

Abstract
Members of the FAD/NAD-linked reductase family are recognized as crucial targets in drug development for cancers, inflammatory disorders, and infectious diseases. However, individual FAD/NAD reductases are difficult to inhibit in a selective manner with off target inhibition reducing usefulness of identified compounds. Thioredoxin glutathione reductase (TGR), a high molecular weight thioredoxin reductase-like enzyme, has emerged as a promising drug target for the treatment of schistosomiasis, a parasitosis afflicting more than 200 million people. Taking advantage of small molecules selected from a high-throughput screen and using X-ray crystallography, functional assays, and docking studies, we identify a critical secondary site of the enzyme. Compounds binding at this site interfere with well-known and conserved conformational changes associated with NADPH reduction, acting as a doorstop for cofactor entry. They selectivity inhibit TGR from Schistosoma mansoni and are active against parasites in culture. Since many members of the FAD/NAD-linked reductase family have similar catalytic mechanisms the unique mechanism of inhibition identified in this study for TGR broadly opens new routes to selectively inhibit homologous enzymes of central importance in numerous diseases.
Keywords: schistosomiasis, redox metabolism, fragment-based drug design, secondary site
Graphical Abstract
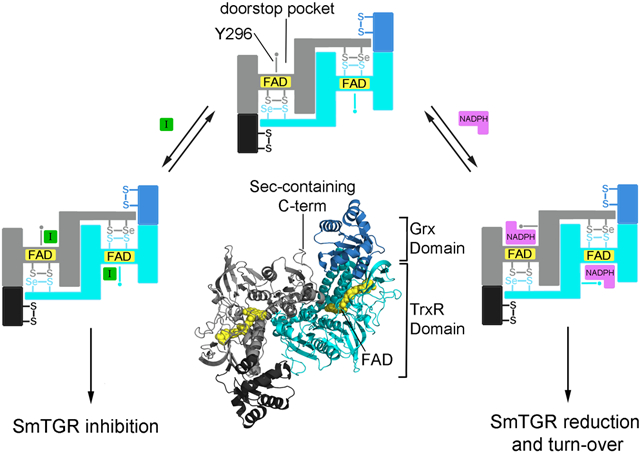
Members of the FAD/NAD-linked reductase family are fundamental enzymes maintaining redox homeostasis in many organisms. For this reason, they are the targets of multiple studies aimed at finding new treatments for several diseases. Trypanothione reductase (TrypR) is considered a promising drug target for leishmaniasis and trypanosomiasis (Krauth-Siegel 1995). The enzyme is closely related to glutathione (GSH) reductase (GR). Both GR and TrypR are dimers. Each subunit is characterized by a single redox active center composed of the FAD cofactor and a pair of proximal cysteine (Cys) residues, which acquire electrons from NADPH and transfer them either to oxidized GSH (Berkholz et al., 2009) or to the parasite-specific reducing substrate trypanothione (Baiocco et al., 2009). More complicated enzymes are the mammalian high molecular weight thioredoxin reductases (hTrxR), which are the subject of several studies aimed at finding compounds active against a plethora of human diseases (Stafford et al., 2018; Zhang et al., 2017; Boumis et al., 2012). For instance, auranofin (Ridaura), clinically used to treat rheumatoid arthritis and which is known to target the selenocysteine (Sec)-dependent human hTrxRs (Gromer et al., 1998), is being repurposed as an anticancer and anti-infective agent (Harbut et al., 2015; Roder et al., 2015). Mammalian hTrxRs share the same FAD redox center as in GR and TrypR but possess an additional redox active C-terminal extension with a Cys-Sec couple, where the Sec residue occupies the penultimate position. This flexible tail of one subunit is capable of transferring electrons from the reduced FAD/Cys site of the other subunit to oxidized thioredoxin (Trx), its primary substrate (Arnér et al., 2009). Thioredoxin glutathione reductase (TGR) is a chimeric enzyme containing a peculiar fusion of glutaredoxin (Grx) and Sec-containing TrxR domains (Alger and Williams, 2002; Angelucci et al., 2008). The N-terminal Grx domain with its redox center, generally characterized by the -CysXXCys- motif, enables the enzyme to also reduce oxidized GSH, a property absent in hTrxRs. In all investigated pathogenic Platyhelminthes, such as Schistosoma, Echinococcus, and Taenia, TGR represents a unique hub that provides electrons to both GSH and Trx systems (Boumis et al., 2011; Williams et al. 2013). These organisms lack both authentic GR and TrxR enzymes and depend entirely on TGR to maintain redox equilibria. TGR is one of most promising drug targets against schistosomiasis (Sayed et al., 2009; Mafud et al., 2016). It has been demonstrated both to be vital for the parasite by RNA interference and to be the target of two anti-schistosomal drugs used previously used to fight the disease, antimony potassium tartrate and oltipraz (Kuntz et al., 2007).
