

Abstract
Background.
Cocaine use disorder (CUD) is a common problem in the United States and worldwide. The mechanisms by which cocaine induces acute cardiovascular toxicity are various. When systemically absorbed through inhaled or intravenous routes, cocaine induces an acute rise in heart rate (HR) and blood pressure (BP) leading to a significant increase in cardiac output (CO) and myocardial oxygen demand. Subjects with chronic CUD represent a special population that has experienced long-term cocaine exposure, often without showing signs of cardiovascular disease. We herein present prospectively collected data on the acute hemodynamic effects of intravenous cocaine in a cohort of non-treatment-seeking individuals with CUD without cardiovascular disease.
Methods and Results.
Baseline physiologic data were collected while participants underwent infusion of escalating doses of cocaine (10 mg, 20 mg, 40 mg administered over 2 minutes) at baseline and after receiving single-blind placebo treatment. Continuous non-invasive hemodynamic monitoring was performed throughout the infusion sessions using the ccNexfin finger cuffs (Edwards Lifesciences Corp, Irvine, CA). The recorded arterial BP tracings allowing measurement of beat-to-beat changes in HR, BP, stroke volume, CO, and systemic vascular resistance (SVR). None of the subjects experienced a treatment related serious adverse event. Cocaine produced significant dose-dependent increases in median HR, BP, CO, and +dP/dt (a measure of cardiac contractility) and a significant dose-dependent reduction in median SVR.
Conclusions.
Intravenous cocaine in a cohort of otherwise healthy subjects with CUD produced dose dependent increases in CO, largely explained by an increase in HR, accompanied by a dose-dependent decrease in SVR.
Keywords: Cocaine, Cardiac Output, Heart Rate, Blood Presure, Systemis Vascular Resistance
Introduction
Cocaine use disorder (CUD) is a common problem in the United States, and worldwide. Recent data estimated that roughly one in five young adults in the US has used cocaine in the past year, and the usage rate may be increasing in certain regions1. Cocaine acts as a potent sympathomimetic agent, preventing the reuptake of catecholamines at synapses in the central and peripheral nervous system2. This property of systemically administered cocaine is thought to contribute to its many cardiovascular complications including acute myocardial infarction, acute ischemic or hemorrhagic stroke, and when used chronically, toxic cardiomyopathy3.
The mechanisms by which cocaine induces acute cardiovascular toxicity are various. When systemically absorbed through inhaled or intravenous routes, cocaine induces an acute rise in heart rate (HR) and blood pressure (BP) leading to a significant increase in cardiac output and myocardial oxygen demand4–7. In some individuals, there is an intense coronary artery spasm that can lead to myocardial ischemia, infarction and even death. Even in the absence of vasospasm, myocardial ischemia may result from increased demand due to increased HR and BP and an imbalance between oxygen demand and supply. A dose-dependent response between cocaine and HR and BP has been reported in cocaine naive individuals4,5 with limited investigation of whether the changes were due to an increase in cardiac output (CO) or systemic vascular resistance (SVR) or both6–9.
Subjects with chronic CUD represent a special population that has experienced long-term cocaine exposure, often without showing signs of cardiovascular disease. We herein present prospectively collected data on the acute hemodynamic effects of intravenous cocaine in a cohort of non-treatment-seeking individuals with CUD without cardiovascular disease.
Methods
Participants
Participant were enrolled as part of a parent study examining the safety of coadministration of lorcaserin—a serotonin (5-HT2C) receptor agonist—and cocaine in non-treatment seeking subjects with CUD. Baseline physiologic data were collected while participants underwent infusion of escalating doses of cocaine at baseline and after receiving single-blind placebo treatment. This manuscript presents only the data gathered from these infusion sessions (i.e. prior to lorcaserin administration). Data concerning the safety of lorcaserin and cocaine coadministration will be reported separately elsewhere.
