

Abstract
Leishmaniasis is a Neglected Tropical Disease caused by the insect-vector borne protozoan parasite, Leishmania species. Infection affects millions of the World's poorest, however vaccines are absent and drug therapy limited. Recently, public-private partnerships have developed to identify new modes of controlling leishmaniasis. Most of these collaborative efforts have relied upon the small molecule synthetic compound libraries held by industry, but the number of New Chemical Entities (NCE) identified and entering development as antileishmanials has been very low. In light of this, here we describe a public-private effort to identify natural products with activity against Leishmania mexicana, a causative agent of cutaneous leishmanaisis (CL). Utilising Hypha Discovery's fungal extract library which is rich in small molecule (<500 molecular weight) secondary metabolites, we undertook an iterative phenotypic screening and fractionation approach to identify potent and selective antileishmanial hits. This led to the identification of a novel oxidised bisabolane sesquiterpene which demonstrated activity in an infected cell model and was shown to disrupt multiple processes using a metabolomic approach. In addition, and importantly, this study also sets a precedent for new approaches for CL drug discovery.
Keywords: Cutaneous leishmaniasis, Leishmania mexicana, Natural product, Screening, Fractionation, Metabolomics
Graphical abstract

1. Introduction
The Neglected Tropical Disease (NTD) leishmaniasis is endemic in over 90 countries worldwide, affecting approximately 12 million people per year with 350 million people living at risk of disease. The causative agent, Leishmania species, are sand fly borne kinetoplastid protozoan parasites (Stuart et al., 2008) and infection leads to a wide spectrum of clinical manifestations in endemic areas, from self-healing but scarring cutaneous leishmaniasis (CL) to fatal visceral disease (VL). Amongst other factors, this diversity of disease is dependent on the parasite species, host immunity and genetic background (Reithinger et al., 2007). Largely due to elimination efforts in south Asia, the global burden of VL has decreased substantially in the past decade. However, due to forced migration, the cases of CL have substantially increased in the same period (0.7–1 million per year) (Burza et al., 2018).
A vaccine to prevent leishmaniasis is not available and treatment relies entirely on a limited arsenal of chemotherapeutics. Recent years have seen the emergence of public-private partnerships which have used industrial scale (>1,000,000) compound libraries to screen for antileishmanials either phenotypically (Khare et al., 2016; Pena et al., 2015) or target-based (Norcliffe et al., 2018). However, all the major initiatives have focused on VL leaving CL, the most common form of leishmaniasis, as a neglected NTD. Current treatment of CL largely relies on the pentavalent antimonials such as sodium stibogluconate (Pentostam) and meglumine antimoniate (Glucantime) (Croft and Coombs, 2003; Kedzierski et al., 2009). Both Pentostam and Glucantime have been in clinical use for over 70 years despite their associated problems, which include severe side-effects such as cardiotoxicity (Chappuis et al., 2007) and the fact that they require parenteral administration (Demicheli et al., 2004). In addition, the use of pentavalent antimonials in the treatment of leishmaniasis is under threat from the emergence of drug resistance (Croft et al., 2006). Amphotericin B (Fungizone) (Thakur et al., 1999) and diamidine pentamidine (Bray et al., 2003) are employed as second-line drugs in the treatment of CL. Like the antimonials, they induce severe side-effects and parasite resistance, although not yet conclusively confirmed in the field, has been observed under laboratory conditions (Di Giorgio et al., 1999). Given the aforementioned issues with both the current first- and second-line drugs used to treat CL there is clearly a need to develop new and effective therapies for this disease.
Natural products have long formed the backbone of traditional therapies, including for leishmaniasis (Charlton et al., 2018; Cockram and Smith, 2018). The active compounds of these medicines include vast numbers of alkaloids (Mishra et al., 2009), flavonoids (da Silva et al., 2012; Mittra et al., 2000), chalcones (Aponte et al., 2010; Boeck et al., 2006; Chen et al., 1994; de Mello et al., 2014) and terpenoids (Arruda et al., 2005; do Socorro et al., 2003). It should also be noted that the second-line CL drug amphotericin B is itself a natural product, originally isolated from Streptomyces nodosus as an antifungal in 1955 (Dutcher, 1968).
Given this history, it is logical to consider the natural world as a potential source of novel antileishmanials. Hypha Discovery's (http://www.hyphadiscovery.co.uk) large-scale extract (HDLSX) library of 1–5 L volume fermentation extracts is derived from the company's unique collection of higher fungal strains, namely the mushroom-producing basidiomycetes and ascomycetes, which are underexplored in the field of antiprotozoal drug discovery. The producing strains included in the library are regarded as being biosynthetically talented since they had all demonstrated production of bioactivity in various whole cell-based assays against microbial pathogens and/or human tumour cell lines. In this paper we describe a collaborative effort to exploit this resource for the discovery of novel antileishmanials. Utilising an iterative process of screening against the CL pathogen Leishmania mexicana and mammalian cells, followed by fractionation and scale up of identified parasite selective hits, following dereplication we identified a known antileishmanial class and, more importantly, a novel oxidised bisabolane sesquiterpene which demonstrated activity in an infected cell model. The mode of action of this latter hit was investigated using a metabolomics approach.
2. Materials and Methods
2.1. HDLSX library generation and format
Scale-up fermentations of fungi selected for inclusion in the HDLSX library were obtained in multiple shake flasks (each 250 mL Erlenmeyer flask containing 100 mL Leatham's medium, contents per litre of water: D-Glucose (25.00 g), L(+)-glutamic acid monosodium salt monohydrate (3.20 g), KH2PO4 (2.00 g), MgSO4.7H2O (2.00 g), mineral solution (10 mL), trace element solution (1.0 mL), vitamin solution (1.0 mL) and 1% aqueous salicylic acid (0.10 mL)) to a total volume of 1–5 L. The fermentations were harvested after the same period of time that yielded the original activity of interest and this could vary from as little as three to as many as one hundred days. Aqueous broth actives were harvested by various methods depending on the polarities of the components of interest including extraction with ethyl acetate or capture using Diaion HP20 resin and elution with methanol/acetonitrile. Biomass actives were extracted using methanol-acetone (1:1). The extracts were concentrated to dryness for storage. The extracts were weighed and portions stored in 96-well blocks as 10 mg/mL solutions in DMSO. All of the purification and bioactive compound identification work described within this manuscript was conducted on the initially available HDLSX library materials without the need for refermentation of the producing fungal strains, as per the original intention of the library. For subsequent analyses, further HD871-1 material was obtained by refermentation.
2.2. Cell culture
Leishmania mexicana (MNYC/BZ/62/M379) were maintained at 26 °C in Schneider's Drosophila media (Sigma Aldrich) supplemented with heat inactivated foetal bovine sera (HIFBS; 15% for promastigotes, and 20% for amastigotes; Biosera). Promastigotes were transformed into axenic amastigotes by a pH and temperature shift as previously described (Bates, 1994). Ceropithecus aethiops kidney (Vero) cells (ATCC CCL-81) and Mus musculus leukaemia virus transformed macrophage (RAW267.4) cells (ATCC TIB-71) were cultured in Dulbecco's Modified Eagles Medium (DMEM; Fisher Scientific) supplemented with 10% HIFBS (Biosera). Cells were counted using a Neubauer Improved Haemocytometer.
