

Abstract
Background:
Heritability estimates (including twin and SNP-based heritability studies) for fibroids have been inconsistent across prior studies ranging between 8.71 and 69%. These inconsistencies are due to variations in study design and included populations. A major design issue has been lack imaging confirmation to identify control, where asymptomatic women without imaging confirmation may be misclassified as controls leading to an attenuation of heritability estimates. To reconcile the differences in prior heritability estimates and the impact of misclassification of controls on heritability we determined SNP-based heritability and characterized the genetic architecture of pelvic image-confirmed fibroid cases and controls.
Methods:
Analyses were performed among women of European American (EA) descent using genome-wide single nucleotide polymorphism (SNP) data from BioVU, a clinical database composed of DNA linked to de-identified electronic health records. We estimated the genetic variance explained by all SNPs using Genome-wide Complex Trait Analysis on imputed data. Fibroid cases and controls were identify using a previously reported phenotyping algorithm, that required pelvic imaging confirmation.
Results:
In total, we used 1,067 image-confirmed fibroid cases and 1,042 image-confirmed fibroid controls. The SNP-based heritability estimate for fibroid risk was h2 = 0.33±0.18, p-value = 0.040. We investigated the relationship between heritability per chromosome and chromosome length (r2 < 1%), with Chromosome 8 explaining the highest proportion of variance for fibroid risk. There was no enrichment for intergenic or genic SNPs for the fibroid SNP-based heritability. Excluding loci previously associated with fibroid risk from genome-wide association study (GWAS) did not attenuate fibroid heritability suggesting that loci associating with fibroid risk are yet to be discovered.
Conclusion(s):
We observed that fibroid SNP-based heritability was higher than the previous estimate using genome-wide SNP data that relied on self-reported outcomes, but within range of prior twin-pair studies. Furthermore, these data support that imprecise phenotyping can significant effect the ability to estimate heritability using genotype data.
Keywords: Leiomyomata, heritability, genome-wide SNP data, uterus, ultrasound
INTRODUCTION
Uterine leiomyoma (fibroids) are benign tumors of the uterus,[1] and up to 70% of women will develop a fibroid by menopause[2]. Symptoms of fibroids include abnormal bleeding during menstruation, increased urinary frequency, and pelvic pressure[3,4]. Likely because of symptoms, fibroids are the leading cause of hysterectomy in the US[5]. Annual US costs for fibroids is estimated to be between 5.9 to 34.4 billion dollars[6].
Heritability estimates for fibroids have been inconsistent across prior studies. One twin study of European American (EA) women, limiting to women who underwent hysterectomies with pelvic imaging used to determine fibroid status, estimated fibroid heritability to be 69%[7].[7] Another twin study randomly selected samples of monozygotic and dizygotic twins from the Finnish Twin Cohort study and evaluated ultrasound characteristics across twin pairs with and without fibroids; this study estimated the heritability for fibroid number to be 26% [8]. [8] Finally, a study using genome-wide single nucleotide polymorphism (SNP) data estimated the SNP-based heritability of fibroids to be 8.71%[9]. Although, well powered (N=57,151) this study had a low fibroid prevalence (2.77%) and relied on self-report fibroid status[9].[9] It is possible that the inconsistencies across heritability estimates across these studies is due to how fibroids were classified and how subjects were recruited. For example, up to 51% of asymptomatic cases may be misclassified as controls if a study relied on self-report alone, thus self-reported studies result in phenotypic heterogeneity[2,9].
There have been three large-scale genetic studies of fibroids that include two genome-wide association studies (GWAS) of fibroids and one genome-wide linkage scan[10–12]. The GWAS by Cha et al. (2011) found three loci, 10q24.33 (rs7913069; p = 8.65×10−14), 22q13.1 (rs12484776; p = 2.79×10−12), and 11p15.5 (rs2280543; p = 3.82×10−12) associated with fibroid risk in a Japanese cohort[11]. These loci were replicated in an EA population[13,14]. In another GWAS performed among African American (AA) women a genome-wide significant association was observed between rs739187 in cytohesin 4 (CYTH4) in 22q13.1 and fibroid risk[12]. Finally, a linkage study identified two significant linkage peaks at 10p11 (logarithm of odds [LOD] = 4.15) and 3p21 (LOD = 3.73). They also identified a SNP (CCDC57, rs4247357, p = 3.05×10−8) associated with fibroids within the linkage regions.