Potential participants for the parent study were screened using inclusion and exclusion criteria as detailed here. Subjects met inclusion criteria if they: (1) were men or non-pregnant women between 18 and 59 years old; (2) met DSM-5 criteria for CUD of at least moderate severity but were not seeking treatment; (3) were currently using cocaine via smoking or intravenous route; (4) were without clinical evidence of heart disease as determined by physical exam, vital signs, and ECG; and (5) provided written informed consent. Subjects were excluded if they: (1) met DSM-5 criteria for diagnosis of bipolar disorder, major depressive disorder, or schizophrenia; (2) had any clinically significant medical disorder as determined by the enrolling physician; (3) had a history of seizures (excluding childhood febrile seizures); (4) had a positive HIV test; or (5) had a score of > 5 on the Clinical Opiate Withdrawal Scale10. The Structured Clinical Interview for DSM-IV was used to collect information to make eligibility determinations. Study subjects were not allowed to smoke in the hospital but could receive a nicotine patch dosed to match their daily cigarette consumption.
Infusion session
Infusion sessions were conducted in the Clinical Research Services Unit at Virginia Commonwealth University. The sessions began one hour after placebo administration and consisted of hourly infusions of cocaine in escalating doses (10 mg, 20 mg, 40 mg), with one double-blind infusion of saline randomly administered after the first dose of cocaine. For examples of dosing protocols, see Table 1. Cocaine was administered intravenously over a two-minute period by a study nurse under the direct supervision of a physician
Table 1.
Dose administration schedules for cocaine infusion sessions
| Dose | Dose Session 1 | Dose Session 2 | Dose Session 3 | Dose Session 4 |
|---|---|---|---|---|
| 1 | 10 mg Cocaine | 10 mg Cocaine | 10 mg Cocaine | 10 mg Cocaine |
| 2 | 20 mg Cocaine | 0 mg Cocaine | 20 mg Cocaine | 0 mg Cocaine |
| 3 | 0 mg Cocaine | 20 mg Cocaine | 40 mg Cocaine | 20 mg Cocaine |
| 4 | 40 mg Cocaine | 40 mg Cocaine | 0 mg Cocaine | 40 mg Cocaine |
Physiological monitoring
Continuous non-invasive hemodynamic monitoring was performed throughout the infusion sessions using the ccNexfin finger cuffs (Edwards Lifesciences Corp, Irvine, CA). The finger cuffs recorded arterial BP tracings allowing measurement of beat-to-beat changes in HR, BP, stroke volume, CO, SVR, and first derivative of left ventricular pressure increase during systole (+dP/dt)11. Hemodynamic data were for stored offline analysis at the end of each day.
Treatment
Cocaine infusions were obtained from National Institute of Drug Abuse (NIDA) contractors in 2 mL vials containing 20 mg/mL. A pharmacist prepared the cocaine according to the instructions supplied by the NIDA contractor and dispensed the drug according to the infusion protocol randomization. Doses were prepared by diluting the cocaine with normal saline. Placebo capsules containing dextrose powder were prepared and given to each patient one hour prior to the infusion sessions.
Data analysis
Hemodynamic response data are reported as either median change (based upon the median value over the first 30 minutes post-infusion) or peak change (the highest 60 second average over the first 30 minutes post-infusion) compared to baseline values which were defined by median parameters from 3 to 6 minutes before beginning the cocaine infusion session. Results for all hemodynamic changes are presented as median and interquartile range. We then evaluated for dose dependent changes by linear regression. Spearman’s correlation analysis was performed all hemodynamic changes. All analyses were conducted using SPSS Statistics 25 (IBM, Armonk, NY) statistical software package. P<0.05 was considered statistically significant.
Regulatory approval
This study was reviewed and approved by the Institutional Review Board at Virginia Commonwealth University. Written informed consent was obtained from each participant prior to beginning the study interventions.
Results
Clinical characteristics of the subjects (n = 13) are found in Table 2. Cocaine produced significant dose-dependent increases in median HR, systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP), CO, and +dP/dt and a significant dose-dependent reduction in median SVR; no significant dose-dependent changes were observed on median SV (Figure 1). Cocaine produced significant dose-dependent increases on peak values of HR, SBP, and CO; no significant dose-dependent changes were observed on peak values of DBP, MAP, SV, SVR, or +dP/dt (Figure 2). Correlations between changes in hemodynamic responses are found in Tables 3 and 4.