2.3. Screening against axenic amastigote forms
Cytotoxicity analyses were performed in 96-well plates (Nunc) using alamarBlue® (Life Technologies) with some modifications to the published, optimized protocol (Chadbourne et al., 2011). Briefly, in initial screens, extracts or isolated compounds were incubated with 100 μL axenic amastigote L. mexicana at 5 × 105 mL−1 for 24 h at 33 °C before the addition of 10 μL of alamarBlue® and incubation for a further for 24 h at the same temperature, all in Parafilm (Fisher Scientific) sealed plates. Counter screening was performed against Vero cells, with plates seeded with 5 × 103 cells per well. Following overnight incubation at 37 °C, 5% CO2, cells were incubated with extracts or isolated compounds in 100 μL of media for 24 h in the same conditions and then for a further 4 h following the addition of 10 μL of alamarBlue®. Subsequently cell viability was assessed using a fluorescent plate reader (Biotek; 560EX nm/600 EM nm), with amphotericin B as positive (10 μM) and DMSO as negative controls.
Initial screening of the HDLSX library (313 extracts) was carried out using a dose response approach, in duplicate against both axenic amastigote L. mexicana and mammalian Vero cells. The Selectivity Index (SI) was calculated from these data (EC50 Vero / EC50 L. mexicana). Subsequent fractions were screened at single concentrations in duplicate against the parasite. Determination of the EC50 of the identified novel antileishmanial against axenic amastigote L. mexicana was carried out using the same assay in triplicate on three separate occasions to ensure a robust data set was collected.
2.4. Screening against infected macrophages
Efficacy against L. mexicana infected macrophages was tested as previously described (Bolt et al., 2016; Eggimann et al., 2015). The RAW264.7 cells were seeded in 96-well plates at 2.5 × 105 mL−1 (200 μL/well) in DMEM (10% HIFBS) and incubated for 24 h at 37 °C, 5% CO2. Following washing with DMEM (2% HIFBS), L. mexicana axenic amastigotes (or not for RAW264.7 cytotoxicity) were added at 25 × 105 mL−1 (200 μL/well) in DMEM (2% HIFBS) and plate incubated for 24 h at 37 °C, 5% CO2. Subsequently, the infected RAW264.7 cells were washed carefully 5× with DMEM (2% HIFBS) before adding 100 μL/well of fresh DMEM (2% HIFBS) to each well. Isolated compounds and control (10 μM amphotericin B and DMSO vehicle) were added at the required concentrations before a further incubation for 24 h at 37 °C, 5% CO2. Finally, the infected macrophages cells were washed 3× with Schneider's Insect medium (serum-free) and then lysed with 20 μL/well of SDS (0.05%, v/v in Schneider's) for 30 s before the addition of 180 μL/well of Schneider's Insect medium (pH7, 15% HIFBS). Plates were Parafilm sealed and incubated for 48 h at 26 °C. Subsequently, 10 μL of alamarBlue® was added to each well before a 4 h incubation at 26 °C prior to assessing parasite viability using a fluorescent plate reader (Biotek; 560EX nm/ 600 EM nm). All of the experiments described above were carried out on a three separate occasions in triplicate to ensure a robust data set was collected. The Selectivity Index (SI) was calculated from these data (EC50 RAW267.4 / EC50 intracellular L. mexicana).
2.5. Assay-guided purification
Preliminary small-scale activity-guided purification studies on hits from 12 organisms that showed promising antiparasitic activity profiles (EC50 < 1 mg/mL; therapeutic index >100) were conducted using semi-preparative HPLC approaches as follows: extract aliquots (0.9 mL) were chromatographed by reversed phase HPLC on an Xbridge Prep Phenyl 5 μm OBD column (19 mm × 100 mm) with a guard column (19 mm × 10 mm), using gradient elution of 10–100% acetonitrile in water in the presence of 0.1% formic acid over 8 min, held at 100% acetonitrile for 3 min before returning to starting conditions over 1 min at a flow rate of 17 mL/min. 24 fractions were collected for each fractionation and concentrated to dryness in a Genevac HT12 centrifugal evaporator. The resulting dried fractions were re-dissolved in DMSO-MeOH (3:1, 0.45 mL) and aliquots (25 μL) transferred to 96 well microtiter plates, lyophilised for shipping.
Analysis of active fractions by HPLC-mass spectrometry employing an orthogonal reversed phase HPLC-MS method was conducted on a SymmetryShield RP8 column (3.5 μm; 4.6 mm × 75 mm) eluted with a linear gradient of 10–95% MeCN in water, containing 10 mM ammonium formate + 0.1% formic acid, at a flow rate of 1 mL/min, held at 95% MeCN for 1.0 min before returning to initial conditions over 0.5min; total run time 12 min. UV–visible spectra were acquired using a Waters 2996 photodiode array detector and positive and negative ion electrospray mass spectra were acquired using a Waters Acquity SQ detector. Putative molecular weights, UV–Visible maxima and producing organism taxonomic data were used to search two natural products databases to identify known compounds, namely Antibase and the Chapman and Hall Dictionary of Natural Products.
Specific chromatographic methods were then developed for the most promising active primary fractions to separate and resolve their constituent components / HPLC peaks using a reversed phase column chemistry orthogonal to that used for the primary fractionation step (usually SymmetryShield RP8). The primary fraction was then fractionated on the semi-preparative scale and individual HPLC peaks collected into secondary fractions that were concentrated to dryness, re-dissolved and shipped for assay as described above.
2.6. Scale-up purification and structure elucidation
The target extracts were purified by preparative HPLC methods scaled-up from the purification methods developed from the small-scale assay guided purification investigations. HD871-1 was purified from the Diaion HP20 extract of a 5.0 L fermentation of Marasmius sp. HFN871. The extract was first chromatographed by preparative HPLC on a Waters Nova-Pak C18 column (40 × 100 mm with a 40 × 10 mm guard column) eluted with a gradient increasing linearly from 10 to 95% acetonitrile in water containing 0.1% formic acid over a period of 8 min at a flow rate of 50 mL/min. HPLC was used to check for the presence of the target metabolite with MH + at m/z 263 and this was present in fractions eluting between 4.5 and 5.5 min. These were combined and further purified by semi-preparative HPLC on a Waters Atlantis T3 ODS column (10 × 250 mm) eluted isocratically with 45% aqueous acetonitrile containing 0.1% formic acid at a flow rate of 4.6 mL/min. HPLC-MS analysis showed that the compound of interest was present in fractions eluting from 8 to 9 min. A final purification step was achieved by semi-preparative HPLC on a Waters X-Select C18 column (19 × 100 mm with a 19 × 10 mm guard column) eluted with a linear gradient increasing from 25 to 30% aqueous acetonitrile containing 0.1% formic acid over a period of 14 min at a flow rate of 20 mL/min. The peak eluting at 10 min was collected and concentrated to dryness to yield pure HD871-1 (6.9 mg) as a colourless residue. 1H and 13C NMR spectra were acquired at 500 MHz and 125 MHz, respectively, at 298K in DMSO-d6, on a Bruker AVANCE III 500 MHz NMR spectrometer at the University of Surrey, Guildford, UK. Datasets for structure elucidation included COSY, HSQC, HMBC and NOESY spectra, which were interpreted at Hypha Discovery using Mestrenova software. High resolution mass spectrometry was conducted on a Bruker micrOTOF-Q instrument.
