

Abstract
Hydrogen sulfide is an antioxidant molecule that has a wide range of biological effects against oxidative stress. Balanced oxidative stress is also vital for maintaining cellular function in biological system, where reactive oxygen species are the main source of oxidative stress. When the normal redox balance is disturbed, deoxyribonucleic acid, lipid, and protein molecules are oxidized under pathological conditions, like diabetes mellitus that leads to diabetic peripheral neuropathy. In diabetes mellitus-induced diabetic peripheral neuropathy, due to hyperglycemia, pancreatic beta cell (β cell) shows resistance to insulin secretion. As a consequence, glucose metabolism is disturbed in neuronal cells which are distracted from providing proper cell signaling pathway. Not only diabetic peripheral neuropathy but also other central damages occur in brain neuropathy. Neurological studies regarding type 1 diabetes mellitus patients with Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis have shown changes in the central nervous system because high blood glucose levels (HbA1c) appeared with poor cognitive function. Oxidative stress plays a role in inhibiting insulin signaling that is necessary for brain function. Hydrogen sulfide exhibits antioxidant effects against oxidative stress, where cystathionine β synthase, cystathionine γ lyase, and 3-mercaptopyruvate sulfurtransferase are the endogenous sources of hydrogen sulfide. This review is to explore the pathogenesis of diabetes mellitus-induced diabetic peripheral neuropathy and other neurological comorbid disorders under the oxidative stress condition and the anti-oxidative effects of hydrogen sulfide.
Keywords: Alzheimer's disease, amyotrophic lateral sclerosis, antioxidant, diabetic peripheral neuropathy, DNA oxidation, hydrogen sulfide, mitochondrial dysfunction, oxidative stress, Parkinson's disease, reactive oxygen species
Introduction
The main source of reactive oxygen species (ROS) is mitochondrial respiratory chain, where oxidative stress generates. Oxidative stress in diabetic patients leads to dysfunction during insulin secretion in the nervous system and thus, neurodegeneration like diabetic peripheral neuropathy (DPN) occurs (Babizhayev et al., 2015). Pancreatic β cell is the most important metabolically active part in the body, where metabolites take place for energy synthesis at the high level of glucose concentration (Gerber and Rutter, 2017). The main role of the pancreatic β cell is glucose mediated insulin secretion. In diabetes mellitus (DM), insulin secretion is hampered by ROS production in the pancreatic cells (Newsholme et al., 2007). Dysfunction of peripheral nerve in DM is observed with symptoms in distal part which contains 75% DPN (Pop-Busui et al., 2017). Down regulation of Schwann cells, aggregation of sorbitol during signaling of polyol pathway, inactivation of Na+/K+ signaling, hyperglycemia-induced oxidative stress are causative factors of neuropathy in the brain (Tian et al., 2016). About 50% of diabetic patients are affected by neurological disorders, which are the most common comorbidities of DM (Karki et al., 2017). Some neurodegenerative diseases, like Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) occur simultaneously in the central nervous system (CNS) in diabetic patients because of oxidative stress (Karki et al., 2017).
Oxidative stress is the resultant effects of over production of ROS and reactive nitrogen species. The most prominent ROS and reactive nitrogen species include hydrogen peroxide (H2O2), superoxide (O•2−), hydroxyl (• OH), nitric oxide (NO•), peroxynitrite (ONOO−), which generate free radicals due to their instability (Zhang et al., 2015). However, oxidative stress is suppressed by antioxidant molecules. Hydrogen sulfide (H2S) mediates antioxidant function through enzymatic pathways. For example, super oxide dismutase (SOD) 1, SOD2, glutathione (GSH), glutathione peroxidase (GPx), and catalase (CAT) are those enzymes that provide H2S mediated antioxidant function (Marengo et al., 2016).
Oxidative stress is assessed in DPN and due to hyperglycemia-induced oxidative stress H2O2 production and lipid peroxidation in protein kinase C (PKC) pathway, accumulation of advanced glycation end products (AGEs) that inhibit axonal regeneration (Dal and Sigrist, 2016).
The effect of hyperglycemia-induced oxidative stress on neurons is varied in both patients with T1DM and type 2 diabetes mellitus (T2DM), and DPN patients with T2DM have low capacity of controlling hyperglycemia (Callaghan et al., 2012). In the peripheral nerve of T2DM patients, oxidative stress is increased from proximal to distal parts, passes from DRG to sciatic nerve , and decreases the metabolism under glycolytic and tricarboxylic acid cycle (TCA) (Hinder et al., 2013). Not only this but also β cells are damaged by ROS, where β cells are vital for insulin secretion from pancreas in T2DM patients. Because of β cell dysfunction, insulin cannot distribute in several parts of the brain and neurological disease appears (Størling and Pociot, 2017). In AD, cholinergic homeostasis is hampered by downregulation of insulin/insulin growth factor (IGF) resistance, leading to downregulation of target genes (Mendiola-Precoma et al., 2016). There is evidence that 40% of patients with DM develop PD and glucose is impaired at an early stage (Garber et al., 2018). IGF-1 is a very important factor, whereas IGF-1 binding protein level increases and insulin level decreases in the serum of ALS patients (Torres-Aleman et al., 1998).
However, H2S can mitigate the effects of oxidative stress on nerve cells. H2S is an endogenous biological mediator that is produced from cystathionine β synthase (CBS), cystathionine gamma-lyase (CSE), cysteine aminotransferase and 3-mercaptopyruvate sulfurtransferase (3MST) enzymes (Calvert et al., 2010). In this review, we will focus on resultant effects of oxidative damage in patients with DM-induced neurological diseases and the activity of H2S as an antioxidant in each neurodegenerative disease against oxidative stress.
Database Search Strategy
We searched PubMed to retrieve studies regarding the protective effects of H2S on neurodegenerative diseases reported before June 8, 2018 (Additional Table 1 (298.9KB, pdf) ). This database search was based on all studies including animal experiments and clinical trials of neurodegenerative patients. “Diabetes mellitus”, “oxidative stress”, “reactive oxygen species”, “hydrogen sulfide”, “glutathione”, “antioxidant”, “neurodegenerative diseases” were used as search terms
Oxidative Stress and Aging
Oxidative stress is the common cause of aging and aging-associated diseases. Generally, cellular damage by oxidative stress is marked by DNA, protein, and lipid damage. When oxidative stress persists for a long time, cellular senescence might arise and ROS is the main mediator in neurodegeneration (Liguori et al., 2018).
Oxidative damage with aging is caused by either increased or decreased generation of electrophiles and oxidants. Various studies have shown that the stimulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, cytochrome p450, monoamine oxidase, and nitric oxide synthase (Zhang et al., 2015) are the sources of oxidant generation that varies according to pathophysiological conditions (Zhang et al., 2015). In aging, protein modification is the common process in which ROS triggers various signaling pathways like DNA damage response, transforming growth factor-β, alternate reading forms or mitogen activated protein kinase3, upregulation of cell cycle inhibitor, p53 (Muñoz-Espín and Serrano, 2014). The protein modification results from 4-hydroxynonenal.
In essence, the increased and decreased level of oxidative stress not only demolishes the antioxidant functions but also leads to generation of brain neuropathy.