Schistosomiasis is a widespread human parasitic disease affecting more than 200 million people. Expanded efforts to control schistosomiasis, based primarily on yearly administration of praziquantel, has resulted in increased use of the drug so that about 61 million people were treated in 2014 (WHO report 2014). Additional attention has been directed to this parasitosis due to the emergence of strains less sensitive to praziquantel, the only drug currently on the market to fight the disease and the certainty that drug resistance will evolve as treatment coverage increases (Vale et al., 2017). Development of inhibitors of TGR with a novel mechanism of action may provide a solution to the poor existing drug armamentarium.
Due to the crucial role of TGR/hTrxR enzymes in several human pathological processes, finding specific inhibitors is urgently needed but, as often reported, challenging (Zhang B et al., 2017; Zhang J et al., 2017). The main reason is that the majority of known inhibitors reported in the literature and patents are electrophilic compounds, such as metal derivatives or Michael acceptors, likely targeting the highly nucleophilic Sec residue or any of the low pKa redox active cysteines. This may result in low specificity and toxicity in vivo, where the high content of thiols may give rise to multiple, undesirable cross-reactions, although very recently a specific irreversible inhibitor of human hTrxR 1 targeting the Sec residue has been found (Stafford et al., 2018). Additionally, inhibitors that target the NADPH binding site are rare and, given the conservation of this site throughout the members of the family, are likely to be nonspecific as was previously demonstrated with indomethacin, an NADPH competitive inhibitor (Chen et al., 1994). This scenario is exacerbated by the lack of structural data for protein-inhibitor complexes. In the protein data bank only two structures with metal inhibitors, i.e., TGR from Schistosoma mansoni (SmTGR) in complex with auranofin (Angelucci et al., 2009) and human hTrxR I (HsTrxR) in complex with terpyridine-platinum (Lo et al., 2009), are present. This reflects the difficulty of crystallizing non-homogenous protein preparations caused by the presence of several redox and nucleophilic centers in each subunit of TGR/hTrxR when the inhibitor is a reactive compound (for a review see Saccoccia et al., 2014).
Allosteric or secondary sites have been suggested as a solution to inhibit difficult targets (Hardy et al., 2004). These sites are, in general, separated from the primary or orthosteric sites, are less conserved in amino acid composition, and drugs designed to target these pockets are likely to display high selectivity (Langmead et al., 2014) and improved resistance profiles (Di Santo, 2014), providing new routes for therapeutic intervention. Even though it was recently demonstrated that secondary sites are present in most proteins, their identification and confirmation of their biological relevance remains a difficult task (Ludlow et al., 2015). Detection of low molecular weight compounds, which are often derived from compound library screenings, in protein structures by X-ray crystallography represents a promising strategy to find new secondary sites, especially when the compounds are characterize by low-affinity interactions (KD > 10 mM; Ludlow et al., 2015; Hardy et al., 2004). In addition, small organic molecular probes derived from the crystallization cocktails may be instrumental in this approach, considering that they can bind as drug-like molecules (Drwal et al., 2017).
Combining high-to-medium resolution crystal structures and functional studies, we identified a novel secondary binding site in SmTGR that we call the “doorstop pocket” (Figure 1). Co-crystallization studies identified two subpockets in this cavity with biological relevance. Small molecules bound at these sites are capable of disrupting the well-known structural transitions conserved throughout the protein family, which are associated with both NADPH binding and enzyme reduction. Chimeric compounds combining the structural features of the initial fragments into single chemical entities are demonstrated by docking to bind in the same site, to have improved TGR inhibition, to be active against ex vivo larval and adult worms at low micromolar concentrations, and to be selective for SmTGR. The doorstop pocket is present in several members of the FAD/NAD-linked reductase family. Its amino acid composition is not conserved and differences in the overall shape and charge topology may present an avenue for the development of selective inhibitors. The results reported here demonstrate that SmTGR can be inhibited via binding to this doorstop site and suggest a new, general strategy to target other important enzymes of the same family.
Figure 1.

a) Crystal structure of the 1,8-naphthyridine-2-carboxylate (in green sticks, compound 1 in Table 1) in complex with SmTGR and bound in the doorstop pocket. The green electron density map represents the Fo – Fc omit map contoured at 3σ. FAD is in yellow sticks and the SmTGR subunit is in grey cartoon. The hydrophobic amino acid residues within 5 Å contacting 1 are displayed as grey sticks. The H-bond with the main chain of Q440 and the face-on H-bond with T471 are shown as dotted lines. b) The solvent exposed surfaces of the doorstop and NADPH pockets are displayed together with the surface of Y296 in the closed conformation at their boundary. FAD is shown in yellow spheres. b) Superposition between the complexes of the SmTGR-1 and SmTGR-NADPH (PDB ID: 2X99). There is not direct overlap between NADPH (in magenta sticks) and 1. By contrast, overlaps are present between NADPH and Y296 (in gray sticks) in the closed conformation and between 1 and Y296 in the open conformation. d) Mixed inhibition of SmTGR by 1. Curves are steady state experiments where SmTGR is in presence of different concentrations of 1 and its activity is tested varying NADPH, ranging from 0.01 to 1 mM, at saturating DTNB concentration (3 mM). There is an apparent lowering of both KM and kcat consistent with a mixed inhibition mechanism. Data represent averages of three experiments.