2.7. Metabolite extraction
The optimal concentration of HD871-1 for this experiment was established by estimation of the half time of L. mexicana axenic amastigotes to death at 4 concentrations (1.25, 2.5, 5 and 10 μM) by microscopic observation at 12, 24, 36 and 48 h. Subsequently the amastigotes, seeded at 1 × 107 parasites/mL, were exposed to 10 μM HD871-1, or a vehicle (DMSO) control, for 36 h at 32 °C. Metabolites were then extracted and analysed as follows:
For LC-MS metabolomic analyses, sample extraction was performed as described (Creek et al., 2011; Kovářová et al., 2018). In summary, 108 cells were rapidly cooled in a dry ice/ethanol bath to 4 °C, centrifuged, washed with 1 × PBS, and resuspended in 200 μL extraction solvent (chloroform:methanol:water, 1:3:1 vol ratio). Samples were centrifuged at 16,000 g (4 °C for 10 min) after shaking for 1 h at 4 °C. The supernatant was stored under argon at −80 °C until running on the mass spectrometer. Samples were first separated using HPLC with a 150 × 4.6 mm ZIC-pHILIC (Merck) column on UltiMate 3000 RSLC (Thermo Scientific). Mass detection used an Orbitrap Q-Exactive mass spectrometer (Thermo Fisher) at Glasgow Polyomics, using positive and negative polarity switching mode, with a 10 μL injection volume and flow rate of 300 μL/min 249 authentic standards were included to assist compound identification. Data were processed and analysed using mzMatch (Scheltema et al., 2011) and IDEOM software (Creek et al., 2012). All the MS analyses were performed in at least triplicate. The Comparison page of the IDEOM file is included as supplementary material (Supplementary Table 1). This contains information on those masses detected as likely metabolites through the software processing. Exact mass is measured and this is converted, where possible to a formula corresponding to that mass. Screening of a database of metabolites with that formula then yields a putative identity. Where an authentic standard has a matching mass and retention time this is taken as a likely hit. Other annotations are tentative (metabolomics standards initiative level 2, Sumner et al., 2007) with this data.
3. Results
3.1. Primary screening
Utilising a 96-well protocol previously developed to identify antileishmanial compounds (Chadbourne et al., 2011) the HDLSX library (313 fungal extracts) was screened against mammalian stage L. mexicana axenic amastigotes in a dose response experiment. Extracts inhibiting with an EC50 < 0.025 mg/mL (Effective Concentration eliciting 50% inhibition of proliferation) against the parasites were selected, and these 38 hits counter screened in the same manner against mammalian Vero cells. These hits represent complex mixtures with non-cytotoxic extracts possibly containing minor cytotoxic components, and conversely, non-active extracts may harbour minor antiparasitic components. However, to narrow down the extracts for further analyses, based on the screening data, potent hits with a selectivity index (SI) >48 were initially selected. After subtracting those with known cytotoxicity and intellectual property issues we were left with 17 hits. Based on Hypha data, replicated extracts were removed leaving 12 hits (HDLSX-9, 14, 18, 21, 22, 33, 35, 67, 79, 127, 159, 171; Table 1), a hit rate of 3.8%.
Table 1.
The 38 potent hits (EC50 < 0.025 mg/ml against axenic amastigote L. mexicana) were counter screened against mammalian Vero cells. Those with a selectivity index (SI) >48 were initially selected. Final selection (12 hits, Y in bold) was made after subtracting those with known cytotoxicity and intellectual property issues, and de-replication.
| Compound number | Amastigote EC50 mg/ml | Vero EC50 mg/ml | Selectivity index (SI) | SI > 48 | Selection |
|---|---|---|---|---|---|
| HDLSX-171 | 3.37E-16 | >1.33E+00 | >3.95E+15 | Y | Y |
| HDLSX-106 | 2.45E-02 | 1.07E-02 | 4.38E-01 | N | N |
| HDLSX-67 | 1.31E-04 | 6.35E-03 | 4.84E+01 | Y | Y |
| HDLSX-33 | 5.22E-04 | >1.33E+00 | >2.55E+03 | Y | Y |
| HDLSX-134 | 6.66E-04 | 3.28E-04 | 4.92E-01 | N | N |
| HDLSX-14 | 6.84E-04 | >1.33E+00 | >1.94E+03 | Y | Y |
| HDLSX-159 | 7.94E-04 | 3.96E-01 | 4.99E+02 | Y | Y |
| HDLSX-127 | 9.26E-04 | >1.33E+00 | >1.44E+03 | Y | Y |
| HDLSX-21 | 2.72E-03 | 1.34E-01 | 4.92E+01 | Y | Y |
| HDLSX-208 | 2.77E-03 | >1.33E+00 | >4.80E+02 | Y | N |
| HDLSX-79 | 3.26E-03 | 1.83E+00 | 5.61E+02 | Y | Y |
| HDLSX-27 | 3.36E-03 | 9.97E-03 | 2.97E+00 | N | N |
| HDLSX-177 | 3.56E-03 | >1.33E+00 | >3.73E+02 | Y | N |
| HDLSX-84 | 4.84E-03 | 1.07E-01 | 2.21E+01 | N | N |
| HDLSX-23 | 5.09E-03 | 2.29E-01 | 4.50E+01 | N | N |
| HDLSX-179 | 5.94E-03 | 1.37E-01 | 2.31E+01 | N | N |
| HDLSX-130 | 6.75E-03 | >1.33E+00 | >1.97E+02 | Y | N |
| HDLSX-209 | 6.86E-03 | 4.40E-02 | 6.41E+00 | N | N |
| HDLSX-18 | 7.15E-03 | >1.33E+00 | >1.86E+02 | Y | Y |
| HDLSX-22 | 7.42E-03 | >1.33E+00 | >1.79E+02 | Y | Y |
| HDLSX-39 | 7.55E-03 | 6.92E-02 | 9.17E+00 | N | N |
| HDLSX-213 | 9.05E-03 | >1.33E+00 | >1.47E+02 | Y | N |
| HDLSX-13 | 9.24E-03 | >1.33E+00 | >1.44E+02 | Y | N |
| HDLSX-72 | 9.53E-03 | 1.69E-01 | 1.77E+01 | N | N |
| HDLSX-126 | 1.02E-02 | >1.33E+00 | >1.30E+02 | Y | N |
| HDLSX-114 | 1.03E-02 | 5.92E-03 | 5.73E-01 | N | N |
| HDLSX-37 | 1.18E-02 | 9.93E-02 | 8.42E+00 | N | N |
| HDLSX-35 | 1.37E-02 | >1.33E+00 | >9.71E+01 | Y | Y |
| HDLSX-163 | 1.44E-02 | 7.71E-01 | 5.34E+01 | Y | N |
| HDLSX-180 | 1.47E-02 | >1.33E+00 | >9.06E+01 | Y | N |
| HDLSX-9 | 1.47E-02 | >1.33E+00 | >9.03E+01 | Y | Y |
