LED power and 20% MST power, “Fluo. Before” time was 5 s, MST on time was 30 s, and “Fluo. After” time was 5 s.
Statistical Analysis
Quantitative data are expressed as the means ± SEM. Statistical analysis was carried out with the GraphPad Prism software. Data analysis was performed with paired Student’s t test (for two means) or one-way ANOVA followed by Dunnett’s test (for >2 means) where appropriate. Sample sizes (n) are reported in the corresponding figure legends. Significance levels of **p < 0.01 and *p < 0.05 were adopted.
Author Contributions
Conceptualization, Y.X., X.Z., and D.Z.; Methodology, Y.X., X.Z., Y.F., J.Q., M.L., M.M., S.W., and S.L.; Investigation, Y.X., X.Z., Y.F., J.Q., and M.L.; Resources, S.L. and D.Z.; Writing – Original Draft, Y.X., X.Z., J.Q., and M.L.; Writing – Review & Editing, X.Z. and D.Z.; Supervision, X.Z. and D.Z.; Funding Acquisition, Y.X., X.Z., and D.Z.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Guangtian Wang for the preparation of R8-Liposome. The PASMCs of iPAH patients were provided by Professor Jian Wang from State Key Laboratory of Respiratory Diseases, Guangzhou Institute of Respiratory Disease, and The First Affiliated Hospital of Guangzhou Medical University. This work was supported by the National Natural Science Foundation of China (31771276 and 31471095 to D.Z., 31701010 to Y.X., 31500936 to X.Z., and 81600034 to S.W.); Specialized Research Fund for the Doctoral Program of Higher Education (20112307110022 to D.Z.); the Natural Science Foundation of Heilongjiang Province (C2016038 to Y.X.); China Postdoctoral Science Foundation (2016M601452 to Y.X.); Postdoctoral Foundation of Heilongjiang Province (LBH-Z16239 to Y.X.); Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education (2013008 to Y.X.); Postdoctoral Career Development Fund of Heilongjiang Province (LBH-Q14119 to X.Z.); Harbin Medical University Scientific Research Innovation Fund (2016JCZX05 to Y.X. and 2017JCZX09 to X.Z.); Foundation of Daqing Department of Science and Technology (Szdy-2015-02 to Y.X.), Specifically Foundation for Returned Scholars in Heilongjiang Province to X.Z., and Wu Liande Youth Science Foundation of Harbin Medical University-Daqing (DQWLD201601 to X.Z.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.07.022.
Contributor Information
Xiaodong Zheng, Email: zhengxiaodong@hmudq.edu.cn.
Daling Zhu, Email: dalingz@yahoo.com.
Supplemental Information
References
- 1.Anderson J.R., Nawarskas J.J. Pharmacotherapeutic management of pulmonary arterial hypertension. Cardiol. Rev. 2010;18:148–162. doi: 10.1097/CRD.0b013e3181d4e921. [DOI] [PubMed] [Google Scholar]
- 2.Welsh D.J., Peacock A.J. Cellular responses to hypoxia in the pulmonary circulation. High Alt. Med. Biol. 2013;14:111–116. doi: 10.1089/ham.2013.1016. [DOI] [PubMed] [Google Scholar]
- 3.Nathan S.D., Hassoun P.M. Pulmonary hypertension due to lung disease and/or hypoxia. Clin. Chest Med. 2013;34:695–705. doi: 10.1016/j.ccm.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Crosswhite P., Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol. Med. 2014;20:191–201. doi: 10.2119/molmed.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bienertova-Vasku J., Novak J., Vasku A. MicroRNAs in pulmonary arterial hypertension: pathogenesis, diagnosis and treatment. J. Am. Soc. Hypertens. 2015;9:221–234. doi: 10.1016/j.jash.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Guo L., Qiu Z., Wei L., Yu X., Gao X., Jiang S., Tian H., Jiang C., Zhu D. The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-α1C. Hypertension. 2012;59:1006–1013. doi: 10.1161/HYPERTENSIONAHA.111.185413. [DOI] [PubMed] [Google Scholar]
- 7.Qian Z., Zhang L., Chen J., Li Y., Kang K., Qu J., Wang Z., Zhai Y., Li L., Gou D. MiR-328 targeting PIM-1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget. 2016;7:54998–55011. doi: 10.18632/oncotarget.10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang Y.N., Zhang K., Hu Z.M., Qi H.X., Shi Z.M., Han X.H., Han Y.W., Hong W. Hypoxia-regulated lncRNAs in cancer. Gene. 2016;575:1–8. doi: 10.1016/j.gene.2015.08.049. [DOI] [PubMed] [Google Scholar]
- 9.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 10.Kour S., Rath P.C. Long noncoding RNAs in aging and age-related diseases. Ageing Res. Rev. 2016;26:1–21. doi: 10.1016/j.arr.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Mercer T.R., Mattick J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 12.Meseure D., Drak Alsibai K., Nicolas A., Bieche I., Morillon A. Long Noncoding RNAs as New Architects in Cancer Epigenetics, Prognostic Biomarkers, and Potential Therapeutic Targets. BioMed Res. Int. 2015;2015:320214. doi: 10.1155/2015/320214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon J.H., Abdelmohsen K., Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014;34:9–14. doi: 10.1016/j.semcdb.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karreth F.A., Tay Y., Perna D., Ala U., Tan S.M., Rust A.G., DeNicola G., Webster K.A., Weiss D., Perez-Mancera P.A. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tay Y., Kats L., Salmena L., Weiss D., Tan S.M., Ala U., Karreth F., Poliseno L., Provero P., Di Cunto F. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferdin J., Nishida N., Wu X., Nicoloso M.S., Shah M.Y., Devlin C., Ling H., Shimizu M., Kumar K., Cortez M.A. HINCUTs in cancer: hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ. 2013;20:1675–1687. doi: 10.1038/cdd.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang F., Huo X.S., Yuan S.X., Zhang L., Zhou W.P., Wang F., Sun S.H. Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol. Cell. 2013;49:1083–1096. doi: 10.1016/j.molcel.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Wu J., Okada T., Fukushima T., Tsudzuki T., Sugiura M., Yukawa Y. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 2012;9:302–313. doi: 10.4161/rna.19101. [DOI] [PubMed] [Google Scholar]