Oxidative Stress and CNS Dysfunction
Nerve cells are very sensitive to hyperglycemia-induced oxidative stress which is based on the uptake of external glucose. In the case of diabetic neuropathy, axons cannot send the signal in the hyperglycemic state because of mitochondrial oxidative damage. As a result, axons loss energy and thus, axonal degeneration occurs in the nervous system (Babizhayev et al., 2015). Excessive generation of ROS is also responsible for resistance of insulin signaling by c-Jun N-terminal kinase (JNK) signaling pathway (Verdile et al., 2015). Moreover, neurotrophic factors like nerve growth factor and brain-derived neurotrophic factor are responsible for central damage of motor neurons. In the sciatic nerve of diabetic rats, expression of brain-derived neurotrophic factor is reduced (Lenart et al., 2019).
The nuclear dysfunction of AD is related to amyloid beta 42 (Aβ42) that drives neurochemical changes. Protein synthesis machinery may be impaired by Aβ42 and the function of tau protein is also impaired (Ahmad et al., 2017). According to a study by Mahmoud et al. (2017), when differentiated SHSY5Y cells are incubated with Aβ42 oligomers, oxidative stress increased, leading to nuclear stress. As a consequence, 45S-pre-ribosomal (45S-pre-rRNA) and heterochromatin compaction production altered, and protein and RNA synthesis decreased (Hernández Ortega et al., 2016). The nuclear dysfunction in PD appears with the mtDNA and nuclear DNA gene mutation in mitochondria, and changes in dynamics of mitochondrial fusion or fission, and transcriptional impairment. Due to oxidative stress, nuclear dysregulation in PD is exerted by the transcription coactivator, peroxisome-proliferator-activated receptor gamma coactivator-1α (Henchcliffe and Beal, 2008). Due to nuclear DNA mutation, overexpression of peroxisome-proliferator-activated receptor gamma coactivator-1α causes sudden degeneration of dopaminergic neuron.
In terms of ALS, nuclear dysfunction due to ROS is exerted by the dislocation of RNA-binding protein, field-effect transistors protein. Field-effect transistors consists of fused in Sarcoma/translocated in liposarcoma, Ewing sarcoma, and TATA-binding protein-associated factor 15, which function upon nuclear processing steps like DNA and RNA processing, gene expressing, and cell signaling (Svetoni et al., 2014). Upon oxidative stress condition, cellular models of ALS, EWS mutant proteins displayed different localization from fused in Sarcoma mutants. Moreover, in cytoplasm DNA binding protein, TDP-43 aggregates due to oxidative stress which causes nucleoplasmic transport deficits as shown by chromosome 9 open reading frame 72 (C9orf72) hexnucleotide repeat expansion (Jovičić et al., 2016). TDP-43 transits from the nucleus to cytoplasm, where pathological aggregation occurs. Furthermore, mutations in TDP-43 or related RNA binding protein fused in Sarcoma activate unfolded protein response and generate oxidative stress which causes upregulation of tap-1 expression with neuronal dysfunction. This indicates chronic induction of nucleic acid binding protein by cellular stress which may contribute to neurodegeneration (Vaccaro et al., 2012).
In short, axonal transport is important to facilitate energy to nerve cells. Oxidative stress is responsible more or less for hampering energy metabolism, energy transportation, and mutation of genetic factors. In most cases, H2O2 plays a role in inhibiting axonal transport. To observe oxidative modification by H2O2 more precisely, further investigation is necessary.
Oxidative Stress-Induced Different Pathways in DPN
Excessive level of ROS causes DNA, protein damage, lipid peroxidation in the peripheral nerve. In particular, in patients with T2DM, antioxidant defense system is impaired by initiating free radicals. These initiated free radicals contribute to nerve damage through several signaling pathways (Sasson, 2017).
Polyol pathway is activated in the peripheral nerve, especially in Schwann cells. Normally in polyol pathway, aldose reductase (AR) converts glucose to fructose by sorbitol dehydrogenase so that 30–50% of body’s glucose can be utilized in the hyperglycemic state (Fantus, 2002). Oxidative damage resulted glucose consumption is reduced by sorbitol dehydrogenase because it competes for GAPDH for NAD+ generation. In this way, the polyol pathway is dysregulated by oxidative stress in the pathogenesis of DPN (Yan, 2014). Moreover, when sorbitol is converted to fructose in the presence of sorbitol dehydrogenase, the oxidaxion of sorbitol causes the Na+/K+ signaling defects which leads to axonal dysfunction. For this reason, myo-inositol uptake is decreased and axonal transport is hampered by abnormal action potential (Hosseini and Abdollahi, 2013). The activity of oxidative stress through the polyol pathway is shown in Figure 1.
Figure 1.
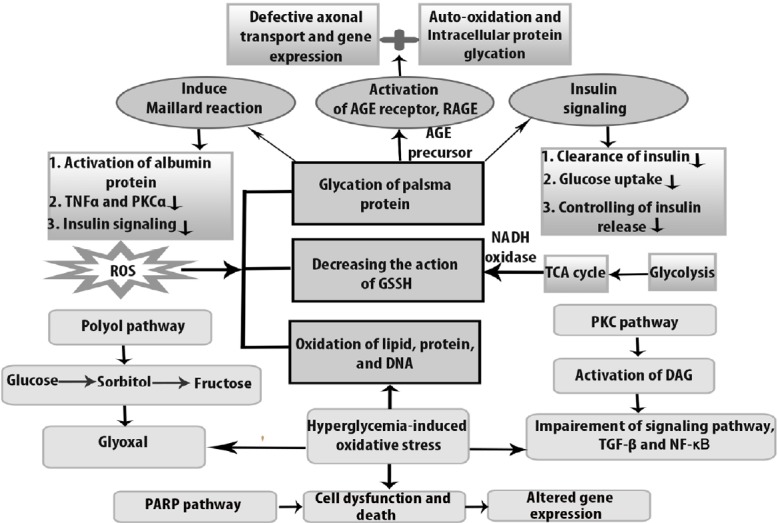
Oxidative stress-induced dysfunctional pathways in diabetic peripheral neuropathy.
The figure illustrates about the mechanism of cells of diabetic peripheral neuropathy through different signaling pathways, which lead to different abnormalities under oxidative stress condition. Hyperglycemia initiates dysfunction by producing metabolite glyoxal by which polyol pathway inhibits cytosolic nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) as well as glutathione (GSH). In the presence of excessive amount of glucose, advanced glycation end products (AGEs) bind with receptor for advanced glycation end products (RAGE) and impairs the activity of functional protein and causes auto-oxidation in diabetic peripheral neuropathy. As a consequence, insulin signaling is downregulated, whereas reactive oxygen species (ROS) acts as a second messenger. At the same time, due to maillard reaction, particularly albumin protein is depleted and tumor necrosis factor (TNFα) and protein kinase C alpha (PKCα) show less activity. PKC activation increases the hyperglycemic level and accelerates dysfunction in transcriptional factor, through nuclear factor kappa B (NF-κB) impairment. On the other hand, hyperglycemia-induced oxidative stress causes necrosis and abnormal gene expression through PARP pathway. GSSH: Oxidized glutathione; TCA: tricarboxylic acid cycle; TGF: transforming growth factor.