| HDLSX-8 | 1.48E-02 | 7.26E-02 | 4.92E+00 | N | N |
| HDLSX-40 | 1.61E-02 | 9.71E-02 | 6.05E+00 | N | N |
| HDLSX-44 | 1.74E-02 | >1.33E+00 | >7.63E+01 | Y | N |
| HDLSX-28 | 1.78E-02 | 1.32E-01 | 7.45E+00 | N | N |
| HDLSX-85 | 1.89E-02 | >1.33E+00 | >7.04E+01 | Y | N |
| HDLSX-211 | 1.90E-02 | >1.33E+00 | >7.00E+01 | Y | N |
| HDLSX-145 | 2.06E-02 | 7.91E-01 | 3.85E+01 | N | N |
3.2. Primary fractionation and secondary screening
As described in Materials and Methods primary fractionation was undertaken of the 12 hits, yielding 24 fractions per sample. 10 of the initial 12 hits had components demonstrating complete cytotoxic activity against axenic amastigotes at 1 mg/mL (fractions from HDLSX-9, 18, 21, 22, 35, 67, 79, 159, 171, 127; Fig. 1). Initial analyses by mass spectrometry indicated the complexity and, in some cases, content of the active fractions. For example, striatals (including striatal B) were present in HDLSX-21 (from HFN65 Cyathus cf. crassimurus). This was confirmed by chromatographic comparison with a pure sample of striatal B previously isolated from a Cyathus sp. fermentation at Hypha. The 1H NMR spectrum of this material is shown in Supplementary Figure 1 and the signals observed are consistent with those previously reported for striatal B (Anke et al., 2002). Notably the striatals have previously demonstrated to be antileishmanial (Inchausti et al., 1997) (Fig. 2). A pure sample of the related compound striatal C was tested and found to have an ED50 of < 0.8 μM, but considerable toxicity to mammalian cells. Other compounds identified at this stage and deprioritised were a glycolipid from HDLSX-18 (HFN67, Dacryopinax sp.), which had low potency with an ED50 > 100 μM, and the known metabolite nitidon from HDLSX-67 (HFN528 Junghuhnia nitida), which is cytotoxic against other cell lines (Gehrt et al., 1998). The active HDLSX-79 fractions were also dereplicated due to chemical overlap with the HDLSX-22 fractions. This dereplication process left 8 active primary extract fractions to take forward to secondary fractionation and analyses - two from HDLSX-9, and one each from HDLSX-21, 22, 35, 127, 159, 171.
Figure 1.

Screening of primary fractions of the 12 hits against against axenic amastigote L. mexicana at 1 mg/ml y-axis shows proliferation (%) relative to vehicle (DMSO) treated control. Each selected extract (HDLSX-9, 14, 18, 21, 22, 33, 35, 67, 79, 127, 159, 171) was fractionated into 24 fractions, e.g. HDSLX9.1, 9.3 etc. For clarity, only odd number fractions are labelled on the x-axis.
Figure 2.

A peak (6.5 min) in fraction 14 of HDLSX-21 had a product MH+ m/z 489.6 and MNH4+ m/z 506.6 in the positive ion spectrum (starred in A); and [M-H]- m/z 487.6 in the negative ion spectrum (starred in B). This corresponded to known antimicrobial striatal B (C).
3.3. Secondary fractionation and tertiary screening
Secondary fractions were undertaken of the 8 remaining active primary fractions demonstrating chemical novelty (two from HDLSX-9, and one each from HDLSX-21, 22, 35, 127, 159, 171). These isolated factions were screened against axenic amastigotes at 1 mg/mL. HDLSX-9, 22, and 127 derived secondary fractions had components demonstrating complete cytotoxic activity against axenic amastigotes, further fractionation of the 5 other primary fractions (including from HDLSX-21 not containing striatal B) did not yield samples with high level activity (Fig. 3). Given that the fractions were screened at equivalent concentrations (1 mg/mL) it is likely that the apparent loss of activity on further fractionation of these 5 samples was due to a loss of the additive antileishmanial efficacy observed in the initial mixture of compounds. Initial analyses by mass spectrometry indicated the complexity and, in some cases, content of the active fractions. Informed by these analyses, secondary fractions were screened against axenic amastigotes if pure (HDLSX9Fr9+10F7 and HDLSX127Fr11Fr9; Fig. 3A and G) or further fractionated (from HDLSX-22; Fig. 3D) before screening. The pure HDLSX-9 (HDLSX9Fr9+10F7) secondary fraction had minimal activity against the L. mexicana amastigotes (EC50 > 100 μM; data not shown). Furthermore, none of the HDLSX-22 tertiary fractions demonstrated complete cytotoxicity against axenic amastigotes at 1 mg/mL (data not shown). In contrast, the pure HDLSX-127 secondary fraction (HD871-1; HDLSX-127Fr11Fr9 in Fig. 3G) showed good activity (EC50 = 4.73 μM) against the parasite. Furthermore, utilising a previous established assay (Bolt et al., 2016; Eggimann et al., 2015), this compound was tested against infected macrophages and for macrophage cytotoxicity, demonstrating a promising profile with respect to efficacy (EC50 = 4.32 μM) and selectivity (5.51) (Fig. 4).
Figure 3.


Screening of secondary fractions of the 8 active primary fractions (two from HDLSX-9, and one each from HDLSX-21, 22, 35, 127, 159, 171) against axenic amastigote L. mexicana at 1 mg/ml x-axis shows the secondary fractions from the following primary extract fractions: A HDLSX9 fractions 9 and 10 (HDLSX9Fr9+10); B HDLSX9 fraction 13 (HDLSX9Fr13); C HDLSX21 fraction 1 (HDLSX21Fr1); D HDLSX22 fractions 13 and 14 (HDLSX22Fr13 + 14); E HDLSX35 fraction 17 (HDLSX35Fr17); F HDLSX159 fractions 12 and 13 (HDLSX159Fr12 + 13); G HDLSX127 fraction 11 (HDLSX127Fr11); H HDLSX171 fraction 7 (HDLSX171Fr7). y-axis shows proliferation (%) relative to vehicle (DMSO) treated control.
Figure 4.