RESULTS AND DISCUSSION
An important question is if new functional protein regions, alternatives to the orthosteric sites and exploitable in drug-design projects, exist in the pharmacologically relevant hTrxR subfamily (for a recent review see Zhang et al., 2017). The structural and functional data reported here identify a unique and druggable secondary site in SmTGR, one of the most promising drug targets against schistosomiasis and against other related parasitic diseases (Williams et al., 2013). We report the first structure of an hTrxR-like enzyme with a non-metal inhibitor bound. The pocket identified in SmTGR is present in the unliganded, inactive conformation of most members of this protein family establishing a route to discover new classes of inhibitors targeting other members of the FAD/NAD linked reductase family.
The Naphthyridine Subpocket and Its Functional Role as a “Doorstop” for NADPH Entry.
The refined structure solved at 2.5 Å of SmTGR in complex with compound 1, selected from a qHTS (Inglese et al., 2006) against SmTGR (bioassay AID: 485364), clearly shows additional electron density in a pocket we call “the doorstop pocket” adjacent to the NADPH binding site (Fig. 1a and 1b). The real space correlation coefficient (RSCC) of 1 is 0.90. In general, values greater than 0.7 indicate a good fit of the ligand atoms in the experimental electron density (Pearce et al., 2017). The cavity is lined both by hydrophobic side chains, such as Y296, and the isoalloxazine ring of the FAD. The compound is anchored with one H-bond between the nitrogen of the main chain of Q440 and one oxygen of its carboxylate moiety (Fig. 2a). It also makes several hydrophobic interactions with 5 amino acid residues whose atoms are within 5 Å (Fig. 1a). The interaction between with the hydroxyl group of T471 and 1 is compatible with a face-on H-bond. The distance between the oxygen of the T471 side chain and the ring centroid is less than 3.8 Å and the angle formed between the oxygen-centroid line and the ring normal is less than 25° as defined by Steiner and collaborators for this kind of interaction (Steiner et al., 2001). The shortest distance between 1 and the isoalloxazine ring of FAD is from the C4X atom of the cofactor and the C5 of the compound (5.5 Å, atom numbering is according to that in the PDB file).
Figure 2.

The HEPE derivatives occupy a different subsite to that of 1 (Figure 1) but both are localized in the same cavity. The green electron density maps represent the Fo – Fc omit map contoured at 3σ. a. b. The HEPE derivatives, i.e., 1,4-bis(2-hydroxyethyl)piperazine (compound 2) and 2-[4-(4-aminobutyl)piperazin-1-yl]ethanol (compound 3), both in light blue sticks, were investigated due to the presence of HEPES in the doorstop pocket of several SmTGR crystal structures (see Supplementary Fig. 1). The arrays of polar interactions are displayed as dotted-lines for both of the compounds. Water molecules are depicted as red spheres. The putative cation-pi interaction with F324 is also shown. c. Cartoon representation of the crystal structure of SmTGR U597C mutant showing the “doorstop pocket” (framed in yellow) and its immediate proximity to the NADPH binding site (circled in magenta). The two Grx domains are shown in cyan, while the two subunits of the dimeric TrxR domain are shown in grey and in light blue. A magnified view of the solvent exposed surface of newly discovered pocket is displayed on the right. The computer-aided analysis indicates that it consists of both the side chain residues displayed as grey sticks (i.e., K162, Y296, E300, R322, G323, F324, P439, A436, G437, Q440, L441, S467, V469, T471, V473, Y479, A481, G483, D488, H538, I570*, P572*) and of FAD (in yellow sticks). Residues belonging to the other subunit are indicated with an asterisk. The HEPE and naphthyridine subpockets present in the doorstop pocket are highlighted in cyan and green, respectively.
Superposition of the SmTGR structures in complex with 1 and NADPH (pdb code: 2X99; Angelucci et al., 2010; 0.3 Å r.m.sd within 583 CA atoms; Fig 1c) shows that the binding of the two ligands is mutually exclusive even though (i) the pocket in which 1 is found is spatially distinct from the NADPH-binding site (Fig. 1b) and (ii) the structural overlap between 1 and NADPH is not evident from the structural superposition. Indeed, 3.0 Å is the closest distance between the atoms of the two molecules. These findings prompted us to define the pocket in which 1 is localized as the “doorstop pocket”. The apparent incompatibility of simultaneous binding of both NADPH and 1 is due to the presence of the aromatic ring of Y296 at the boundary of their individual binding sites (Fig. 1b and 1c). In the presence of NADPH, the ring of Y296 is in the open conformation partially occupying the doorstop pocket allowing the nicotinamide moiety of NADPH to be proximal to the FAD. In this conformation binding of 1 is sterically not possible. By contrast, the nicotinamide moiety of NADPH cannot stack with FAD in presence of the inhibitor due to Y296 projecting into the NADPH-binding pocket in the closed conformation (Fig. 1c).