Table 2.
Clinical characteristics of the subjects
| Number | 13 |
|---|---|
| Age (years) | 49 [46-56] |
| Gender (males) | 11 (85%) |
| Race (African American) | 11 (85%) |
| Weight (kg) | 160 [158.5-182] |
| BMI (kg/m2) | 22.5 [22.2-26.7] |
| eGFR (mL/min/1.73m2) | 93.5 [78.8-104.3] |
| HR (bpm) | 71 [63-78] |
| SBP (mmHg) | 117 [108-134] |
| DBP (mmHg) | 70 [66-82] |
| MAP (mmHg) | 87 [83-100] |
| CO (L/min) | 5.5 [5.3-6.6] |
| SV (mL) | 88 [71-95] |
| SVR (dyn/s/cm5) | 1303 [1142-1424] |
| +dP/dt (mmHg/s) | 752 [589-976] |
| Cocaine use disorder (%) | 13 [100%] |
| Duration of use (years) | 21 [7.0-26.3] |
| Moderate grade (%) | 9 (69%) |
| Severe grade (%) | 4 (31%) |
| Tobacco use disorder (%) | 12 [92%] |
| pack-day-year use | 12 [3.3-26.3] |
| Alcohol use disorder (%) | 12 [92%] |
| drinks-day-years use (g) | 165984 [0-319284] |
| Marijuana use disorder (%) | 9 [69%] |
| years of use | 20 [5.0-29.8] |
Figure 1. Effects of cocaine.

The y-axis represents median changes in the hemodynamic parameter during the 30 minutes after cocaine infusion. Short horizontal lines identify the median, boxes identify the interquartile range (IQR), and whiskers identify an additional 1.5 times the IQR range. Asterices and circles represent outlying data points beyond 1.5 times outside the IQR. +dP/dt = first derivative of left ventricular pressure increase during systole.
Figure 2.

The y-axis represents peak changes (sustained over a minimum of 60 seconds) in the hemodynamic parameter during the 30 minutes after cocaine infusion. Short horizontal lines identify the median, boxes identify the interquartile range (IQR), and whiskers identify an additional 1.5 times the IQR range. Asterices and circles represent outlying data points beyond 1.5 times outside the IQR. +dP/dt = first derivative of left ventricular pressure increase during systole.
Table 3.
Correlations between median hemodynamic changes (R-value/p-value)
| Change in HR | Change in SBP | Change in DBP | Change in MAP | Change in SV | Change in CO | Change in SVR | Change in +dP/dt | |
|---|---|---|---|---|---|---|---|---|
| Change in HR | -- | +0.404 (<0.001) | +0.412 (<0.001) | +0.441 (<0.001) | −0.057 (0.581) | +0.674 (<0.001) | −0.433 (<0.001) | +0.357 (<0.001) |
| Change in SBP | +0.404 (<0.001) | -- | +0.757 (<0.001) | +0.922 (<0.001) | +0.300 (0.003) | +0.526 (<0.001) | +0.113 (0.271) | +0.792 (<0.001) |
| Change in DBP | +0.412 (<0.001) | +0.757 (<0.001) | -- | +0.913 (<0.001) | −0.200 (0.051) | +0.203 (0.048) | +0.426 (<0.001) | +0.509 (<0.001) |
| Change in MAP | +0.441 (<0.001) | +0.922 (<0.001) | +0.913 (<0.001) | -- | +0.050 (0.631) | +0.396 (<0.001) | +0.279 (0.006) | +0.685 (<0.001) |
| Change in SV | −0.057 (0.581) | +0.300 (0.003) | −0.200 (0.051) | +0.050 (0.631) | -- | +0.591 (<0.001) | −0.513 (<0.001) | +0.445 (<0.001) |
| Change in CO | +0.674 (<0.001) | +0.526 (<0.001) | +0.203 (0.048) | +0.396 (<0.001) | +0.591 (<0.001) | -- | −0.673 (<0.001) | +0.612 (<0.001) |
| Change in SVR | −0.433 (<0.001) | +0.113 (0.271) | +0.426 (<0.001) | +0.279 (0.006) | −0.513 (<0.001) | −0.673 (<0.001) | -- | −0.077 (0.456) |
| Change in +dP/dt | +0.357 (<0.001) | +0.792 (<0.001) | +0.509 (<0.001) | +0.685 (<0.001) | +0.445 (<0.001) | +0.612 (<0.001) | −0.077 (0.456) | -- |
HR: Heart rate; SBP: Systolic Blood Pressure; DBP: Diastolic Blood Pressure; MAP: Mean Arterial Blood Pressure; SV: Stroke Volume; CO: Cardiac Output; SVR: Systemic Vascular Resistance; +dP/dt: Rate of pressure increase.