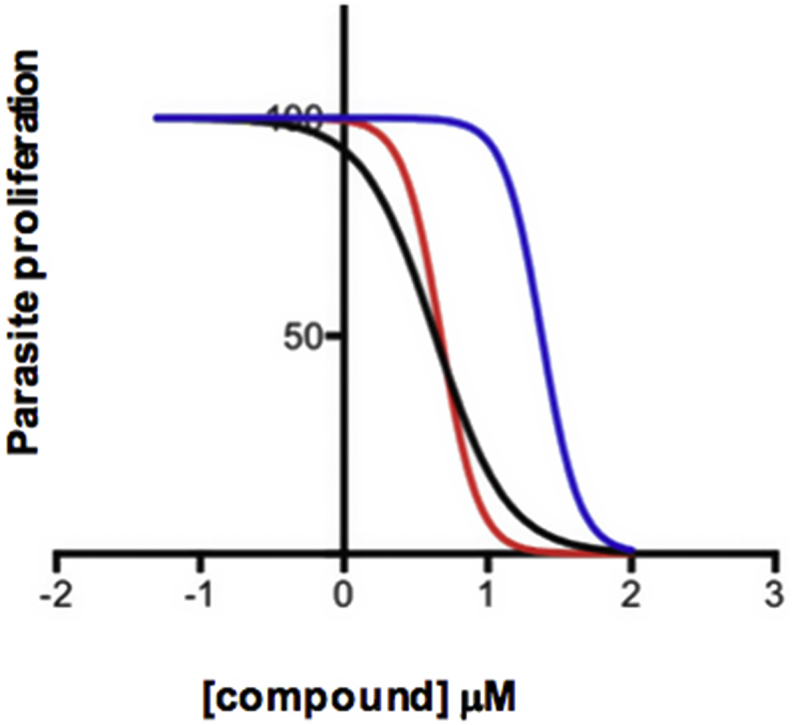
HD871-1 activity against L. mexicana axenic amastigotes (red plot line: EC50 = 4.73 μM; 95% Confidence Interval [CI] 3.65–6.12 μM); intramacrophage amastigotes with RAW264.7 cells (black plot line: EC50 = 4.32 μM; 95% CI 3.03–6.15 μM); uninfected RAW264.7 macrophages (blue plot line: EC50 = 23.8 μM; 95% CI 20.9–27.1 μM). From these data the compound was calculated to have a selectivity index of 5.51.
3.4. Structure elucidation of HD871-1
The ESI-MS spectrum of HD871-1 showed a molecular ion [MH]+ at m/z 263, indicating a molecular weight of 262. High resolution mass measurement of the sodiated molecular ion yielded an m/z value of 285.1095 Da, consistent with a molecular formula of C15H18O4 (the calculated value for C15H18NaO4 is 285.1097 Da). The UV spectrum of HD871-1 showed a maximum absorbance at 226 nm, suggesting the presence of an α,β-unsaturated carbonyl moiety. The 1H and 13C NMR data for HD871-1 are summarised in Table 2 and the 1H, COSY, HSQC, HMBC and NOESY NMR spectra are presented in Supplementary Figures 2-6 respectively. The 13C signals were detected indirectly through the heteronuclear shift correlation HSQC and HMBC spectra and were found to comprise two methyl carbons, four methylene carbons including an olefinic methylene group, four methine carbons including an olefinic methine and five quaternary carbons. Inspection of the 1H–1H COSY spectrum indicated the presence of two fragments comprising C6–C1–C2–C3 and C9–C10–C11–C13 as shown in Fig. 5. HMBC correlations from the methyl proton signal at 1.80 ppm (15-CH3) with carbon signals at 143.3, 135.9 and 198.4 ppm (corresponding to positions 2, 3 and 4, respectively) and from the methylene proton signals at 2.55 and 2.44 ppm (5-CH2) with the carbon signals at 198.4, 36.1 and 31.5 ppm (positions 4, 6 and 1) established the presence of a cyclohexenone ring. HMBC correlations from the olefinic methylene protons at 6.23 and 6.00 ppm to carbons at 36.1, 148.0 and 194.5 ppm (positions 6, 7 and 8) established the connectivity of these with the cyclohexenone moiety. HMBC correlations the methyl proton signal at 1.31 ppm with carbon signals at 33.1, 34.7 and 178.2 ppm were consistent with the presence of a butanolide ring. Although there were no HMBC correlations to connect this with the rest of the molecule, its linkage as shown in Fig. 5 was supported by NOE correlations in the NOESY spectrum between the olefinic methylene protons at 6.23 and 6.00 ppm (14-CH2) and the 9-oxymethine proton at 5.31 ppm. HD871-1 is therefore proposed to be a novel bisabolane sesquiterpene with a high degree of oxygenation and unsaturation.
Table 2.
1H and 13C NMR assignments for HD871-1 in CDCl3, referenced to internal TMS at 0.00 ppm
| Position | HD871-1 |
|
|---|---|---|
| δH/ppm, m (J/Hz) | δC/ppm | |
| 1 | 2.51, m; 2.27, ddt (18.0, 10.5, 2.5) | 31.5 |
| 2 | 6.73, m | 143.3 |
| 3 | 135.9 | |
| 4 | 198.4 | |
| 5 | 2.55, ddd (16.0, 3.9, 1.4); 2.44, dd (16.0, 12.8) | 42.8 |
| 6 | 3.35, m | 36.1 |
| 7 | 148.0 | |
| 8 | 194.5 | |
| 9 | 5.31, dd (9.2, 7.1) | 77.0 |
| 10 | 2.62, m; 2.02, m | 33.1 |
| 11 | 2.75, m | 34.7 |
| 12 | 178.2 | |
| 13 | 1.31, d (7.1) | 15.5 |
| 14 | 6.23, s; 6.00, s | 127.0 |
| 15 | 1.80, s | 16.1 |
Figure 5.