Structural data suggests that binding of 1 and NADPH are mutually exclusive, each compound preventing or at least severely impairing the binding of the other. Thus a competitive inhibition mechanism is predicted. To explore this point, steady state experiments were carried out to determine the effect of 1 on the KM for NADPH (Fig. 1d). The experimental data are compatible with a mixed competitive inhibition mechanism in which 1 changes both KM and kcat of the enzyme. This mechanism predicts binding of the inhibitor to both the NADPH-free enzyme (with an estimated KI = 0.57 μM at 3 mM DTNB) and the NADPH-bound enzyme (with an estimated KIS = 4.8 mM at 3 mM DTNB). The effect of 1 on the apparent KM for NADPH proves that its binding is reversible on the time scale of the activity assay. Indeed, for the inhibitor to compete with the substrate it is necessary not only that binding is reversible, but also that association and dissociation rates are as fast as, or faster than, those of the substrate (Bellelli and Carey, 2018; Saccoccia et al., 2014). Irreversible inactivation was observed after extended incubation of reduced TGR with 1 (results not shown), but our data clearly rule out this possibility over the time window of the experiments described in this paper. Although simultaneous binding of 1 and NADPH may appear unlikely in view of the structural data, the effect of 1 on the kcat for NADPH requires that at high concentrations both compounds bind to the enzyme, presumably by inducing a distortion of the respective binding pockets. Indeed, the perturbation of the native protein conformation may require only a few kcal, which the binding energies of the substrate and inhibitor, both at high concentrations, could easily provide. This effect would not be apparent in the crystal structure because of the strong lattice forces of the packed protein crystal. Moreover, in a two-substrate enzyme the apparent values of KM and kcat one measures for the first substrate at fixed concentration of the second depend on the concentration of the second substrate used, especially if not saturating. Thus, the effect of 1 on kcat may stem from two nonexclusive contributions: (i) a fraction of the enzyme is trapped in the inactive or less active complex with both 1 and NADPH; and (ii) the affinity and saturation of TGR for the oxidizing substrate, kept at constant concentration (DTNB) may be reduced. Compound 1 and its analogs (the first series in Table 1) display IC50s in the low millimolar range. Importantly, 1 does not inhibit mammalian hTrxR.
Table 1.
Structures and activities of compounds investigated in this study.
| compound | Chemical structure | SmTGR IC50 (mM) | Mammalian TrxR IC50 (mM)* | Structural information | |
|---|---|---|---|---|---|
| Derivatives of 1,8-naphthyridine-2-carboxylate (1) | 1 |
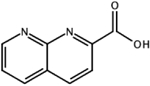 CID: 735156 |
1.1 | Inactive | pdb ID 6FP4 (2.5 Å) |
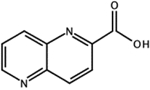 CID: 12426408 |
2.6 | n.d.ⱡ | n.d. | ||
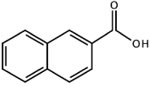 CID: 7123 |
1.1 | n.d. | n.d. | ||
| 1-(2-hydroxyethyl)piperazine (HEPE) derivatives | 2 |
CID: 67151 |
inactive | n.d. | pdb ID 6FMU (1.8 Å) |
| 3 |
CID: 7138432 |
>10 | n.d. | pdb ID 6FMZ (1.8 Å) |
|
| 4 |
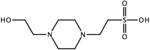 CID: 23831 |
3.3 | n.d. | pdb ID 6FTC (1.8 Å) |
|
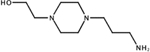 CID: 255283 |
0.76 | n.d. | n.d. | ||
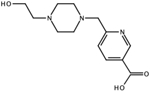 CID: 80159879 |
4.4 | n.d. | n.d. | ||
| Putative chimeras between 1 and HEPE | 5 |
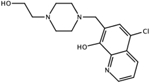 CID: 606080 |
0.12 | Inactive | Docking |
| 6 |
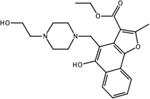 CID: 1523396 |
0.00006 | 0.001122 | Docking |
From Bioassays AID 588453 and AID 588456;
= not determined
The HEPE Subpocket.