Table 4.
Correlations between peak hemodynamic changes (R-value/p-value)
| Change in peak HR | Change in peak SBP | Change in peak DBP | Change in peak MAP | Change in peak SV | Change in peak CO | Change in peak SVR | Change in trough SVR | Change in peak +dP/dt | |
|---|---|---|---|---|---|---|---|---|---|
| Change in peak HR | -- | +0.204 (0.045) | +0.381 (<0.001) | +0.323 (0.001) | −0.199 (0.050) | +0.524 (<0.001) | +0.290 (0.004) | −0.234 (0.021) | +0.314 (0.002) |
| Change in peak SBP | +0.204 (0.045) | -- | +0.764 (<0.001) | +0.885 (<0.001) | +0.106 (0.303) | +0.182 (0.074) | +0.316 (0.002) | +0.386 (<0.001) | +0.677 (<0.001) |
| Change in peak DBP | +0.381 (<0.001) | +0.764 (<0.001) | -- | +0.902 (<0.001) | −0.214 (0.035) | +0.112 (0.276) | +0.431 (<0.001) | +0.447 (<0.001) | +0.389 (<0.001) |
| Change in peak MAP | +0.323 (0.001) | +0.885 (<0.001) | +0.902 (<0.001) | -- | −0.097 (0.346) | +0.136 (0.183) | +0.413 (<0.001) | +0.433 (<0.001) | +0.550 (<0.001) |
| Change in peak SV | −0.199 (0.050) | +0.106 (0.303) | −0.214 (0.035) | −0.097 (0.346) | -- | +0.427 (<0.001) | −0.208 (0.041) | −0.483 (<0.001) | +0.235 (0.020) |
| Change in peak CO | +0.524 (<0.001) | +0.182 (0.074) | +0.112 (0.276) | +0.136 (0.183) | +0.427 (<0.001) | -- | −0.147 (0.152) | −0.638 (<0.001) | +0.459 (<0.001) |
| Change in peak SVR | +0.290 (0.004) | +0.316 (0.002) | +0.431 (<0.001) | +0.413 (<0.001) | −0.208 (0.041) | −0.147 (0.152) | -- | +0.375 (<0.001) | +0.124 (0.227) |
| Change in trough SVR | −0.234 (0.021) | +0.386 (<0.001) | +0.447 (<0.001) | +0.433 (<0.001) | −0.483 (<0.001) | −0.638 (<0.001) | +0.375 (<0.001) | -- | +0.028 (0.789) |
| Change in peak +dP/dt | +0.314 (0.002) | +0.677 (<0.001) | +0.389 (<0.001) | +0.550 (<0.001) | +0.235 (0.020) | +0.459 (<0.001) | +0.124 (0.227) | +0.028 (0.789) | -- |
HR: Heart rate; SBP: Systolic Blood Pressure; DBP: Diastolic Blood Pressure; MAP: Mean Arterial Blood Pressure; SV: Stroke Volume; CO: Cardiac Output; SVR: Systemic Vascular Resistance; +dP/dt: Rate of pressure increase.
Discussion
The findings in this study both corroborate and expand upon the existing literature on the acute hemodynamic effects of cocaine by including subjects with chronic cocaine use disorder. A clear dose-dependent effect of intravenous cocaine was observed on median HR, SBP, DBP, CO, and +dP/dt when measured over a period of 30 minutes. We also observed dose-dependent effects of cocaine on peak HR, SBP, and CO during the same observation period. These changes are consistent with the sympathomimetic effects of cocaine reported in other populations. However, the unique observation in this study was the dose-dependent decrease in median SVR. The findings suggest that the sympathomimetic effects of systemically administered cocaine in subjects with CUD are driven primarily by HR (and possibly contractility)— without any notable increase in systemic vascular resistance —and therefore establish HR as a key biomarker of acute response to intravenous cocaine in this setting and population.
