

Abstract
Background
Spinal muscular atrophy (SMA) is caused by a homozygous deletion of the survival motor neuron 1 (SMN1) gene on chromosome 5, or a heterozygous deletion in combination with a point mutation in the second SMN1 allele. This results in degeneration of anterior horn cells, which leads to progressive muscle weakness. By definition, children with SMA type I are never able to sit without support and usually die or become ventilator dependent before the age of two years. There have until very recently been no drug treatments to influence the course of SMA. We undertook this updated review to evaluate new evidence on emerging treatments for SMA type I. The review was first published in 2009 and previously updated in 2011.
Objectives
To assess the efficacy and safety of any drug therapy designed to slow or arrest progression of spinal muscular atrophy (SMA) type I.
Search methods
We searched the Cochrane Neuromuscular Specialised Register, CENTRAL, MEDLINE, Embase, and ISI Web of Science conference proceedings in October 2018. We also searched two trials registries to identify unpublished trials (October 2018).
Selection criteria
We sought all randomised controlled trials (RCTs) or quasi‐RCTs that examined the efficacy of drug treatment for SMA type I. Included participants had to fulfil clinical criteria and have a genetically confirmed deletion or mutation of the SMN1 gene (5q11.2‐13.2).
The primary outcome measure was age at death or full‐time ventilation. Secondary outcome measures were acquisition of motor milestones, i.e. head control, rolling, sitting or standing, motor milestone response on disability scores within one year after the onset of treatment, and adverse events and serious adverse events attributable to treatment during the trial period.
Treatment strategies involving SMN1 gene replacement with viral vectors are out of the scope of this review.
Data collection and analysis
We followed standard Cochrane methodology.
Main results
We identified two RCTs: one trial of intrathecal nusinersen in comparison to a sham (control) procedure in 121 randomised infants with SMA type I, which was newly included at this update, and one small trial comparing riluzole treatment to placebo in 10 children with SMA type I.
The RCT of intrathecally‐injected nusinersen was stopped early for efficacy (based on a predefined Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) response). At the interim analyses after 183 days of treatment, 41% (21/51) of nusinersen‐treated infants showed a predefined improvement on HINE‐2, compared to 0% (0/27) of participants in the control group. This trial was largely at low risk of bias.
Final analyses (ranging from 6 months to 13 months of treatment), showed that fewer participants died or required full‐time ventilation (defined as more than 16 hours daily for 21 days or more) in the nusinersen‐treated group than the control group (hazard ratio (HR) 0.53, 95% confidence interval (CI) 0.32 to 0.89; N = 121; a 47% lower risk; moderate‐certainty evidence). A proportion of infants in the nusinersen group and none of 37 infants in the control group achieved motor milestones: 37/73 nusinersen‐treated infants (51%) achieved a motor milestone response on HINE‐2 (risk ratio (RR) 38.51, 95% CI 2.43 to 610.14; N = 110; moderate‐certainty evidence); 16/73 achieved head control (RR 16.95, 95% CI 1.04 to 274.84; moderate‐certainty evidence); 6/73 achieved independent sitting (RR 6.68, 95% CI 0.39 to 115.38; moderate‐certainty evidence); 7/73 achieved rolling over (RR 7.70, 95% CI 0.45 to 131.29); and 1/73 achieved standing (RR 1.54, 95% CI 0.06 to 36.92; moderate‐certainty evidence). Seventy‐one per cent of nusinersen‐treated infants versus 3% of infants in the control group were responders on the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) measure of motor disability (RR 26.36, 95% CI 3.79 to 183.18; N = 110; moderate‐certainty evidence).
Adverse events and serious adverse events occurred in the majority of infants but were no more frequent in the nusinersen‐treated group than the control group (RR 0.99, 95% CI 0.92 to 1.05 and RR 0.70, 95% CI 0.55 to 0.89, respectively; N = 121; moderate‐certainty evidence).
In the riluzole trial, three of seven children treated with riluzole were still alive at the ages of 30, 48, and 64 months, whereas all three children in the placebo group died. None of the children in the riluzole or placebo group developed the ability to sit, which was the only milestone reported. There were no adverse effects. The certainty of the evidence for all measured outcomes from this study was very low, because the study was too small to detect or rule out an effect, and had serious limitations, including baseline differences. This trial was stopped prematurely because the pharmaceutical company withdrew funding.
Various trials and studies investigating treatment strategies other than nusinersen, such as SMN2 augmentation by small molecules, are ongoing.
Authors' conclusions
Based on the very limited evidence currently available regarding drug treatments for SMA type 1, intrathecal nusinersen probably prolongs ventilation‐free and overall survival in infants with SMA type I. It is also probable that a greater proportion of infants treated with nusinersen than with a sham procedure achieve motor milestones and can be classed as responders to treatment on clinical assessments (HINE‐2 and CHOP INTEND). The proportion of children experiencing adverse events and serious adverse events on nusinersen is no higher with nusinersen treatment than with a sham procedure, based on evidence of moderate certainty. It is uncertain whether riluzole has any effect in patients with SMA type I, based on the limited available evidence. Future trials could provide more high‐certainty, longer‐term evidence to confirm this result, or focus on comparing new treatments to nusinersen or evaluate them as an add‐on therapy to nusinersen.
Plain language summary
Drug treatment for spinal muscular atrophy type I
What is the aim of this review?
The aim of this Cochrane Review was to look at the effects of drug treatments on spinal muscular atrophy (SMA) type I, in terms of age at death or full‐time ventilation and the ability to reach motor milestones, e.g. rolling, sitting or standing, within one year after beginning treatment, and any adverse events.
Key messages
Nusinersen probably increases ventilation‐free and overall survival in children with SMA type I. Nusinersen may improve the proportion of infants achieving motor milestones. Adverse events and serious adverse events are probably not more common with nusinersen injection than with a sham procedure.
It is uncertain whether riluzole has any effect in SMA type I, based on the limited available evidence.
What was studied in the review?
SMA is a disorder with onset in childhood and adolescence and leads to increasing muscle weakness. SMA type I, also known as Werdnig‐Hoffman disease, is the most severe form of SMA and begins before the age of six months. Untreated children with SMA type I will never be able to sit without support and in general die or develop respiratory insufficiency and need non‐invasive ventilation before they reach the age of two years.
At the time of the previous versions of the review there was no known treatment to slow down or cure SMA type I. We updated the review to include emerging evidence.
Cochrane review authors collected relevant studies and found two trials. Both studies were funded by pharmaceutical companies. One trial, in 121 infants with SMA type I, studied nusinersen, which is an antisense oligonucleotide drug, given by injection into the spinal canal. The researchers compared the effects of nusinersen with a sham procedure in the control group. This trial stopped early because results showed that nusinersen improved the proportion of infants achieving motor milestones. The other trial compared riluzole to placebo and involved 10 infants with SMA type I. This trial was stopped prematurely because the pharmaceutical company withdrew funding.
What are the main results of the review
Results were not all reported at the same follow‐up point, as the trial was stopped before some participants completed the planned follow‐up. Nusinersen probably reduced the risk of death or progression to full‐time ventilation (assisted breathing) by 47% compared to the control group. The evidence is also moderately certain that the percentage of children with a response on objective clinical assessments of motor function was higher in the nusinersen‐treated group than the sham procedure group (51% versus 0% on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) and 71% versus 3% on the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND)). Infants treated with nusinersen are probably also more likely to reach developmental milestones: 16 of the 73 treated infants achieved head control, six achieved independent sitting, seven achieved the ability to roll over, and one achieved the ability to stand; none of the 37 infants in the control group achieved any of these milestones.
There was probably little difference between the nusinersen‐treated and control group in the number of infants with adverse events; the majority experienced adverse events. Serious adverse events were probably no more common with nusinersen than with the sham procedure.
One study compared riluzole treatment to placebo (an identical, but inactive treatment) in 10 children with SMA type I. The certainty of the evidence was very low, mainly because the study was too small to detect or rule out an effect. In this trial, all three children in the placebo group and four of the seven children in the riluzole group died within 12 months of the study. Three of the seven children treated with riluzole were still alive at the ages of 30, 48, and 64 months. None of the children in the riluzole or placebo group developed the ability to sit. The evidence can neither confirm nor rule out an effect of riluzole in children with SMA type I because of its small size and severe limitations.
How up‐to‐date is this review?
We searched for studies that had been published up to October 2018.
Summary of findings
Background
Description of the condition
Spinal muscular atrophy (SMA) is a genetic anterior horn cell disorder with onset ranging from infancy to adolescence and even adulthood. It is caused by the homozygous deletion or heterozygous deletion in combination with a point mutation in the second allele of the survival motor neurone 1 (SMN1) gene that has been mapped to chromosome 5q11.2‐13.3 (Brzustowicz 1990; Gilliam 1990; Lefebvre 1995; Melki 1990a; Melki 1990b). SMA has an annual incidence of 1 in 6000 to 12,000 (Arkblad 2009; Cobben 2001; Nicole 2002). It is clinically characterised by muscle weakness, proximal more than distal and in the legs more than the arms, that progresses over time (Iannaccone 2001; Talbot 1999). There are indications that other structures than anterior horn cells, including the neuromuscular junction and muscle may also be sensitive to the deficiency of SMN protein due to the homozygous deletion of the SMN1 gene (Braun 1995; Cifuentes‐Diaz 2002; Kariya 2008; Murray 2008).
Weakness shows a particular pattern, with the best known example, axial and proximal weakness with weakened intercostal muscles and sparing of the diaphragm (Kroksmark 2001; Thomas 1994). Survival depends primarily on respiratory function (Dubowitz 1995; Russman 1992; Talbot 1999). Although the face is often spared, tongue fasciculations and facial weakness are not unusual findings (Iannaccone 1993). The cognitive function of SMA patients is normal and in infantile cases there is often a striking discrepancy between alertness and the ability to move (Iannaccone 1998; Thomas 1994).
Classification of SMA according to the International SMA Collaboration distinguishes five SMA types (0 to IV) and is based on age of onset and maximal acquired motor function (Finkel 2015; Mercuri 2012; Munsat 1992). SMA type 0, and IV represent the two ends of the spectrum of SMA, which are out of the scope of this review. SMA types II and III are the topic of a separate Cochrane Review (Wadman 2018).
SMA type I is the most common form and is also known as Werdnig‐Hoffmann disease, acute SMA, and infantile‐onset SMA. The age of onset is before six months and it is further characterised by severe progressive muscle weakness and hypotonia (Iannaccone 1998). Children with SMA type I will never be able to sit without support, and respiratory insufficiency usually occurs before the age of two years (Cobben 2008; Finkel 2014; Iannaccone 1993; Oskoui 2007; Parker 2008; Thomas 1994). It is one of the most important causes of death due to a genetic disease in childhood (Nicole 2002).
The SMN region contains two SMN genes, the telomeric SMN gene (SMN1 or SMNt) and the centromeric SMN gene (SMN2 or SMNc) (Iannaccone 1998; Nicole 2002). The SMN1 and SMN2 gene are almost identical, but a crucial C to T nucleotide difference in exon 7 results in its exclusion from most SMN2 mRNA copies (Lefebvre 1995; Lorson 1999). Consequently, there is no transcription of stable SMN protein from the SMN1 gene and the SMN2 gene is not able to produce enough stable SMN protein (Cobben 1995; Lefebvre 1995; Nicole 2002). The clinical severity of the disease is related to the number of copies of the SMN2 gene (Feldkotter 2002; Harada 2002; Piepers 2008; Swoboda 2005; Wadman 2017a).
The cellular functions of the SMN protein are multiple, but its vital role in motor neurons is not known (Sumner 2007). SMN protein is important for ribonucleoprotein (RNP) assembly (Burghes 2009; Gendron 1999; Jablonka 2000; Lefebvre 1998; Pellizzoni 1998), motor axon outgrowth and axonal transport (McWhorter 2003; Rossoll 2003), protection against superoxide dismutase 1 (SOD1) toxicity (Zou 2007), endocytosis (Hosseinibarkooie 2016; Riessland 2017), and ubiquitination homeostasis (Wishart 2014).
Description of the intervention
Drug treatment for SMA type I is urgently needed. Management of SMA consists of preventing or treating the complications (Finkel 2018; Iannaccone 1998; Mercuri 2018; Wang 2007). Administration of agents capable of increasing the expression of SMN protein levels may improve the outcome in SMA (Feldkotter 2002; Gavrilina 2008; Lorson 1999). Transcriptional SMN2 activation, facilitation or correction of SMN2 splicing, translational activation, and stabilisation of the full‐length SMN protein are possible therapeutic strategies for SMA. Other strategies are improvement of motor neuron viability by neuroprotective or neurotrophic agents (Lunn 2008; Thurmond 2008; Wirth 2006). Recently, trials with splice site modulators (EMBRACE 2015; Finkel 2016; SHINE 2015), or RNA‐degradation inhibitors (Butchbach 2010; Gogliotti 2013; Van Meerbeke 2013), and compounds that replace the SMN1 gene have started (Mendell 2017).
Drugs that have been tested in open and uncontrolled studies of children with SMA type I are riluzole (Abbara 2011; ASIRI 2008), valproate (CARNIVAL Type I 2008; Conceicao 2010; SMART01; Swoboda 2009; SMART02 2016), recombinant human ciliary neurotrophic factor (CNTF) (Franz 1995), sodium phenylbutyrate or phenylbutyrate (NPTUNE02 2007; STOPSMA 2007), hydroxyurea (Chang 2002; NCT00568698), SMN2 antisense oligonucleotides (Finkel 2017 (ENDEAR); Finkel 2016; SHINE 2015; EMBRACE 2015), and small molecules (FIREFISH 2016; NCT02268552; MOONFISH 2015). SMN1 gene therapy and stem cell treatment are outside the scope of this review but are also the subject of ongoing trials (see Appendix 1; Mendell 2016; Mendell 2017 and Appendix 2; Villanova 2015; NCT02855112).
Below, we describe the working mechanisms and preclinical studies in SMA models of the various drugs tested in trials with patients with SMA type I. Several other compounds have been shown to have an effect on SMN expression in vivo and in vitro SMA studies, but have not (yet) been tested in studies or trials with patients with SMA and are therefore outside the scope of this review (see Appendix 3).
Antisense oligonucleotides
Antisense oligonucleotides or 'morpholinos', are synthetic strands of nucleic acids which are able to interfere with (stimulate or inhibit) mRNA products of the target DNA sequence. In this way, antisense oligonucleotides can modify potential splice sites and interfere with splicing (Porensky 2013). Multiple antisense oligonucleotides for the SMN2 gene have been developed and investigated (Bogdanik 2015; Keil 2014; Nizzardo 2014; Osman 2014; Shababi 2012; Skordis 2003; Staropoli 2015; Zhou 2013; Zhou 2015). The intronic splice silencer in intron 7 of SMN2, called nusinersen (formerly known as SMN Rx 39443 or IONIS SMN Rx or ISIS‐SMN Rx), specifically targets the splice silencer in intron 7 and ensures the inclusion of SMN2 exon 7. This results in increased SMN2 full‐length mRNA and protein production (Hua 2010), and subsequently SMA animal models have shown improved performance and survival (Hua 2011; Passini 2011). Nusinersen is an intrathecally‐injected therapy.
Ciliary neurotrophic factor (CNTF)
CNTF is thought to support the survival of motor neurons, but its working mechanism is unknown (ACTS 1996; Bongioanni 2004; Miller 1996).
Hydroxyurea
Hydroxyurea is a histone deacetylase inhibitor and a few studies have suggested a therapeutic role for these agents in SMA, as they appeared to activate SMN2 expression (Darras 2007; Kernochan 2005; Wirth 2006). In vitro, hydroxyurea increased SMN2 gene expression and production of SMN protein in cultured lymphocytes of SMA patients (Grzeschik 2005; Liang 2008).
Phenylbutyrate
Phenylbutyrate is another histone deacetylase inhibitor. In fibroblast cultures and leucocytes of patients with SMA treated with phenylbutyrate, the drug was able to increase SMN transcript expression (Also‐Rallo 2011; Andreassi 2004; Brahe 2005).
Riluzole
Riluzole is thought to have a neuroprotective effect on the motor neuron by blocking the presynaptic release of glutamate. Glutamate is released after presynaptic depolarisation and if the amino acid is not efficiently cleared it leads to increased levels of free radicals and eventually degeneration of motor neurons (Bryson 1996; Merlini 2003). In a mouse model of SMA, riluzole attenuated the disease progression (Haddad 2003).
Small molecules
RO6885247 or RG7800
The small molecule RO6885247/RG7800 selectively modulates SMN2 splicing toward the inclusion of exon 7 and thereby stimulates production of full‐length SMN2 messenger RNA. Administration of RO6885247/RG7800 improved and almost rescued motor function and survival of SMA mice (Naryshkin 2014).
RO7034067 or RG7916
The small molecule RO7034067/RG7916 modulates SMN2 splicing, but exact details on structure and pharmacology are not available.
LMI070
LMI070 reportedly modulates SMN2 function, but the precise working mechanism of LMI070 is unknown (NCT02268552).
Valproate
Valproate is a histone deacetylase inhibitor that increases SMN protein in vitro by increasing transcription of SMN2 (Kernochan 2005; Weihl 2006). It also has an antiglutamatergic effect (Kim 2007). Valproate has been tested in SMA and has shown positive results on SMN expression in vitro (Brichta 2003; Brichta 2006; Sumner 2003), and in vivo (Piepers 2011).
Why it is important to do this review
There is no treatment to slow down or cure SMA type I (Bosboom 2009; Wadman 2012).
Over the past decades, many studies explored various drug treatments in SMA animal models and/or patients with SMA. Currently, there are several drugs and compounds tested in uncontrolled, unblinded and non‐randomised settings, showing possible positive effects on SMA disease course through neuroprotection (e.g. cardiotrophin‐1, creatine, gabapentin, lamotrigine, riluzole), SMN2‐inducing activity by histone deacetylase inhibitors (e.g. valproate, phenylbutyrate, hydroxyurea), improvement of neuromuscular transmission function (e.g. pyridostigmine), SMN2 RNA modification by antisense oligonucleotides (e.g. nusinersen), genetic restoration of SMN1 through viral vectors, improvement of muscle metabolism and strength (e.g. creatine) and other (unknown) factors (e.g. somatotropin, salbutamol, thyrotropin releasing hormone). Overall, these studies show conflicting evidence about their effects on muscle strength, motor function and survival.
In the last couple of years the number of studies and trials for drug treatment in SMA has rapidly expanded. Maintaining an up‐to‐date systematic review of the effects of interventions for SMA type I as they emerge is important.
This is an update of a review first published in 2009 and updated in 2011. Drug treatment for SMA types II and III is the subject of a separate Cochrane Review (Wadman 2018).
Objectives
To assess the efficacy and safety of any drug therapy designed to slow or arrest progression of spinal muscular atrophy (SMA) type I.