We identify an additional subpocket exploitable as an anchoring site for fragment improvement at the level of the carboxylate of 1 where the doorstop cavity becomes wider and opens to the solvent. HEPES (4) binds in this region at a site close to but non-overlapping with 1. To rule out the possibility nonspecific binding of 4, structural studies were carried out with 1-(2-hydroxyethyl)piperazine (HEPE) analogs compounds 2 and 3. The structures of the complexes between SmTGR and 2 or 3 were solved at 1.8 Å. Compounds 2, 3, and 4 bind similarly and in the same pocket as 1 but to a different subsite (Fig. 2a and 2b and Supplementary Fig. 1). Compounds 2 and 3 make the same array of interactions and retain the same binding mode as 4 but are characterized by different chemical substituents on the piperazine rings. Their RSCC values are 0.78, 0.86 and 0.89, respectively. These lower values with respect to that of 1 likely reflect the multiple conformations adopted by their piperazine rings, as reflected by the higher B-factors of the ring atoms with respect to the 2-hydroxyethyl moiety. Independent of the chemical nature of the substituent at the N4 of the piperazine (N1 is the nitrogen bonded to the hydroxyethyl group), the HEPE derivatives bind in the same manner with the substituent at N4 positions not involved in the interactions with the protein. Indeed, 2 is characterized by a neutral tail at N4, while 3 has a longer aliphatic chain that ends with a primary amine and 4 bears a sulfate at its end (Table 1). In all the cases, the OH of the ethyl group at the N1 of the piperazine is involved in H-bonds with the O of the G483 main chain and with two water molecules, kept in place by the phenolic oxygen of Y479 and the O of G323 (Fig. 2a and 2b). Both the N1 and N4 of the piperazine can be protonated in a mutually exclusive manner at physiological pH (Sledz et al., 2009; Long et al., 2010) and the network of interactions present in the SmTGR structures are compatible with both protonation states. The water molecules kept in place by the main chain of G483 are at H-bond distances with the N1 of the piperazine (3.0 Å). The N4 atom of the piperazine is 4.4 Å from the ring centroid of F324, a distance compatible with a cation-pi interaction (Meyer et al., 2003). Structural superposition of SmTGR with the HEPE derivatives and NADPH (0.3 Å r.m.sd within 583 common CA atoms) reveals no evident steric clashes between the ligands except for the 3-TGR complex, where the primary amine of its long aliphatic chain is found at 2.6 Å from the oxygen of the NADPH ribose (not shown). The close vicinity of the HEPE binding site to that of NADPH and the inhibitor effect of different chemical nature of the substituent of the piperazine rings strongly suggests that ligand binding in this subpocket may interfere both with the binding of NADPH and with the structural change associated with it (Fig. 2c).
Bridging the Naphthyridine and HEPE Subpockets: Chimeras Between 1 and HEPE.
The consistent presence in SmTGR structures of HEPES and analogs in the same cavity but in a different site of that of 1 (Fig. 2c) suggested an anchoring site exploitable for drug development. HEPES is a weak inhibitor of SmTGR with an IC50 = 3.3 mM (Table 1) and the closely related analogs (the second series in Table 1) have IC50s ranging from 0.7 to more than 10 mM. Encouraged by the findings that (i) the binding modes of the HEPE moiety of compounds 2-4 to SmTGR are very similar in the X-ray structures (Fig. 2), ii) the inhibition effect of the HEPE derivatives (see Table 1) is in some way dependent on the different chemical nature of the substituents at the N4 of the piperazine, and (iii) 1 and HEPE are proximal and non-overlapping (Fig. 2c), a chimeric ligand combining the structural features of 1 and HEPE was constructed using the corresponding X-ray structures (see Supplementary Fig. 2). Compounds similar to the resulting putative compound were selected from the list of active hits from bioassay AID: 485364 and retested in the enzymatic assay to confirm their activity. Compound 5 and 6 (the third series in Table 1) were confirmed to be selective inhibitors of SmTGR with no or lower activity against mammalian hTrxR. These compounds exhibit significantly increased inhibition of SmTGR and worm-killing activity compared to those of the initial fragments. These compounds were advanced for structural analyses.
Docking Studies
Both compounds 5 and 6 did not co-crystallize with SmTGR likely due to their limited solubility in the crystallization conditions, a situation often encountered in hit-to-lead studies. To gain insights into their putative interaction with SmTGR, compounds 5 and 6 were docked to the X-ray structure of SmTGR co-crystallized with 1 using the docking module in the MOE modeling software (MOE 2016). First, the docking procedure was validated by re-docking compounds 1-4 to SmTGR. In all the cases, the docking procedure was able to reproduce the binding modes found in X-ray structures, indicating that the method is robust. The top docked poses of 5 and 6 are shown in Fig. 3a and 3b. Both compounds display binding modes similar to those of 1 and HEPE, i.e., with the piperazine ring close/identical to the position of the HEPE derivatives and with the aryl portions close to that of 1 (Fig. 3c). The quinoline ring of 5 and the aryl portion of 6 form hydrophobic interactions with the same residues involved in the binding of 1. Additionally, the chlorine atom of the former makes a repulsive polar interaction with the hydroxyl of the T471 side chain (Cl-O distance 3.0 Å), while the carbonyl oxygen atom of the carboxyethyl moiety of 6 forms hydrogen bond with the same residue. The aryl portion of 5 is somewhat smaller than that of 6, preventing the quinoline ring of 5 from penetrating as deeply in the doorstop pocket as the aryl of 6. In 6, the ethoxy group interacts with the sidechains of V473, Y296, and L441 and is overall bulkier than the chlorine atom in 5, resulting in an overall larger area of contact with the hydrophobic pocket at the bottom of the doorstop pocket in SmTGR. These differences are likely to account for the substantially better potency of 6 compared to that of 5. The HEPE moiety in both the compounds make interactions similar to those observed in the X-ray structures of the parent compounds. The MOE protonation procedure suggests protonation of the N1 piperazine ring, which is close to structurally conserved two water molecules interacting with a cluster of negatively charged carboxylic acid sidechains of D488, E487, and D325. Protonation of another basic nitrogen in the piperazine ring is less likely considering that the interaction energy with the negatively charged sidechains of D488, E487, and D325 would be much weaker in this case. Most importantly, when the top docked poses of 5 and 6 are superposed with that of SmTGR in complex with NADPH (pdb id: 2X99), their hydrophobic groups are again in steric clash with the open conformation of Y296, suggesting the same inhibition mechanism as that of 1.