PKC is a group of serine kinases that act as an intracellular signaling molecule (Qiao et al., 2016). Activation of diacylglycerol by hyperglycemia stimulation is the common phenomenon in the PKC pathway for maintaining nerve function and in the pathogenesis of DPN. Generally, intracellular signaling cascade such as nuclear factor kappa-B and transforming growth factor-β is overexpressed due to PKC activation (Figure 1). In addition to this, PKC activation in DPN reduces nerve conduction velocity and also PKCβ reduces Na+/K+ ATPase activity (Farmer et al., 2012). β and δ isoforms are also activated by PKC in the hyperglycemic state in DPN (Giacco and Brownlee, 2010). Some lipid molecules like arachidonic acid, phosphatidylinositol diphosphate, and phosphatidylinositol diphosphate enhance PKC activity but as a result of lipid peroxidation, the PKC pathway obtains destabilized conformation (Xiao et al., 2018).
RAGE induces ROS formation using signalling conductions like JNK and p38, where NADPH oxidase and NF-κB gene are expressed (Oyenihi et al., 2015a). In general, glycation occurs at the N-terminal of valine in β-chain, which is known as the maillard reaction (Nowotny et al., 2015). During the maillard reaction, AGEs increase and accumulate within cells, contributing to oxidative stress (Nowotny et al., 2015). Tan et al. (2011) reported that insulin resistance, as evaluated by homeostasis model assessment index, was associated with AGE level, which was indirectly related to T2DM. Insulin level in the fasting state in the non-diabetic models was independently correlated with AGEs (Tahara et al., 2012).
Generally, poly ADP ribose polymerase (PARP) is a NAD+ dependent nuclear enzyme which repairs DNA but its excessive activation due to oxidative stress causes cell death. Normally, PARP is expressed in Schwann cells and endothelial cells, but due to occurrence of NAD+ depletion, glycolytic pathways are closed. When ADP ribose molecule attaches to the nuclear protein molecule, PARP is found to play a role in converting NAD+ to nicotinamide that may alter transcription factors, and make changes in the glycolytic pathway during AGEs formation and in the PKC pathway (Giri et al., 2018).
In a nutshell, PARP is involved in neural cell damage by exhibiting damaged inflammation protein. To avoid the dysfunction, the combined action of antioxidant and nicotinamide may provide therapeutic action. Antioxidants in rodent and xanthine oxide inhibitor may also take part.
Oxidative Damage of β-cell and Insulin Signaling
As hyperglycemia-induced oxidative stress has been marked as an inhibitor of insulin secretion, it has consecutive effects on β-cell dysfunction (Oyenihi et al., 2015b). In β-cells, protons are getting leaked by the activation of uncoupling protein 2 and lowering the ratio of adenosine diphosphate/ATP (ADP/ATP). ROS are mainly observed in cell membrane by recognizing oxidative markers like malondialdehyde and 8-hydroxyguanosine. The death of cell is also marked by low signaling of nuclear factor-κB (NF-κB).
However, due to hyperglycemia in DPN, additional lipid level is increased and ROS are also increased that are found in β-cell dysfunction (Gerber and Rutter, 2017). Glucose also stimulates ROS in β-cells through glucose transportation because glucose transport is higher than the glucose phosphorylation. According to glucose fluxing the TCA is enhanced that shows higher level of ADP/ATP ratio (Thallas-Bonke et al., 2008). Consequently, ATP sensitive K+ channel is hyperpolarized and thus, Ca2+ is raised in intracellular fluid (Cameron et al., 2005). Finally, insulin secretion occurs. Apart from this, in mammalian cells, rapid fluxing of glucose increases the mitochondrial oxidative activity because 100% of glucose carbon is oxidized to CO2 (Nishikawa et al., 2000). Although both processes, TCA cycle and glycolytic flux enhance insulin secretion, they are responsible for ROS generation in pancreatic β cells and activation of PKC pathway (Fadupin et al., 2007). This PKC pathway causes O2•− formation (Hong and Garvin, 2014). Additionally, lower glucose level leads to ROS formation in β cells (Sampson et al., 2010). At 0–5 mM glucose concentration, O•2− is formed. Moreover, consistent studies show that glucose leads to reduced GSH oxidation ratio in pancreatic β cells (Gerber and Rutter, 2017).
On the other hand, oxidative stress enhances resistance in insulin signaling. T2DM pathogenesis is proceeding as the consequence of declining insulin secretion. Normally, insulin resistance is associated with physical inactivity, excessive adipokines, and abdominal obesity (Tangvarasittichai, 2015). It also causes impaired glucose tolerance in T2DM. Mostly, overproduction of ROS in liver and adipose tissue causes insulin resistance in obese mice. Many researchers have found that antioxidants reduce triglycerides and glucose levels and retard insulin resistance in KKAy mice (Furukawa et al., 2017). Also, high levels of insulin become reduced by supplying coenzyme, Q10 in SHR/cp rats, which is an antioxidant (Tangvarasittichai, 2015). As a result of insulin resistance, the TCA cycle that increases NADH through ETC of mitochondria and increases O•2− formation (Tangvarasittichai, 2015).
Although the impairment in islet β cell production by substantial generation of mitochondrial oxidative stress, elevated antioxidant can demolish ROS-induced β cell death. Also, antioxidants provide functions against insulin resistance. For initiating function of CAT, Gpx, and SOD diabetic GK/Par rat models can be used experimentally which show increased expression of antioxidant but STZ mouse models only show SOD expression.
Oxidative Stress and Diabetes-Associated Other Neurodegenerative Diseases
Oxidative stress and diabetes-associated AD
Due to insulin function disorder in nerve, AD and T2DM are overwhelming diseases and they are also age-associated diseases (Chatterjee and Mudher, 2018). The impaired control of blood glucose occurs by insulin producing pancreatic β cells (Chatterjee and Mudher, 2018). According to clinical, epidemiological, and genetic data, T2DM and AD including impaired glucose metabolism were interacted. According to pathophysiological mechanisms, function of AGEs and mitochondrial damage result in T2DM and AD (Ahmad et al., 2017).
For insulin supplying in the brain, the regulation of glucose utilization and metabolism is important. In the early stage of AD, cognitive dysfunction occurs if there are any deficits in energy metabolism. For glucose metabolism, glucose transporter 4 provides function of insulin receptors (Hoyer, 2004). Thus, glucose metabolism glucose transporter 4 has an important role in the generation of energy for cognitive disorder. On the other hand, ROS causes Aβ-induced membrane lipid peroxidation which hampers significant decrease in glucose transport (Zhu et al., 2012). As a result, DNA, RNA, and protein β molecules functionally become inactive in AD patients (Hoyer, 2004).
From another point of view, aging is the most common risk factor in AD. According to growing evidence, insulin resistance in T2DM causes brain insulin/IGF resistance and thus mild cognitive impairment is associated with AD (Chatterjee and Mudher, 2018). What so ever, only several fields are broadly expanded to study the relationship between deficiency of brain insulin resistance and cognitive impairment in AD. An experimental study shows that insulin resistance by oxidative stress promotes Aβ aggregation. Insulin resistance causes Aβ deposition, accelerates trafficking of amyloid beta precursor protein (AβPP) from the trans Golgi network to the plasma membrane and also inhibits its intracellular degradation. Where amyloid beta protein precursor-amyloid beta causes GSK-3β and hyper phosphorylation of tau protein (Chang et al., 2018).