Methods
Criteria for considering studies for this review
Types of studies
All randomised or quasi‐randomised (alternate or other systematic treatment allocation) studies examining the effect of drug treatment designed to slow or arrest disease progression in children with spinal muscular atrophy (SMA) type I. Effects of drug treatment need to be examined compared to placebo or any other proven, efficacious treatment and/or standard of care. We imposed no limitations by language or publication status.
Types of participants
Children with SMA type I fulfilling the criteria outlined in Table 6.
1. Diagnostic criteria for SMA type I.
| Primary criteria |
| Age of onset before six months and have never been able to sit independently |
| Genetic analysis to confirm the diagnosis, with homozygous deletion or heterozygous mutation of the SMN1 gene (5q11.2‐13.3) |
| Supporting criteria |
| Symmetrical muscle weakness of limb and trunk |
| Proximal muscles more affected than distal muscles and lower limbs more than upper limbs |
| No abnormality of sensory function |
| Serum creatine kinase (CK) activity not more than five times the upper limit of normal |
| Denervation on electrophysiological examination, and no nerve conduction velocities below 70% of the lower limit of normal. There are no abnormal sensory nerve action potentials |
| Muscle biopsy showing atrophic fibres of both types, hypertrophic fibres of one type (usually type I), and in chronic cases type grouping |
| No involvement of the central neurological systems, like hearing or vision |
| No involvement of non‐neurological organs |
SMN1 gene ‐ survival motor neurone 1 gene Zerres 1999
Types of interventions
Any drug treatment, alone or in combination, designed to slow or arrest the progress of the disease compared to placebo, with no restrictions on the route of administration.
Types of outcome measures
These are the outcomes of interest within whichever studies are included, and are not part of the criteria for including studies in a review.
Primary outcomes
Time from birth until death or full‐time ventilation (a requirement for 16 hours of ventilation per day regardless of whether this is with a tracheostomy, tube or mask)
Secondary outcomes
Acquisition of head control, within one year after the onset of treatment
Acquisition of the ability to roll, within one year after the onset of treatment
Acquisition of the ability to sit, within one year after the onset of treatment
Acquisition of the ability to stand, within one year after the onset of treatment
Change in motor disability score (e.g. Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2), Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND)), as determined by the original study authors
Adverse events attributable to treatment, during the whole study period, separated into severe (requiring or lengthening hospitalisation, life‐threatening, or fatal), and others
Search methods for identification of studies
Electronic searches
We searched the following databases on 22 October 2018.
The Cochrane Neuromuscular Specialised Register via the Cochrane Register of Studies (CRS‐Web) (22 October 2018: Appendix 4).
The Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies (CRS‐Web) (22 October 2018; Appendix 5).
MEDLINE (1991 to 19 October 2018; Appendix 6)
Embase (1991 to 19 October 2018; Appendix 7).
ISI Web of Science proceedings (1991 to October 2018; Appendix 8).
We searched the following trials registries in October 2018 to identify additional trials that had not yet been published.
Clinical trials registry of the US National Institute of Health (www.clinicaltrials.gov; Appendix 9).
WHO International Clinical Trials Registry Platform (ICTRP; apps.who.int/trialsearch; Appendix 10).
We limited searches to 1991 onwards, because at that time, genetic analysis of the survival motor neurone 1 (SMN1) gene became widely available and could be used to establish the diagnosis of SMA.
Searching other resources
We handsearched relevant cited references, published studies, reviews, textbooks and conference proceedings. Readers are invited to suggest studies, particularly in other languages, which should be considered for inclusion.
Data collection and analysis
Selection of studies
For this updated review, two review authors (RW and AV) independently checked titles and abstracts obtained from literature searches to identify potentially relevant trials for full review. From the full texts, the review authors independently selected the trials that met the selection criteria for inclusion. Review authors were not blinded to the trial author and source institution. The review authors resolved disagreements by reaching consensus. We presented an adapted PRISMA flowchart of study selection (Moher 2009).
Data extraction and management
Two review authors (RW and AV) extracted data independently using a specially designed data extraction form and entered data into Review Manager 5 for analysis (Review Manager 2014). We obtained missing data from the trial authors or pharmaceutical company whenever possible. Disagreement did not occur, but would have been resolved by reaching consensus or with third party adjudication if necessary.
If any reports had required translation, the translator would have extracted data directly using a data extraction form, or the review authors would have extracted data from the translation. Where possible a review author would have checked numerical data in the translation against the study report.
Assessment of risk of bias in included studies
The 'Risk of bias' assessment took into account allocation concealment, security of randomisation, intention‐to‐treat analysis, participant blinding (parent blinding), blinding of outcome assessment, incomplete outcome data, selective reporting and 'other bias'. We also looked for explicit inclusion and exclusion criteria, explicit outcome criteria and how studies dealt with baseline differences between treatment groups. We scored each bias item according to the Cochrane Handbook for Systematic Reviews of Interventions as 'low', 'high' or 'unclear' (Higgins 2011).
Statistical considerations involved a trade‐off between bias and precision. We assessed the risk of bias as 'unclear' when too few details were available to make a judgement of ‘high’ or ‘low' risk, when the risk of bias was genuinely unknown despite sufficient information about the conduct of the study, or when an entry was not relevant to a study.
Two review authors (RW and AV) graded the risk of bias in included studies independently. In case of disagreement, the review authors reassessed studies and reached agreement by consensus.
Measures of treatment effect
We intended to analyse continuous outcomes using mean differences (MDs) and 95% confidence intervals (CIs) and dichotomous outcomes (such as ability to roll, sit or stand and adverse events) using risk ratios (RRs) with 95% CI. We planned to calculate MDs for pooled data if studies were sufficiently comparable. We would have reported standardised mean differences (SMDs) with 95% CIs if studies assessed an outcome using different but comparable scales. We would have used the standard Review Manager generic inverse variance analysis using treatment effect differences with their standard errors if available data had not been sufficiently comparable between studies. We would have re‐expressed SMDs in units on a known scale, or provided a rule of thumb guide to aid interpretation (Cohen 1988; Higgins 2011).
For time to death or full‐time ventilation, we obtained results from the Kaplan‐Meier survival analyses.
Unit of analysis issues
We did not anticipate cluster‐randomised trials, cross‐over trials or multiple observations for the same outcome and anticipated that any included studies would not present these unit of analysis issues.
If we had identified multiarm studies, we would have analysed them so as to avoid double‐counting of participants, for example, we would have combined intervention groups if clinically appropriate, or halved a control group.
Dealing with missing data
We carefully evaluated important numerical data such as screened, randomised participants as well as intention‐to‐treat (ITT), and as‐treated and per protocol populations. We investigated attrition rates, e.g. dropouts, losses to follow‐up and withdrawals, and critically appraised issues of missing data and imputation methods (e.g. last observation carried forward). In case of missing outcome data we would have performed an ITT analysis. If standard deviations (SDs) for outcomes had not been reported, we would have imputed these values by assuming the SD of the missing outcome to be the average of the SDs from studies that provided this information.
In the event of missing data, we would have contacted the trial investigators to provide additional data.
Assessment of heterogeneity
We planned to identify heterogeneity by visual inspection of the forest plots and a standard Chi² test with a significance level of alpha = 0.1, in view of the low power of this test.
We would also have considered the I² statistic, using the rule of thumb thresholds described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity; and over 75% may indicate considerable heterogeneity. We would have interpreted the importance of I² in relation to the significance of the Chi² test and the magnitude and direction of effects.
Assessment of reporting biases
We carried out a thorough search, and searched trials registries for this update to identify completed but unpublished studies and study protocols, to minimise reporting bias. The review included too few studies for the use of funnel plots.
Data synthesis
We would have pooled results of studies with the same class of drug treatment. If Chi² analysis showed the data to be heterogeneous, we would have used a random‐effects model with a maximum likelihood estimation, carrying out a sensitivity analysis with a fixed‐effect model (Mantel‐Haenszel RR method). We would have performed formal comparisons of intervention effects according to risk of bias by meta‐regression. The major approach to incorporating 'Risk of bias' assessments would have been to incorporate such studies in the review and restrict meta‐analyses to studies at low (or lower) risk of bias.
Where studies included in meta‐analyses had different follow‐up periods we planned to make appropriate adjustments, if necessary using Poisson regression allowing for the aggregate person‐time‐at‐risk in the study groups.
'Summary of findings' table
We created 'Summary of findings' tables with the following outcomes.
Time from birth until death or full‐time ventilation (a requirement for 16 hours of ventilation per day regardless of whether this is with a tracheostomy, tube or mask)
Acquisition of head control, within one year after the onset of treatment
Acquisition of the ability to sit, within one year after the onset of treatment
Acquisition of the ability to stand, within one year after the onset of treatment
Change in motor disability score, as predefined by the authors of the trial
Adverse effects attributable to treatment
Severe adverse effects attributable to treatment
According to the Cochrane guidelines, we included a maximum of seven outcomes in the 'Summary of findings' tables (Higgins 2011).
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of a body of evidence (studies that contribute data for the prespecified outcomes). We followed methods and recommendations described in Chapter 11 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b), using GRADEproGDT software (GRADEpro GDT 2015). We justified in footnotes all decisions to downgrade or upgrade the certainty of evidence and made comments to aid readers' understanding of the review where necessary.
For future updates, two or more review authors will independently grade the certainty of the evidence in 'Summary of findings' tables and arrive at an agreed assessment by consensus. We will include 'Summary of findings' tables for all comparisons for which any data are available. If comparisons are all of equivalent importance, we will include them all as 'Additional tables'; if one comparison is of greater clinical importance we will choose it for presentation at the start of the review.
Subgroup analysis and investigation of heterogeneity
In the event of substantial clinical, methodological or statistical heterogeneity, we would not have reported study results as the pooled effect estimate in a meta‐analysis.
If we had found heterogeneity, we would have attempted to determine potential reasons for it by examining individual study and subgroup characteristics in sensitivity analyses.
We would have performed sensitivity analyses in order to explore the influence of the SMN2 copy number (when applicable) on effect sizes and would have used the formal tests for subgroup interactions in Review Manager 5 (Review Manager 2014).
Sensitivity analysis
We would have performed sensitivity analyses as follows, in order to explore the influence of the following factors (if applicable) on effect sizes. We would have restricted the analysis:
by taking into account risk of bias;
to outlier studies (very long, very large, very short or very small) to establish the extent to which they dominate the results.
We would also have tested the robustness of the results by repeating the analysis using different measures of effect size (RR, odds ratio (OR) etc.) and different statistical models (fixed‐effect and random‐effects models).
We planned formal evaluations of intervention effects according to risk of bias by meta‐regression, as noted above.
Non‐randomised evidence
We did not include non‐randomised studies in our final analysis. In the Discussion section, we reviewed the results from open and uncontrolled studies.
Results
Description of studies
For this updated review, the number of new references found by the searches were: Cochrane Neuromuscular Specialised Register 37, CENTRAL 90, MEDLINE 351, Embase 123, and ISI Web of Knowledge 277.
We named studies with no published data and no acronym after their trial register code (www.clinicaltrials.gov; apps.who.int/trialsearch).
Results of the search
We identified and assessed the full‐text reports of 24 studies (13 new) for possible inclusion in this updated review (Brichta 2006; CARNIVAL Type I 2008; Chang 2002; Conceicao 2010; EMBRACE 2015; Finkel 2016; Finkel 2017 (ENDEAR); FIREFISH 2016; Franz 1995; JPRN‐JapicCTI‐163450 2016; Mendell 2017; MOONFISH 2015; NCT00568698; NCT02268552; NCT02855112; NCT02865109; NPTUNE02 2007; Prufer de Queiroz Campos Araujo 2010; Russman 2003; SHINE 2015; SMART01; SMART02 2016; Swoboda 2009; Villanova 2015). We excluded 18 studies.
We included two studies (Finkel 2017 (ENDEAR); Russman 2003). We could not obtain the results of two completed trials, which investigated RO6885247 and hydroxyurea (MOONFISH 2015; NCT00568698).
See Figure 1 for a flow diagram of the study selection process.
1.
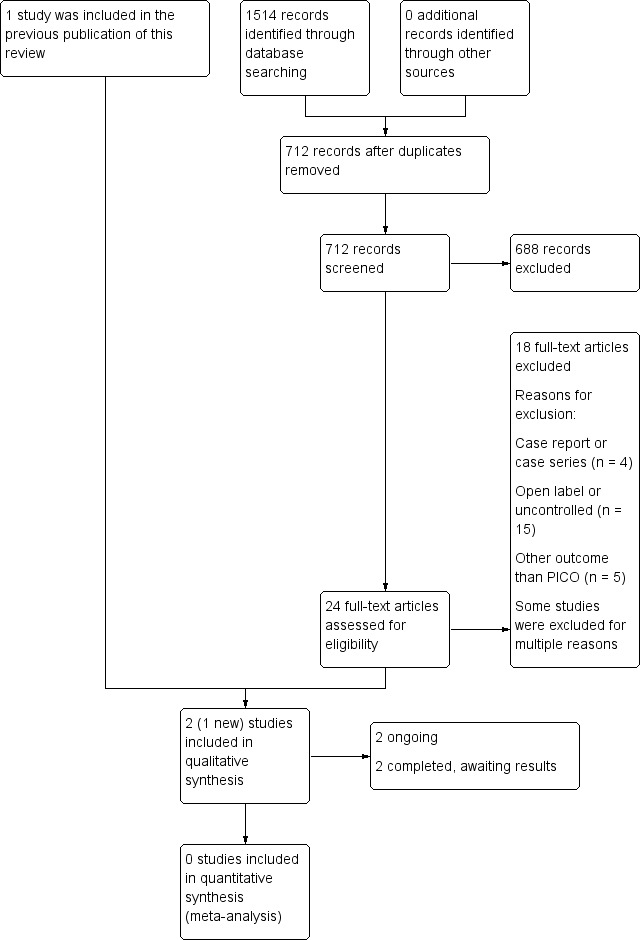
Study flow diagram.
Included studies
Two studies fulfilled the selection criteria (Finkel 2017 (ENDEAR); Russman 2003). Details of the studies are shown in Characteristics of included studies.
Nusinersen versus sham procedure
This study was a phase III, randomised, double‐blind, sham procedure‐controlled study with nusinersen in infants with spinal muscular atrophy (SMA) with a genetic confirmation of a deletion of SMN1 and 2 copies of SMN2. All participants (n = 121) were less than seven months old at the time of inclusion. Randomisation was in a 2:1 ratio (nusinersen:sham procedure). The planned treatment period was 13 months, with a prespecified interim analysis at six months (183 days).
Nusinersen was administered intrathecally by a lumbar puncture (at an age‐scaled equivalent dose of 12 mg in 5 mL: 0 to 3 months old, 9.6 mg in 4.0 mL; 3 to 6 months old, 10.3 mg in 4.3 mL; 6 to 12 months old, 10.8 mg in 4.5 mL; 12 to 24 months old, 11.3 mg in 4.7 mL). Participants received their treatment or sham procedure at days 1, 15, 29, and 64, followed by dosing at day 183 and 302. The sham procedure consisted of a small needle prick on the lower back at the location where the lumbar puncture injection is normally made. The needle broke the skin but no lumbar puncture injection or needle insertion occurred. The needle prick was covered with the same bandage that was used to cover the lumbar puncture injection in the treatment group.
The primary outcome in the trial was time to death or permanent assisted ventilation (defined as at least 16 hours per day for more than 21 continuous days). The investigators added a second primary outcome, the percentage of motor milestone responders on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2), after the results of the phase II trial became available (Finkel 2016). A response on the HINE‐2 was defined as improvement in at least one category (i.e. an increase in the score for head control, rolling, sitting, crawling, standing, or walking of at least 1 point, an increase in the score for kicking of at least 2 points, or achievement of the maximal score for kicking) and more categories with improvement than categories with worsening (i.e. a decrease was defined as at least 1 point decrease in the score for head control, rolling, sitting, crawling, standing, or walking and a decrease in the score for kicking was defined as a decrease of at least 2 points).
The trial also reported the proportion of responders on the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) (a responder was defined as at least a 4‐point score increase from baseline at day 183, 302, or 394 assessments), overall survival rate, percentage of infants not requiring permanent ventilation, proportion of compound muscle action potential (CMAP) responders (peroneal CMAP amplitude increasing to or maintained at 1 mV or more versus baseline at day 183, 302, or 394 assessments), subgroup analyses of time to death or permanent ventilation in patients with a higher or lower disease duration compared to the median disease duration, and adverse events (see Characteristics of included studies).
All participants who had a day 183 visit (78 participants; 51 treated with intrathecal nusinersen and 27 who underwent the sham procedure) were included in the interim analysis of motor milestones (HINE‐2). The study was stopped after the interim analysis showed significant benefit from nusinersen compared to the sham procedure. Participants were at that time invited to attend for a final visit for end of trial assessments at least two weeks after their most recent dose of nusinersen or sham procedure.
Oral riluzole versus placebo
Russman 2003 was a randomised, placebo‐controlled study with riluzole in 10 children with SMA type I. The main outcome measure was the occurrence of adverse events and the secondary outcome was mortality, under the assumption that the life expectancy without treatment would be no more than 24 months. All children fulfilled clinical criteria for SMA type I, namely onset before the age of six months, never acquiring the ability to sit independently, and genetic confirmation of the diagnosis of SMA. The study investigators had planned to include 30 children with randomisation in a 2:1 ratio (riluzole:placebo). Unfortunately, support from the pharmaceutical industry was withdrawn when Rhone‐Poulenc was taken over by Aventis. From then on no more children were enrolled in the study and therefore the total number of included children was only 10.
Excluded studies
We excluded 18 (13 new) studies because they were not randomised or controlled (Brichta 2006; CARNIVAL Type I 2008; Chang 2002; Conceicao 2010; FIREFISH 2016; Franz 1995; JPRN‐JapicCTI‐163450 2016; Finkel 2016; Mendell 2017; NCT02268552; NCT02855112; NCT02865109; NPTUNE02 2007; Prufer de Queiroz Campos Araujo 2010; SHINE 2015; SMART01; Swoboda 2009; Villanova 2015; see Characteristics of excluded studies).
Six of these excluded studies were not yet completed at the time of our search but we excluded them because of an open‐label, non‐controlled design (FIREFISH 2016; JPRN‐JapicCTI‐163450 2016; NCT02268552; NCT02855112; NCT02865109; SHINE 2015). Two of the excluded studies were recently completed or terminated and results were pending at the time of writing, but we excluded them because of their non‐randomised study design (NPTUNE02 2007; SMART01).
Risk of bias in included studies
The 'Risk of bias' assessments for the included studies of Finkel 2017 (ENDEAR) and Russman 2003 can be seen in the Characteristics of included studies table and Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study. Red (‐) indicates high risk of bias, green (+) indicates low risk of bias and yellow (?) indicates unclear risk of bias.