Figure 3.

Docking of the compounds 5 and 6. a) Top docked pose of 5 (orange sticks). b) Top docked pose of 6 (pink sticks). c) Top poses of 5 (orange sticks) and the chimeric compound (Supplementary Fig. 2). The picture highlights the relative positions of the compounds in the doorstop pocket and the interacting side-chain residues as found in the x-ray structures of the initial fragments HEPE and 1.
Activity of Chimeric Compounds Against ex vivo Worms.
Compounds 1, 5, and 6 were tested for schistosomicidal activity. Adult worms were obtained from mice seven weeks after infection. Schistosomula were prepared by mechanical transformation of cercariae obtained from infected snails. Compounds were tested on adult worms and schistosomula at concentrations of 10 μM and 50 μM, respectively and show substantially improved worm-killing activity over 1. Against adult worms at 50 μM 6 killed 100% of the worms in four days, 5 killed 88% in five days and 1 killed 13% in five days (Fig. 4a). At 10 μM 5 killed 27%, 6 killed 9%, and 1 killed 0% of the worms in five days. At 50 μM both 5 and 6 killed all schistosomula in 24 hr and 1 killed 11% in five days. At 10 μM 5 killed 80%, 6 killed 100%, and 1 killed 0% in five days (Fig. 4b).
Figure 4.

Activity of compounds 1, 5, and 6 against ex vivo parasites. Compounds were added to cultures of A. adult worms and B. schistosomula, and worm survival was followed by monitoring worm movement. Compounds 1 (♦), 5 (■), and 6 (•) were tested at 50 μM against adult worms and 10 μM against schistosomula; control, untreated worms (▲). C. TGR activity in treated worms. Worms were cultured in the presence of 50 μM of 5 (■) or 6 (•). At the indicated times worms were collected and tested for residual TrxR activity (closed symbols) or GR activity (open symbols). All worms were alive when collected. Activity is expressed as compared to carrier (DMSO) alone treated worms cultured ex vivo for the same length of time.
Compounds 5 and 6 both kill both the adult worms and schistosomula. The effect on schistosomula is particularly relevant given that this earlier parasite developmental stage is less susceptible to praziquantel (Pica-Mattoccia and Cioli, 2004), which, at present, is the only clinically available drug against schistosomiasis. Treating worms with either 5 or 6 resulted in significant inhibition of both TrxR (DTNB reduction) and GR (GSH disulfide reduction) activities in worms (Fig. 4c). Since TGR provides all of both TrxR and GR activities in the worms, this indicates that the worm killing activity of 5 and 6 was due at least part to inhibition of TGR. Although IC50s of 5 and 6 are very different, the latter is 2000-fold more potent than the former, they both affect worm survival in the low micromolar range. It should be noted that these compounds represent only the first iteration of active and highly promising HTS hits. In the course of the experiments with worms, we noted that compound 6 initially precipitated in the culture medium then slowly dissolved over the following hours. Schistosomes possess a nonspecific esterase activity localized in the tegument (Doenhoff et al. 1988). We speculate that this activity may transform 6 in the more soluble but less active or less bioavailable carboxylate derivative, thus accounting for the comparable ex vivo activity of 5 and 6. Additional SAR studies would be necessary not only to optimize activity and solubility of these compounds but also to exclude factors such as potential metabolic instability and off-target activity.
Shape and Electrostatic Features of the Doorstop Pocket in SmTGR and Comparison with Other Drug Targets of the FAD/NAD Linked Reductase Family.
The two subpockets of 1 and HEPE can be virtually recognized in the doorstop pocket if one considers the narrow gap approximately positioned at the level of the carbon-carbon bond that joins the two rings of the naphthyridine. In this region the pocket has a maximum diameter of about 8 Å measured between the two furthest amino acids (Fig. 2c). The more solvent exposed HEPE subpocket is wider and hosts the carboxylate of 1 and the HEPE derivatives. The naphthyridine subpocket, where the unsubstituted aromatic ring of naphthyridine lies, is mainly hydrophobic. It is constituted of the entire aromatic ring of Y296 and ends with hydrophobic side chains of I570 and P572 belonging to the other subunit of the physiological dimer. This latter cavity is also lined by the terminal side chain atoms of K162 and E300, two residues involved in the stabilization of the terminal amide of bound NADPH (Fig. 2c; Angelucci et al., 2010).