Current GWAS of fibroids have identified only a few variants that explain a small proportion of fibroid risk, as expected based on the available sample sizes. Better characterization of fibroid heritability, with well phenotyped populations, is needed to understand the genetic architecture influencing risk for fibroids. Our objective was to reconcile differences reported across heritability estimates from twin studies and those using GWAS data and to assess the role of phenotyping on heritability estimates by estimating and characterizing the SNP-based heritability of image-confirmed fibroids in EA women using GWAS data.
MATERIALS AND METHODS
Study Population
Detailed descriptions of the biorepository at Vanderbilt University Medical Center (BioVU) have been reported previously[15,16]. BioVU (2007-present) consists of DNA linked to de-identified EHRs. The EHRs include clinical and demographic data for each patient at the Vanderbilt University Medical Center[15]. Studies using the BioVU are classified as non-human subjects research by the Institutional Review Boards and the Office of Human Research Protections[16].
Fibroid cases and controls were identified using a validated phenotyping algorithm with a positive predictive value of 96% and a negative predictive value of 98% [17]. The algorithm classified an individual as a case if the individual was listed as white, was at least 18 years old, and had international classification of diseases, ninth revision, (ICD-9) or current procedural terminology (CPT) codes for at least one pelvic imaging or surgery for treating fibroids and for a fibroid diagnosis[17]. Controls were at least 18 years old, had at least two ICD-9 or CPT codes for pelvic imaging, and had no history of myomectomy, hysterectomy, or uterine artery embolization[17] and were group matched by ancestry to cases. We also conducted a case-only analysis to evaluate the SNP-based heritability of fibroid size. To determine fibroid size, we manually abstracted fibroid measurements from imaging and surgical reports. Patient demographic information was recorded at the time of diagnosis.
Outcome measurements include fibroid presence (case vs control), largest fibroid dimension of all measurements, and largest fibroid volume. Fibroid volume was calculated using the equation of an ellipsoid: (Length × Width × Height × 0.523). For the outcome to have a normal distribution, fibroid largest dimension and volume were log10 transformed. Approximately a third of individuals with volume measurements had their third fibroid dimension measurement imputed by taking the mean of the first two measurements. This study was evaluated and approved by the Vanderbilt University Medical Center Institutional Review Board. In addition, all methods were carried out in accordance with relevant regulations and guidelines.
Genotyping
Genotyping of BioVU EA individuals was performed on the Affymetrix Axiom Biobank array (Affymetrix, Inc., Santa Clara, CA) using PicoGreen (Invitrogen, Inc., Grand Island, NY) to purify and to quantify the DNA.
Quality Control (QC) of Genome-Wide Genotype Data
Genetic data had standard QC prior to imputation using PLINK 1.07 software[18]. Pre-imputed QC included: removing subjects with low genotyping efficiency (<95%), removing related individuals (both individuals from a pair with a probability of identify by descent (IBD) > 0.95 and one from a pair with a probability of IBD from 0.2–0.95), removing individuals with inconsistency between genetic and reported sex. Additionally, we removed SNPs out of Hardy-Weinberg equilibrium (HWE) (p≤10−6), SNPs with a low genotyping efficiency (<95%), SNPs with a minor allele frequency (MAF) < 0.01, and SNPs without a chromosome location. Finally, SNPs were aligned to the + strand of the 1000 Genomes (build 37, 2013).
We imputed non-genotyped SNPs using IMPUTE2.3.0 software[19] and subsequently converted the imputed genetic to PLINK 1.07 format[18] after filtering SNPs with a info score ≤0.95 and removing insertion/deletion polymorphisms. We then applied the Genome-wide Complex Trait Analysis (GCTA)-specific QC that was performed by Hong Lee et al. (2011)[20] which included: removing SNPs with a MAF < 0.05, removing related individuals (one from a pair with a probability of IBD >0.05, and removing SNPs out of HWE (≥0.05). Post-QC SNP numbers included 4,500,362 for fibroid risk, 4,522,829 for fibroid volume, and 4,518,340 for largest fibroid dimension (Supplemental Table 1). In addition, post-QC numbers of individuals for each outcome are shown in Supplemental Table 1.