The current study employed two different approaches to evaluate the time course of responses to intravenous cocaine infusion. We defined “peak response” as the changes in value that was sustained for 60 seconds and “median response” as the median value over the entire 30-minute observation period. Given the rapid onset of euphoric effects of cocaine administration, it was notable to observe that median response values proved to be more sensitive to cocaine effects than peak response values. This may be reflective of sustained physiologic effects of cocaine over the course of 30 minutes following intravenous infusion or may suggest that the natural variance in hemodynamic parameters render minute-to-minute values less reliable. It remains to be seen whether larger cocaine doses might provoke more dramatic peak effects in this or other populations. In either case, we saw no evidence of significant fluctuations of hemodynamic parameters following cocaine 10-40 mg intravenous infusion in subjects with CUD.
Acute rises in HR and BP following systemic cocaine administration both in regular cocaine users and in cocaine naive individuals are well documented. An early landmark study conducted by Fischman and colleagues found that the dose of intravenous cocaine was linearly associated with median peak changes in HR among a small cohort of volunteers with prior cocaine use6. This finding is corroborated by the data presented in our manuscript. Another small study in subjects currently using cocaine found a significant increase in HR and cardiac index, but not in stroke volume in response to an average weight-adjusted intravenous cocaine dose of 36.5 mg7. The results of the present manuscript are remarkably similar, demonstrating an increase in HR and CO but not stroke volume in a similar patient population in response to a similar dose of cocaine. However, the method for echocardiographically determining cardiac index and stroke volume used in that study has notable limitations12,13, and the continuous collection of data using the Nexfin system in our study add a layer of validation to those earlier findings. Boehrer and colleagues collected extensive hemodynamic data in a group of 15 cocaine naive men and women undergoing cardiac catheterization for chest pain. Intranasal cocaine (2 mg/kg) administration led to increases in HR, mean arterial pressure, cardiac index, and both positive and negative dP/dt as measured by catheter devices using transcardiopulmonary thermodilution5. Overall, our data are consistent with these earlier findings and provide independent validation through the use of a different method for hemodynamic monitoring.
Perhaps the most interesting and novel finding of this study is the negative correlation between increasing doses of intravenous cocaine and changes in SVR. Cocaine is well known for its sympathomimetic and local vasoconstrictor activity, which leads to the inference that cocaine might also induce systemic vasoconstriction resulting in increased SVR. This seemingly paradoxical property of cocaine may be explained by the route of administration and relative activation of reflex nervous responses to changes in arterial pressure. A well conducted experiment by Tuncel and colleagues studied the effects of intra-brachial artery (local) cocaine and intra-nasal (systemic) cocaine administration on blood pressure, heart rate, and systemic vascular resistance in 15 cocaine naive subjects9. While intrabrachial cocaine infusion resulted in increased forearm venous concentration of norepinephrine and in forearm vascular resistance, intranasal (systemic) cocaine administration resulted in a decrease forearm and systemic vascular resistance, despite the same dose of cocaine reaching the forearm venous system. Tellingly, mean arterial pressure increased with intranasal (systemic) cocaine but not with intrabrachial (local) infusion. The increased mean arterial pressure induced by intranasal cocaine triggered a reflex decrease sympathetic nerve activity and decreased forearm venous norepinephrine, with a resulting decrease in forearm vascular resistance both compared to intrabrachial infusion and compared to baseline vascular resistance9. The changes in SVR observed in the present study align well with those findings and provide some evidence of a similar mechanism influencing SVR in cocaine using individuals, and reflect that the increase in blood pressure and cardiac output due to systemic cocaine are not due to an increase in SVR.