Oxidative stress and diabetes-associated PD
Generally, PD is one of the age-related neurodegenerative diseases, where it is marked by the loss of dopamine producing substantia nigra pars compacta cell (Schernhammer et al., 2011). As a consequence, loss of dopamine causes the broad spectrum of motor disorder like sensory dysfunction, cognitive impairment, and behavioral abnormalities in older adult patients (Schernhammer et al., 2011). PD has also appeared with adult onset diabetes which was firstly noted two decades ago. Diabetes has been associated with dysfunction of motor neuron in PD patients. A study of American people showed that about 40% diabetic patients have the risk of PD. Another study of Finish people showed that 324 out of 591 men and 309 out of 507 women have the risk of PD with diabetes (Schernhammer et al., 2011). Comparatively, men have the higher risk of PD with diabetes than woman.
Although the relationship between PD and DM has not been established yet, some hypothetical issues are related to diabetes and PD. For instance, dysregulated insulin signaling and mitochondrial oxidative stress are well documented in DM and PD signaling pathways.
Oxidative stress and diabetes-associated ALS
The biological mechanism of T2DM is linked to ALS. T2DM patients at the age of 30 years have the risk of developing ALS. An animal study showed that the gene, TAR DNA-binding protein 43 was mutated in 4% of familial ALS which induces glucose and energy metabolism (Mariosa et al., 2015). Furthermore, in zebra fish models of ALS, the overexpression of progranulin (multifunctional protein) mediates insulin resistance and rescues mutant TDP-43-induced axonopathy (Mariosa et al., 2015). The differences between ALS with T1DM and T2DM are based on two pathophysiological conditions. The one is identified by insulin resistance and the other is destruction of β-cells in the pancreas. At the age of 50, T2DM increases the risk of sporadic ALS.
According to another hypothesis, accumulation of glutamine is associated with diabetes in ALS (Trudeau et al., 2004). Due to the aggregation of glutamate, the functional activity of neurotransmitter receptors becomes altered in diabetes. Additionally, over-stimulation of glutamate causes neural cell death in ALS and is named glutamate excitotoxicity. As a consequence, increased intracellular Ca2+ causes ROS generation (Shah et al., 2016). On the other hand, under the normal physiological condition, excessive Ca2+ causes cellular dysfunction (Shah et al., 2016). Increased oxidative stress might be the cause of insulin resistance, β-cell dysfunction and impaired glucose tolerance in T2DM (Evans et al., 2003). At the same time, oxidative damage in ALS patients causes mutations in antioxidant enzymes, SOD1 which initiates 20% of familial ALS. In addition to this, in the presence of excessive ROS, the morphological and functional defects are found in mitochondria of ALS patients which show axonopathy in ALS.
The over-stimulation of glutamate causes neuronal cell death in ALS and is named oxidative glutamate excitotoxicity. The increased extracellular glutamate level stimulates ionotropic glutamate receptors. As a result, NMDA receptors increase massive Ca2+ influx into the cells which causes nerve cell death (Lewerenz and Maher, 2015). In addition to this, when glutamate inhibits cysteine import through cysteine/glutamate antiporter system, GSH level is depleted (Wallis et al., 2018). When consumption of molecular oxygen is greatly reduced, ROS is formed (Munro and Treberg, 2017).
In summary, the increased risk factors of diabetes in ALS are still unknown. For further investigations about the relationship between diabetes and ALS, clinical studies are necessary in diabetes patients. However, antioxidant function of H2S can mitigate oxidative stress in neurodegenerative diseases.
H2S
As an endogenous biological compound, H2S is produced by CBS, CSE, and 3MST, where CBS and CSE are primary precursors of H2S production. CBS, as a cytosolic enzyme, shows highest expression in the brain. CBS is also a pyridoxal-5′-phosphate and S-adenosyl methionine-dependent enzyme, where pyridoxal-5-phosphate and S-adenosyl methionine act as cofactors in physiological system. CBS is also regulated by post-transcriptional modification in the liver and kidney and located in mitochondrial HCT116 cells, and CBS has the stimulatory effects on GAPDH (Qu et al., 2008). Homocysteine is catalyzed by CBS.
The enzymes, 3MST and CAT also take part in H2S synthesis whereas 3MST is mostly found in astrocytes and CAT is found in vascular endothelial cells. 3MST helps to produce H2S by forming sulfane sulfur, whereas 3MST undergoes transulfuration via 3-mercaptopyruvate in the mitochondria and minority of 3MST is present in the cytoplasm (Shefa et al., 2018). To observe the bound sulfane sulfur, the experimental study showed that cys247 (C247S) mutant remained at the basal level but arginine (R187G) mutant increased from the basal level which denotes 50% of H2S activity compared to wild type (Balasubramanian et al., 2019). As 3MST is a pyridoxal-5′-phosphate independent enzyme, it acts as an alternative source of H2S generation instead of CBS (Balasubramanian et al., 2019).
Every enzyme including CBS, CSE, 3MST, and CAT contributes to H2S production enzymatically and endogenously.
GSH mediated neuroregenerative potential of H2S in the brain
The GSH system is especially vital for the protection of cellular damage by ROS. The antioxidant function of H2S is measured by the enhancing levels of GSH. GSH is formed cysteine transport, where cysteine is reduced in the extracellular space. For the synthesis of GSH, the interaction between astroglial and neuronal cells occurs. When cysteine can determine neuronal GSH, in the presence of astroglial cells, the increased GSH levels indicates that astrocytes provide cysteine to neurons by releasing GSH (Yin et al., 2016). Under oxidative stress condition, glutamate stimulates H2S to produce GSH. Glutamate is responsible for oxidative damage of neurons. In this case, glutamyl cysteine synthatase enzyme is inhibited by H2S in the CNS (Yin et al., 2016). GSH synthesis is associated with cysteine molecule via the transulfuration pathway. Moreover, free radicals are scavenged by the GSH levels through activating KATP channels. GSH also protects HT22 cells from glutamate oxidative toxicity (Jia et al., 2013).
GSH maintains thiol redox potential by sulfhydryl group and regenerates from oxidized group, GSSH. Apart from this, GSH is consumed by GST or by release from cells. Moreover, GSH detoxifies H2O2-induced oxidative stress via the action of catalyzing enzyme, Gpx (Rahman et al., 1999). As a result, astrocytes can recruit the function of CAT by H2O2 disposal but GSH does not fully compensate for the loss of CAT reaction (Dringen et al., 2000). Thus, GSH provides rapid function by removing peroxides from astroglial cells (Sekar et al., 2018). Dringen et al. (2000) reported that GSH protected cells against H2O2 at the ratio of astroglial cells to neurons of 1:20. However, H2S has the great importance in brain cells to demolish free radicals as well as vulnerable stage of neurons.