Statistical Analyses
Covariate and demographic data were summarized by Stata/SE (College Station, Texas). Principal components (PCs) were created using EIGENSTRAT4.2 software[21]. To reduce admixture within BioVU, we removed all samples whose first PC (PC1) and second PC (PC2) were more than four standard deviations from the mean of PC1 and PC2 of the European (EUR) individuals (Supplemental Figures 1–2).
We estimated the genetic variance explained by all SNPs using GCTA software[22]. GCTA creates a genetic relationship matrix (GRM) between pairs of individuals within a cohort using all SNPs. GCTA uses a mixed linear model (MLM) that estimates the genetic variance explained by all SNPs for a trait using the restricted maximum likelihood approach (REML) [22].[22] The final model for fibroid risk included the GRM which was modeled as a random effects term and the fixed effects terms age at diagnosis, BMI, and top five PCs. We transformed the observed estimate of SNP-based heritability to the liability scale using a fibroids prevalence estimate of 70%. A prevalence estimate of 70% was used in models on fibroid risk because the mean age of our cohort was 50.8 and previous cumulative incidence estimates were ~70% for women near 51 years of age[2].[2] SNP-based heritability estimates for a range of prevalences are shown in Supplemental Table 2. Secondary analyses included estimating SNP-based heritability of fibroid size, namely volume and largest dimension. Final models for fibroid volume and max dimension adjusted for age at diagnosis, body max index (BMI), and five PCs without a prevalence adjustment. To show that potential residual population stratification did not lead to spurious SNP-based heritability estimates, we performed sensitivity analyses where SNP-based heritability estimates were calculated adjusting for age at diagnosis, BMI, and varying numbers of PCs.
To characterize the genetic architecture of fibroids, we partitioned the genetic variance into separate GRMs by chromosomes, by genic and intergenic regions. We annotated SNP location into genic/intergenic regions using ANNOVAR software[23] We used a distance of one kilobase (kb) from a transcript as the boundary threshold for genic and intergenic SNPs. Additional subanalyses involved excluding genomic regions previously implicated in fibroid risk based on GWAS. Exclusion criteria included selecting SNPs from final meta-analyses of GWAS[11,12] with a p-value ≤ 1.00×10−6 and subsequently removing all loci ± 0.5 mega bases (Mb) of these selected SNPs, to account for linkage disequilibrium in the region. Lastly, there is a common inversion that spans approximately 12 cM on chromosome 8 encompassing 8p23.1–8p22 with a frequency of approximately 21% in European populations[24]. We determined the effect that this inversion has on fibroid risk by removing it from chromosome-specific SNP-based heritability estimates and comparing the difference in point estimates.
RESULTS
Demographic Data
There were 2,109 EA women (1,067 cases and 1,042 controls) included in these analyses (Table 1). The mean age was 50.8±16 years, and most women were overweight or obese (mean BMI: 28.6±7 km/m2). There were 373 cases with fibroid volume measurements (median of largest volume: 9.6 cm3) and 551 cases with fibroid dimension measurements (median of largest dimension: 2.6 cm).
Table 1.
Demographics table of BioVU European American women
| Demographic Characteristics | N | BioVU |
|---|---|---|
| Age (mean±SD) | 2,109 | 50.8±16 |
| BMI (kg/m2) (mean±SD) | 2,109 | 28.6±7 |
| Fibroid Risk | 2,109 | |
| Control (%) | 1,042 | 49 |
| Case (%) | 1,067 | 51 |
| Fibroid Volume (cm3) median (IQR) | 373 | 9.6 (2.3–39.0) |
| Largest Fibroid Dimension (cm) median (IQR) |
551 | 2.6 (1.6–4.4) |
BMI-body mass index; kg/m2-kilograms per meters squared; cm3-cubic centimeters; cm-centimeters; SD-standard deviation; IQR-interquartile range.
SNP-Based Heritability Estimation
The SNP-based heritability estimate for fibroid risk, or the proportion of phenotypic variance that was explained by genetic variance, was h2 = 0.33±0.18, p-value 0.040 (Table 2), and this estimate was robust to a MAF threshold of 1%. The SNP-based heritability estimates for fibroid largest dimension was h2 = 0.35±0.47, p-value 0.238, and for volume was h2 = 0.14±0.66, p-value 0.417. These SNP-based heritability estimates for each outcome were similar after adjusting for a range of PCs (risk h2: 0.29–0.38; largest dimension h2: 0.34–0.44; volume h2: 0.12–0.35) (Supplemental Tables 3–5).