Correlational analyses between changes in hemodynamic parameters confirmed the dose-response relationships observed with linear regression and provide further evidence for the role of HR in driving the observed increase in CO and BP. While changes in SV do correlate with increasing BP and CO, these effects do not appear to be cocaine dose-dependent, perhaps due to the observed reduction in preload as expected with an increase in HR and a reduction in SVR. Conversely, changes in +dP/dt, a load-independent measure of contractility, show positive correlations and dose-responsiveness with cocaine.
This study has several limitations, including the small sample size of 13 subjects. The small sample size may have prevented to detect associations and correlations due to limited power. It is worth noting, however, the patient population enrolled in our study was highly selected based on stringent inclusion and exclusion criteria, and therefore this may represent an advantage by constructing homogeneous conditions to test the hypothesis. However, the strict selection criteria may also represent a limitation. Indeed, due to safety concerns, individuals with existing health conditions were excluded, leading to a healthy cohort of subjects with CUD that is unlikely to be representative of the broader population of individuals with CUD. External validation with a less selected group of subjects with CUD is necessary before extrapolating these findings to the cocaine using population at large. The parent study from which these data were gathered and analyzed was not designed to examine the pharmacologic mechanism by which cocaine exerts effects on SVR. Finally, the Nexfin device may not always perfectly agree with the gold standard of CO measurement11 and those disagreements are likely to have more influence in a smaller patient population.
In conclusion, intravenous cocaine infusions produced dose dependent increases in HR, systolic and diastolic BP, CO, dP/dt, and a dose-dependent decrease in SVR in a cohort of healthy subjects with CUD and free of cardiovascular disease (Figure 3). This protocol also corroborates the safety of intravenous cocaine administration and validates HR as a key pharmacodynamic response in this population.
Figure 3.
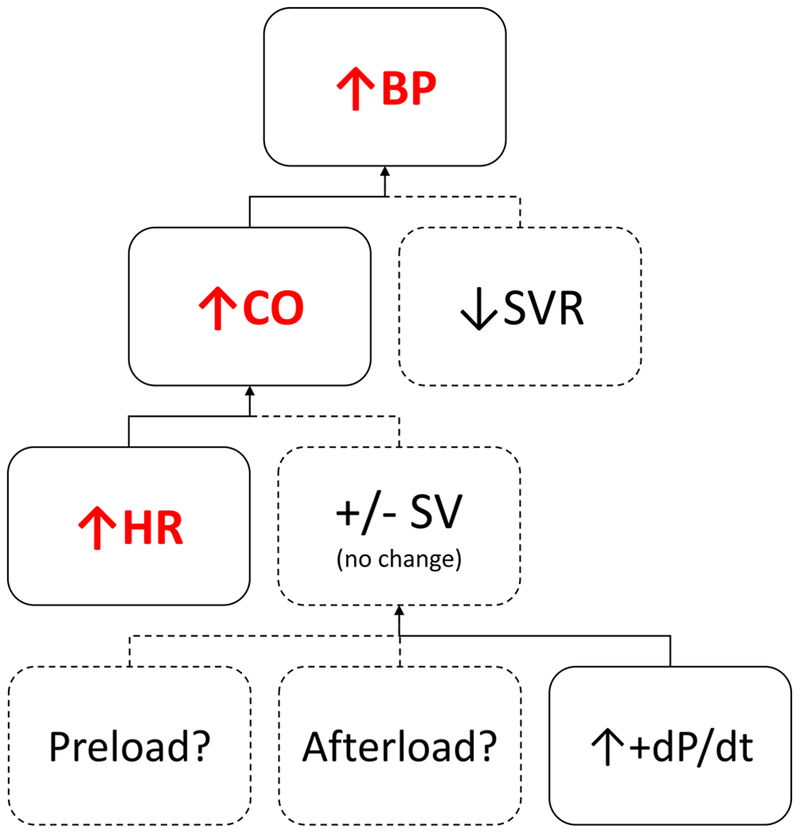
CO = Cardiac Output; HR = Heart Rate; MAP = Mean Arterial Pressure; SV = Stroke Volume; SVR = Systemic Vascular Resistance; +dP/dt = first derivative of left ventricular pressure increase during systole.
Acknowledgments.