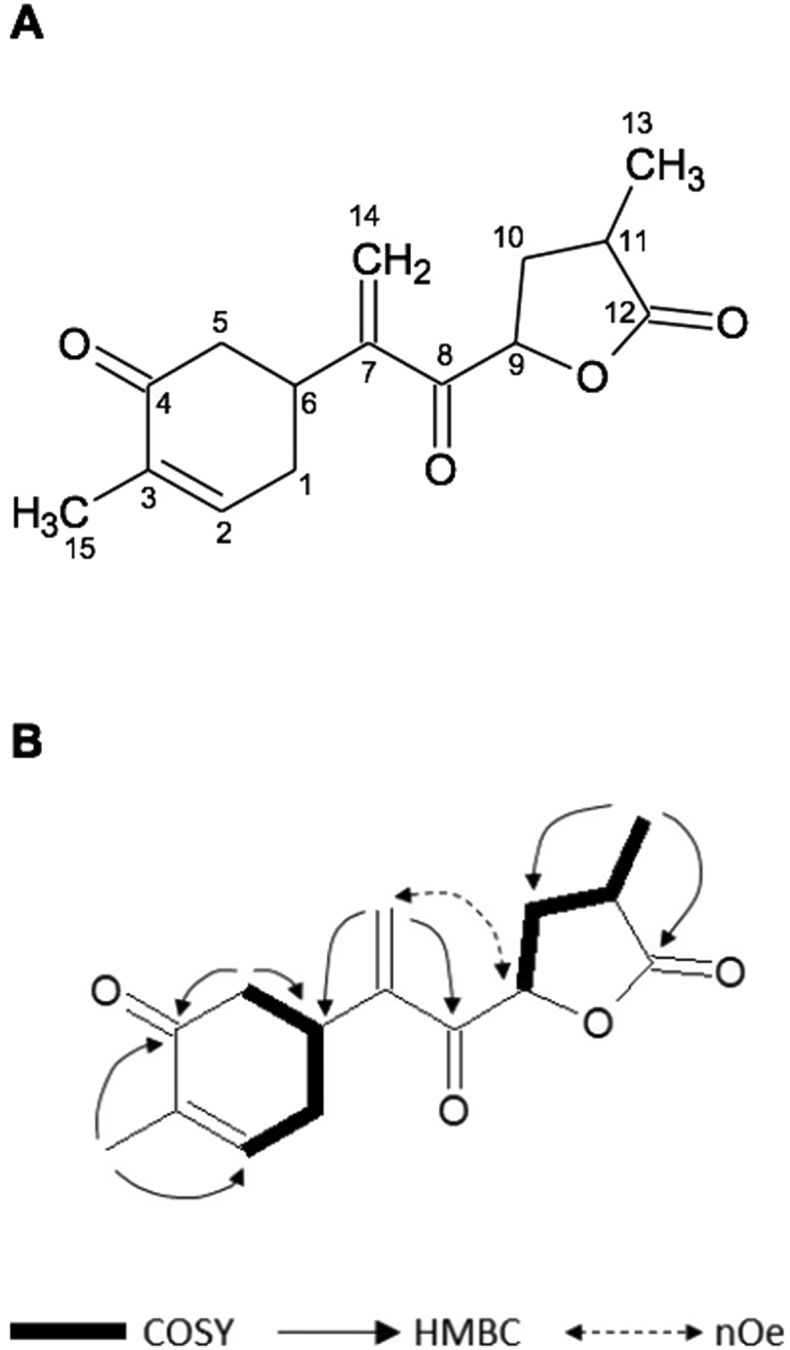
Structure of HD871-1 (A); key COSY, HMBC and NOE correlations (B).
3.5. Untargeted metabolomics assessment of metabolic perturbation induced by HD871-1
Metabolomics can enable identification of drug targets where these targets represent metabolic enzymes (Vincent and Barrett, 2015). For example, treatment of African trypanosomes with the drug eflornithine (difluoromethylornithine; DFMO) lead to profound increases in ornithine and loss of putrescine and spermidine, which relates to the drug being an inhibitor of the enzyme ornithine decarboxylase (Vincent et al., 2012). The untargeted metabolomics approach we took does not identify all metabolites within the metabolome, but it does probe many pathways and is able to indicate key areas of perturbed metabolism. The data is reported in Supplementary Table 1. Although multiple peptides and also lipids were noted to change profoundly, we discarded these from further consideration. Peptides often reveal profound changes in kinetoplastids exposed to different drugs as protein turnover is influenced in unpredictable ways. Lipids are not resolved using the hydrophilic chromatography platform used here, hence they tend to elute at the solvent front during chromatography and suffer poor peak shapes and multitudinous ion suppression effects. Substantial changes did, however, occur to metabolism across many pathways under the conditions used (Fig. 6). However, a clear, individual target could not be identified. Of note was an increase in deoxynucleotide monophosphates and a decrease in deoxynucleotide diphosphates (Fig. 7). These changes point to alterations in nucleotide homeostasis and metabolism, but the impact on cell viability is not certain. Profound decreases in orotate and dihydroorotate also point to an impact on pyrimidine biosynthesis, but pyrimidines themselves are not diminished, presumably due to the parasite's ability to transport and interconvert pyrimidines (Alzahrani et al., 2017). In the absence of the ability to synthesize the purine ring, salvage is essential for Leishmania viability (Boitz et al., 2012; Martin et al., 2016). The increase in dAMP and dGMP (Fig. 7) indicated this process was unaffected by HD871-1 treatment. However, dADP was substantially decreased suggesting that further phosphorylation maybe inhibited (Fig. 7). It was also noteworthy that a mass consistent with trypanothione, the bis(glutathionyl) spermidine adduct that performs key redox roles in Leishmania spp and other trypanosomatids, was greatly diminished in treated cells. The peak of this compound (m/z = 361.6527 corresponding to the doubly positively charged trypanothione) was poorly resolved by chromatography, hence quantitative comparison is not possible, but treated cells contained profoundly less signal for this mass than untreated cells. In addition to reduced trypanothione, its oxidised disulphide derivative, (m/z = 361.6441), was also 3–4 fold less abundant in treated cells, and its thiol precursor glutathione (mz = 307.0837) also diminished in abundance by >60% in the treated cells. Another poorly resolved peak consistent with the oxidised cysteine derivative, cystine (mz = 240.0238) was identified in treated, but not untreated cells. This accumulated data points to the cells having been exposed to oxidative stresses over the treatment regime.
Figure 6.

Principle Component Analysis (PCA) plot showing the global differences between treated and untreated cells. Five replicates of the measured metabolites from untreated (blue dots) and treated with HD871 (red dots). The green dots show pooled samples from both types and indicate good reproducibility across the machine run. PC1 and 2 = Principle Components.
Figure 7.

Effect on nucleotide metabolism in Leishmania mexicana axenic amastigotes treated with HD871-1, 10 μM for 36 h at 32 °C. Compared with untreated samples (DMSO, U), levels of dAMP, dTMP, dCMP and dGMP all climbed significantly during the course of treatment while the dinucleotides dADP and dTDP diminished (dGDP and dCDP were not identified). Orotate and dihydroorotate were both significantly diminished by treatment. y-axis is arbitrary units (AU).
4. Discussion
Given the paucity of safe and effective therapies for CL, and a rising number of cases in an increasing geographic area, the search for novel antileishmanials is as urgent as ever and natural product discovery forms part of this global effort (Charlton et al., 2018; Cockram and Smith, 2018). In this study, using an iterative screening and fractionation approach, we ‘mined’ the HDLSX library (313 fungal extracts, enriched in small molecule secondary metabolites) for compounds showing activity against Leishmania mexicana. A pure secondary fractionation component, HD871-1 (molecular weight of 262), was identified as a potent antileishmanial which demonstrated activity in an infected cell model (Fig. 4). This compound was characterised as a novel bisabolane sesquiterpene with a high degree of oxygenation and unsaturation (Fig. 5, Table 2). HD871-1 has structural similarities to cheimonophyllons A-E, bisabolane sesquiterpenes isolated from fermentations of the basidiomycete Cheimonophyllum candidissimum, which have been shown to demonstrate nematicidal properties (Stadler and Anke, 1994). Taken together, these studies indicate that this class of compounds could represent a new class of broad-spectrum of anti-parasitics.