Table 2.
SNP-based heritability estimates for fibroid risk using European Americans within BioVU
| Outcome | SNP-Based Heritability | P-value |
|---|---|---|
| Estimate ± (Std. Err.) | ||
| Fibroid Risk a | 0.33 ± (0.18) | 0.040a |
| Largest Fibroid Dimension | 0.35 ± (0.47) | 0.238 |
| Largest Fibroid Volume | 0.14 ± (0.66) | 0.417 |
Model: Adjusted for age, BMI, and 5 PCs.
SNPs are limited to ≥5% MAF.
a: Prevalence estimates of fibroid risk is set at 70%.
Statistically significant
The correlation (r2) between chromosome length in base pairs and SNP-based heritability per chromosome was 0.89% (p = 0.669) (Figure 1). Chromosome 8 explained the greatest proportion of variance for fibroid risk (8.76%), followed by chromosome 10 (5.57%), 11 (5.11%), and 7 (2.65%). The summation of the SNP-based heritability estimate of each chromosome was 34%, which is not significantly different from the univariate estimate suggesting that residual population stratification and cryptic relatedness are well controlled[25]. In addition, there is an approximately 12 cM common inversion present at a frequency of 21% in EA populations on chromosome 8 spanning 8p23.1–8p22[24]. It is possible that this inversion could affect the disproportionate contribution of SNP-based heritability for chromosome 8. Because of this, we estimated fibroid SNP-based heritability on chromosome 8 excluding 8p23.1–8p22, 8p, and 8q, respectively (Supplemental Figure 3). We found that excluding 8p23.1–8p22 did not change the contribution of SNP-based heritability from chromosome 8. We also found that most of the SNP-based heritability from chromosome 8 was located in 8q. Even though fibroids are a sex-linked disease and have a hormonal component especially with estrogen,[26] we did not observe enrichment of SNP-based heritability on chromosome X which contributed about 1% to the overall estimate (proportional to the number of X chromosome SNPs included in the analysis).
Figure 1.
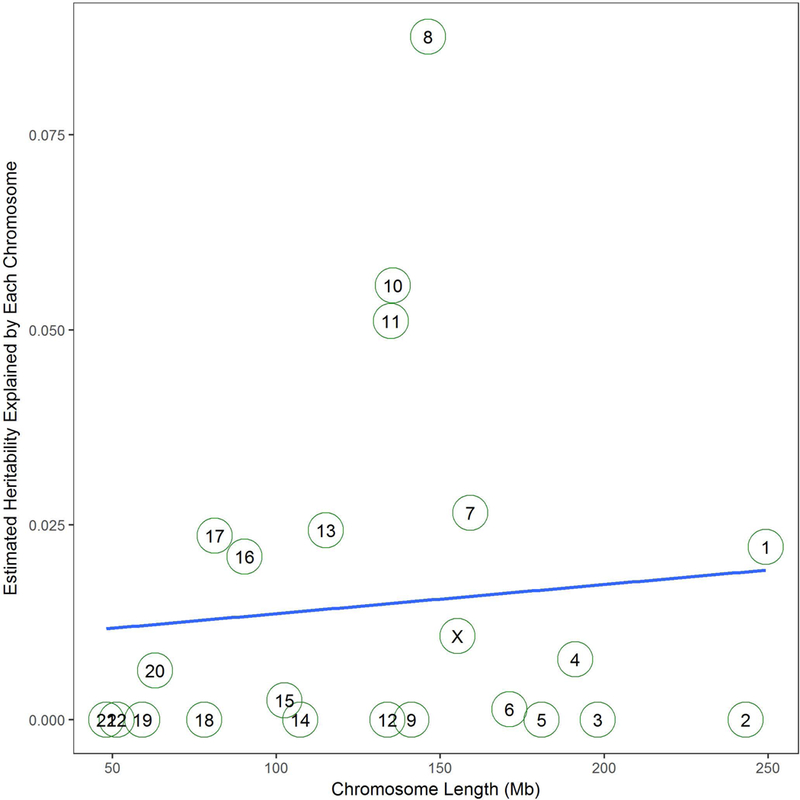
Estimated SNP-based heritability of each chromosome by chromosome length in Mb. R2 is 0.89%, and the p-value of the model is 0.669.