- 20.Michalik K.M., You X., Manavski Y., Doddaballapur A., Zörnig M., Braun T., John D., Ponomareva Y., Chen W., Uchida S. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 21.Manji S.S., Sørensen B.S., Klockars T., Lam T., Hutchison W., Dahl H.H. Molecular characterization and expression of maternally expressed gene 3 (Meg3/Gtl2) RNA in the mouse inner ear. J. Neurosci. Res. 2006;83:181–190. doi: 10.1002/jnr.20721. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Y., Zhang X., Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J. Mol. Endocrinol. 2012;48:R45–R53. doi: 10.1530/JME-12-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braconi C., Kogure T., Valeri N., Huang N., Nuovo G., Costinean S., Negrini M., Miotto E., Croce C.M., Patel T. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–4756. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu K.H., Li W., Liu X.H., Sun M., Zhang M.L., Wu W.Q., Xie W.P., Hou Y.Y. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461. doi: 10.1186/1471-2407-13-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X., Ji G., Ke X., Gu H., Jin W., Zhang G. MiR-141 Inhibits Gastric Cancer Proliferation by Interacting with Long Noncoding RNA MEG3 and Down-Regulating E2F3 Expression. Dig. Dis. Sci. 2015;60:3271–3282. doi: 10.1007/s10620-015-3782-x. [DOI] [PubMed] [Google Scholar]
- 26.Bonnet S., Provencher S., Guignabert C., Perros F., Boucherat O., Schermuly R.T., Hassoun P.M., Rabinovitch M., Nicolls M.R., Humbert M. Translating Research into Improved Patient Care in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017;195:583–595. doi: 10.1164/rccm.201607-1515PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potus F., Ruffenach G., Dahou A., Thebault C., Breuils-Bonnet S., Tremblay È., Nadeau V., Paradis R., Graydon C., Wong R. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation. 2015;132:932–943. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 28.Schuster-Gossler K., Bilinski P., Sado T., Ferguson-Smith A., Gossler A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev. Dyn. 1998;212:214–228. doi: 10.1002/(SICI)1097-0177(199806)212:2<214::AID-AJA6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 29.Sun Z., Nie X., Sun S., Dong S., Yuan C., Li Y., Xiao B., Jie D., Liu Y. Long Non-Coding RNA MEG3 Downregulation Triggers Human Pulmonary Artery Smooth Muscle Cell Proliferation and Migration via the p53 Signaling Pathway. Cell Physiol. Biochem. 2017;42:2569–2581. doi: 10.1159/000480218. [DOI] [PubMed] [Google Scholar]
- 30.Zhu B., Gong Y., Yan G., Wang D., Qiao Y., Wang Q., Liu B., Hou J., Li R., Tang C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem. Biophys. Res. Commun. 2018;495:2125–2132. doi: 10.1016/j.bbrc.2017.11.185. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J., Zhang X., Zhou Y., Ansell P.J., Klibanski A. Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. Int. J. Biochem. Cell Biol. 2006;38:1808–1820. doi: 10.1016/j.biocel.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 32.Armoskus C., Moreira D., Bollinger K., Jimenez O., Taniguchi S., Tsai H.W. Identification of sexually dimorphic genes in the neonatal mouse cortex and hippocampus. Brain Res. 2014;1562:23–38. doi: 10.1016/j.brainres.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi L., Kojonazarov B., Elgheznawy A., Popp R., Dahal B.K., Böhm M., Pullamsetti S.S., Ghofrani H.A., Gödecke A., Jungmann A. miR-223-IGF-IR signalling in hypoxia- and load-induced right-ventricular failure: a novel therapeutic approach. Cardiovasc. Res. 2016;111:184–193. doi: 10.1093/cvr/cvw065. [DOI] [PubMed] [Google Scholar]
- 34.Paulin R., Meloche J., Jacob M.H., Bisserier M., Courboulin A., Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1798–H1809. doi: 10.1152/ajpheart.00654.2011. [DOI] [PubMed] [Google Scholar]
- 35.Renard S., Paulin R., Breuils-Bonnet S., Simard S., Pibarot P., Bonnet S., Provencher S. Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm. Circ. 2013;3:74–81. doi: 10.4103/2045-8932.109917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015;6:7743. doi: 10.1038/ncomms8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin Y., Wu X., Yang Z., Zhao J., Wang X., Zhang Q., Yuan M., Xie L., Liu H., He Q. The potential efficacy of R8-modified paclitaxel-loaded liposomes on pulmonary arterial hypertension. Pharm. Res. 2013;30:2050–2062. doi: 10.1007/s11095-013-1058-8. [DOI] [PubMed] [Google Scholar]
- 38.Spizzo R., Almeida M.I., Colombatti A., Calin G.A. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–4587. doi: 10.1038/onc.2011.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Provencher S., Archer S.L., Ramirez F.D., Hibbert B., Paulin R., Boucherat O., Lacasse Y., Bonnet S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018;122:1021–1032. doi: 10.1161/CIRCRESAHA.117.312579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.