However, to best take forward and develop any antileishmanial/antiparasitic the identification of the mode of action is key. To begin deconvolution of the target of HD871-1, in this study metabolomics was used to identify changes to the metabolic network of L. mexicana amastigotes exposed to HD871-1. Using a 36 h timepoint and compound dose 2-fold higher than the EC50, a number of clear changes to metabolism occurred (Supplementary Table 1), although a single clear hit pointing to a specific enzyme targeted by the drug was not apparent (as had been found, for example, using eflornithine against T. brucei, Vincent et al., 2012). Changes in the deoxynucleotide pool indicated possible changes in nucleotide homeostasis and metabolism, although whether this is a cause of cell death, or a secondary effect due to other changes is not clear. Loss of the chief redox reactive thiol trypanothione and accumulation of cystine pointed to oxidative stress being induced by HD871-1. The presence of two α,β-unsaturated ketone moieties in the structure of HD871-1 with the potential to act as Michael acceptors suggests a likely reactive nature towards appropriate nucleophiles such as trypanothione and glutathione. Since both reduced and oxidised trypanothione were diminished, as was glutathione, a possible role in drug binding prior to efflux from the cell, as occurs in the parasite response to antimony (Wyllie et al., 2004), would be worthy of future investigation.
5. Conclusions
Using an iterative process of screening and fractionation a novel bisabolane sesquiterpene antileishmanial was identified from a fungal extract library. Metabolomic analyses indicated that the compound induced oxidative stress in the parasite. Further analyses are required to fully define the mode of action, however the process described sets a precedent for the discovery of much needed novel antileishmanials.
Declarations of interest
None.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ijpddr.2019.05.003.
Acknowledgements and Funding
This project was supported by the Biotechnology and Biological Sciences Research Council through a Sparking Impact Award (Durham University) and a Proof of Concept grant (NPRONET, the natural products and bioengineering network). The research leading to these results has, in part, received funding from UK Research and Innovation via the Global Challenges Research Fund under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- Alzahrani K.J.H., Ali J.A.M., Eze A.A., Looi W.L., Tagoe D.N.A., Creek D.J., Barrett Michael P., de Koning H.P. Functional and genetic evidence that nucleoside transport is highly conserved in Leishmania species: implications for pyrimidine-based chemotherapy. Int J Parasitol Drugs Drug Resist. 2017;7 doi: 10.1016/j.ijpddr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anke T., Rabe U., Schu P., Eizenhofer T., Schrage M., Steglich W. Studies on the biosynthesis of striatal-type diterpenoids and the biological activity of herical. Z Natursforsch. 2002;57c:263–271. doi: 10.1515/znc-2002-3-411. [DOI] [PubMed] [Google Scholar]
- Aponte J.C., Castillo D., Estevez Y., Gonzalez G., Arevalo J., Hammond G.B., Sauvain M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010;20:100–103. doi: 10.1016/j.bmcl.2009.11.033. [DOI] [PubMed] [Google Scholar]
- Arruda D.C., D'Alexandri F.L., Katzin A.M., Uliana S.R. Antileishmanial activity of the terpene nerolidol. Antimicrob. Agents Chemother. 2005;49:1679–1687. doi: 10.1128/AAC.49.5.1679-1687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates P.A. Complete developmental cycle of Leishmania mexicana in axenic culture. Parasitology. 1994;108(Pt 1):1–9. doi: 10.1017/s0031182000078458. [DOI] [PubMed] [Google Scholar]
- Boeck P., Bandeira Falcao C.A., Leal P.C., Yunes R.A., Filho V.C., Torres-Santos E.C., Rossi-Bergmann B. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg. Med. Chem. 2006;14:1538–1545. doi: 10.1016/j.bmc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Boitz J.M., Ullman B., Jardim A., Carter N.S. Purine salvage in Leishmania: complex or simple by design? Trends Parasitol. 2012;28:345–352. doi: 10.1016/j.pt.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt H.L., Eggimann G.A., Denny P.W., Cobb S.L. Enlarging the chemical space of anti-leishmanials: a structure-activity relationship study of peptoids against Leishmania mexicana, a causative agent of cutaneous leishmaniasis. Med.Chem.Comm. 2016;7:799–805. [Google Scholar]
- Bray P.G., Barrett M.P., Ward S.A., de Koning H.P. Pentamidine uptake and resistance in pathogenic protozoa: past, present and future. Trends Parasitol. 2003;19:232–239. doi: 10.1016/s1471-4922(03)00069-2. [DOI] [PubMed] [Google Scholar]
- Burza S., Croft S.L., Boelaert M. Leishmaniasis. Lancet. 2018;392:951–970. doi: 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- Chadbourne F.L., Raleigh C., Ali H.Z., Denny P.W., Cobb S.L. Studies on the antileishmanial properties of the antimicrobial peptides temporin A, B and 1Sa. J. Pept. Sci. 2011;17:751–755. doi: 10.1002/psc.1398. [DOI] [PubMed] [Google Scholar]
- Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R.W., Alvar J., Boelaert M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- Charlton R.L., Rossi-Bergmann B., Denny P.W., Steel P.G. Repurposing as a strategy for the discovery of new anti-leishmanials: the-state-of-the-art. Parasitology. 2018;145:219–236. doi: 10.1017/S0031182017000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Christensen S.B., Theander T.G., Kharazmi A. Antileishmanial activity of licochalcone A in mice infected with Leishmania major and in hamsters infected with Leishmania donovani. Antimicrob. Agents Chemother. 1994;38:1339–1344. doi: 10.1128/aac.38.6.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockram P.E., Smith T.K. Active natural product scaffolds against trypanosomatid parasites: a review. J. Nat. Prod. 2018;81:2138–2154. doi: 10.1021/acs.jnatprod.8b00159. [DOI] [PubMed] [Google Scholar]
- Creek D.J., Jankevics A., Burgess K.E., Breitling R., Barrett M.P. IDEOM: an Excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics. 2012;28:1048–1049. doi: 10.1093/bioinformatics/bts069. [DOI] [PubMed] [Google Scholar]
- Creek D.J., Jankevics A., Rainer B., Watson D.G., Barrett M.P., Burgess K.E. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: improved metabolite identification by retention time prediction. Anal. Chem. 2011;83:8703–8710. doi: 10.1021/ac2021823. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Coombs G.H. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Sundar S., Fairlamb A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva E.R., Maquiaveli Cdo C., Magalhaes P.P. The leishmanicidal flavonols quercetin and quercitrin target Leishmania (Leishmania) amazonensis arginase. Exp. Parasitol. 2012;130:183–188. doi: 10.1016/j.exppara.2012.01.015. [DOI] [PubMed] [Google Scholar]