We analyzed conservation of the doorstop pocket residues in SmTGR homologues that (i) have an aromatic side chain at the re-face of the FAD known to swing upon NADPH entry, (ii) are recognized as potential or validated drug targets for human diseases, and (iii) whose structure is known (Supplementary Fig. 3). We carried out the analysis of the side chain residues (omitting the residues contributing to the main chain) present in this secondary site, which reveal a degree of conservation ranging from 48% (for TrypR from Trypanosoma brucei and T. cruzi) to 96% (for TGR from S. japonicum) sequence identity. The closest related human homolog, i.e., HsTrxR1, displays 74% sequence identity in the pocket residues. Notably, the charge distribution and shape of HsTrxR1 in this site is remarkably different with respect to SmTGR due to the presence of 4 acidic residues, i.e. E337, D338, E341, E368, in place of A436, G437, Q440, S467 (Fig. 5 and Supplementary Fig. 3). Three of these four mutations (G437 is conserved) are also observed in human mitochondrial TrxR2 (NP_006431.2) and Rattus norvegicus TrxR (AAD13801.1; data not shown). This pocket in HsGR has 56% amino acid conservation with that of SmTGR. The absence of a proline residue in HsGR changes the conformation of the loop responsible for binding the carboxylate of 1 and the presence of a lysine results in a partial positive charge to the pocket (Fig. 5).
Figure 5.

Coulombic surfaces of SmTGR (at the top), human hTrxR1 (HsTrxR1, in the middle; pdb ID: 2CFY) and human GR (HsGR, at the bottom; pdb code: 3DK8) around the re-face of the FAD (in yellow sticks) calculated with the program CHIMERA are shown. The surface of the conserved tyrosine residue (Y296 in SmTGR, Y200 in HsTrxR 1 and Y197 in HsGR) is indicated in the three enzymes occupying the same relative position, i.e., pointing with its phenolic oxygen towards the isoalloxazine of the FAD and being at the boundary between the NADPH and the doorstop pocket. The NADPH binding sites on the right of the tyrosine are characterized by an overall positive charge (blue), necessary to bind the phosphates of the cofactor. The doorstop pockets in SmTGR, HsTrxR and HsGR, on the left, present different electrostatic and shape features.
In view of the above model, we speculate that is possible to develop selective inhibitors of enzymes of the FAD/NAD linked reductase family by specifically targeting the doorstop pocket. The aromatic side-chain swing is a well-known mechanism (Voet & Voet, 2010). It is conserved in a majority FAD-NAD linked reductase family members in which an aromatic side-chain residue with its ring perpendicular to the re-face of the isoalloxazine of the FAD is present (Arnér et al., 2009 and Biterova et al., 2005 for hTrxR and Berkholz et al., 2008 for GR). The pocket is present in homologous enzymes considered to be crucial drug-targets against human cancers and infectious diseases (Supplementary Fig. 3), but it displays different chemical features due to differences in amino acid composition. This is consistent with the lower degree of conservation in the secondary site with respect to the orthosteric/primary sites as observed by Ludlov and collaborators (2015). Here we show that the electrostatic features of the NADPH binding site are maintained in SmTGR, HsTrxR and HsGR, while there are significant differences between doorstop pockets (Fig. 5). Therefore, 1, 5, and 6 show high selectivity for SmTGR over hTrxR. The undetectable inhibition of hTrxR by 1 and 5 may be due to a mismatch with the electrostatic potential at the doorstop pocket of the mammalian enzyme. The negative surface formed by E337, D338, E341, E368 in hTrxR (numbering according to the HsTrxR 1) should repel the negatively charged carboxylate of 1 and the electronegative chlorine and the ester oxygen atoms of 5 and 6, respectively. Furthermore, 1 has no detectable activity against hGR (results not shown), likely due to a different turn of the loop necessary to bind the carboxylate of the inhibitor resulting from the absence of a proline residue (P439) in SmTGR (Supplementary Fig. 3).
Model for Inhibition.
Compound 1 is an inhibitor of the enzyme machinery working as a doorstop for NADPH entry: it prevents Y296 from rotating, a process necessary for NADPH binding and enzyme reduction. Therefore, 1 inhibits SmTGR by stabilizing a protein conformation whose affinity for NADPH is greatly reduced. In this sense it can be considered similar to an allosteric effector. The effect of 1 on the apparent KM of NADPH is complete and unequivocal proof of functional competition between the two compounds, consistent with the structural information. The SmTGR catalytic cycle can be divided into reductive and oxidative half reactions (Angelucci et al., 2010; Huang et al., 2011; Fig. 6). In the reductive half-reaction, electrons from NADPH are transferred to the isoalloxazine of FAD and then used to reduce the cysteine couple lying at its si-face, i.e. C154 and C159, forming a charge transfer complex between C159 and FAD and leading to the so-called EH2 form of the enzyme (Angelucci et al., 2010; Berkholz et al., 2008). Then another NADPH molecule transfers two more electrons to the enzyme, leading to a reduction of the mobile Sec-containing carboxyl-terminal arm and giving rise to the EH4 form. This is able to reduce the downstream substrates in the oxidative half-reaction, i.e., oxidized Trx and the Cys couple (C28 and C31) in the Grx domain, where reduction of GSH disulfide occurs (Huang et al., 2011). Y296 orchestrates NADPH entry and thus enzyme activity: in the oxidized apo structure, the aromatic ring of Y296 is perpendicular to the re-face of the isoalloxazine ring (in closed conformation). In this position it shields the isoalloxazine from the solvent, possibly, preventing unwanted loss of electrons from the flavin when the enzyme is reduced (Rice et al., 1984). Upon NADPH binding, Y296 changes conformation in order to make room for the nicotinamide moiety of NADPH, swinging its aromatic ring towards the doorstop cavity and lying almost parallel to those of nicotinamide and FAD (in open conformation), thus allowing electron transfer (Fig. 1c and Fig. 6).