Russman 2003 was at high risk of bias overall. The trial reached only one‐third of the intended enrolment and there were baseline differences between the treatment groups that may have influenced the results: in the placebo group, the age at onset of symptoms, age at diagnosis and age at enrolment in the study were younger than in the riluzole group. The randomisation method, allocation concealment and blinding of parents and observers were not clear; and we assessed randomisation at high risk of bias because of the baseline imbalance. Diagnostic criteria for SMA type I were adequate. The primary outcome was clear and follow‐up of the 10 included children was complete.
The overall risk of bias in Finkel 2017 (ENDEAR) was low. Baseline characteristics were slightly different concerning age at time of onset and diagnosis, use of ventilator support, and the presence of respiratory and bulbar problems. However, children in the nusinersen‐treated group had an earlier onset and were more severely affected by respiratory and bulbar problems, and with the final results on the efficacy of nusinersen, these baseline differences seemed not to affect the final results. Allocation concealment and random sequence generation were adequate. Blinding procedures were adequate, including the sham procedure. Data reporting was adequate.
Effects of interventions
Summary of findings for the main comparison. Intrathecal injected nusinersen compared to sham procedure for infants with SMA and 2 SMN2 copies.
| Intrathecal injected nusinersen compared to sham procedure for infants with SMA and 2 SMN2 copies | ||||||
| Patient or population: infants with SMA and 2 SMN2 copies Setting: in‐hospital treatment for outpatient clinic Intervention: intrathecal injected nusinersen Comparison: sham procedure | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with sham procedure | Risk with intrathecal injected nusinersen | |||||
| Time from birth until death or full‐time ventilationa Follow‐up: range 6 months to 13 monthsc | Study population | HR 0.53 (0.32 to 0.89) | 121 (1 RCT) | ⊕⊕⊕⊝ Moderateb | This represents a 47% lower risk of death or full‐time ventilation with nusinersen than with the sham procedure | |
| 68 per 100 | 46 per 100 (31 to 64) | |||||
| Acquisition of head control within one year after the onset of treatment Follow‐up: range 6 months to 13 monthsc | 0 of 37 participants | 16 of 73 participants in the nusinersen‐treated group achieved head control | RR 16.95 (1.04 to 274.84) | 110 (1 RCT) | ⊕⊕⊝⊝ Moderated | |
| Acquisition of the ability to sit within one year after the onset of treatment Follow‐up: range 6 months to 13 monthsc | 0 of 37 participants | 6 of 73 participants in the nusinersen‐treated group achieved the ability to sit independently |
RR 6.68 (0.39 to 115.38) |
110 (1 RCT) | ⊕⊕⊝⊝ Moderated | |
| Acquisition of the ability to stand within one year after the onset of treatment Follow‐up: range 6 months to 13 monthsc | 0 of 37 participants in the sham procedure group | 1 of 73 participants in the nusinersen‐treated group achieved the ability to stand |
RR 1.54 (0.06 to 36.92) |
110 (1 RCT) | ⊕⊕⊝⊝ Moderated | |
| Change in motor disability score ‐ response on HINE‐2 within one year after the onset of treatmente Follow‐up: range 6 months to 13 months | 0 of 37 participants in the sham procedure group | 37 of 73 participants in the nusinersen‐treated group showed a motor milestone response on the HINE‐2 | RR 38.51 (2.43 to 610.14) | 110 (1 RCT) | ⊕⊕⊝⊝ Moderated | |
|
Adverse events attributable to treatment Measured as adverse events (all) Follow‐up: range 6 months to 13 months |
Study population |
RR 0.99 (0.92 to 1.05) |
121 (1 RCT) | ⊕⊕⊕⊝ Moderatef | Including bleeding risk from thrombocytopenia, renal toxicity, hyponatraemia, reduced growth, rash and possible (cerebral) vasculitis, hepatotoxicity, QTc interval prolongation on electrocardiogram, aspiration, infections, gastrointestinal problems | |
| 976 per 1000 |
966 per 1000 (898 to 1000) |
|||||
|
Severe adverse events attributable to treatment Measured as severe adverse events (all) Follow‐up: range 6 months to 13 months |
Study population |
RR 0.70 (0.55 to 0.89) |
121 (1 RCT) | ⊕⊕⊕⊝ Moderatef | Including respiratory problems, cardiorespiratory arrest, death, brain injury, hypoxic ischaemic encephalopathy | |
| 805 per 1000 |
563 per 1000 (443 to 716) |
|||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CHOP INTEND: Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE‐2: Hammersmith Infant Neurological Examination‐Section 2; CI: confidence interval; HR: hazard ratio; RCT: randomised controlled trial; RR: risk ratio; SMA: spinal muscular atrophy | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDefined as a requirement for 16 hours of ventilation per day regardless of whether via tracheostomy, tube or mask. bWe downgraded the certainty of the evidence once for risk of bias and imprecision (not sufficient to downgrade once for each). A slight baseline imbalance meant that children in the nusinersen‐treated group had an earlier onset and were more severely affected by respiratory and bulbar problems. This baseline imbalance in factors related to respiratory decline would tend to favour the control intervention for this outcome. Although the effect of nusinersen is large, there is some degree of uncertainty in the effect estimate arising from imprecision in a single study of this size. cBased on the final analysis. An interim analysis of motor milestones (HINE‐2) was performed on all participants who had a day 183 visit. The study was then stopped for significant benefit from nusinersen. Final analysis was performed on data including participants fulfilling at least six months of trial enrolment. dWe downgraded the certainty of the evidence once for risk of bias and imprecision (not sufficient to downgrade once for each). There was slight baseline imbalance and there is some degree of uncertainty in the effect estimate arising from imprecision in a single study of this size. We did not downgrade the motor milestone outcome results further for imprecision, in spite of wide CI. The absence of events in the control group is consistent with the natural history of SMA type 1 and a response represents a large treatment effect. eResponse was defined according to scores on the HINE‐2, which assesses the development of motor function through the achievement of motor milestones; in this trial, the scores accounted for 7 of the 8 motor milestone categories, excluding voluntary grasp. Infants were considered to have a motor milestone response if they met the following two criteria: improvement in at least one category (i.e. an increase in the score for head control, rolling, sitting, crawling, standing, or walking of ≥ 1 point, an increase in the score for kicking of ≥ 2 points, or achievement of the maximal score for kicking) and more categories with improvement than categories with worsening (i.e. a decrease was defined as ≥ 1 point decrease in the score for head control, rolling, sitting, crawling, standing, or walking and a decrease in the score for kicking was defined as a decrease of ≥ 2 points). fWe downgraded one level for imprecision because the small sample size and shortened study duration mean that the study is unlikely to have captured uncommon adverse events.
Summary of findings 2. Riluzole compared to placebo for children with SMA type I.
| Riluzole compared to placebo for children with SMA type I | ||||
| Patient or population: children with SMA type I Setting: outpatient clinic Intervention: riluzole Comparison: placebo | ||||
| Outcomes | Impact | № of participants (studies) | Certainty of the evidence (GRADE) | Commments |
| Time from birth until death or full‐time ventilation Follow‐up: range 1 month to 64 months | In the 3 children in the placebo group, the median age at death was 8 months (range 6 to 13 months). 4/7 children treated with riluzole died during the trial, at a median age of 17 months (range 5 to 25 months); the remaining 3 children treated with riluzole were still alive at 30, 48 and 64 months. | 10 (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| Acquisition of head control | ‐ | ‐ | ‐ | Not measured |
| Acquisition of the ability to sit Follow‐up: range 1 month to 64 months | None of the children in the riluzole or the placebo group acquired the ability to sit (follow‐up was extended up to 30 to 64 months in the children treated with riluzole). | 10 (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| Acquisition of the ability to stand | ‐ | ‐ | ‐ | Not measured |
| Change in motor disability score | ‐ | ‐ | ‐ | Not measured |
| Adverse events attributable to treatment Follow‐up: range 1 month to 9 months | No adverse events occurred in children treated with placebo or riluzole. | 10 (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| Severe adverse events attributable to treatment Follow‐up: range 1 months to 9 months | No adverse events occurred in children treated with placebo or riluzole. | 10 (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). RCT: randomised controlled trial; SMA: spinal muscular atrophy | ||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||
aWe downgraded the certainty of the evidence three levels: twice for serious imprecision, because of the small cohort, and once for risk of bias as there were baseline differences between treatment and control groups.
We could not perform a meta‐analysis as the two included trials investigated different drug treatments. Outcomes and study designs also varied (Finkel 2017 (ENDEAR); Russman 2003; Table 3; Table 4).
2. Oral riluzole versus placebo (Russman 2003).
| Riluzole | Placebo | |
| Number of participants randomised | 7 | 3 |
| Number (%) of participants evaluable for analysis | 7 (100%) | 3 (100%) |
| Number of participants (%) who died during the 12‐month study | 4 (57%) | 3 (100%) |
| Median age at death (months)* | 17 | 8 |
| Range of ages at death (months)* | 5‐25 | 6‐13 |
| Adverse events | 0 | 0 |
*Among the children who died during the 12‐month study. Three of the seven children treated with riluzole were alive at 30, 48 and 64 months' follow‐up.
3. Intrathecally‐injected nusinersen versus sham procedure (Finkel 2017 (ENDEAR)).
| Nusinersen | Sham procedure | Risk ratio (95% CI) | Hazard ratio (95% CI) | P value | |
| Number of participants randomised and included in time‐to‐event analysis | 80 (100%) | 41 (100%) | |||
| Number (%) of participants who died or had received permanent (> 16 hours per day) ventilation | 21 (39%) | 28 (68%) | ‐ | 0.53 (0.32 to 0.89) | 0.005 |
| Number (%) of participants evaluable for interim analysisa | 51 (63%) | 27 (66%) | |||
| Number (%) of participants achieving motor milestone response on HINE‐2 (interim analysis)bc | 21 (41%) | 0 (0%) | 23.15 (1.46 to 368.01) | ‐ | 0.03 |
| Number (%) of participants evaluable in final analysisb | 73 (91%) | 37 (90%) | |||
| Number (%) of participants who survived until the end of trialb | 67 (91%) | 25 (68%) | ‐ | 0.37 (0.18 to 0.77) | 0.004 |
| Number (%) of participants who did not use permanent (> 16 hours per day) assisted ventilation until the end of trialb | 62 (85%) | 28 (76%) | ‐ | 0.66 (0.3 to 1.37) | 0.13 |
| Number (%) of participants achieving motor milestone response on HINE‐2 (final analysis)bc | 37 (51%) | 0 (0%) | 38.51 (2.43 to 610.14) | ‐ | 0.010 |
| Number (%) of participants achieving head controlb | 16 (22%) | 0 (0%) | 16.95 (1.04 to 274.84) | ‐ | 0.05 |
| Number (%) of participants able to roll overb | 7 (10%) | 0 (0%) | 7.7 (0.45 to 131.29) | ‐ | 0.16 |
| Number (%) of participants able to sit independentlyb | 6 (8%) | 0 (0%) | 6.68 (0.39 to 115.38) | ‐ | 0.19 |
| Number (%) of participants able to standb | 1 (1%) | 0 (0%) | 1.54 (0.06 to 36.92) | ‐ | 0.79 |
| Number (%) of participants with response on the CHOP INTENDbd | 52 (71%) | 1 (3%) | 26.36 (3.79 to 183.18) | ‐ | <0.001 |
| Nusinersen | Sham procedure | Risk ratio (95% CI) | P value | ||
| Number (%) of adverse eventse | 77 (96%) | 40 (98%) | 0.99 (0.92 to 1.05) | ‐ | 0.68 |
| Number (%) of participants with adverse events occurring < 72 hours of treatment or sham procedure | 51 (64%) | 24 (59%) | 1.08 (0.81 to 1.43) | ‐ | 0.58 |
| Number (%) of participants with severe adverse events | 45 (56%) | 33 (80%) | 0.70 (0.55 to 0.89) | ‐ | 0.004 |
aInterim analysis included all participants that had a day 183 visit at the time of cut‐off (15 June 2016). bFinal analysis was performed on data, including participants fulfilling at least six months of trial enrolment. cResponse was defined according to scores on the HINE‐2, which assesses the development of motor function through the achievement of motor milestones; in this trial, the scores accounted for seven of the eight motor milestone categories, excluding voluntary grasp. Infants were considered to have a motor milestone response if they met the following two criteria: improvement in at least one category (i.e. an increase in the score for head control, rolling, sitting, crawling, standing, or walking of ≥ 1 point, an increase in the score for kicking of ≥ 2 points, or achievement of the maximal score for kicking) and more categories with improvement than categories with worsening (i.e. a decrease was defined as ≥ 1 point decrease in the score for head control, rolling, sitting, crawling, standing, or walking and a decrease in the score for kicking was defined as a decrease of ≥ 2 points). d Response was defined as an increase of at least 4 points from baseline in the CHOP INTEND score at the end of trial visit (day 183, 302, or 394). e In case a participant had more than one event, only the event with the highest severity was counted.
HINE: Hammersmith Infant Neurological Examination; CHOP INTEND: Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders
Russman 2003 did not perform statistical analysis because the trialists had enrolled only 10 of the intended 30 participants before the withdrawal of funding. The results are therefore descriptive (see Table 3).
Intrathecal nusinersen versus sham injection procedure
Data analysis was performed on different subgroups. The prespecified interim analysis included the 78 infants (51 in the nusinersen group and 27 in the sham procedure group) who had been enrolled for at least six months (183 days). The time‐to‐event analysis included all 121 infants (80 in the nusinersen group and 41 in the sham procedure group) who had undergone randomisation and the assigned procedure at least once. All other end points were tested in the final analysis in 110 patients (73 in the nusinersen group and 37 in the sham procedure group) who had been enrolled at least six months before the last participant's final visit (See Characteristics of included studies tables).
Primary outcome measure: time from birth until death or full‐time ventilation
By the time of the final analysis (when follow‐up ranged from 6 months to 13 months), 39% of infants in the nusinersen group and 68% of infants in the sham procedure group had died or required full‐time ventilation (at least 16 hours daily for 21 days or more). The trialists calculated a hazard ratio (HR) with the use of a Cox proportional hazards model that was adjusted for disease duration at screening in each infant: HR 0.53, 95% confidence interval (CI) 0.32 to 0.89; moderate‐certainty evidence. This represents a 47% lower risk of death or full‐time ventilation with nusinersen than with the sham procedure.
Secondary outcome measures
Acquisition of the ability to have head control, roll, sit, or stand within one year after the onset of treatment
In the nusinersen‐treated group 16/73 infants (22%) achieved head control (risk ratio (RR) 16.95, 95% CI 1.04 to 274.84), 7/73 infants (10%) achieved the ability to roll over (RR 7.70, 95% CI 0.45 to 131.29), 6/73 infants (8%) achieved the ability to sit independently (RR 6.68, 95% CI 0.39 to 115.38) and 1/73 infants (1%) achieved the ability to stand (RR 1.54, 95% CI 0.06 to 36.92). None of the 37 infants in the sham procedure group achieved any of these milestones (Analysis 1.1; Analysis 1.2; Analysis 1.3; Analysis 1.4). We judged the certainty of evidence to be moderate.
1.1. Analysis.
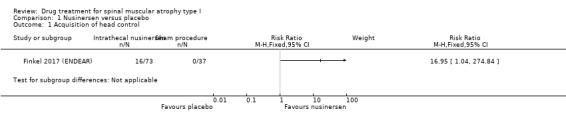
Comparison 1 Nusinersen versus placebo, Outcome 1 Acquisition of head control.
1.2. Analysis.
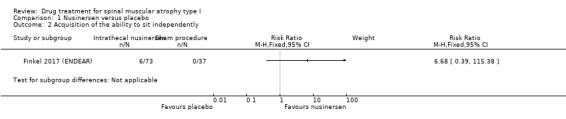
Comparison 1 Nusinersen versus placebo, Outcome 2 Acquisition of the ability to sit independently.
1.3. Analysis.
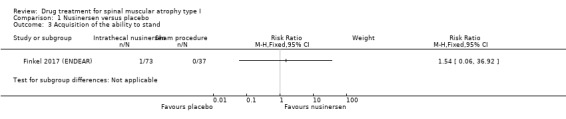
Comparison 1 Nusinersen versus placebo, Outcome 3 Acquisition of the ability to stand.
1.4. Analysis.
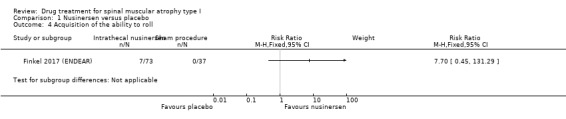
Comparison 1 Nusinersen versus placebo, Outcome 4 Acquisition of the ability to roll.
Change in motor disability score
Motor milestone response (based on predefined criteria) on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) within one year after the onset of treatment
The responder analysis on motor milestones in 110 participants showed an improvement on the HINE‐2 (according to the predefined response definition) in 37 (51%) participants treated with nusinersen, while none of the participants treated with the sham procedure reached this endpoint (RR 38.51, 95% CI 2.43 to 610.14; N = 110; moderate‐certainty evidence).
Response (based on predefined criteria) on CHOP INTEND within one year after the onset of treatment
A responder analysis on Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) showed 71% of nusinersen‐treated infants responded versus 3% of sham procedure‐treated infants (RR 26.36, 95% CI 3.79 to 183.18; N = 110; moderate‐certainty evidence).
Adverse effects attributable to treatment during the whole study period, separated into severe (requiring or lengthening hospitalisation, life‐threatening, or fatal), and others
Reporting included all infants who were randomised and received at least one dose of nusinersen or sham procedure (N = 121).
The proportion of participants experiencing adverse events was similar in the nusinersen group (77/80 participants, 96%) and the control group (40/41 participants, 98%) (RR 0.99, 95% CI 0.92 to 1.05; N = 121; moderate‐certainty evidence).
The proportion experiencing adverse events, defined as serious, was slightly lower in the nusinersen group (45/80, 56%) than in the placebo group (33/41, 80%) (RR 0.70, 95% CI 0.55 to 0.89; N = 121; moderate‐certainty evidence). We downgraded the certainty of evidence for any adverse events and serious adverse events because the trial cohort and period were small and are unlikely to have captured uncommon adverse events. Adverse events included urinary and respiratory tract infections, respiratory failure, gastrointestinal disorders, pyrexia, intrathecal procedural complications (pain, swelling, site reactions), electrolyte imbalance, cardiac rhythm disorders, rash, and death.