Neuroprotective Role of H2S in Neurodegenerative Diseases
Antioxidant function of H2S in DPN
Antioxidant can act as oxidative stress scavenger through decomposing H2O2 and inhibiting enzymes. Except CAT, Gpx, and SOD, GST, peroxiredoxin, flavonoids, lipoic acids, thioredoxin, glutaredoxins also paly the important role in the attenuation of ROS (Marengo et al., 2016). GSH gives function with GST to produce hydrophilic substances. The cellular function of H2S as an antioxidant in DPN is shown in Figure 2.
Figure 2.

Cellular function of H2S as an antioxidant in DPN.
The beneficial action of H2S through mediating superoxide dismutase (SOD), glutathione (GSH), and catalase (CAT) in hyperglycemia-induced oxidative stress. Superoxide dismutase (SOD) binds with alpha lipoic acid (ALA) to exert antioxidant effects by increasing GSH level. On the other hand, GSH is reduced by H2O2 formation and it is converted into oxidized GSSH. The ratio of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) to nicotinamide adenine dinucleotide phosphate (NADP) is altered by polyol pathway. Glutathione peroxidase (Gpx) can attenuate the balance to provide antioxidant function. In the case of CAT, it also provides similar antioxidant mechanism by removing H2O2 formation. As a consequence, oxidized GSSH is decreased and GSH is increased. DPN: Diabetic peripheral neuropathy; H2S: hydrogen sulfide; NADPH: nicotinamide adenine dinucleotide phosphate; PKC: protein kinase C; ROS: reactive oxygen species.
H2S provides antioxidant function through enzymatic activity against oxidative stress. SOD is one of them. The total amount of SOD depends on the generation of O•2− and antioxidant capacity (Fujita et al., 2009). Generally, SOD converts O•2− into H2O2 and oxygen. Then H2O2 is converted to water molecule by CAT and GPx. In the case of DPN, the activity of both extracellular SOD1 and SOD2 is reduced in peripheral blood. Moreover, transgenic mice expressing SOD1 and SOD2 genes are resistant to diabetes-induced neuropathy. A previous study invovling mouse models revealed that low dose of STZ induces low grade of β cell damage. On the other hand, low dose of STZ leads to a decrease in podocyte marker in C57BL/6 mice rather than in the wild-type mice (Breyer et al., 2007).
Loss of GSH due to redox modification has also negative impact on gene expression. GSH has a negative expression due to oxidative stress. Additionally, as a result of reduced GSH, sorbitol started to accumulate because of hyperglycemic vascular damage (Sandireddy et al., 2014). The oxidized form of GSH is GSSH and in the presence of NADPH and glutathione reductase, whereas NADPH is oxidized into NADP+. Several lines of evidence have shown that in STZ-induced diabetic rats, GSH provides antioxidant function twice a week and protects against motor and sensory nerve conduction velocity where, motor and sensory nerve conduction velocity reduce the endoneural blood flow (Chen et al., 2018).
CAT is another kind of antioxidant that provides protection against pancreatic β cell damage from H2O2 oxidation. In the hyperglycemic condition, enzymatic deficiency leads to damage of β cells. The CAT deficiency causes the deficiency of glucose-6-phosphate dehydrogenase. In the case of DPN, the combined action of antioxidant mostly effective as SOD provides antioxidant effects by binding with alpha lipoic acid. Not only that but also SOD, GSH, and CAT play combined roles in other neurological disorders.
Antioxidant function of H2S in AD, PD, and ALS
H2S and AD
Oxidative damage of DNA in AD mostly occurs in the cerebral cortex and cerebellum. H2S protects AD brain from oxidative damage by the activity of GSH (Figure 3). In this case, N-acetylcysteine acts as a precursor of GSH because Aβ shows impaired memory functions in AD patients compared with controls. In addition to this, H2S inhibits Aβ-induced cell injury through MAPK and JNK signaling pathway (Zhang et al., 2013). In addition to this, AD is exacerbated by 4-hydroxynonenal in the absence of aldehyde dehydrogenase (ALDH2) but the upregulation of ALDH2 by FA is protective or toxic remains unclear. Michel et al. (2010) demonstrated that mitochondrial ALDH2 activity was significantly increased in the putamen of AD patients. Upregulated ALDH2 increases 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress in dopaminergic neuron by heme oxygenase interaction. This upregulation of ALDH2 expression by FA is also blocked by H2S (Chen et al., 2017). Apart from this, NaHS downregulates glucose regulated protein 78 expression, suppresses hippocampal chomologous protein expression, reduces hippocampal cleaved caspase-12 expression, and enhances the generation of hippocampal endogenous H2S in STZ-exposed diabetic rats (Zou et al., 2017).
Figure 3.

Antioxidant function of H2S against oxidative stress in diabetes-induced neurodegenerative diseases.
Oxidative stress increases cellular dysfunction in type 2 diabetes mellitus (T2DM), Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). At the stage of hyperglycemia and in the early stage of insulin resistance, AD develops because of β cell failure, and lower level of insulin shows impaired central nervous system (CNS) signaling. As a consequence, brain glucose metabolism becomes reduced and neurodegenerative disease AD develops. Insulin resistance is also associated with the elevated level of amyloid beta (Aβ) accumulation and hyperphosphorylation of tau protein which are decreased by increased level of superoxide dismutase (SOD) in the mitochondria. Similarly, oxidative stress is responsible for diminishing nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) in T2DM. On the other hand, reactive oxygen species (ROS) generates in substantia nigra compacta. The level of DA neurons is higher than that of other neurons because oxidative metabolism of dopamine (DA) takes place in axon terminal. Antioxidant function of H2S is mediated by increased level of SOD which suppresses the 6-hydroxydopamine (6-OHDA)-induced ROS formation that ultimately reduces the risk of PD. At the same time, H2S donor, sodium hydrogen sulfide (NaHS) increases β-cell oxidation to decrease dopaminergic loss but reduced glutathione (GSH) is produced weakly in DA neurons. In the case of ALS, oxidative damage to ribonucleic acid (RNA) is seen in the CNS and SOD1 is over-expressed in mutant SOD1G93A. CAT: Catalase; H2S: hydrogen sulfide; OS: oxidative stress; RNS: reactive nitrogen species.
H2S and PD
H2S also plays a role as an antioxidant in PD. PD is very common in the population of over 65 years of age that is characterized by nigral dopaminergic loss (Babizhayev et al., 2015). ROS are largely responsible for dopaminergic loss. The striatal dopamine and the dysfunctional neurons show a variety of motor symptoms as a result of oxidative insults (Yue et al., 2016). To overcome the motor neuron disorder in PD, an experimental study showed that H2S protects MPP+-induced oxidative damage in PC12 cells where NaHS acts as a donor of H2S (Yin et al., 2009). Additionally, upregulated ALDH2 increases MPP+-induced oxidative stress in dopaminergic neuron by heme oxygenase interaction while H2S can attenuate this toxicity (Tang et al., 2012). The endogenous reduced level of H2S was observed in PD with loss of tyrosine-hydroxylase positive nerve. 6-OHDA-induced dopamine nerve shows the reduced level of endogenous H2S. In rotenone-induced rats, H2S donor, NaHS treatment inactivates microglia in the SN and increases proinflammatory factors, tumor necrosis factor and nitric oxide (Hu et al., 2010). Moreover, H2S mediates cell survival through S-glutathionylation by oxidant-sensitive cysteine molecule. Increased level of GSH is not only retard dopaminergic loss but also helps to identify the mutant proteins which are linked to PD progression (Figure 3).