We also partitioned the genetic data into genic and intergenic regions resulting in 2,801,797 intergenic SNPs (62.3%) and 1,694,891 genic SNPs (37.7%) (Table 3). There was no enrichment for intergenic or genic SNPs for the SNP-based heritability of fibroid risk (Figure 2).
Table 3.
SNP-based heritability estimates from partitioned SNPs into ntergenic and genic regions
| Location | SNP-based Heritability Estimate ± (Sth. Err.) |
Number of SNPsa |
|---|---|---|
| Intergenic | 0.21 ± 0.15 | 2.80 |
| Genic | 0.12 ± 0.14 | 1.69 |
Model: Adjusted for age, BMI, and 5 PCs.
SNPs are limited to ≥5% MAF.
Prevalence estimates of fibroids are set at 70%.
a: In 1,000,000 SNPs
Figure 2.
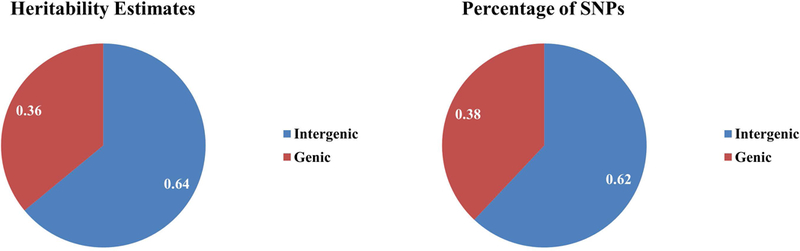
Proportions of SNP-based heritability estimates and percentage of SNPs pie charts for SNPs that were partitioned into intergenic and genic SNPs.
SNP-Based Heritability Explained by Known Fibroid GWAS Loci
The genomic regions that met the exclusion criteria were 10q24.33–10q25.1, 11p15.5, and 22q13.1–22q13.2 from the Cha et al. (2011) GWAS[11] and 3p26.1, 8p23.2, 8q21.11, and 22q12.3–22q13.1 from the Hellwege et al. (2017) GWAS[12]. Excluding genetic loci previously associated with fibroid risk from all previous GWAS[11,12] did not attenuate the estimated SNP-based heritability of fibroid risk (h2 from all studies = 0.34) (Supplemental Table 6).
DISCUSSION
This was the first study to estimate and characterize the SNP-based heritability of fibroids using a dataset of EA women with image-confirmed fibroid status. Estimated SNP-based heritability of fibroid risk was 0.33±0.18. Chromosome 8 contributed the greatest proportion of SNP-based heritability and we observed little correlation between chromosome length and chromosomal SNP-based heritability. There was no enrichment of fibroid SNP-based heritability for intergenic or genic regions. Finally, censoring genetic loci previously implicated in fibroid risk did little to attenuate the estimated SNP-based heritability.
Our SNP-based heritability estimate for fibroid risk was within the range of estimates previously reported in twin studies (26–69%)[7,8]. The first study by Snieder et al. (1998) conducted in a sample of 98 MZ and 125 DZ twin pairs from the United Kingdom found the heritability of fibroids to be 69%. At least one twin from each pair underwent a hysterectomy[7]. The high heritability from this study could be because they used hysterectomy as a criteria for selecting participants, which may results in overrepresentation of severe fibroids and thus greater genetic liability. The second study conducted ultrasounds on randomly selected twin pairs (17 MZ and 16 DZ) from the Finnish Twin Cohort. They estimated heritability of fibroid number at 26%, closer to our fibroid risk heritability estimate[8].
Our SNP-based heritability estimate was higher than previous SNP-based heritability estimates from genome-wide SNP data by Ge et al. (2017) [9]. This study had a sample size of 57,151 composed of women from the UK Biobank aged 40 to 69 with self-reported outcomes[9] and a fibroid prevalence of only 2.77%. When compared to the general population this prevalence rate is low suggesting potential misclassification in phenotype definition[9]. For example, a study by Baird et al. (2003) observed that the cumulative incidence of fibroids to be approximately 70% for white women aged 49 years in a study where women were systematically screened by ultrasounds [2]. The large discrepancy between prevalence estimates between the two studies could signal potential misclassification for fibroid controls resulting in a lower SNP-based heritability estimate in their study.