Oral riluzole versus placebo
Primary outcome measure: time from birth until death or full‐time ventilation
In the group treated with riluzole, four of seven (57%) children died during the study at a median age of 17 (range 5 to 25) months (mean age 15.75 months). In the placebo group all three children (100%) died at a median age of eight (range 6 to 13) months (mean age 9 months). Three children treated with riluzole were still alive at the age of 30, 48, and 64 months, which was 23, 39, and 49 months after starting the therapy. These children used bilevel positive airway pressure ventilation (BiPAP) only at night.
Secondary outcome measures
Acquisition of the ability to control the head, roll, sit, or stand within one year after the onset of treatment
None of the children in the riluzole or placebo group developed the ability to sit. It is unclear if clinical measurements included the acquisition of the ability to roll. The acquisition of the ability to stand was not included in this study.
Change in motor disability score
Not measured.
Adverse effects attributable to treatment during the whole study period, separated into severe (requiring or lengthening hospitalisation, life‐threatening, or fatal), and others
There were no adverse side effects in either the riluzole‐treated or the placebo‐treated group.
Discussion
Summary of main results
This review includes two published randomised controlled trials (RCTs) on drug treatment for spinal muscular atrophy (SMA) type I (total of 131 patients) (Finkel 2017 (ENDEAR); Russman 2003). The treatments investigated were intrathecally‐injected nusinersen and oral riluzole.
Intrathecally‐injected nusinersen was shown to be an effective treatment for the improvement of motor milestone achievement and survival or time to require full‐time ventilation in SMA type I, with moderate‐certainty evidence of improvement on the motor milestone outcome (Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2)) and moderate‐certainty evidence of improvement in survival or time to full‐time ventilation in the treatment group, compared to the sham procedure group (Finkel 2017 (ENDEAR)). Risk of bias in this trial was low.
Baseline characteristics were slightly different concerning age at time of onset and diagnosis, use of ventilator support and the presence of respiratory and bulbar problems, but these differences might have even underestimated the effect of nusinersen since participants in the nusinersen‐treated group had an earlier onset and were more severely affected by respiratory and bulbar problems.
It is uncertain whether oral riluzole has any effect in patients with SMA type I. The certainty of the evidence for all measured outcomes from this study was very low, because the study was too small to detect or rule out an effect, and had serious limitations (Russman 2003). The trial was not free of bias because of possible baseline differences, and methods of randomisation and blinding were not described.
Four additional RCTs investigating oral RO6885247, oral hydroxyurea, intrathecally‐injected nusinersen and oral valproic acid were either ongoing or completed, but no data for analysis were available at the time of writing and they could not be included in the final assessment (EMBRACE 2015; MOONFISH 2015; NCT00568698; SMART02 2016).
Evidence from other studies in SMA type I
Drugs that have been tested in open and uncontrolled studies of children with SMA type I are riluzole (Abbara 2011; ASIRI 2008), valproate (Conceicao 2010; CARNIVAL Type I 2008; SMART01; SMART02 2016; Swoboda 2009), recombinant human ciliary neurotrophic factor (CNTF) (Franz 1995), sodium phenylbutyrate or phenylbutyrate (NPTUNE02 2007; STOPSMA 2007), hydroxyurea (Chang 2002; NCT00568698), SMN2 antisense oligonucleotides (Finkel 2017 (ENDEAR); Finkel 2016; SHINE 2015; EMBRACE 2015), and small molecules (FIREFISH 2016; MOONFISH 2015; NCT02268552). We here discuss the results of treatment with each of these drugs from non‐randomised studies and trials in participants with SMA, especially type I. Drug treatment in SMA types II and III are the topic of another Cochrane Review (Wadman 2018).
Antisense oligonucleotides
A phase II open‐label, dose‐finding study in participants with SMA type I investigating two different dosages of nusinersen (multiple intrathecal doses 6 mg and/or 12 mg respectively), showed significant improvement in outcomes (including achievement of motor milestones, Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) motor function, compound muscle action potential (CMAP) in two investigated nerves and survival) in the 12 mg treated group compared to natural history data (Finkel 2016). A RCT investigating one dose level of nusinersen compared to a sham procedure is ongoing and includes participants with atypical SMA type I, excluding patients with two SMN2 copies and under six months old at onset (EMBRACE 2015).
Histone deacetylase inhibitors
Valproate
Valproate has been tested in various open‐label studies (Conceicao 2010; Darbar 2011; Kissel 2011; Saito 2014; Swoboda 2009; Tsai 2007; Weihl 2006). One retrospective study in 15 infants with SMA type I showed stable motor function over months, suggesting an effect of valproate treatment. However, analysis of effect was done only in five participants who survived the two‐year study period (Conceicao 2010). An open‐label, uncontrolled trial in 37 infants with SMA did not show effects on survival or respiratory function after treatment with valproate and L‐carnitine compared to an untreated, matched retrospectively analysed cohort.
(CARNIVAL Type I 2008). One open‐label uncontrolled trial in infants with SMA type I investigating valproate is ongoing (SMART01). An open‐label trial with valproate in 42 children and adults with SMA types I, II and III showed slight improvement in gross motor function in younger non‐ambulatory type II children and variable responses of the survival motor neurone (SMN) transcripts in blood (Swoboda 2009).
There are two ongoing studies with valproate in SMA type I: one RCT in children with SMA types I and II aged one to seven years old (SMART02 2016), and one open‐label trial of sodium valproate in 16 children with SMA types I, II and III (JPRN‐JapicCTI‐163450 2016).
Hydroxyurea
In an uncontrolled open‐label trial in two participants with SMA type I, five participants with SMA type II and two participants with SMA type III, hydroxyurea showed an improvement in muscle strength without side effects (Chang 2002).
Phenylbyturate
A multicentre, open‐label phase I/II trial in children with SMA type I on treatment with multiple dosage levels of phenylbutyrate has been terminated due to extremely slow enrolment (NPTUNE02 2007). The results of a trial including 14 presymptomatic infants genetically confirmed to have SMA are pending (STOPSMA 2007).
Recombinant ciliary neurotrophic factor
A non‐randomised uncontrolled study on the safety and tolerability of recombinant ciliary neurotrophic factor (CNTF) included 10 children with SMA type I (Franz 1995). Six children died; no change in muscle function and strength was noted and there was no difference in disease course and side effects between placebo and treatment groups.
Small molecules
RO6885247 or RG7800
A phase I randomised, double‐blind, placebo‐controlled, multiple‐dose study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of RO6885247/RG7800 in patients with SMA types I, II and III started in November 2014, but the trial was terminated in December 2016 for safety reasons (MOONFISH 2015).
RO7034067 or RG7916
An open‐label, dose‐escalating trial with RO7034067 or RG7916, also called risdiplam, in infants with SMA type I is currently ongoing (FIREFISH 2016). An open‐label trial with risdaplam once daily in presymptomatic infants with SMA and two SMN2 copies is planned (RAINBOWFISH).
LMI070
A phase I, open‐label study with the small molecule 'LMI070', also called branaplam, has started in patients with SMA type I (NCT02268552).
Other experimental factors
From studies on coenzyme Q10, lithium carbonate and guanidine hydrochloride, it was not clear on clinical grounds whether the participant population consisted only of patients with SMN1 deleted SMA, partially because SMN gene analysis was not possible prior to 1991 (Angelini 1980; Folkers 1995; Il'ina 1980). Therefore, we have not discussed the therapeutic effects of these drugs.
In vitro and animal studies have found several other compounds to have an effect on SMN expression, but they are as yet untested in patients with SMA. They are therefore outside the scoop of this review. See Appendix 1 for a brief description of these compounds.
SMN1 gene therapies are outside the scope of this review. We have added some information on trials in Appendix 2 for overall completeness.
Overall completeness and applicability of evidence
We are confident that we identified all clinically relevant trials, as we conducted a comprehensive search of all published literature and clinical trials registers and four of the review authors regularly attend international conferences on SMA.
Nusinersen is the first treatment effective in SMA.
A major issue in SMA, irrespective of the investigated therapy, is the timing of the treatment in relation to its potential effect. Previous experimental studies suggest that there is a limited window of opportunity to rescue or stabilise motor neuron function in the early or presymptomatic stages of the disease. Two trials are currently investigating the efficacy of treatment in presymptomatic SMA patients (NURTURE 2015; STOPSMA 2007). A phase I/II study with phenylbutyrate in presymptomatic infants genetically confirmed to have SMA, and suspected to have SMA type I or II according to family history and SMN2 copy number, has been completed and results are pending (STOPSMA 2007). Another trial was recently started with nusinersen treatment in presymptomatic infants with genetically confirmed SMA (NURTURE 2015).
For future (international) trials it is important that the level of supportive care is explicitly described to avoid baseline differences in the treatment arms, since practice guidelines of supportive care, e.g. pulmonary, nutritional and orthopaedic supportive therapy in children and adults with SMA probably differ between centres and countries (Bladen 2014). Global practice guidelines for the clinical care of children and adults with SMA are given in the consensus statement for standard care in SMA (Finkel 2018; Mercuri 2018).
Certainty of the evidence
We concluded from the one RCT in 10 infants with SMA type I that there is no evidence for or against efficacy of nine months' treatment with oral riluzole in SMA type I (Russman 2003). The included trial was not free of bias because possible baseline differences between intervention and control groups occurred and the methods of randomisation and blinding were not described. We performed an analysis of risk of bias, heterogeneity, imprecision and publication bias using GRADE (Atkins 2004). We downgraded the certainty of evidence two levels for imprecision (very small sample size) and one level for study limitations (baseline differences between groups), which meant that our assessment of the certainty of evidence was very low.
See Table 2.
The study of nusinersen, Finkel 2017 (ENDEAR), was at low risk of bias. A slight baseline imbalance meant that children in the nusinersen‐treated group had an earlier onset and were more severely affected by respiratory and bulbar problems. This baseline imbalance in factors related to respiratory decline would tend to favour the control intervention for this outcome. We downgraded the certainty of evidence on survival and motor milestone responses once to moderate because of the risk of bias from baseline imbalance and imprecision (not sufficient to downgrade once for each). Although the effect of nusinersen is large, there is some degree of uncertainty in the effect estimate arising from imprecision in a single study of this size. We did not further downgrade the motor milestone response evidence for imprecision in spite of very wide CIs. The low event rate in the control group is consistent with the natural history of SMA, and responses represented a large effect. We also downgraded the certainty of adverse event findings because small sample size and short trial periods are unlikely to have captured uncommon adverse events. See Table 1.
Potential biases in the review process
There may be some potential for bias in this review process as we made changes to the protocol. These included additions and deletions to the outcomes, as reported in Differences between protocol and review. None of these changes were made as a result of the findings of the included studies, but rather to improve the structure of the review.
We are confident that we identified all clinically relevant trials, as we conducted a comprehensive search of all published literature and clinical trials registers and four of the review authors regularly attend international conferences on SMA.
The results of our review might be biased by publication since the results of two completed trials on hydroxyurea (NCT00568698), and RO6885247 (MOONFISH 2015), were not available for analysis and review, and two trials investigating nusinersen (EMBRACE 2015), and valproic acid (SMART01), are still ongoing.
One of the authors of this review (SI) is participating as investigator of different trials on drug treatment in SMA type I. In the next update of the Cochrane Review on SMA type I, an independent analyst will check the data analysis of these trials to avoid the suggestion of bias.
Agreements and disagreements with other studies or reviews
To the best of our knowledge, there are no other systematic reviews investigating the whole spectrum of drug treatment in SMA type I. Several reviews have also identified and discussed various drug treatments in SMA (Anderton 2015; Arnold 2013; Darras 2007; Lewelt 2012; Nurputra 2013; Stavarachi 2007; Swoboda 2007; Tisdale 2015), with some focusing specifically on preclinical studies (Seo 2013), genetic therapies (Donelly 2012; Zanetta 2014), solely HDACI therapies (Mohseni 2013), SMN‐inducing therapies (Kaczmarek 2015), or small molecule and molecular therapies (Zanetta 2014). Our conclusions are in line with other reviews.
Although we have tried to give an overview of the efficacy of drug treatment with riluzole, hydroxyurea, valproate, phenylbutyrate, recombinant ciliary neurotrophic factor, SMN2 antisense oligonucleotides and small molecules in SMA (see Description of the intervention and Discussion), we did not systematically review non‐randomised and preclinical trials and studies and might have missed potential studies.
Authors' conclusions
Implications for practice.
Evidence of moderate certainty shows that the antisense oligonucleotide therapy, nusinersen, shows efficacy in patients with spinal muscular atrophy (SMA) type I by prolonging survival and improving motor function. Moderate‐certainty evidence also indicates that a greater proportion of infants treated with nusinersen than with a sham procedure achieve motor milestones and may be classed as responders to treatment on clinical assessments (Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) and Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND)). The evidence is also of moderate certainty that the proportion of children with adverse events and serious adverse events on intrathecal nusinersen is no higher with nusinersen than with a sham procedure.
There is no evidence for or against the use of riluzole, the only other drug studied in a randomised trial, as the certainty of evidence is too low for conclusions to be drawn.
Other trials are ongoing or awaiting results.
Implications for research.
Since antisense oligonucleotide therapy with nusinersen has been found to be an efficacious drug therapy for SMA type I in a clinical trial, future trials should preferably compare new treatments to nusinersen or be evaluated as an add‐on therapy to nusinersen.
Most trials investigating new therapies are focused on the early phases of the disease, since motor improvement or lack of decline is the simplest way to establish drug efficacy. However, therapies should also be sought to prevent disease progression, conserve motor function, and improve quality of life for those who have a longer disease duration.
What's new
| Date | Event | Description |
|---|---|---|
| 22 October 2018 | New search has been performed | Search updated to October 2018. One author (J Wokke) withdrew. Fay‐Lynn Asselmann joined the review. We added two new outcomes (head control and motor development scores), which are newly developed outcome measures for infants with type 1 SMA. We revised the Methods to meet current Cochrane standards. Authors made a protocol modification to Table 6 (diagnostic criteria), included 'Summary of findings' tables and re‐evaluated the 'Risk of bias' assessments. |
| 15 September 2016 | New citation required and conclusions have changed | This update includes one new trial, which compared intrathecal nusinersen and sham injection. |
History
Protocol first published: Issue 4, 2006 Review first published: Issue 1, 2009
| Date | Event | Description |
|---|---|---|
| 15 February 2012 | Amended | 'Declaration of interest' and 'Contributions of authors' updated; no other changes to text. |
| 15 February 2012 | New citation required but conclusions have not changed | Change in listing of authors to include Dr WL van der Pol. This corrects an error in the authorship of this update. |
| 31 March 2011 | New citation required but conclusions have not changed | RI Wadman included as new lead author. |
| 8 March 2011 | New search has been performed | Databases were searched and review was updated. No new trials were found. |
| 30 July 2008 | Amended | Converted to new review format. |
Acknowledgements
This project was supported by the National Institute for Health Research (NIHR) via Cochrane Infrastructure funding to Cochrane Neuromuscular. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health. Cochrane Neuromuscular is also supported by the MRC Centre for Neuromuscular Diseases.
We used a standard template provided by Cochrane Neuromuscular to complete some of the mandatory sections of the methods.
Appendices
Appendix 1. SMN1 gene therapies
SMN1 gene therapy
Studies that aim to study the repair of SMN1 deletion by the introduction of the SMN1 gene have recently started. Viral vectors, such as the self‐complementary adeno‐associated virus (scAAV9) are used to incorporate the SMN1 gene. Recent in vitro studies in fibroblasts of people with SMA (Azzouz 2004; Dominguez 2011), and in vivo studies in mice (Benkhelifa‐Ziyyat 2013; Dominguez 2011), primates, and pigs with SMA phenotype have shown promising results on SMN1‐expression with effect on motor function and survival (Duque 2015; Foust 2010; Glascock 2012a; Glascock 2012b; Passini 2011; Robbins 2014; Valori 2010). These studies also indicate that intramuscular or intravenous injection of the AAV results in widespread dissemination of the gene, including penetration of the central nervous system (Benkhelifa‐Ziyyat 2013; Foust 2010; Glascock 2012a; Meyer 2015).
A phase I study with intravenous AVXS‐101 (scAAV9.CB.SMN) in infants with SMA type I has been completed and showed all 15 participants to be alive and event‐free at 20 months of age, as compared with a rate of survival of 8% in a historical cohort. Participants included in the study also showed improvement on the CHOP‐INTEND (0‐66 scale) (increase of 9.8 points at 1 month and 15.4 points at 3 months, as compared with a decline in this score in a historical cohort) (Mendell 2016; Mendell 2017).
Appendix 2. Stem cell treatment
Stem cell treatment in SMA is outside the scope of this review, but for reasons of completeness we here summarise the (ongoing) study and trial. A case series of three patients with SMA type I treated with multiple intrathecal and intravenous infusions of allogeneic mesenchymal stem cells described no negative effects and reported a potential beneficial effect on the CHOP‐INTEND but data was very limited. The benefits of the treatment were lost when the therapy was withdrawn (Villanova 2015). An open‐label phase I trial in children with SMA type I to investigate the effects of multiple intrathecal injections of allogeneic adipose derived mesenchymal stem cell transplants has started (NCT02855112).
Appendix 3. Other experimental factors studied in vivo and vitro
Several compounds have been shown to have an effect on SMN expression in vivo and in vitro. These include 2,4‐diaminoquinazolines (RG3039 and D156844) (Butchbach 2010; Gogliotti 2013; Jarecki 2005; Singh 2008; Thurmond 2008, Van Meerbeke 2013), 2,4‐diaminoquinazoline inhibitors of the decapping scavenger enzyme DcpS (DAQ‐DcpSi) (Cherry 2017), 2‐(4,6‐Dimethylpyrazolo[1,5‐a]pyrazin‐2‐yl)‐7‐(4‐methylpiperazin‐1‐yl)‐4H‐pyrido[1,2‐a]pyrimidin‐4‐one (Woll 2016), 3‐(6,8‐Dimethylimidazo[1,2‐a]pyrazin‐2‐yl)‐7‐(4‐methylpiperazin‐ 1‐yl)‐1H‐isochromen‐1‐one (Woll 2016), 4PBA (Butchbach 2016), 5‐(N‐ethyl‐N‐isopropyl)‐amiloride (Yuo 2008), AAV8.LSP.dnMstn (Liu 2016), AAV8.LSP.sActRIIB (Lui 2016), aclarbicin (Andreassi 2004; Ting 2007), amikacin (Wolstencroft 2005), BAY 55‐9837 (Hadwen 2014), bortezomib (Kwon 2011), butyrate prodrug pivaloyloxymethyl butyrate (AN9) (Edwards 2016), dacinstat (Mohseni 2016), E1mov11 (Osman 2016), edavarone (Ando 2017); fasudil (Bowerman 2012), Genetics/G418 (Heier 2009; Heier 2015), indoprofen (Lunn 2004), ioganin (Tseng 2016), M344 (Riessland 2006), ML372 (Abera 2017), panobinostat/LBH589 (Garbes 2009), pip6a‐PMO (Hammond 2016), PTK‐SMA1 (Hastings 2009), quercetin (Uzunallı 2015; Wishart 2014), quisinostat/JNJ‐26481585 (Schreml 2013), romidepsin (Hauke 2009), scAAV9‐siPTEN (Ning 2010; Little 2015), securinine (Chen 2017), SMN‐AS1 (d'Ydewalle 2017; Woo 2017), sodium vanadate (Liu 2014; Ting 2007; Zhang 2001), suberoylanilide hydroxamic acid (Hahnen 2006; Mohseni 2016; Riessland 2010), TC007 (Mattis 2009a; Mattis 2009b; Mattis 2012), (S)‐3‐(6,8‐dimethylimidazo[1,2‐a]pyrazin‐2‐yl)‐7‐(4‐ethyl‐3‐ methylpiperazin‐1‐yl)‐2H‐chromen‐2‐one (Woll 2016), tobramycin (Wolstencroft 2005), trichostatin A (Avila 2007; Liu 2014; Ting 2007), triptolode (Hsu 2012), VK563 (Butchbach 2016) and Y‐27632 (Bowerman 2010). Every compound has its own potential and way of interacting with the SMN complex. Description of the working mechanism of each compound goes behind the scope of this review, since none of the compounds have yet been investigated in human studies.