H2S and ALS
ALS is one of the factors that affect motor neuron degeneration which shows neuronal damage in the upper and lower motor neurons. In ALS, oxidized proteins and SOD1 mutations are responsible factors and the mutated genes are Gly37, Gly85, and Gly93 (Johnson et al., 2012). The high level of H2S in SOD1G93A showed altered metabolism in familial ALS (Shefa et al., 2018). Increased H2S level was found in ALS patients and it was identified as glia-released inflammatory factor that contributes to motor neuron death (Davoli et al., 2015). Although GSH suppresses oxidative stress, an in vitro study showed that depletion of GSH causes motor neuron death in ALS (Islam, 2017). Moreover, according to the study by Basso et al. (2009), NSC-34 cells containing Gly93Ala SOD1 showed 30% increased level of cellular GSH in ALS through short term expression and on the other hand, prolonged expression showed 30% decreased GSH levels (Johnson et al., 2012). To remove the oxidative modification of SOD1 in ALS (Figure 3), the S-glutathionylation of cys111 residue was essential because it suppressed protein damage and accumulation.
Conclusions
Diabetes-induced peripheral as well as motor neuron disorders are related to metabolic pathways that are associated with oxidative stress in the mitochondria because ROS is generated from the mitochondria. This oxidative stress is increased when antioxidant function is reduced. In DPN, hyperglycemia-induced oxidative stress causes the accumulation of ROS through auto-oxidation of protein, lipid, and glucose by utilizing cellular pathways that involved polyol, PKC, AGEs, and PARP. Reduction of ROS by antioxidant, H2S reduces the hyperglycemic effect which minimizes diabetic complications including other neurodegenerative motor neuron diseases. At a large extent, oxidative stress is the fundamental factor for neural damage in DM. For obtaining therapeutic outcome of different functions of antioxidant, H2S might be an effective source. Also, for proper maintaining of H2S against oxidative stress, the metabolic function of H2S should proceed under neurodegenerative disease condition through mediating proper cell signaling.
Additional file:
Additional Table 1 (298.9KB, pdf) : Database search strategy.
Database search strategy
Footnotes
Conflicts of interest: The authors reported no potential conflicts of interests.
Financial support: This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, No. 2018R1A2B6001123 (to NYJ), No. 2018R1D1A1B07040282 (to JJ).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, No. 2018R1A2B6001123 (to NYJ), No. 2018R1D1A1B07040282 (to JJ)..
C-Editor: Zhao M; S-Editor: Li CH; L-Editor: Song LP; T-Editor: Jia Y
References
- 1.Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S. Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/ RNS generation. J Biomed Sci. 2017;24:76. doi: 10.1186/s12929-017-0379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babizhayev MA, Strokov IA, Nosikov VV, Savel’yeva EL, Sitnikov VF, Yegorov YE, Lankin VZ. The role of oxidative stress in diabetic neuropathy: generation of free radical species in the glycation reaction and gene polymorphisms encoding antioxidant enzymes to genetic susceptibility to diabetic neuropathy in population of type I diabetic patients. Cell Biochem Biophys. 2015;71:1425–1443. doi: 10.1007/s12013-014-0365-y. [DOI] [PubMed] [Google Scholar]
- 3.Balasubramanian S, Pandranki L, Maupin S, Ramasamy K, Taylor AB, Hart PJ, Baseman JB, Kannan TR. Disulfide bond of Mycoplasma pneumoniae community acquired respiratory distress syndrome toxin is essential to maintain the ADP‐ribosylating and vacuolating activities. Cell Microbiol. 2019:e13032. doi: 10.1111/cmi.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basso M, Samengo G, Nardo G, Massignan T, D’Alessandro G, Tartari S, Cantoni L, Marino M, Cheroni C, De Biasi S. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One. 2009;4:e8130. doi: 10.1371/journal.pone.0008130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breyer MD, Tchekneva E, Qi Z, Takahashi T, Fogo AB, Zhao HJ, Harris RC. Genetics of diabetic nephropathy: Lessons from mice. In: Young JPW, Cinelli P, Corominas M, et al., editors. Seminars in nephrology. Basel Postfach, Switzerland: Elsevier; 2007. pp. 237–247. [DOI] [PubMed] [Google Scholar]
- 6.Callaghan BC, Hur J, Feldman EL. Diabetic neuropathy: one disease or two? Curr Opin Neurol. 2012;25:536. doi: 10.1097/WCO.0b013e328357a797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide–mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cameron NE, Gibson TM, Nangle MR, Cotter MA. Inhibitors of advanced glycation end product formation and neurovascular dysfunction in experimental diabetes. Ann N Y Acad Sci. 2005;1043:784–792. doi: 10.1196/annals.1333.091. [DOI] [PubMed] [Google Scholar]
- 9.Chang JK, Leso A, Subaiea GM, Lahouel A, Masoud A, Mushtaq F, Deeb R, Eid A, Dash M, Bihaqi SW. Tolfenamic acid: a modifier of the tau protein and its role in cognition and tauopathy. Curr Alzheimer Res. 2018;15:655–663. doi: 10.2174/1567205015666180119104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee S, Mudher A. Alzheimer’s disease and type 2 diabetes: A critical assessment of the shared pathological traits. Front Neurosci. 2018;12:383. doi: 10.3389/fnins.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Liu W, Yi H, Hu X, Peng L, Yang F. The natural rotenoid deguelin ameliorates diabetic neuropathy by decreasing oxidative stress and plasma glucose levels in rats via the Nrf2 signalling pathway. Cell Physiol Biochem. 2018;48:1164–1176. doi: 10.1159/000491983. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Zhou CF, Xiao F, Huang HL, Zhang P, Gu HF, Tang XQ. Inhibition of ALDH2 protects PC12 cells against formaldehyde-induced cytotoxicity: involving the protection of hydrogen sulphide. Clin Exp Pharmacol Physiol. 2017;44:595–601. doi: 10.1111/1440-1681.12741. [DOI] [PubMed] [Google Scholar]
- 13.Dal S, Sigrist S. The protective effect of antioxidants consumption on diabetes and vascular complications. Diseases. 2016;4:24. doi: 10.3390/diseases4030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davoli A, Greco V, Spalloni A, Guatteo E, Neri C, Rizzo GR, Cordella A, Romigi A, Cortese C, Bernardini S. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann Neurol. 2015;77:697–709. doi: 10.1002/ana.24372. [DOI] [PubMed] [Google Scholar]
- 15.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. FEBS J. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 16.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes. 2003;52:1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 17.Fadupin GT, Akpoghor AU, Okunade KA. A comparative study of serum ascorbic acid level in people with and without type 2 diabetes in Ibadan Nigeria. Afr J Med Med Sci. 2007;36:335–339. [PubMed] [Google Scholar]
- 18.Fantus IG. The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinol Rounds. 2002;2:1–8. [Google Scholar]