This project was supported by a National Institute on Drug Abuse grant (No. U54 DA038999) and a CTSA award (No. UL1 TR002649) from the National Center for Advancing Translational Sciences to F. Gerard Moeller, MD. Its contents are solely the responsibility of the authors and do not necessarily represent official views of National Institutes of Health.
Disclosures. Dr Moeller discloses grant funding from Indivior and Nektar therapeutics. All other authors have no disclosures to report.
Works Cited
- 1.Hughes A, Williams MR, Lipari RN, Van Horn S. State Estimates of Past Year Cocaine Use among Young Adults: 2014 and 2015. Substance Abuse and Mental Health Services Administration (US); 2013. [PubMed] [Google Scholar]
- 2.Lange RA, Hillis LD. Cardiovascular Complications of Cocaine Use. N Engl J Med. 2001;345(5):351–358. doi: 10.1056/NEJM200108023450507 [DOI] [PubMed] [Google Scholar]
- 3.Havakuk O, Rezkalla SH, Kloner RA. The Cardiovascular Effects of Cocaine. J Am Coll Cardiol. 2017;70(1):101–113. doi: 10.1016/j.jacc.2017.05.014 [DOI] [PubMed] [Google Scholar]
- 4.Lange RA, Cigarroa RG, Yancy CW, et al. Cocaine-Induced Coronary-Artery Vasoconstriction. N Engl J Med. 1989;321(23):1557–1562. doi: 10.1056/NEJM198912073212301 [DOI] [PubMed] [Google Scholar]
- 5.Boehrer JD, Moliterno DJ, Willard JE, et al. Hemodynamic effects of intranasal cocaine in humans. J Am Coll Cardiol. 1992;20(1):90–93. [DOI] [PubMed] [Google Scholar]
- 6.Fischman MW, Schuster CR, Resnekov L, et al. Cardiovascular and subjective effects of intravenous cocaine administration in humans. Arch Gen Psychiatry. 1976;33(8):983–989. [DOI] [PubMed] [Google Scholar]
- 7.Kleerup EC, Wong M, Marques-Magallanes JA, Goldman MD, Tashkin DP. Acute Effects of Intravenous Cocaine on Pulmonary Artery Pressure and Cardiac Index in Habitual Crack Smokers. Chest. 1997;111(1):30–35. doi: 10.1378/chest.111.1.30 [DOI] [PubMed] [Google Scholar]
- 8.Yoon JH, Shah RS, Arnoudse NM, De La Garza R. Remote physiological monitoring of acute cocaine exposure. J Med Eng Technol. 2014;38(5):244–250. doi: 10.3109/03091902.2014.902513 [DOI] [PubMed] [Google Scholar]
- 9.Tuncel M, Wang Z, Arbique D, Fadel PJ, Victor RG, Vongpatanasin W. Mechanism of the blood pressure--raising effect of cocaine in humans. Circulation. 2002;105(9):1054–1059. [DOI] [PubMed] [Google Scholar]
- 10.Wesson DR, Ling W. The Clinical Opiate Withdrawal Scale (COWS). J Psychoactive Drugs. 2003;35(2):253–259. doi: 10.1080/02791072.2003.10400007 [DOI] [PubMed] [Google Scholar]
- 11.Broch O, Renner J, Gruenewald M, et al. A comparison of the Nexfin ® and transcardiopulmonary thermodilution to estimate cardiac output during coronary artery surgery. Anaesthesia. 2012;67(4):377–383. doi: 10.1111/j.1365-2044.2011.07018.x [DOI] [PubMed] [Google Scholar]
- 12.Chin CWL, Khaw HJ, Luo E, et al. Echocardiography Underestimates Stroke Volume and Aortic Valve Area: Implications for Patients With Small-Area Low-Gradient Aortic Stenosis. Can J Cardiol. 2014;30(9):1064–1072. doi: 10.1016/j.cjca.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeder MT, Karapanagiotidis S, Dewar EM, Kaye DM. Accuracy of Echocardiographic Cardiac Index Assessment in Subjects with Preserved Left Ventricular Ejection Fraction. Echocardiography. 2015;32(11):1628–1638. doi: 10.1111/echo.12928 [DOI] [PubMed] [Google Scholar]