At the time of writing, it is unclear whether treatment with any of these drugs has a beneficial clinical effect on the disease course of SMA types I.
Appendix 4. Cochrane Neuromuscular Specialised Register via the Cochrane Register of Studies (CRS‐Web) search strategy
Search run on 22 October 2018
#1 MeSH DESCRIPTOR Muscular Atrophy, Spinal Explode All AND INREGISTER #2 MeSH DESCRIPTOR Muscular Disorders, Atrophic AND INREGISTER #3 spinal NEAR3 "muscular atroph*" AND INREGISTER #4 Werdnig NEXT Hoffman* AND INREGISTER #5 Kugelberg next Welander AND INREGISTER #6 #1 or #2 or #3 or #4 or #5 AND INREGISTER #7 (#1 or #2 or #3 or #4 or #5) AND (INREGISTER)
Appendix 5. Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies (CRS‐Web) search strategy
Search run on 22 October 2018
#1 MeSH DESCRIPTOR Muscular Atrophy, Spinal Explode All AND CENTRAL:TARGET #2 MeSH DESCRIPTOR Muscular Disorders, Atrophic AND CENTRAL:TARGET #3 spinal NEAR3 "muscular atroph*" AND CENTRAL:TARGET #4 Werdnig NEXT Hoffman* AND CENTRAL:TARGET #5 Kugelberg next Welander AND CENTRAL:TARGET #6 #1 or #2 or #3 or #4 or #5 AND CENTRAL:TARGET
Appendix 6. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations and Daily <1946 to October 19, 2018> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (469969) 2 controlled clinical trial.pt. (92707) 3 randomized.ab. (424303) 4 placebo.ab. (192545) 5 drug therapy.fs. (2055263) 6 randomly.ab. (299003) 7 trial.ab. (442071) 8 groups.ab. (1843132) 9 or/1‐8 (4300960) 10 exp animals/ not humans.sh. (4506554) 11 9 not 10 (3717979) 12 exp Muscular Atrophy, Spinal/ (4440) 13 muscular disorders, atrophic/ (389) 14 spinal muscular atroph$.mp. (4831) 15 (Werdnig adj Hoffman$).mp. (386) 16 (Kugelberg adj Welander).mp. (188) 17 or/12‐16 (6771) 18 11 and 17 (745) 19 remove duplicates from 18 (741) 20 limit 19 to yr="1991 ‐Current" (676)
Appendix 7. Embase (OvidSP) search strategy
Database: Embase <1980 to 2018 Week 43> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (56721) 2 double‐blind procedure.sh. (150846) 3 single‐blind procedure.sh. (32618) 4 randomized controlled trial.sh. (513165) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (1530476) 6 trial.ti. (246665) 7 or/1‐6 (1693508) 8 (animal/ or nonhuman/ or animal experiment/) and human/ (1690778) 9 animal/ or nonanimal/ or animal experiment/ (3370354) 10 9 not 8 (2793470) 11 7 not 10 (1554234) 12 limit 11 to (conference abstracts or embase) (1320265) 13 spinal muscular atrophy/ or hereditary spinal muscular atrophy/ (6717) 14 (Werdnig adj Hoffman$).mp. (638) 15 (Kugelberg adj Welander).mp. (280) 16 spinal muscul$ atroph$.mp. (7858) 17 or/13‐16 (8130) 18 12 and 17 (203) 19 limit 18 to yr="1991 ‐Current" (199) 20 remove duplicates from 19 (196)
Appendix 8. ISI Web of Science proceedings search strategy
WOS 22 October 2018 Indexes=CPCI‐S, CPCI‐SSH Timespan=1991‐2018 #6 8 #5 AND #4 #5 211,588 TS=(random* or placebo or "single blind" or "double blind*" or crossover or "cross‐over" ) #4 543 #1 or #2 or #3 #3 3 TS=(Kugelberg AND Welander) #2 14 TS=(Werdnig AND Hoffman*) #1 536 TS=("muscular atrophy" NEAR spinal)
Appendix 9. ClinicalTrials.gov search strategy
1 spinal muscular atrophy
2 treatment
Appendix 10. WHO International Clinical Trials Registry Platform
1 spinal muscular atrophy
2 SMA
Data and analyses
Comparison 1. Nusinersen versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Acquisition of head control | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Acquisition of the ability to sit independently | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Acquisition of the ability to stand | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Acquisition of the ability to roll | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Motor milestone response on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) (final analysis) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6 Motor milestone response on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) (interim analysis) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7 Response on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) (final analysis) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9 Severe adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
1.5. Analysis.
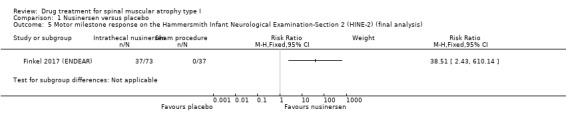
Comparison 1 Nusinersen versus placebo, Outcome 5 Motor milestone response on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) (final analysis).
1.6. Analysis.

Comparison 1 Nusinersen versus placebo, Outcome 6 Motor milestone response on the Hammersmith Infant Neurological Examination‐Section 2 (HINE‐2) (interim analysis).
1.7. Analysis.
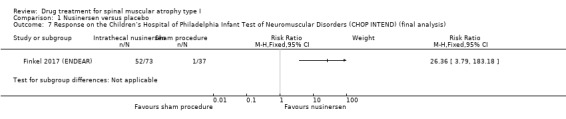
Comparison 1 Nusinersen versus placebo, Outcome 7 Response on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) (final analysis).
1.8. Analysis.
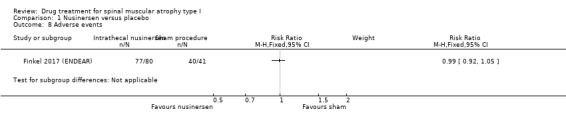
Comparison 1 Nusinersen versus placebo, Outcome 8 Adverse events.
1.9. Analysis.
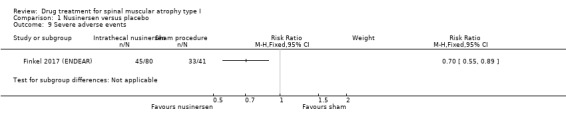
Comparison 1 Nusinersen versus placebo, Outcome 9 Severe adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Finkel 2017 (ENDEAR).
| Methods | Phase III, randomised, double‐blind, sham procedure‐controlled study Randomisation 2:1 |
|
| Participants | 121 participants with infantile‐onset SMA < 7 months (210 days) of age at time of inclusion, with genetically confirmed deletion or mutation of SMN1 and 2 copies of SMN2. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Participants received loading doses on day 1, 15, 29, and 64, followed by maintenance doses on day 183 and 302 (i.e. every 4 months) Prespecified interim analysis 78 infants: 51 nusinersen, 27 sham procedure Time‐to‐event analysis 121 infants: 80 nusinersen, 41 sham procedure Final analysis 110 participants: 73 nusinersen, 37 sham procedure |
|
| Outcomes | Primary
Secondary
*"A response on the HINE‐2 was defined as improvement in at least one category (i.e. an increase in the score for head control, rolling, sitting, crawling, standing, or walking of ≥1 point, an increase in the score for kicking of ≥ 2 points, or achievement of the maximal score for kicking) and more categories with improvement than categories with worsening (i.e. a decrease in the score for head control, rolling, sitting, crawling, standing, or walking of ≥ 1 point or a decrease in the score for kicking of ≥ 2 points." The lowest possible score for the HINE is 0 (zero), and the highest possible score is 28. **"A participant was considered a CHOP INTEND responder if the change from baseline in CHOP INTEND total score is ≥ 4 points based on assessment at the end of trial visit at day 183, day 302, or day 394 study visits." Total CHOP INTEND scores range from 0 to 64, with higher scores indicating better movement functioning. |
|
| Funding | "Supported by Biogen and Ionis Pharmaceuticals" | |
| Conflicts of interest | "The sponsors, Biogen and Ionis Pharmaceuticals, designed the trial in collaboration with clinicians who had experience in the treatment of spinal muscular atrophy. An independent data and safety monitoring board provided trial oversight in collaboration with the sponsors. Investigators collected the data, which was analyzed by the sponsors. All the authors contributed to data interpretation and manuscript development, approved the manuscript for submission, and vouch for the accuracy and completeness of the reported data. All the principal investigators agreed to follow the protocol and protocol amendments (available with the full text of this article at NEJM.org). The first draft of the manuscript was written by the first author and the senior industry author (penultimate author); medical writing assistance was paid for by Biogen. The sponsors reviewed the manuscript and provided feedback to the authors, who had full editorial control." | |
| Notes | Study stopped after predefined interim analysis and participants transitioned to the open‐label SHINE study (SHINE 2015) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned by central and electronic procedure. Within each randomisation, subjects were also stratified for disease duration (participant's age at screening ‐ age at symptom onset): ≤ 12 weeks versus > 12 weeks |
| Allocation concealment (selection bias) | Low risk | Computer‐generated allocation. Permutated block randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study personnel who delivered the treatment were not involved in the assessments of participants. Key personnel for assessments and parents of participants were not present during the procedure. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study personnel who delivered the treatment were not involved in the assessments of participants. Key personnel for assessments and parents of participants were not present during the procedure. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 149 infants screened, 27 excluded following screening (reasons reported) 122 infants randomised, 1 infant (nusinersen group) withdrawn before treatment 78 infants in interim analysis (43 infants were "without at least day 183 assessment at data cut‐off date for interim analysis") 110 participants in final analysis (11 infants were "without at least day 183 assessment at data cut‐off date for final analysis") 121 infants in safety analysis Comment: adequate, stopped early for efficacy |
| Selective reporting (reporting bias) | Low risk | A protocol exists; changes to outcome measurement related to interim analyses were documented. Motor milestone response became the primary outcome and analysed as response rates rather than change from baseline, with rationale: "Change informed by Phase 2 data that suggest that a functional response could provide early evidence of efficacy and thus allow for an earlier interim analysis" CHOP‐Intend analysed as a responder analysis rather than change from baseline, with reason: "to assess the magnitude of response in individual subjects while accounting for the potential influence of different survival rates in treated and sham subjects" Comment: there is clear evidence (through examination of the trial protocol) that reported results correspond to all intended outcome measurements in the revised protocol. |
| Other bias | Unclear risk | Baseline characteristics were slightly different concerning age at time of onset and diagnosis, respiratory complications and the presence of respiratory and bulbar problems. Children in the nusinersen‐treated group had an earlier onset and were more severely affected by respiratory and bulbar problems. These baseline differences seem not to have affected the final results. |
Russman 2003.
| Methods | Randomised (2:1 ratio), placebo‐controlled trial The investigational review boards of the participating centres approved the protocol and consent forms |
|
| Participants | 10 children (mean age at inclusion 10 months; range 3 to 15 months) who fulfilled international classification criteria for SMA type I and have a homozygous deletion of the SMN1 gene | |
| Interventions |
Duration of treatment 9 months, follow‐up 12 months Power was calculated on 30 included participants (2:1 ratio) |
|
| Outcomes |
|
|
| Funding | The study was funded in part by Rhone‐Poulenc‐Rorer, the manufacturer of riluzole, who provided the medication. Support was withdrawn when the company was taken over by Aventis. | |
| Conflicts of interest | Not stated | |
| Notes | Enrolment goal was 30 children with SMA type I, but only 10 children were included until funding was withdrawn | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomly assigned (2:1), no method reported. Infants receiving riluzole were older at diagnosis (5.2 months) than those who received placebo (1.2 months) and older at enrolment (9.3 versus 4.3 months) |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unknown |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unknown |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All 10 participants recruited provided outcome data |
| Selective reporting (reporting bias) | Low risk | There is only one possible way in which the outcome domains can be measured (hence there is no opportunity to select from multiple measures). |
| Other bias | Low risk | Trial was terminated prematurely |
CHOP INTEND: Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CSF: cerebrospinal fluid; ECG: electrocardiogram: HINE: Hammersmith Infant Neurological Examination; SMA: spinal muscular atrophy; SMN: survival motor neuron
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Brichta 2006 | Valproate. Not randomised. Study was on the effect of valproate on human survival motor neuron (SMN) expression in blood |
| CARNIVAL Type I 2008 | Valproate and carnitine. Open‐label trial, no placebo was given |
| Chang 2002 | Hydroxyurea. Not randomised, not controlled. Only 2 participants |
| Conceicao 2010 | Valproate. Not randomised, not controlled. No placebo was given |
| Finkel 2016 | Nusinersen. Not randomised, not controlled (no placebo was given) |
| FIREFISH 2016 | RO7034067/RG7916. Open‐label, not controlled |
| Franz 1995 | Recombinant human ciliary neurotrophic factor. Not randomised, not controlled |
| JPRN‐JapicCTI‐163450 2016 | Sodium valproate. Open‐label, non‐randomised |
| Mendell 2017 | AVXS‐101. Phase I trial. Not controlled (no placebo was given). Gene therapy |
| NCT02268552 | LMI070. Open‐label, first‐in‐human study. No placebo was given for control The study is ongoing, but not recruiting |
| NCT02855112 | Allogeneic adipose derived stem cells. Pilot trial |
| NCT02865109 | Nusinersen. Expanded Access Program to address a high unmet medical need. No placebo was given |
| NPTUNE02 2007 | Sodium phenylbutyrate. Dose‐escalating study. Non‐randomised, not controlled. No placebo was given. Trial terminated due to extremely slow enrolment |
| Prufer de Queiroz Campos Araujo 2010 | Salbutamol. Pilot trial. Not controlled (no placebo was given), not randomised |
| SHINE 2015 | Nusinersen. Open‐label, not randomised. No placebo or sham procedure was given |
| SMART01 | Valproate. Open‐label trial. Not randomised, not controlled (no placebo was given) Completed study, no published data as yet |
| Swoboda 2009 | Valproate and carnitine. Not randomised, not controlled; open‐label |
| Villanova 2015 | Allogeneic mesenchymal stem cells. Case series |
Characteristics of studies awaiting assessment [ordered by study ID]
MOONFISH 2015.
| Methods | Phase I, randomised, double‐blind, placebo‐controlled trial |
| Participants | 64 participants with SMA types I, II or III aged 2 to 55 years or below 7 months |
| Interventions | RO6885247 orally once daily for 12 weeks versus placebo orally once daily for 12 weeks |
| Outcomes | Safety (incidence of adverse events), pharmacokinetics (plasma concentrations of RO6885247 and RO6885247 exposure), pharmacodynamics (SMN protein levels in blood and in vivo splicing of SMN2 mRNA in blood), effect on compound muscle action potential, effect on electrical impedance myography |
| Notes | Recruitment of participants suspended since April 2015 for safety reasons. In parallel to the Moonfish trial, Hoffmann‐La Roche have been investigating the effects of the long‐term use of RG7800 in animals. These animal studies are a standard requirement in the development of new medicines. In this study, they observed an unexpected safety finding in the eye of animals and subsequently immediately suspended dosing in the Moonfish trial as a precautionary measure. The trial in participants with SMA was therefore terminated in December 2016. Last update received December 22, 2016 (www.clinicaltrials.gov), but results have not yet been published. |
NCT00568698.
| Methods | A pilot therapeutic, randomised, placebo‐controlled, double‐blind trial using hydroxyurea in children with SMA type I |
| Participants | Children with SMA type I who never achieved independent sitting with an age of onset before 6 months and confirmation of a homozygous deletion or mutation of the SMN1 gene. Enrolment in study within 6 months after diagnosis up to 2 years of age and not requiring continuous respiratory support before the inclusion |
| Interventions | Hydroxyurea or placebo (dose route and duration of treatment not mentioned) |
| Outcomes | Adverse events; length of survival and age of ventilator dependence; motor unit number estimation; biomarker assays: SMN protein and SMN mRNA |
| Notes | This study is completed (last update received July 8, 2009 (www.clinicaltrials.gov), but results have not yet been published. |
mRNA: messenger ribonucleic acid; SMA: spinal muscular atrophy; SMN: survival motor neuron
Characteristics of ongoing studies [ordered by study ID]
EMBRACE 2015.
| Trial name or title | A phase 2, randomized, double‐blind, sham procedure‐controlled study to assess the safety and tolerability and explore the efficacy of IONIS 396443 (BIIB058) administered intrathecally in subjects with spinal muscular atrophy who are not eligible to participate in the clinical studies IONIS 396443‐CS3B or IONIS 396443‐CS4 |
| Methods | Phase 2, randomised, double‐blind, sham procedure‐controlled study |
| Participants | 21 participants with genetically confirmed SMA with onset of clinical signs and symptoms consistent with SMA at ≤ 6 months of age and have documentation of 3 SMN2 copies or onset of clinical signs and symptoms consistent with SMA at ≤ 6 months of age, > 7 months of age (211 days) at screening, and have documentation of 2 SMN2 copies or onset of clinical signs and symptoms consistent with SMA at > 6 months of age, are ≤ 18 months of age at screening, and have documentation of 2 or 3 SMN2 copies |
| Interventions | Multiple intrathecal injections of nusinersen (IONIS‐SMNRx or 396443) versus multiple sham procedures |
| Outcomes | Number of adverse events and serious adverse events, change from baseline in clinical laboratory parameters, change from baseline in electrocardiogram, change from baseline in vital signs, change from baseline in neurological exam, including motor function, change in plasma concentration of IONIS 396443 and change in cerebrospinal fluid concentration of IONIS 396443 |
| Starting date | June 2015 |
| Contact information | Biogen |
| Notes | The study is ongoing, but not recruiting participants |
SMART02 2016.
| Trial name or title | Multicenter cooperative and investigator initiated clinical trial using valproic acid in childhood onset spinal muscular atrophy: confirmatory trial (SMART02) |
| Methods | Phase IIB, placebo‐controlled, randomised, double‐blind trial |
| Participants | 28 patients with SMA types I and II, age 1 to 7 years |
| Interventions | Oral valproic acid or placebo, 12.5 mg/kg or 25 mg/kg once a day after supper. Treatment period is 40 weeks |
| Outcomes | HFMSE, HFMS, motor function, World Health Organization motor milestones |
| Starting date | January 2016 |
| Contact information | Kayoko Saito, Institute of Medical Genetics, Tokyo Women's Medical University |
| Notes | None |
HFMS(E): Hammersmith Functional Motor Scale (Expanded); SMA: spinal muscular atrophy
Differences between protocol and review
We updated the 'Risk of bias' methodology according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). In the 2019 update, we expanded the methods according to current standards (MECIR 2018), as meta‐analysis may be possible in future updates.