While we estimated the SNP-based heritability for fibroid largest dimension and volume, our same sizes were too small to get reliable estimates. Future studies with larger sample sizes are needed to evaluate the SNP-based heritability of fibroid size. In addition, while our phenotyping algorithm is improved with regard to outcome classification of cases and controls [17], our sample size was on the minimum threshold for performing such SNP-based heritability estimates.
We present the first estimate of fibroid SNP-based heritability using a dataset of EA women with image-confirmed fibroid status using genotype data. We observed that fibroid SNP-based heritability in our study was higher than a previous genetic estimate using GWAS data which we believe is due to including only image-confirmed cases and controls in our analysis. In addition, we observed that our fibroid SNP-based heritability estimate was not significantly attenuated when censoring genetic loci previously associated with fibroid risk, suggesting that many of the genetic loci that are associated with fibroid risk in EA women are yet to be discovered. Larger future studies on fibroid risk are needed to understand the genetic underpinnings of fibroid SNP-based heritability in EA women.
Supplementary Material
ACKNOWLEDGEMENTS
This study was funded by the National Institutes of Health (NIH) grants (R01HD074711, R03HD078567, and R01HD093671) to Digna R. Velez Edwards and by the Human Genetic Training Grant (5T32GM080178) and the VICTR Training Grant (6TL1TR000447) to Michael J. Bray.
The publication described was supported by CTSA award No. UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
FUNDING SOURCES
This study was funded by the National Institutes of Health (NIH) grants (R01HD074711, R03HD078567, and R01HD093671) to Digna R. Velez Edwards and by the Human Genetic Training Grant (5T32GM080178) and the VICTR Training Grant (6TL1TR000447) to Michael J. Bray.
The publication described was supported by CTSA award No. UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Footnotes
STATEMENT OF ETHICS
The authors have no ethical conflicts to disclose.
DISCLOSURE STATEMENT
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Stewart EA: Uterine fibroids. Lancet 2001;357:293–298. [DOI] [PubMed] [Google Scholar]
- 2.Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM: High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol 2003;188:100–107. [DOI] [PubMed] [Google Scholar]
- 3.De La Cruz MS, Buchanan EM: Uterine Fibroids: Diagnosis and Treatment. American family physician 2017;95:100–107. [PubMed] [Google Scholar]
- 4.Zimmermann A, Bernuit D, Gerlinger C, Schaefers M, Geppert K: Prevalence, symptoms and management of uterine fibroids: an international internet-based survey of 21,746 women. BMC women’s health 2012;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farquhar CM, Steiner CA: Hysterectomy rates in the United States 1990–1997. Obstetrics and gynecology 2002;99:229–234. [DOI] [PubMed] [Google Scholar]
- 6.Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH: The estimated annual cost of uterine leiomyomata in the United States. American journal of obstetrics and gynecology 2012;206:211 e211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snieder H, MacGregor AJ, Spector TD: Genes control the cessation of a woman’s reproductive life: a twin study of hysterectomy and age at menopause. The Journal of clinical endocrinology and metabolism 1998;83:1875–1880. [DOI] [PubMed] [Google Scholar]
- 8.Luoto R, Kaprio J, Rutanen EM, Taipale P, Perola M, Koskenvuo M: Heritability and risk factors of uterine fibroids--the Finnish Twin Cohort study. Maturitas 2000;37:15–26. [DOI] [PubMed] [Google Scholar]
- 9.Ge T, Chen CY, Neale BM, Sabuncu MR, Smoller JW: Phenome-wide heritability analysis of the UK Biobank. PLoS genetics 2017;13:e1006711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eggert SL, Huyck KL, Somasundaram P, Kavalla R, Stewart EA, Lu AT, Painter JN, Montgomery GW, Medland SE, Nyholt DR, Treloar SA, Zondervan KT, Heath AC, Madden PA, Rose L, Buring JE, Ridker PM, Chasman DI, Martin NG, Cantor RM, Morton CC: Genome-wide linkage and association analyses implicate FASN in predisposition to Uterine Leiomyomata. American journal of human genetics 2012;91:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha PC, Takahashi A, Hosono N, Low SK, Kamatani N, Kubo M, Nakamura Y: A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nature genetics 2011;43:447–450. [DOI] [PubMed] [Google Scholar]