- de Mello T.F., Bitencourt H.R., Pedroso R.B., Aristides S.M., Lonardoni M.V., Silveira T.G. Leishmanicidal activity of synthetic chalcones in Leishmania (Viannia) braziliensis. Exp. Parasitol. 2014;136:27–34. doi: 10.1016/j.exppara.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Demicheli C., Ochoa R., da Silva J.B., Falcao C.A., Rossi-Bergmann B., de Melo A.L., Sinisterra R.D., Frezard F. Oral delivery of meglumine antimoniate-beta-cyclodextrin complex for treatment of leishmaniasis. Antimicrob. Agents Chemother. 2004;48:100–103. doi: 10.1128/AAC.48.1.100-103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio C., Faraut-Gambarelli F., Imbert A., Minodier P., Gasquet M., Dumon H. Flow cytometric assessment of amphotericin B susceptibility in Leishmania infantum isolates from patients with visceral leishmaniasis. J. Antimicrob. Chemother. 1999;44:71–76. doi: 10.1093/jac/44.1.71. [DOI] [PubMed] [Google Scholar]
- do Socorro S.R.M.S., Mendonca-Filho R.R., Bizzo H.R., de Almeida Rodrigues I., Soares R.M., Souto-Padron T., Alviano C.S., Lopes A.H. Antileishmanial activity of a linalool-rich essential oil from Croton cajucara. Antimicrob. Agents Chemother. 2003;47:1895–1901. doi: 10.1128/AAC.47.6.1895-1901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher J.D. The discovery and development of amphotericin B. Dis. Chest. 1968;54(Suppl. 1):296–298. doi: 10.1378/chest.54.supplement_1.296. [DOI] [PubMed] [Google Scholar]
- Eggimann G.A., Bolt H.L., Denny P.W., Cobb S.L. Investigating the anti-leishmanial effects of linear peptoids. ChemMedChem. 2015;10:233–237. doi: 10.1002/cmdc.201402416. [DOI] [PubMed] [Google Scholar]
- Gehrt A., Erkel G., Anke T., Sterner O. Nitidon, a new bioactive metabolite from the basidiomycete Junghuhnia nitida (Pers. Fr.) Ryv. Z. Naturforsch. C. 1998;53:89–92. doi: 10.1515/znc-1998-1-216. [DOI] [PubMed] [Google Scholar]
- Inchausti A., Yaluff G., deArias A.R., Torres S., Ferreira M.E., Nakayama H., Schinini A., Lorenzen K., Anke T., Fournet A. Leishmanicidal and trypanocidal activity of extracts and secondary metabolites from basidiomycetes. Phytother Res. 1997;11:193–197. [Google Scholar]
- Kedzierski L., Sakthianandeswaren A., Curtis J.M., Andrews P.C., Junk P.C., Kedzierska K. Leishmaniasis: current treatment and prospects for new drugs and vaccines. Curr. Med. Chem. 2009;16:599–614. doi: 10.2174/092986709787458489. [DOI] [PubMed] [Google Scholar]
- Khare S., Nagle A.S., Biggart A., Lai Y.H., Liang F., Davis L.C., Barnes S.W., Mathison C.J., Myburgh E., Gao M.Y. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovářová J., Pountain A.W., Wildridge D., Weidt S., Bringaud F., Burchmore R.J.S., Achcar F., Barrett M.P. Deletion of transketolase triggers a stringent metabolic response in promastigotes and loss of virulence in amastigotes of Leishmania mexicana. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1006953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J.L., Yates P.A., Boitz J.M., Koop D.R., Fulwiler A.L., Cassera M.B., Ullman B., Carter N.S. A role for adenine nucleotides in the sensing mechanism to purine starvation in Leishmania donovani. Mol. Microbiol. 2016;101:299–313. doi: 10.1111/mmi.13390. [DOI] [PubMed] [Google Scholar]
- Mishra B.B., Singh R.K., Srivastava A., Tripathi V.J., Tiwari V.K. Fighting against Leishmaniasis: search of alkaloids as future true potential anti-Leishmanial agents. Mini Rev. Med. Chem. 2009;9:107–123. doi: 10.2174/138955709787001758. [DOI] [PubMed] [Google Scholar]
- Mittra B., Saha A., Chowdhury A.R., Pal C., Mandal S., Mukhopadhyay S., Bandyopadhyay S., Majumder H.K. Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Mol. Med. 2000;6:527–541. [PMC free article] [PubMed] [Google Scholar]
- Norcliffe J.L., Mina J.G., Alvarez E., Cantizani J., de Dios-Anton F., Colmenarejo G., Valle S.G., Marco M., Fiandor J.M., Martin J.J. Identifying inhibitors of the Leishmania inositol phosphorylceramide synthase with antiprotozoal activity using a yeast-based assay and ultra-high throughput screening platform. Sci. Rep. 2018;8:3938. doi: 10.1038/s41598-018-22063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena I., Pilar Manzano M., Cantizani J., Kessler A., Alonso-Padilla J., Bardera A.I., Alvarez E., Colmenarejo G., Cotillo I., Roquero I. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci. Rep. 2015;5:8771. doi: 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithinger R., Dujardin J.C., Louzir H., Pirmez C., Alexander B., Brooker S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007;7:581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- Scheltema R.A., Jankevics A., Jansen R.C., Swertz M.A., Breitling R. PeakML/mzMatch: a file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal. Chem. 2011;83:2786–2793. doi: 10.1021/ac2000994. [DOI] [PubMed] [Google Scholar]
- Stadler M., Anke H. New nematicidal and antimicrobial compounds from the basidiomycete Cheimonophyllum candidissimum (Berk & Curt.) sing. I. Producing organism, fermentation, isolation, and biological activities. J. Antibiot. (Tokyo) 1994;47:1284–1289. doi: 10.7164/antibiotics.47.1284. [DOI] [PubMed] [Google Scholar]
- Stuart K., Brun R., Croft S., Fairlamb A., Gurtler R.E., McKerrow J., Reed S., Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Investig. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner L.W., Amberg A., Barrett D., Beale M.H., Beger R., Daykin C.A., Fan T.W., Fiehn O., Goodacre R., Griffin J.L. Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI) Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C.P., Singh R.K., Hassan S.M., Kumar R., Narain S., Kumar A. Amphotericin B deoxycholate treatment of visceral leishmaniasis with newer modes of administration and precautions: a study of 938 cases. Trans. R. Soc. Trop. Med. Hyg. 1999;93:319–323. doi: 10.1016/s0035-9203(99)90037-8. [DOI] [PubMed] [Google Scholar]
- Vincent I.M., Barrett M.P. Metabolomic-based strategies for anti-parasite drug discovery. J. Biomol. Screen. 2015;20:44–55. doi: 10.1177/1087057114551519. [DOI] [PubMed] [Google Scholar]
- Vincent I.M., Creek D.J., Burgess K., Woods D.J., Burchmore R.J., Barrett M.P. Untargeted metabolomics reveals a lack of synergy between nifurtimox and eflornithine against Trypanosoma brucei. PLoS NTD. 2012;6:e1618. doi: 10.1371/journal.pntd.0001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S., Cunningham M.L., Fairlamb A.H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004;279:39925–39932. doi: 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.