We included a PRISMA flow chart to illustrate the study selection process, clarified that searches were not limited by publication status or language, and the role of outcomes.
We described methods used in 'Summary of findings' tables.
We added two new outcomes: achieving head control as a new motor milestone and change in motor disability score, which have been integrated in spinal muscular atrophy (SMA) trials since publication of the protocol for this review (Bosboom 2006).
We adjusted the definition on SMA types I in Table 6 and added the highest achieved motor milestones (sitting) as discriminator of the SMA type I. This was not stated in Table 1 of the original protocol and the 2011 update, although it was mentioned in the main text.
We adjusted the search strategy. We performed searches from 1991 onwards because at that time genetic analysis of the survival motor neurone 1 (SMN1) gene became widely available and could establish the diagnosis of SMA.
We revised the 'Adverse events' section and we did not discuss the adverse events from the included trials in relation to the side effects of drug treatment in non‐randomised literature. We have outlined the most common adverse events of treatments in the 'Summary of findings' tables (see Table 1; Table 2).
John Wokke withdrew from authorship at this update.
Contributions of authors
All authors contributed substantially to the concept and design of the review. Dr Bosboom and Dr Vrancken performed data extraction and analyses for the original review. Dr Bosboom wrote the first draft of the original review, and the other co‐authors contributed to subsequent revisions for important intellectual content. Drs Wadman, Vrancken and Van der Pol updated the review in 2011 and 2019 and the other authors approved the revisions.
Sources of support
Internal sources
University Medical Center Utrecht, Department of Neurology and Neuromuscular diseases, Utrecht, Netherlands.
University of Texas Southwestern Medical Center, Department of Pediatrics, Dallas, Texas, USA.
Onze Lieve Vrouwe Gasthuis West, Department of Neurology, Amsterdam, Netherlands.
External sources
No sources of support supplied
Declarations of interest
Dr Wadman was involved as an investigator in the investigator‐initiated trial on the efficacy of pyridostigmine for children and adults with SMA types II, III and IV (SPACE 2014). She was involved as an investigator at a participating centre in the trial on the safety and efficacy of cholest‐4‐en‐3‐one, oxime for children with SMA type II and IIIa (Bertini 2017; NCT02628743).
Dr van der Pol receives research support from the Prinses Beatrix Spierfonds, Stichting Spieren voor Spieren, Netherlands ALS foundation. His employer receives fees for consultancy services to Biogen, Avexis and Novartis. He was involved as an investigator at a participating centre in trials on the safety and efficacy of olesoxime for children with SMA types II and IIIa (Bertini 2017; NCT02628743), and is involved as an investigator of the mono‐centre placebo‐controlled trial on pyridostigmine in children and adults with SMA types II‐IV (SPACE 2014).
Dr Bosboom: none known.
Fay‐Lynn Asselman: none known.
Dr van den Berg serves on scientific advisory boards for Biogen, Cytokinetics, Orion for amyotrophic lateral sclerosis (ALS)‐related studies, and Shire for multifocal motor neuropathy; received an educational grant from Shire; serves on the editorial board of Amyotrophic Lateral Sclerosis and the Journal of Neurology, Neurosurgery and Psychiatry; and receives research support from the Netherlands ALS Foundation, The Netherlands Organization for Health Research and Development (Vici Scheme, JPND). He was involved as an investigator at a participating centre in trials on the safety and efficacy of olesoxime for children with SMA types II and IIIa (Bertini 2017; NCT02628743), and is involved as an investigator of the mono‐centre placebo‐controlled trial on pyridostigmine in children and adults with SMA types II‐IV (SPACE 2014).
Dr Iannaccone was involved in the trial of riluzole as one of the investigators and authors (Russman 2003). She was involved in a trial of the efficacy of creatine for children with spinal muscular atrophy types II and III as investigator and author (Wong 2007) and she was involved in a trial of the efficacy of riluzole (not published). She has received support for research from AveXis, Biogen and Scholar Rock for clinical trials in SMA patients and from Sarepta, Reveragen, Mallinckrodt, Fibrogen, and PTC Therapeutics for clinical trials in muscular dystrophy. She has been a consultant for AveXis, Biogen, Sarepta, Audentes, Catabasis and Genentech/Roche.
Dr Vrancken: none known. He was involved as an investigator at a participating centre in trials on the safety and efficacy of olesoxime for children with SMA types II and IIIa (Bertini 2017; NCT02628743), and is involved as an investigator of the mono‐centre placebo‐controlled trial on pyridostigmine in children and adults with SMA types II‐IV (SPACE 2014).
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Finkel 2017 (ENDEAR) {published and unpublished data}
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. New England Journal of Medicine 2017;377(18):1723‐32. [DOI] [PubMed] [Google Scholar]
- NCT02193074. A study to assess the efficacy and safety of IONIS‐SMN rx in infants with spinal muscular atrophy [A phase 3, randomized, double‐blind, sham‐procedure controlled study to assess the clinical efficacy and safety of IONIS 396443 administered intrathecally in patients with infantile‐onset spinal muscular atrophy]. clinicaltrials.gov/show/NCT02193074 (first received 14 July 2014). [IONIS 396443‐CS3B ; NCT02193074]
Russman 2003 {published data only}
- Russman BS, Iannaccone ST, Samaha FJ. A phase 1 trial of riluzole in spinal muscular atrophy. Archives of Neurology 2003;60(11):1601‐3. [PUBMED: 14623733] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Brichta 2006 {published data only}
- Brichta L, Holker I, Haug K, Klockgether T, Wirth B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Annals of Neurology 2006;59(6):970‐5. [DOI] [PubMed] [Google Scholar]
CARNIVAL Type I 2008 {published data only}
- Krosschell KJ, Kissel JT, Townsend EL, Simeone SD, Zhang RZ, Reyna SP, et al. Project Cure SMA Investigator's Network. Clinical trial of L‐Carnitine and valproic acid in spinal muscular atrophy type I. Muscle & Nerve 2018;57(2):193‐9. [CTG: NCT00661453; 25409 IND 79276] [DOI] [PubMed] [Google Scholar]
Chang 2002 {published data only}
- Chang JG, Tsai FJ, Wang WY, Jong YJ. Treatment of spinal muscular atrophy by hydroxyurea. American Journal of Human Genetics 2002;71(Suppl 4):2402. [Google Scholar]
Conceicao 2010 {published data only}
- Conceicao E, Silva T, Umbertine R, Maria Joaquina D. Analysis of motor skill acquisition among children with type I spinal muscular atrophy submitted to medication with valproic acid. Amyotrophic Lateral Sclerosis 2010;11(Suppl 1):63‐71. [Google Scholar]
Finkel 2016 {published data only}
- Finkel R, Chiriboga C, Vajsar J, Day J, Montes J, Vivo D, et al. Interim results of a phase 2 clinical study of nusinersen (ISIS‐SMNRx) in patients with infantile‐onset spinal muscular atrophy. Annals of Neurology 2016;80(Suppl 20):S371‐2. [Google Scholar]
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, Vivo DC, et al. Treatment of infantile‐onset spinal muscular atrophy with nusinersen: a phase 2, open‐label, dose‐escalation study. Lancet 2016;388(10063):3017‐26. [DOI] [PubMed] [Google Scholar]
- NCT01839656. A study to assess the safety and pharmacokinetics of ionis SMNRX in infants with spinal muscular atrophy [A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile‐onset spinal muscular atrophy]. www.clinicaltrials.gov/show/NCT01839656 (first received 22 April 2013). [CTG: NCT01839656; IONIS SMNRx 396443 ‐ CS3A]
FIREFISH 2016 {unpublished data only}
- NCT02913482. A study to Investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of RO7034067 in infants with type1 spinal muscular atrophy (Firefish) [A two part seamless, open‐label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of RO7034067 in infants with type 1 spinal muscular atrophy]. clinicaltrials.gov/show/NCT02913482 (first received 21 September 2016). [ClinicalTrials.go: NCT02913482]
Franz 1995 {published data only}
- Franz DN, Tudor CA, Samaha FJ. A phase I trial of recombinant human ciliary neurotrophic factor in spinal muscular atrophy. Annals of Neurology 1995; Vol. 38, issue 3:546.
JPRN‐JapicCTI‐163450 2016 {unpublished data only}
- JPRN‐JapicCTI‐163450. Phase 3 study of K‐828‐SP [Phase 3 study of K‐828‐SP (Long‐term study of sodium valproate in child patients with spinal muscular atrophy)]. apps.who.int/trialsearch/Trial2.aspx?TrialID=JPRN‐JapicCTI‐163450 (first received November 2016).
Mendell 2017 {published and unpublished data}
- Mendell JR, Al‐Zaidy S, Shell R, Arnold WD, Rodino‐Klapac L, Kissel JT, et al. Gene therapy for spinal muscular atrophy type 1 shows potential to improve survival and motor functional outcomes. Molecular Therapy 2016;24(Suppl 1):S190. [EMBASE: 72276590; WOS:000375264200475] [Google Scholar]
- Mendell JR, Al‐Zaidy S, Shell R, Arnold WD, Rodino‐Klapac LR, Prior TW, et al. Gene‐Replacement Therapy for Spinal Muscular Atrophy. New England Journal of Medicine 2017;377(18):1713‐22. [DOI] [PubMed] [Google Scholar]
- NCT02122952. Gene transfer clinical trial for spinal muscular atrophy type 1 [Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS‐101]. www.clinicaltrial.gov/show/NCT0122952 (first received 23 April 2014). [CTG: NCT02122952; AVXS‐101‐CL‐101]
- Sproule D, Al‐Zaidy S, Shell R, Arnold D, Rodino‐Klapac L, Kissel J, et al. Interim safety, efficacy and achievement of developmental milestones in this phase 1, first‐in‐human study of the systemic delivery of AVXS‐101, an AAV9‐mediated gene therapy for children with spinal muscular atrophy (SMA) type 1. Annals of Neurology 2016;80(Suppl 20):S368. [Google Scholar]
NCT02268552 {unpublished data only}
- NCT02268552. An open label study of LMI070 in type 1 spinal muscular atrophy (SMA) [An open label multi‐part first‐in‐human study of oral LMI070 in infants with type 1 spinal muscular atrophy]. www.clinicaltrials.gov/show/NCT02268552 (first received 1 October 2014). [CTG: NCT02268552; Sponsor's Protocol Code Number: CLMI070X2201]
NCT02855112 {unpublished data only}
- NCT02855112. Allogeneic adipose derived stem cells for Werdnig Hoffman patients [The effectiveness of allogeneic adipose derived mesenchymal stem cells (ADMSCs) in the phenotypic changes of Werdnig Hoffman patients]. clinicaltrials.gov/show/NCT02855112 (first received 22 July 2016. [CTG: NCT02855112]
NCT02865109 {unpublished data only}
- NCT02865109. To provide access to nusinersen to eligible patients with Infantile‐onset Spinal Muscular Atrophy (SMA) (consistent with Type 1) to address a high‐unmet medical need. clinicaltrials.gov/show/NCT02865109 (first received10 August 2016).
NPTUNE02 2007 {unpublished data only}
- NCT00439218. Clinical trial of sodium phenylbutyrate in children with spinal muscular atrophy type I [Phase I/IIa clinical trial of sodium phenylbutyrate in pediatric subjects with type I spinal muscular atrophy]. clinicaltrials.gov/show/NCT00439218 (first received 22 February 2007). [CTG: NCT00439218; HHSN265200423611C; N01NS42361_NPTUNE02]
Prufer de Queiroz Campos Araujo 2010 {published data only}
- Prufer de Queiroz Campos Araujo A. Long‐term open salbutamol trial in spinal muscular atrophy. Journal of Neurology 2010;257(Suppl 1):S101. [Google Scholar]
SHINE 2015 {unpublished data only}
- NCT02594124. An open‐label study (SHINE) for patients with spinal muscular atrophy (sma) who participated in studies with IONIS‐SMNRX [An open‐label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443]. clinicaltrials.gov/show/NCT02594124 (first received 30 October 2015). [CTG: NCT02594124; EudraCT: EUCTR2015‐001870‐16‐DE; Sponsor's Protocol Code Number: ISIS 396443‐CS11 ]
SMART01 {unpublished data only}
- SMART01. Multicenter cooperative and investigator initiated clinical trial using valproic acid in childhood onset spinal muscular atrophy. dbcentre3.jmacct.med.or.jp/JMACTR/App/JMACTRS06/JMACTRS06.aspx?seqno=4602 (first received July 2014). [JPRN‐SMA‐IIA00190]
Swoboda 2009 {published data only}
- Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One 2009;4(5):e5268. [PUBMED: 19440247] [DOI] [PMC free article] [PubMed] [Google Scholar]
Villanova 2015 {published data only}
- Villanova M, Bach JR. Allogeneic mesenchymal stem cell therapy outcomes for three patients with spinal muscular atrophy type 1. American Journal of Physical Medicine & Rehabilitation 2015;94(5):410‐5. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
MOONFISH 2015 {unpublished data only}
- MOONFISH ‐ NCT02240355. A study of RO6885247 in adult and pediatric patients with spinal muscular atrophy (MOONFISH) [A multicenter, randomized, double blind, placebo controlled, multiple dose study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of RO6885247 following 12 weeks of treatment in adult and pediatric patients with spinal muscular atrophy (MOONFISH)]. clinicaltrials.gov/show/NCT02240355 (first received 11 September 2014). [CTG: NCT02240355; 2014‐002246‐41; BP29420]
NCT00568698 {published data only}
- NCT00568698. A pilot therapeutic trial using hydroxyurea in type I spinal muscular atrophy patients. clinicaltrials.gov/show/NCT00568698 (first received 4 December 2007). [78811; NCT00083746; NCT00568698; SU‐11012007‐783]
References to ongoing studies
EMBRACE 2015 {unpublished data only}
- NCT02462759. A study to assess the safety and tolerability of ISIS 396443 (ISIS SMNRx) in participants with spinal muscular atrophy (SMA). (EMBRACE) [A phase 2, randomized, double‐blind, sham‐procedure controlled study to assess the safety and tolerability and explore the efficacy of ISIS 396443 (BIIB058) administered intrathecally in subjects with spinal muscular atrophy who are not eligible to participate in the clinical studies ISIS 396443‐CS3B or ISIS 396443‐CS4]. clinicaltrials.gov/show/NCT02462759 (first received 14 May 2015). [CTG: NCT02462759; EudraCT : EUCTR2014‐003657‐33‐DE; 232SM202]
SMART02 2016 {unpublished data only}
- SMART02. Multicenter cooperative and investigator initiated clinical trial using valproic acid in childhood onset spinal muscular atrophy: confirmatory trial (SMART02). dbcentre3.jmacct.med.or.jp/JMACTR/App/JMACTRS06/JMACTRS06.aspx?seqno=5544 (first received January 2016). [27‐3594; JPRN‐JMA‐IIA00231]
Additional references
Abbara 2011
- Abbara C, Estournet B, Lacomblez L, Lelièvre B, Ouslimani A, Lehmann B, et al. Riluzole pharmacokinetics in young patients with spinal muscular atrophy. British Journal of Clinical Pharmacology 2011;71(3):403‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Abera 2017
- Abera MB, Xiao J, Nofziger J, Titus S, Southall N, Zheng W, et al. ML372 blocks SMN ubiquitination and improves spinal muscular atrophy pathology in mice. JCI Insight 2017;1(19):e88427. [DOI] [PMC free article] [PubMed] [Google Scholar]
ACTS 1996
- ALS CNTF Treatment Study Group. A double‐blind placebo‐controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology 1996;46(5):1244‐9. [DOI] [PubMed] [Google Scholar]
Also‐Rallo 2011
- Also‐Rallo E, Alias L, Martinez‐Hernandez R, Caselles L, Barcelo MJ, Baiget M, et al. Treatment of spinal muscular atrophy cells with drugs that upregulate SMN expression reveals inter‐ and intra‐patient variability. European Journal of Human Genetics 2011;19:1059‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderton 2015
- Anderton RS, Mastaglia FL. Advances and challenges in developing a therapy for spinal muscular atrophy. Expert Review of Neurotherapeutics 2015;15(8):895‐908. [DOI] [PubMed] [Google Scholar]
Ando 2017
- Ando S, Funato M, Ohuchi K, Kameyama T, Inagaki S, Seki J, et al. Edaravone is a candidate agent for spinal muscular atrophy: In vitro analysis using a human induced pluripotent stem cells‐derived disease model. European Journal of Pharmacology 2017;814:161‐8. [DOI] [PubMed] [Google Scholar]
Andreassi 2004
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, Vincenzi E, Boninsegna A, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. European Journal of Human Genetics 2004;12(1):59‐65. [DOI] [PubMed] [Google Scholar]
Angelini 1980
- Angelini C, Micaglio GF, Trevisan C. Guanidine hydrochloride in infantile and juvenile spinal muscular atrophy. A double blind controlled study. Acta Neurologica 1980;2(6):460‐5. [PubMed] [Google Scholar]
Arkblad 2009
- Arkblad E, Tulinius M, Kroksmark AK, Henricsson M, Darin N. A population‐based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatrica 2009;98(5):865‐72. [DOI] [PubMed] [Google Scholar]
Arnold 2013
- Arnold WD, Burghes AHM. Spinal Muscular Atrophy: the development and implementation of potential treatments running head. Annals of Neurology 2013;74(3):348‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
ASIRI 2008
- ASIRI ‐ NCT00774423. Study to evaluate the efficacy of riluzole in children and young adults with spinal muscular atrophy (SMA) (ASIRI) [Multicentric, randomized, double‐blind study versus placebo, with two parallel groups treated to evaluate the efficacy and the tolerance of riluzole in children and young adults (6 to 20 years of age) with SMA. (Type II and Type III)]. www.clinicaltrials.gov/show/NCT00774423 (first received 16 October 2008). [NCT00774423; P040904]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck‐Ytter Y, Flottorp S, et al. GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Avila 2007
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. Journal of Clinical Investigation 2007;117(3):659‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Azzouz 2004
- Azzouz M, Le T, Ralph GS, Walmsley L, Monani UR, Lee DC, et al. Lentivector‐mediated SMN replacement in a mouse model of spinal muscular atrophy. Journal of Clinical Investigation 2004;114(12):1726‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Benkhelifa‐Ziyyat 2013
- Benkhelifa‐Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R, et al. Intramuscular scAAV9‐SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Molecular Therapy 2013;21(2):282‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bertini 2017
- Bertini E, Dessaud E, Mercuri E, Muntoni F, Kirschner J, Reid C, et al. Safety and efficacy of olesoxime in patients with type 2 or non‐ambulatory type 3 spinal muscular atrophy: a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Neurology 2017;S1474‐4422(17):30085‐6. [NCT01302600; TRO19622 CLEQ 1275‐1] [DOI] [PubMed] [Google Scholar]