- 19.Farmer KL, Li C, Dobrowsky RT. Diabetic peripheral neuropathy: should a chaperone accompany our therapeutic approach? Pharmacol Rev. 2012;64:880–900. doi: 10.1124/pr.111.005314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita H, Fujishima H, Chida S, Takahashi K, Qi Z, Kanetsuna Y, Breyer MD, Harris RC, Yamada Y, Takahashi T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol. 2009;20:1303–1313. doi: 10.1681/ASN.2008080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2017;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, Dagogo-Jack S, DeFronzo RA, Einhorn D, Fonseca VA. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91–120. doi: 10.4158/CS-2017-0153. [DOI] [PubMed] [Google Scholar]
- 23.Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26:501–518. doi: 10.1089/ars.2016.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giri B, Dey S, Das T, Sarkar M, Banerjee J, Dash SK. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction infection cancer progression and other pathophysiological consequences: an update on glucose toxicity. Biomed Pharmacother. 2018;107:306–328. doi: 10.1016/j.biopha.2018.07.157. [DOI] [PubMed] [Google Scholar]
- 26.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 27.Hernández‐Ortega K, Garcia‐Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 2016;26:593–605. doi: 10.1111/bpa.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinder LM, Vivekanandan-Giri A, McLean LL, Pennathur S, Feldman EL. Decreased glycolytic and tricarboxylic acid cycle intermediates coincide with peripheral nervous system oxidative stress in a murine model of type 2 diabetes. J Endocrinol. 2013;216:1–11. doi: 10.1530/JOE-12-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong NJ, Garvin JL. Endogenous flow-induced superoxide stimulates Na/H exchange activity via PKC in thick ascending limbs. Am J Physiol Physiol. 2014;307:F800–805. doi: 10.1152/ajprenal.00260.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosseini A, Abdollahi M. Diabetic neuropathy and oxidative stress: therapeutic perspectives. Oxid Med Cell Longev. 2013;2013:168039. doi: 10.1155/2013/168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoyer S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. In: Segev I, Abbott L, Kaas JH, et al., editors. Frontiers in Clinical Neuroscience. Lausanne, Switzerland: Springer; 2004. pp. 135–152. [DOI] [PubMed] [Google Scholar]
- 32.Hu L, Lu M, Tiong CX, Dawe GS, Hu G, Bian J. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell. 2010;9:135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- 33.Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39:73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 34.Jia J, Xiao Y, Wang W, Qing L, Xu Y, Song H, Zhen X, Ao G, Alkayed NJ, Cheng J. Differential mechanisms underlying neuroprotection of hydrogen sulfide donors against oxidative stress. Neurochem Int. 2013;62:1072–1078. doi: 10.1016/j.neuint.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4:1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jovičić A, Paul JW, Gitler AD. Nuclear transport dysfunction: a common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J Neurochem. 2016;138:134–144. doi: 10.1111/jnc.13642. [DOI] [PubMed] [Google Scholar]
- 37.Karki R, Hofmann-Apitius M. Comorbidity analysis between Alzheimer’s disease and type 2 diabetes mellitus (T2DM) based on shared pathways and the role of T2DM drugs. J Alzheimer’s Dis. 2017;60:721–731. doi: 10.3233/JAD-170440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenart L, Balogh DB, Lenart N, Barczi A, Hosszu A, Farkas T, Hodrea J, Szabo AJ, Szigeti K, Denes A. Novel therapeutic potential of angiotensin receptor 1 blockade in a rat model of diabetes-associated depression parallels altered BDNF signalling. Diabetologia. 2019 doi: 10.1007/s00125-019-4888-z. doi: 101007/s00125-019-4888-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewerenz J, Maher P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front Neurosci. 2015;9:469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757. doi: 10.2147/CIA.S158513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahmoud AM, Alexander MY, Tutar Y, Wilkinson FL, Venditti A. Oxidative stress in metabolic disorders and drug-induced injury: the potential role of Nrf2 and PPARs activators. Oxid Med Cell Longev. 2017;2017:2508909. doi: 10.1155/2017/2508909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marengo B, Nitti M, Furfaro AL, Colla R, Ciucis CD, Marinari UM, Pronzato MA, Traverso N, Domenicotti C. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid Med Cell Longev. 2016;2016:6235641. doi: 10.1155/2016/6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mariosa D, Kamel F, Bellocco R, Ye W, Fang F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur J Neurol. 2015;22:1436–1442. doi: 10.1111/ene.12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G. Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int. 2016;2016:2589276. doi: 10.1155/2016/2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michel TM, Gsell W, Käsbauer L, Tatschner T, Sheldrick AJ, Neuner I, Schneider F, Grünblatt E, Riederer P. Increased activity of mitochondrial aldehyde dehydrogenase (ALDH) in the putamen of individuals with Alzheimer’s disease: a human postmortem study. J Alzheimers Dis. 2010;19:1295–1301. doi: 10.3233/JAD-2010-1326. [DOI] [PubMed] [Google Scholar]
- 46.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol cell Biol. 2014;15:482. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 47.Munro D, Treberg JR. A radical shift in perspective: mitochondria as regulators of reactive oxygen species. J Exp Biol. 2017;220:1170–1180. doi: 10.1242/jeb.132142. [DOI] [PubMed] [Google Scholar]
- 48.Newsholme P, Haber EP, Hirabara SM, Rebelato ELO, Procopio J, Morgan D, Oliveira‐Emilio HC, Carpinelli AR, Curi R. Diabetes associated cell stress and dysfunction: role of mitochondrial and non‐mitochondrial ROS production and activity. J Physiol. 2007;583:9–24. doi: 10.1113/jphysiol.2007.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 50.Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oyenihi AB, Ayeleso AO, Mukwevho E, Masola B. Antioxidant strategies in the management of diabetic neuropathy. Biomed Res Int. 2015;2015:515042. doi: 10.1155/2015/515042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pop-Busui R, Boulton AJ, Feldman EL, Bril V, Freeman R, Malik RA, Sosenko JM, Ziegler D. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2017;40:136–154. doi: 10.2337/dc16-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiao H, Chen H, Dong Y, Ma H, Zhao G, Tang F, Li Z. Polydatin attenuates H2O2-induced oxidative stress via PKC pathway. Oxid Med Cell Longev. 2016;2016:5139458. doi: 10.1155/2016/5139458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qu K, Lee SW, Bian JS, Low CM, Wong PTH. Hydrogen sulfide: Neurochemistry and neurobiology. Neurochem Int. 2008;52:155–65. doi: 10.1016/j.neuint.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 55.Rahman Q, Abidi P, Afaq F, Schiffmann D, Mossman BT, Kamp DW, Athar M. Glutathione redox system in oxidative lung injury. Crit Rev Toxicol. 1999;29:543–568. doi: 10.1080/10408449991349276. [DOI] [PubMed] [Google Scholar]