- 12.Hellwege JN, Jeff JM, Wise LA, Gallagher CS, Wellons M, Hartmann KE, Jones SF, Torstenson ES, Dickinson S, Ruiz-Narvaez EA, Rohland N, Allen A, Reich D, Tandon A, Pasaniuc B, Mancuso N, Im HK, Hinds DA, Palmer JR, Rosenberg L, Denny JC, Roden DM, Stewart EA, Morton CC, Kenny EE, Edwards TL, Velez Edwards DR: A multi-stage genome-wide association study of uterine fibroids in African Americans. Human genetics 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards TL, Hartmann KE, Velez Edwards DR: Variants in BET1L and TNRC6B associate with increasing fibroid volume and fibroid type among European Americans. Hum Genet 2013;132:1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards TL, Michels KA, Hartmann KE, Velez Edwards DR: BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum Genet 2013;132:943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, Masys DR: Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clinical pharmacology and therapeutics 2008;84:362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR: Principles of human subjects protections applied in an opt-out, de-identified biobank. Clinical and translational science 2010;3:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feingold-Link L, Edwards TL, Jones S, Hartmann KE, Velez Edwards DR: Enhancing uterine fibroid research through utilization of biorepositories linked to electronic medical record data. J Womens Health (Larchmt) 2014;23:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howie BN, Donnelly P, Marchini J: A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SH, Wray NR, Goddard ME, Visscher PM: Estimating missing heritability for disease from genome-wide association studies. American journal of human genetics 2011;88:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Lee SH, Goddard ME, Visscher PM: GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broman KW, Matsumoto N, Giglio S, Martin CL, Roseberry JA, Zuffardi O, Ledbetter DH, Weber JL: Common long human inversion polymorphism on chromosome 8p. Lecture Notes-Monograph Series 2003:237–245. [Google Scholar]
- 25.Davis LK, Yu D, Keenan CL, Gamazon ER, Konkashbaev AI, Derks EM, Neale BM, Yang J, Lee SH, Evans P, Barr CL, Bellodi L, Benarroch F, Berrio GB, Bienvenu OJ, Bloch MH, Blom RM, Bruun RD, Budman CL, Camarena B, Campbell D, Cappi C, Cardona Silgado JC, Cath DC, Cavallini MC, Chavira DA, Chouinard S, Conti DV, Cook EH, Coric V, Cullen BA, Deforce D, Delorme R, Dion Y, Edlund CK, Egberts K, Falkai P, Fernandez TV, Gallagher PJ, Garrido H, Geller D, Girard SL, Grabe HJ, Grados MA, Greenberg BD, Gross-Tsur V, Haddad S, Heiman GA, Hemmings SM, Hounie AG, Illmann C, Jankovic J, Jenike MA, Kennedy JL, King RA, Kremeyer B, Kurlan R, Lanzagorta N, Leboyer M, Leckman JF, Lennertz L, Liu C, Lochner C, Lowe TL, Macciardi F, McCracken JT, McGrath LM, Mesa Restrepo SC, Moessner R, Morgan J, Muller H, Murphy DL, Naarden AL, Ochoa WC, Ophoff RA, Osiecki L, Pakstis AJ, Pato MT, Pato CN, Piacentini J, Pittenger C, Pollak Y, Rauch SL, Renner TJ, Reus VI, Richter MA, Riddle MA, Robertson MM, Romero R, Rosario MC, Rosenberg D, Rouleau GA, Ruhrmann S, Ruiz-Linares A, Sampaio AS, Samuels J, Sandor P, Sheppard B, Singer HS, Smit JH, Stein DJ, Strengman E, Tischfield JA, Valencia Duarte AV, Vallada H, Van Nieuwerburgh F, Veenstra-Vanderweele J, Walitza S, Wang Y, Wendland JR, Westenberg HG, Shugart YY, Miguel EC, McMahon W, Wagner M, Nicolini H, Posthuma D, Hanna GL, Heutink P, Denys D, Arnold PD, Oostra BA, Nestadt G, Freimer NB, Pauls DL, Wray NR, Stewart SE, Mathews CA, Knowles JA, Cox NJ, Scharf JM: Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS genetics 2013;9:e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker CL, Stewart EA: Uterine fibroids: the elephant in the room. Science 2005;308:1589–1592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.