Bladen 2014
- Bladen CL, Thompson R, Jackson JM, Garland C, Wegel C, Ambrosini A, et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. Jounal of Neurology 2014;261(1):152‐63. [DOI] [PubMed] [Google Scholar]
Bogdanik 2015
- Bogdanik LP, Osborne MA, Davis C, Martin WP, Austin A, Rigo F, et al. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proceedings of the National Academy of Sciences of United States of America 2015;112(43):E5863‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bongioanni 2004
- Bongioanni P, Reali C, Sogos V. Ciliary neurotrophic factor (CNTF) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database of Systematic Reviews 2004, Issue 3. [DOI: 10.1002/14651858.CD004302.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bowerman 2010
- Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho‐kinase inactivation prolongs survival of an intermediate SMA mouse model. Human Molecular Genetics 2010;19(8):1468‐78. [DOI] [PubMed] [Google Scholar]
Bowerman 2012
- Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Medicine 2012;7(10):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brahe 2005
- Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. European Journal of Human Genetics 2005;13(2):256‐9. [DOI] [PubMed] [Google Scholar]
Braun 1995
- Braun S, Croizat B, Lagrange MC, Warter JM, Poindron P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet 1995;345(8951):694‐5. [PUBMED: 7741893] [DOI] [PubMed] [Google Scholar]
Brichta 2003
- Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, et al. Valproic acid increases the SMN2 protein level: a well‐known drug as a potential therapy for spinal muscular atrophy. Human Molecular Genetics 2003;12(19):2481‐9. [DOI] [PubMed] [Google Scholar]
Bryson 1996
- Bryson HM, Fulton B, Benfield P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 1996;52(4):549‐63. [DOI] [PubMed] [Google Scholar]
Brzustowicz 1990
- Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, et al. Genetic mapping of chronic childhood‐onset spinal muscular atrophy to chromosome 5q11.2‐13.3. Nature 1990;344(6266):540‐1. [DOI] [PubMed] [Google Scholar]
Burghes 2009
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?. Nature Reviews Neuroscience 2009;10(8):597‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Butchbach 2010
- Butchbach ME, Singh J, Thorsteinsdottir M, Saieva L, Slominski E, Thurmond J, et al. Effects of 2,4‐diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Human Molecular Genetics 2010;19(3):454‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Butchbach 2016
- Butchbach ME, Lumpkin CJ, Harris AW, Saieva L, Edwards JD, Workman E, et al. Protective effects of butyrate‐based compounds on a mouse model for spinal muscular atrophy. Experimental Neurology 2016;279:13‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2017
- Chen YC, Chang JG, Liu TY, Jong YJ, Cheng WL, Yuo CY. Securinine enhances SMN2 exon 7 inclusion in spinal muscular atrophy cells. Biomedicine & Pharmacotherapy 2017;88:708‐14. [DOI] [PubMed] [Google Scholar]
Cherry 2017
- Cherry JJ, DiDonato CJ, Androphy EJ, Calo A, Potter K, Custer SK, et al. In vitro and in vivo effects of 2,4 diaminoquinazoline inhibitors of the decapping scavenger enzyme DcpS: Context‐specific modulation of SMN transcript levels. PLoS One 2017;12(9):eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cifuentes‐Diaz 2002
- Cifuentes‐Diaz C, Frugier T, Melki J. Spinal muscular atrophy. Seminars in Pediatric Neurology 2002;9(2):145‐50. [PUBMED: 12138998] [DOI] [PubMed] [Google Scholar]
Cobben 1995
- Cobben JM, Steege, Grootscholten P, Visser M, Scheffer H, Buys CH. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. American Journal of Human Genetics 1995;57(4):805‐8. [PMC free article] [PubMed] [Google Scholar]
Cobben 2001
- Cobben JM, Visser M, Scheffer H. From gene to disease; 'survival' motor neuron protein and hereditary proximal spinal muscle atrophy. Nederlands Tijdschrift voor Geneeskunde 2001;145(52):2525‐7. [PubMed] [Google Scholar]
Cobben 2008
- Cobben JM, Lemmink HH, Snoeck I, Barth PA, Lee JH, Visser M. Survival in SMA type I: a prospective analysis of 34 consecutive cases. Neuromuscular Disorders 2008;18(7):541‐4. [DOI] [PubMed] [Google Scholar]
Cohen 1988
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd Edition. Routledge, 1988. [Google Scholar]
d'Ydewalle 2017
- d'Ydewalle C, Ramos DM, Pyles NJ, Ng SY, Gorz M, Pilato CM, et al. The antisense transcript SMN‐AS1 regulates SMN expression and Is a novel therapeutic target for spinal muscular atrophy. Neuron 2017;93(1):66‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Darbar 2011
- Darbar IA, Plaggert PG, Resende MB, Zanotelli E, Reed UC. Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurology 2011;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Darras 2007
- Darras BT, Kang PB. Clinical trials in spinal muscular atrophy. Current Opinion in Pediatrics 2007;19(6):675‐9. [DOI] [PubMed] [Google Scholar]
Dominguez 2011
- Dominguez E, Marais T, Chatauret N, Benkhelifa‐Ziyyat S, Duque S, Ravassard P, et al. Intravenous scAAV9 delivery of a codon‐optimized SMN1 sequence rescues SMA mice. Human Molecular Genetics 2011;20(4):681‐93. [DOI] [PubMed] [Google Scholar]
Donelly 2012
- Donnelly EM, Boulis NM. Update on gene and stem cell therapy approaches for spinal muscular atrophy. Expert Opinion on Biological Therapy 2012;11:1463‐71. [DOI] [PubMed] [Google Scholar]
Dubowitz 1995
- Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscular Disorders 1995;5(1):3‐5. [DOI] [PubMed] [Google Scholar]
Duque 2015
- Duque SI, Arnold WD, Odermatt P, Li X, Porensky PN, Schmelzer L, et al. A large animal model of spinal muscular atrophy and correction of phenotype. Annals of Neurology 2015;77(3):399‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edwards 2016
- Edwards JD, Butchbach ME. Effect of the butyrate prodrug pivaloyloxymethyl butyrate (AN9) on a mouse model for spinal muscular atrophy. Journal of Neuromuscular Disorders 201629;3(4):511‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Feldkotter 2002
- Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real‐time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. American Journal of Human Genetics 2002;70(2):358‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Finkel 2014
- Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014;83(9):810‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Finkel 2015
- Finkel R, Bertini E, Muntoni F, Mercuri E, ENMC SMA Workshop Study Group. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7‐9 November 2014, Heemskerk, The Netherlands. Neuromuscular Disorders 2015;25(7):593‐602. [DOI] [PubMed] [Google Scholar]
Finkel 2018
- Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromusculular Disordorders 2018;28(3):197‐207. [DOI] [PubMed] [Google Scholar]
Folkers 1995
- Folkers K, Simonsen R. Two successful double‐blind trials with coenzyme Q10 (vitamin Q10) on muscular dystrophies and neurogenic atrophies. Biochimica et Biophysica Acta 1995;1271(1):281‐6. [DOI] [PubMed] [Google Scholar]
Foust 2010
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature Biotechnology 2010;28(3):271‐4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Garbes 2009
- Garbes L, Riessland M, Hölker I, Heller R, Hauke J, Tränkle C, et al. LBH589 induces up to 10‐fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non‐responsive to valproate. Human Molecular Genetics 2009;18(19):3645‐58. [DOI] [PubMed] [Google Scholar]
Gavrilina 2008
- Gavrilina TO, McGovern VL, Workman E, Crawford TO, Gogliotti RG, DiDonato CJ, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle‐specific SMN expression has no phenotypic effect. Human Molecular Genetics 2008;17(8):1063‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gendron 1999
- Gendron NH, MacKenzie AE. Spinal muscular atrophy: molecular pathophysiology. Current Opinion in Neurology 1999;12(2):137‐42. [DOI] [PubMed] [Google Scholar]
Gilliam 1990
- Gilliam TC, Brzustowicz LM, Castilla LH, Lehner T, Penchaszadeh GK, Daniels RJ, et al. Genetic homogeneity between acute and chronic forms of spinal muscular atrophy. Nature 1990;345(6278):823‐5. [DOI] [PubMed] [Google Scholar]
Glascock 2012a
- Glascock JJ, Shababi M, Wetz MJ, Krogman MM, Lorson CL. Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochemical and Biophysical Research Communications 2012;417:376‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glascock 2012b
- Glascock JJ, Osman EY, Wetz MJ, Krogman MM, Shababi M, Lorson CL. Decreasing disease severity in symptomatic, SMN(–/–);SMN2(+/+), spinal muscular atrophy mice following scAAV9‐SMN delivery. Human Gene Therapy 2012;23(3):330‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gogliotti 2013
- Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Human Molecular Genetics 2013;22(20):4048‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed prior to 2 September 2019. Hamilton (ON): McMaster University (developed by Evidence Prime).
Grzeschik 2005
- Chen TH, Chang JG, Yang YH, Mai HH, Liang WC, Wu YC, et al. Randomized, double‐blind, placebo‐controlled trial of hydroxyurea in spinal muscular atrophy. Neurology 2010;75(24):2190‐7. [DOI] [PubMed] [Google Scholar]
Haddad 2003
- Haddad H, Cifuentes‐Diaz C, Miroglio A, Roblot N, Joshi V, Melki J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle & Nerve 2003;28(4):432‐7. [DOI] [PubMed] [Google Scholar]
Hadwen 2014
- Hadwen J, MacKenzie D, Shamim F, Mongeon K, Holcik M, MacKenzie A, et al. VPAC2 receptor agonist BAY 55‐9837 increases SMN protein levels and moderates disease phenotype in severe spinal muscular atrophy mouse models. Orphanet Journal of Rare Diseases 2014;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hahnen 2006
- Hahnen E, Eyüpoglu IY, Brichta L, Haastert K, Tränkle C, Siebzehnrübl FA, et al. In vitro and ex vivo evaluation of second‐generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. Journal Neurochemistry 2006;98(1):193‐202. [DOI] [PubMed] [Google Scholar]
Hammond 2016
- Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, et al. Systemic peptide‐mediated oligonucleotide therapy improves long‐term survival in spinal muscular atrophy. Proceedings of the National Academy of Sciences in the United States of America 2016;113(39):10962‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harada 2002
- Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, et al. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. Journal of Neurology 2002;249(9):1211‐9. [DOI] [PubMed] [Google Scholar]
Hastings 2009
- Hastings ML, Berniac J, Liu YH, Abato P, Jodelka FM, Barthel L, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Science Translational Medicine 2009;1(5):5ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hauke 2009
- Hauke J, Riessland M, Lunke S, Eyüpoglu IY, Blümcke I, El‐Osta A, et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Human Molecular Genetics 2009;18(2):304‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heier 2009
- Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Human Molecular Genetics 2009;18(7):1310‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heier 2015
- Heier CR, DiDonato CJ. ECG in neonate mice with spinal muscular atrophy allows assessment of drug efficacy. Frontiers in Bioscience (Elite Edition) 2015;1(7):107‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. The Cochrane Collaboration: Available from www.cochrane‐handbook.org.
Hosseinibarkooie 2016
- Hosseinibarkooie S, Peters M, Torres‐Benito L, Rastetter RH, Hupperich K, Hoffmann A, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. American Journal of Human Genetics 2016;99(3):647‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hsu 2012
- Hsu YY, Jong YJ, Tsai HH, Tseng YT, An LM, Lo YC. Triptolide increases transcript and protein levels of survival motor neurons in human SMA fibroblasts and improves survival in SMA‐like mice. British Journal of Pharmacology 2012;166(3):1114‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hua 2010
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes & Development 2010;24(15):1634‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hua 2011
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al. Peripheral SMN restoration is essential for long‐term rescue of a severe SMA mouse model. Nature 2011;478(7367):123‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Iannaccone 1993
- Iannaccone ST, Browne RH, Samaha FJ, Buncher CR. Prospective study of spinal muscular atrophy before age 6 years. DCN/SMA Group. Pediatric Neurology 1993;9(3):187‐93. [DOI] [PubMed] [Google Scholar]
Iannaccone 1998
- Iannaccone ST. Spinal muscular atrophy. Seminars in Neurology 1998;18(1):19‐26. [DOI] [PubMed] [Google Scholar]
Iannaccone 2001
- Iannaccone ST, Burghes AH. Spinal muscular atrophies. In: Rahman Pourmand, Yadollah Harati editor(s). Neuromuscular Disorders. Philadelphia: Lippincott Williams and Wilkins, 2001:83‐98. [Google Scholar]
Il'ina 1980
- Il'ina NA, Antipova RI, Khokhlov AP. Use of lithium carbonate to treat Kugelberg‐Welander spinal amyotrophy [Primenenie uglekislogo litiia dlia lecheniia spinal'noi amiotrofii Kugel'berga‐Velandera]. Zhurnal Nevropatologii i Psikhiatrii Imeni S.S. Korsakova 1980;80(11):1657‐60. [PubMed] [Google Scholar]
Jablonka 2000
- Jablonka S, Rossoll W, Schrank B, Sendtner M. The role of SMN in spinal muscular atrophy. Journal of Neurology 2000;247(Suppl 1):I37‐42. [DOI] [PubMed] [Google Scholar]
Jarecki 2005
- Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, et al. Diverse small‐molecule modulators of SMN expression found by high‐throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Human Molecular Genetics 2005;14(14):2003‐18. [DOI] [PubMed] [Google Scholar]
Kaczmarek 2015
- Kaczmarek A, Schneider S, Wirth B, Riessland M. Investigational therapies for the treatment of spinal muscular atrophy. Expert Opinion on Investigational Drugs 2015;24(7):967‐81. [DOI] [PubMed] [Google Scholar]
Kariya 2008
- Kariya S, Park GH, Maeno‐Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Human Molecular Genetics 2008;17(16):2552‐69. [PUBMED: 18492800] [DOI] [PMC free article] [PubMed] [Google Scholar]
Keil 2014
- Keil JM, Seo J, Howell MD, Hsu WH, Singh RN, DiDonato CJ. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Molecular Therapy ‐ Nucleic Acids 2014;3:e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kernochan 2005
- Kernochan LE, Russo ML, Woodling NS, Huynh TN, Avila AM, Fischbeck KH, et al. The role of histone acetylation in SMN gene expression. Human Molecular Genetics 2005;14(9):1171‐82. [DOI] [PubMed] [Google Scholar]
Kim 2007
- Kim JE, Kim DS, Kwak SE, Choi HC, Song HK, Choi SY, et al. Anti‐glutamatergic effect of riluzole: comparison with valproic acid. Neuroscience 2007;147(1):136‐45. [DOI] [PubMed] [Google Scholar]
Kissel 2011
- Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, et al. SMA carni‐val trial part II: a prospective, single‐ armed trial of L‐carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE 2011;6(7):e21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kroksmark 2001
- Kroksmark AK, Beckung E, Tulinius M. Muscle strength and motor function in children and adolescents with spinal muscular atrophy II and III. European Journal of Paediatric Neurology 2001;5(5):191‐8. [DOI] [PubMed] [Google Scholar]
Kwon 2011
- Kwon DY, Motley WW, Fischbeck KH, Burnett BG. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Human Molecular Genetics 2011;20(18):3667‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 1995
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80(1):155‐65. [DOI] [PubMed] [Google Scholar]
Lefebvre 1998
- Lefebvre S, Burglen L, Frezal J, Munnich A, Melki J. The role of the SMN gene in proximal spinal muscular atrophy. Human Molecular Genetics 1998;7(10):1531‐6. [DOI] [PubMed] [Google Scholar]
Lewelt 2012
- Lewelt A, Newcomb TM, Swoboda KJ. New therapeutic approaches to spinal muscular atrophy. Current Neurology and Neuroscience Reports 2012;12(1):42‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liang 2008
- Liang WC, Yuo CY, Chang JG, Chen YC, Chang YF, Wang HY, et al. The effect of hydroxyurea in spinal muscular atrophy cells and patients. Journal of the Neurological Sciences 2008;268(1‐2):87‐94. [DOI] [PubMed] [Google Scholar]
Little 2015
- Little D, Valori CF, Mutsaers CA, Bennett EJ, Wyles M, Sharrack B, et al. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Molecular Therapy 2015;23(2):270‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2014
- Liu H, Yazdani A, Murray LM, Beauvais A, Kothary R. The sm‐independent beneficial effects of trichostatin A on an intermediate mouse model of spinal muscular atrophy. PLoS One 2014;9(7):e101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2016
- Liu M, Hammers DW, Barton ER, Sweeney HL. Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS One 2016;11(11):e0166803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lorson 1999
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America 1999;96:6307‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lui 2016
- Liu M, Hammers DW, Barton ER, Sweeney HL. Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS One 2016;11(11):e0166803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lunn 2004
- Lunn MR, Root DE, Martino AM, Flaherty SP, Kelley BP, Coovert DD, et al. Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase‐independent mechanism. Chemistry & Biology 2004;11(11):1489‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lunn 2008
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371(9630):2120‐33. [DOI] [PubMed] [Google Scholar]
Mattis 2009a
- Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read‐through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Human Molecular Genetics 2009;18(20):3906‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattis 2009b
- Mattis VB, Fosso MY, Chang CW, Lorson CL. Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neuroscience 2009;30(10):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattis 2012
- Mattis VB, Tom Chang CW, Lorson CL. Analysis of a read‐through promoting compound in a severe mouse model of spinal muscular atrophy. Neuroscience Letters 2012;525(1):72‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
McWhorter 2003
- McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. The Journal of Cell Biology 2003;162(5):919‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
MECIR 2018
- Higgins JP, Lasserson T, Chandler J, Tovey D, Churchill R. Methodological Expectations of Cochrane Intervention Reviews. Vol. 1.02, London: Cochrane, 2018. [Google Scholar]