- 56.Sampson SR, Bucris E, Horovitz-Fried M, Parnas A, Kahana S, Abitbol G, Chetboun M, Rosenzweig T, Brodie C, Frankel S. Insulin increases H2O2-induced pancreatic beta cell death. Apoptosis. 2010;15:1165–1176. doi: 10.1007/s10495-010-0517-5. [DOI] [PubMed] [Google Scholar]
- 57.Sandireddy R, Yerra VG, Areti A, Komirishetty P, Kumar A. Neuroinflammation and oxidative stress in diabetic neuropathy: Futuristic strategies based on these targets. Int J Endocrinol. 2014;2014:674987. doi: 10.1155/2014/674987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sasson S. Nutrient overload, lipid peroxidation and pancreatic beta cell function. Free Radic Biol Med. 2017;111:102–109. doi: 10.1016/j.freeradbiomed.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care. 2011;34:1102–1108. doi: 10.2337/dc10-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sekar S, Mani S, Rajamani B, Manivasagam T, Thenmozhi AJ, Bhat A, Ray B, Essa MM, Guillemin GJ, Chidambaram SB. Telmisartan ameliorates astroglial and dopaminergic functions in a mouse model of chronic parkinsonism. Neurotox Res. 2018;34:597–612. doi: 10.1007/s12640-018-9921-3. [DOI] [PubMed] [Google Scholar]
- 61.Shah SA, Amin FU, Khan M, Abid MN, Rehman SU, Kim TH, Kim MW, Kim MO. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J Neuroinflammation. 2016;13:286. doi: 10.1186/s12974-016-0752-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shefa U, Kim MS, Jeong NY, Jung J. Antioxidant and cell-signaling functions of hydrogen sulfide in the central nervous system. Oxid Med Cell Longev. 2018;2018:1873962. doi: 10.1155/2018/1873962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Størling J, Pociot F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes (Basel) 2017;8:72. doi: 10.3390/genes8020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Svetoni F, Caporossi D, Paronetto MP. Oxidative stress affects FET proteins localization and alternative pre-mRNA processing in cellular models of ALS. Free Radic Biol Med. 2014;75(Suppl 1):S51. doi: 10.1016/j.freeradbiomed.2014.10.820. [DOI] [PubMed] [Google Scholar]
- 65.Tahara N, Yamagishi S, Matsui T, Takeuchi M, Nitta Y, Kodama N, Mizoguchi M, Imaizumi T. Serum levels of advanced glycation end products (AGEs) are independent correlates of insulin resistance in nondiabetic subjects. Cardiovasc Ther. 2012;30:42–48. doi: 10.1111/j.1755-5922.2010.00177.x. [DOI] [PubMed] [Google Scholar]
- 66.Tan KC, Shiu SW, Wong Y, Tam X. Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes Metab Res Rev. 2011;27:488–492. doi: 10.1002/dmrr.1188. [DOI] [PubMed] [Google Scholar]
- 67.Tang XQ, Ren YK, Zhou CF, Yang CT, Gu HF, He JQ, Chen RQ, Zhuang YY, Fang HR, Wang CY. Hydrogen sulfide prevents formaldehyde-induced neurotoxicity to PC12 cells by attenuation of mitochondrial dysfunction and pro-apoptotic potential. Neurochem Int. 2012;61:16–24. doi: 10.1016/j.neuint.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 68.Tangvarasittichai S. Oxidative stress, insulin resistance dyslipidemia and type 2 diabetes mellitus. World J Diabetes. 2015;6:456–480. doi: 10.4239/wjd.v6.i3.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FYT, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM. Inhibition of NADPH oxidase prevents advanced glycation end product–mediated damage in diabetic nephropathy through a protein kinase C-α–dependent pathway. 2008 doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 70.Tian R, Yang W, Xue Q, Gao L, Huo J, Ren D, Chen X. Rutin ameliorates diabetic neuropathy by lowering plasma glucose and decreasing oxidative stress via Nrf2 signaling pathway in rats. Eur J Pharmacol. 2016;771:84–92. doi: 10.1016/j.ejphar.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 71.Torres-Aleman I, Barrios V, Berciano J. The peripheral insulin-like growth factor system in amyotrophic lateral sclerosis and in multiple sclerosis. Neurology. 1998;50:772–776. doi: 10.1212/wnl.50.3.772. [DOI] [PubMed] [Google Scholar]
- 72.Trudeau F, Gagnon S, Massicotte G. Hippocampal synaptic plasticity and glutamate receptor regulation: influences of diabetes mellitus. Eur J Pharmacol. 2004;490:177–186. doi: 10.1016/j.ejphar.2004.02.055. [DOI] [PubMed] [Google Scholar]
- 73.Vaccaro A, Tauffenberger A, Ash PE, Carlomagno Y, Petrucelli L, Parker JA. TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012;8:e1002806. doi: 10.1371/journal.pgen.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verdile G, Keane KN, Cruzat VF, Medic S, Sabale M, Rowles J, Wijesekara N, Martins RN, Fraser PE, Newsholme P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. 2015;2015:105828. doi: 10.1155/2015/105828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wallis N, Lau CL, Farg MA, Atkin JD, Beart PM, O’Shea RD. SOD1 mutations causing familial amyotrophic lateral sclerosis induce toxicity in astrocytes: evidence for bystander effects in a continuum of astrogliosis. Neurochem Res. 2018;43:166–179. doi: 10.1007/s11064-017-2385-7. [DOI] [PubMed] [Google Scholar]
- 76.Xiao Y, He X, Han Q, Yang F, Zhou S. Atorvastatin prevents glomerular extracellular matrix formation by interfering with the PKC signaling pathway. Mol Med Rep. 2018;17:6441–6448. doi: 10.3892/mmr.2018.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan LJ. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J Diabetes Res. 2014;2014 doi: 10.1155/2014/137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yin J, Ren W, Yang G, Duan J, Huang X, Fang R, Li C, Li T, Yin Y, Hou Y. l‐Cysteine metabolism and its nutritional implications. Mol Nutr Food Res. 2016;60:134–146. doi: 10.1002/mnfr.201500031. [DOI] [PubMed] [Google Scholar]
- 79.Yin WL, He JQ, Hu B, Jiang ZS, Tang XQ. Hydrogen sulfide inhibits MPP(+)-induced apoptosis in PC12 cells. Life Sci. 2009;85(7-8):269–275. doi: 10.1016/j.lfs.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 80.Yue X, Li H, Yan H, Zhang P, Chang L, Li T. Risk of Parkinson disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. Medicine (Baltimore) 2016;95:e3549. doi: 10.1097/MD.0000000000003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang H, Davies KJA, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol Med. 2015;88:314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH, Liu LS, Jiang ZS. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol Cell Biol. 2013;33:1104–1113. doi: 10.1128/MCB.01215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu X, Chen C, Ye D, Guan D, Ye L, Jin J, Zhao H, Chen Y, Wang Z, Wang X. Diammonium glycyrrhizinate upregulates PGC-1α and protects against Aβ1–42-induced neurotoxicity. PLoS One. 2012;7:e35823. doi: 10.1371/journal.pone.0035823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zou W, Yuan J, Tang ZJ, Wei HJ, Zhu WW, Zhang P, Gu HF, Wang CY, Tang XQ. Hydrogen sulfide ameliorates cognitive dysfunction in streptozotocin-induced diabetic rats: involving suppression in hippocampal endoplasmic reticulum stress. Oncotarget. 2017;8:64203–64216. doi: 10.18632/oncotarget.19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Database search strategy