Melki 1990a
- Melki J, Abdelhak S, Sheth P, Bachelot MF, Burlet P, Marcadet A, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 1990;344(6268):767‐8. [DOI] [PubMed] [Google Scholar]
Melki 1990b
- Melki J, Sheth P, Abdelhak S, Burlet P, Bachelot MF, Lathrop MG, et al. Mapping of acute (type I) spinal muscular atrophy to chromosome 5q12‐q14. The French Spinal Muscular Atrophy Investigators. Lancet 1990;336(8710):271‐3. [DOI] [PubMed] [Google Scholar]
Mendell 2016
- Mendell JR, Al‐Zaidy S, Shell R, Arnold WD, Rodino‐Klapac L, Kissel JT, et al. Gene therapy for spinal muscular atrophy type 1 shows potential to improve survival and motor functional outcomes. Molecular Therapy 2016;24(Suppl 1):S190. [WOS:000375264200475] [Google Scholar]
Mercuri 2012
- Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurology 2012;11(5):443‐52. [DOI] [PubMed] [Google Scholar]
Mercuri 2018
- Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscular Disorders 2018;28(2):103‐15. [DOI] [PubMed] [Google Scholar]
Merlini 2003
- Merlini L, Solari A, Vita G, Bertini E, Minetti C, Mongini T, et al. Role of gabapentin in spinal muscular atrophy: results of a multicenter, randomized Italian study. Journal of Child Neurology 2003;18(8):537‐41. [DOI] [PubMed] [Google Scholar]
Meyer 2015
- Meyer K, Ferraiuolo L, Schmelzer L, Braun L, McGovern V, Likhite S, et al. Improving single injection CSF delivery of AAV9‐mediated gene therapy for SMA: a dose–response study in mice and nonhuman primates. Molecular Therapy 2015;23(3):477‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miller 1996
- Miller RG, Petajan JH, Bryan WW, Armon C, Barohn RJ, Goodpasture JC, et al. A placebo‐controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Annals of Neurology 1996;39(2):256‐60. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mohseni 2013
- Mohseni J, Zabidi‐Hussin ZA, Sasongko TH. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genetics and Molecular Biololgy 2013;36(3):299‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mohseni 2016
- Mohseni J, Al‐Najjar BO, Wahab HA, Zabidi‐Hussin ZA, Sasongko TH. Transcript, methylation and molecular docking analyses of the effects of HDAC inhibitors, SAHA and Dacinostat, on SMN2 expression in fibroblasts of SMA patients. Journal of Human Genetics 2016;61:823‐30. [DOI] [PubMed] [Google Scholar]
Munsat 1992
- Munsat TL, Davies KE. International SMA consortium meeting (26‐28 June 1992, Bonn, Germany). Neuromuscular Disorders 1992;2(5‐6):423‐8. [DOI] [PubMed] [Google Scholar]
Murray 2008
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre‐ and post‐synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Human Molecular Genetics 2008;17(7):949‐62. [PUBMED: 18065780] [DOI] [PubMed] [Google Scholar]
Naryshkin 2014
- Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014;345(6197):688‐93. [DOI] [PubMed] [Google Scholar]
NCT02628743
- NCT02628743. A study to evaluate long term safety, tolerability, and effectiveness of olesoxime in patients with spinal muscular atrophy [Multicenter, open‐label, single arm study to evaluate long‐term safety, tolerability, and effectiveness of 10 mg/kg olesoxime in patients with SMA]. clinicaltrials.gov/show/NCT02628743 (first received 1 December 2015). [2015‐001589‐25; BN29854; EUCTR2015‐001589‐25‐GB; NCT02628743]
Nicole 2002
- Nicole S, Diaz CC, Frugier T, Melki J. Spinal muscular atrophy: recent advances and future prospects. Muscle & Nerve 2002;26(1):4‐13. [DOI] [PubMed] [Google Scholar]
Ning 2010
- Ning K, Drepper C, Valori CF, Ahsan M, Wyles M, Higginbottom A, et al. PTEN depletion rescues axonal growth defect and improves survival in SMN‐deficient motor neurons. Human Molecular Genetics 2010;19(16):3159‐68. [DOI] [PubMed] [Google Scholar]
Nizzardo 2014
- Nizzardo M, Simone C, Salani S, Ruepp MD, Rizzo F, Ruggieri M, et al. Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Δ7 mouse model phenotype. Clinical Therapeutics 2014;36(3):340‐56. [DOI] [PubMed] [Google Scholar]
Nurputra 2013
- Nurputra DK, Lai PS, Harahap NI, Morikawa S, Yamamoto T, Nishimura N, et al. Spinal muscular atrophy: from gene discovery to clinical trials. Annals of Human Genetics 2013;77(5):435‐63. [DOI] [PubMed] [Google Scholar]
NURTURE 2015
- NCT02386553. A study of multiple doses of ionis smnrx (ISIS 396443) delivered to infants with genetically diagnosed and presymptomatic spinal muscular atrophy (NURTURE) [An open‐label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of IONIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy.]. www.clinicaltrials.gov/show/NCT02386553 (first received 27 February 2015). [2014‐002098‐12; 232SM201; NCT02386553]
Oskoui 2007
- Oskoui M, Levy G, Garland CJ, Gray JM, O'Hagen J, Vivo DC, et al. The changing natural history of spinal muscular atrophy type I. Neurology 2007;69:1931‐6. [DOI] [PubMed] [Google Scholar]
Osman 2014
- Osman EY, Miller MR, Robbins KL, Lombardi AM, Atkinson AK, Brehm AJ, et al. Morpholino antisense oligonucleotides targeting intronic repressor element 1 improve phenotype in SMA mouse models. Human Molecular Genetics 2015;23(18):4832‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Osman 2016
- Osman EY, Washington CW, Kaifer KA, Mazzasette C, Patitucci TN, Florea KM, et al. Optimization of morpholino antisense oligonucleotides targeting the intronic repressor element1 in spinal muscular atrophy. Molecular Therapy 2016;24(9):1592‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parker 2008
- Parker GC, Li X, Anguelov RA, Toth G, Cristescu A, Acsadi G. Survival motor neuron protein regulates apoptosis in an in vitro model of spinal muscular atrophy. Neurotoxicity Research 2008;13(1):39‐48. [DOI] [PubMed] [Google Scholar]
Passini 2011
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Science Translational Medicine 2011;3(72):72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pellizzoni 1998
- Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre‐mRNA splicing. Cell 1998;95(5):615‐24. [DOI] [PubMed] [Google Scholar]
Piepers 2008
- Piepers S, Berg LH, Brugman F, Scheffer H, Ruiterkamp‐Versteeg M, Engelen BG, et al. A natural history study of late onset spinal muscular atrophy types 3b and 4. Journal of Neurology 2008;255(9):1400‐4. [DOI: 10.1007/s00415-008-0929-0] [DOI] [PubMed] [Google Scholar]
Piepers 2011
- Piepers S, Cobben JM, Sodaar P, Jansen MD, Wadman RI, Meester‐Delver A, et al. Quantification of SMN protein in leucocytes from spinal muscular atrophy patients: effects of treatment with valproic acid. Journal of Neurology, Neurosurgery, and Psychiatry 2011;82(8):850‐2. [DOI] [PubMed] [Google Scholar]
Porensky 2013
- Porensky PN, Burghes AH. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Human Gene Therapy 2013;24(5):489‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
RAINBOWFISH
- NCT03779334. A study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy (Rainbowfish) [An open‐label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy]. www.clinicaltrial.gov (first date recieved 18 December 2018). [BN40703 ; NCT03779334]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riessland 2006
- Riessland M, Brichta L, Hahnen E, Wirth B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Human Genetics 2006;120(1):101‐10. [DOI] [PubMed] [Google Scholar]
Riessland 2010
- Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Human Molecular Genetics 2010;19(8):1492‐1506. [DOI] [PubMed] [Google Scholar]
Riessland 2017
- Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Löhr H, Bradler C, et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. American Journal of Human Genetics 2017;100(2):297‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Robbins 2014
- Robbins KL, Glascock JJ, Osman EY, Miller MR, Lorson CL. Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Human Molecular Genetics 2014;23(17):4559‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rossoll 2003
- Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, et al. Smn, the spinal muscular atrophy‐determining gene product, modulates axon growth and localization of beta‐actin mRNA in growth cones of motoneurons. Journal of Cell Biology 2003;163(4):801‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Russman 1992
- Russman BS, Iannacone ST, Buncher CR, Samaha FJ, White M, Perkins B, et al. Spinal muscular atrophy: new thoughts on the pathogenesis and classification schema. Journal of Child Neurology 1992;7(4):347‐53. [DOI] [PubMed] [Google Scholar]
Saito 2014
- Saito T, Nurputra DK, Harahap NI, Harahap IS, Yamamoto H, Muneshige E, et al. A study of valproic acid for patients with spinal muscular atrophy. Neurology and Clinical Neuroscience 2015;3:49‐57. [Google Scholar]
Schreml 2013
- Schreml J, Riessland M, Paterno M, Garbes L, Roßbach K, Ackermann B, et al. Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ‐26481585. European Journal of Human Genetics 2013;21(6):643‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, Guyatt GH. Chapter 12: Interpreting results and drawing conclusions.. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Seo 2013
- Seo J, Howell MD, Singh NN, Singh RN. Spinal muscular atrophy: an update on therapeutic progress. Biochimica Biophysica Acta 2013;1832(12):2180‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shababi 2012
- Shababi M, Lorson CL. Optimization of SMN trans‐splicing through the analysis of SMN introns. Journal Molecular Neuroscience 2012;46(3):459‐69. [DOI] [PubMed] [Google Scholar]
Singh 2008
- Singh J, Salcius M, Liu SW, Staker BL, Mishra R, Thurmond J, et al. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chemical Biology 2008;3(11):711‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Skordis 2003
- Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans‐acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proceedings of National Acadamy of Sciences of the United States of America April 2003;100(7):4114‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
SPACE 2014
- SPACE. SPACE trial SMA and Pyridostigmine in Adults and Children; Experimental trial to assess effect of pyridostigmine compared to placebo in patients with spinal muscular atrophy types 2, 3 and 4 [SPACE trial SMA and Pyridostigmine in Adults and Children; Efficacy trial Phase II, mono‐center, doubleblind, placebo‐controlled, crossover trial to assess efficacy of pyridostigmine in patients with spinal muscular atrophy types 2, 3 and 4 ‐ SPACE trial]. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2011‐004369‐34‐NL (first received 3 November 2014). [EUCTR2011‐004369‐34‐NL]
Staropoli 2015
- Staropoli JF, Li H, Chun SJ, Allaire N, Cullen P, Thai A, et al. Rescue of gene‐expression changes in an induced mouse model of spinal muscular atrophy by an antisense oligonucleotide that promotes inclusion of SMN2 exon 7. Genomics 2015;105(4):220‐8. [DOI] [PubMed] [Google Scholar]
Stavarachi 2007
- Stavarachi M, Apostol P, Toma M, Cimponeriu D, Gavrila L. Spinal muscular atrophy disease: a literature review for therapeutic strategies. Journal of Medicine and Life January‐March 2010;3(1):3‐9. [PMC free article] [PubMed] [Google Scholar]
STOPSMA 2007
- STOPSMA ‐ NCT00528268. Study to evaluate sodium phenylbutyrate in pre‐symptomatic infants with spinal muscular atrophy (STOPSMA) [Prospective phase I/II study to evaluate effects of sodium phenylbutyrate in pre‐symptomatic infants with spinal muscular atrophy]. www.clinicaltrials.gov/show/NCT00528268 (first received 10 September 2007). [1R01HD054599‐01; 22183; NCT00528268]
Sumner 2003
- Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Annals of Neurology 2003;54(5):647‐54. [DOI] [PubMed] [Google Scholar]
Sumner 2007
- Sumner CJ. Molecular mechanisms of spinal muscular atrophy. Journal of Child Neurology 2007;22(8):979‐89. [DOI] [PubMed] [Google Scholar]
Swoboda 2005
- Swoboda KJ, Prior TW, Scott CB, McNaught TP, Wride MC, Reyna SP, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Annals of Neurology 2005;57(5):704‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Swoboda 2007
- Swoboda KJ, Kissel JT, Crawford TO, Bromberg MB, Acsadi G, D'Anjou G, et al. Perspectives on clinical trials in spinal muscular atrophy. Journal of Child Neurology 2007;22(8):957‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Talbot 1999
- Talbot K. Spinal muscular atrophy. Journal of Inherited Metabolic Diseases 1999;22(4):545‐54. [DOI] [PubMed] [Google Scholar]
Thomas 1994
- Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy. Neuromuscular Disorders 1994;4(5‐6):497‐502. [DOI] [PubMed] [Google Scholar]
Thurmond 2008
- Thurmond J, Butchbach ME, Palomo M, Pease B, Rao M, Bedell L, et al. Synthesis and biological evaluation of novel 2,4‐diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. Journal of Medicinal Chemistry 2008;51(3):449‐69. [DOI] [PubMed] [Google Scholar]
Ting 2007
- Ting CH, Lin CW, Wen SL, Hsieh‐Li HM, Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Human Molecular Genetics 2007;16(5):499‐514. [DOI] [PubMed] [Google Scholar]
Tisdale 2015
- Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. Journal of Neuroscience 2015;35(23):8691‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tsai 2007
- Tsai LK, Yang CC, Hwu WL, Li H. Valproic acid treatment in six patients with spinal muscular atrophy. European Journal of Neurology 2007;14(12):e8‐9. [DOI] [PubMed] [Google Scholar]
Tseng 2016
- Tseng YT, Chen CS, Jong YJ, Chang FR, Lo YC. Loganin possesses neuroprotective properties, restores SMN protein and activates protein synthesis positive regulator Akt/mTOR in experimental models of spinal muscular atrophy. Pharmacological Research 2016;111:58‐75. [DOI] [PubMed] [Google Scholar]
Uzunallı 2015
- Uzunallı G, Bora‐Tatar G, Dayangaç‐Erden D, Erdem‐Yurter H. Effects of flavonoid quercetin on survival of motor neuron gene expression. Cell Biology International 2015;39(3):350‐4. [DOI] [PubMed] [Google Scholar]
Valori 2010
- Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Science Translational Medicine 2010;35:35‐42. [DOI] [PubMed] [Google Scholar]
Van Meerbeke 2013
- Meerbeke JP, Gibbs RM, Plasterer HL, Miao W, Feng Z, Lin MY, et al. The DcpS inhibitor RG3039 improves motor function in SMA mice. Human Molecular Genetics 2013;22(20):4074‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wadman 2017a
- Wadman RI, Stam M, Gijzen M, Lemmink HH, Snoeck IN, Wijngaarde CA, et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0‐4. Journal of Neurology, Neurosurgery and Psychiatry 2017;88(4):365‐7. [DOI] [PubMed] [Google Scholar]
Wadman 2018
Wang 2007
- Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, et al. Consensus statement for standard of care in spinal muscular atrophy. Journal of Child Neurology 2007;22(8):1027‐49. [DOI] [PubMed] [Google Scholar]
Weihl 2006
- Weihl CC, Connolly AM, Pestronk A. Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology 2006;67(3):500‐1. [DOI] [PubMed] [Google Scholar]
Wirth 2006
- Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy: from gene to therapy. Seminars in Pediatric Neurology 2006;13(2):121‐31. [DOI] [PubMed] [Google Scholar]
Wishart 2014
- Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, et al. Dysregulation of ubiquitin homeostasis and β‐catenin signaling promote spinal muscular atrophy. Journal of Clinical Investigation 2014;124(4):1821‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Woll 2016
- Woll MG, Qi H, Turpoff A, Zhang N, Zhang X, Chen G, et al. Discovery and Optimization of Small Molecule Splicing Modifiers of Survival Motor Neuron 2 as a Treatment for Spinal Muscular Atrophy. Journal of Medicinal Chemistry 2016;59(13):6070‐85. [DOI] [PubMed] [Google Scholar]
Wolstencroft 2005
- Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non‐sequence‐specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Human Molecular Genetics 2005;14(9):1199‐1210. [DOI] [PubMed] [Google Scholar]
Wong 2007
- Wong BL, Hynan LS, Iannaccone ST, AmSMART Group. A randomized, placebo‐controlled trial of creatine in children with spinal muscular atrophy. Journal of Clinical Neuromuscular Disease 2007;8(3):101‐10. [Google Scholar]
Woo 2017
- Woo CJ, Maier VK, Davey R, Brennan J, Li G, Brothers J, et al. Gene activation of SMN by selective disruption of lncRNA‐mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America 2017;114(8):E1509‐E1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yuo 2008
- Yuo CY, Lin HH, Chang YS, Yang WK, Chang JG. 5‐(N‐ethyl‐N‐isopropyl)‐amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells. Annals of Neurology 2008;63(1):26‐34. [DOI] [PubMed] [Google Scholar]
Zanetta 2014
- Zanetta C, Nizzardo M, Simone C, Monguzzi E, Bresolin N, Comi GP, et al. Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clinical Therapeutics 2014;36(1):128‐40. [DOI] [PubMed] [Google Scholar]
Zerres 1999
- Zerres K, Davies KE. 59th ENMC International Workshop: Spinal Muscular Atrophies: recent progress and revised diagnostic criteria 17‐19 April 1998, Soestduinen, The Netherlands. Neuromuscular Disorders 1999;9(4):272‐8. [DOI] [PubMed] [Google Scholar]
Zhang 2001
- Zhang ML, Lorson CL, Androphy EJ, Zhou J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA. Gene Therapy 2001;8(20):1532‐8. [DOI] [PubMed] [Google Scholar]
Zhou 2013
- Zhou H, Janghra N, Mitrpant C, Dickinson RL, Anthony K, Price L, et al. A novel morpholino oligomer targeting ISS‐N1 improves rescue of severe spinal muscular atrophy transgenic mice. Human Gene Therapy 2013;24(3):331‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhou 2015
- Zhou H, Meng J, Marrosu E, Janghra N, Morgan J, Muntoni F. Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Human Molecular Genetics 2015;24(22):6265‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zou 2007
- Zou T, Ilangovan R, Yu F, Xu Z, Zhou J. SMN protects cells against mutant SOD1 toxicity by increasing chaperone activity. Biochemical and Biophysical Research Communication 2007;364(4):850‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Bosboom 2006
- Bosboom WM, Berg LH, Iannaccone ST, Vrancken AF, Wokke JHJ. Drug treatment for spinal muscular atrophy type I. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD006281] [DOI] [Google Scholar]
Bosboom 2009
- Bosboom WM, Vrancken AF, Berg LH, Wokke JH, Iannaccone ST. Drug treatment for spinal muscular atrophy type I. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD006281.pub2] [DOI] [PubMed] [Google Scholar]
Wadman 2012
- Wadman RI, Bosboom WM, Berg LH, Wokke JH, Iannaccone ST, Vrancken AF. Drug treatment for spinal muscular atrophy type I. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD006281.pub3] [DOI] [PubMed] [Google Scholar]