Figure 6.

Cartoon representation showing the proposed inhibition mechanism as assessed by both the structural superposition between the relative SmTGR complexes and steady state experiments. In one subunit of the dimeric unliganded TGR (in gray, pdb ID: 2V6O) two pockets, one on the left (the doorstop pocket) and one on the right of Y296 (the NADPH pocket), facing the isoalloxazine ring of the FAD (in yellow spheres) are present. Upon NADPH binding (NADPH is in magenta spheres, pdb ID of the TGR-NADPH complex: 2X99) the right pocket is filled by the cofactor, while the doorstop pocket completely disappears due to a conformational switch of Y296, necessary to make room to the nicotinamide moiety of the reductant. In the presence of 1 (in green spheres, at the bottom of the figure) the doorstop pocket is occupied hampering the tyrosine switch and thus enzyme reduction and turn-over. Several intermediates of the oxidative half reaction are omitted for simplicity (See Angelucci et al., 2010 and Prast-Nielsen et al., 2011 for a comprehensive description of the TGR mechanism).
Prospective.
Through an integrated approach of structural and functional studies, we have identified a new secondary site and elucidated its functional role in the binding of inhibitors of SmTGR. Our studies provide a solid basis to undertake a structure-based drug discovery approach against this challenging and crucial parasite target. SmTGR is particularly suitable to carry out hit-to-lead studies in comparison to other members of the hTrxR sub-family, given the possibility to solve its structures at ≈ 1.6–2.5Å of resolution (Deller et al., 2015; Angelucci et al., 2010). Interestingly, other members of the FAD/NAD linked reductase family share structural features similar to those described here for SmTGR, suggesting that it is possible to exploit the same strategy for their inhibition in order to generate new therapies for other important human diseases.
Methods:
Chemicals and Reagents.
Tris(Hydroxymethyl)aminomethane (Tris-HCl), 2-Hydroxy-3-(4-(2-hydroxyethyl)piperazin-1-yl)propane-1-sulfonic acid (Heppso), 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), KI, 1,4-Dithiothreitol (DTT), 1,8-naphthyridine-2-carboxylate, 2-[4-(4-aminopropyl)piperazin-1-yl]ethan-1-ol, β-mercaptoethanol, dimethyl sulfoxide, 1,4-bis(2-hydroxyethyl)piperazine, 2-[4-(4-aminobutyl)piperazin-1-yl]ethan-1-ol, 6-((4-(2-Hydroxyethyl)piperazin-1-yl)methyl)nicotinic acid (CID: 80159879), 1,5-Naphthyridine-2-carboxylic acid (CID: 12426408), and 2-Naphthoic acid (CID: 7123) were from Sigma-Aldrich. Nicotinamide adenine dinucleotide phosphate reduced (NADPH) was from Sigma and Cayman Chemicals. Polyethylene glycol 3350 (PEG 3350), potassium thiocyanate, bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane (bis-Tris) were from Molecular Dimension. 5-chloro-7-((4-(2-hydroxyethyl)piperazin-1-yl)methyl)quinolin-8-ol (CID: 606080, compound 5) and ethyl 5-hydroxy-4-{[4-(2-hydroxyethyl)piperazin-1-yl]methyl}−2-methylnaphtho[1,2-b]furan-3-carboxylate (CID: 1523396, compound 6) were purchased from Enamine.
Protein expression and purification.
Three different protein preparations have been used in this study. The untagged Sec597Cys mutant was prepared as described (Kuntz et al. 2007; Huang et al, 2011; Angelucci et al., 2010). Untagged, wild type SmTGR (SmTGRwt) was produced by cloning the wild-type TGR sequence into pABC2-rTRSUAG replacing rat TrxR, expressed in C321.ΔA, and purified as described (Cheng and Arnér, 2017). Six-His-tagged wild type SmTGR was prepared by transferring the protein coding sequence and selenocysteine insertion sequence (SECIS) from a previously described system in pET-24a (Kuntz et al., 2007) into pET-100 (Invitrogen). 6-His-SmTGRwt protein was expressed in BL21 cells co-transformed with pSUABC as described (Kuntz et al., 2007). Bacterial cell pellets were resuspended in 50 mM potassium phosphate (pH 7.8), 500 mM NaCl (buffer 1) with 30 mM imidazole, 1 mg/ml lysozyme, 100 μM FAD, and 1 mM phenylmethanesulfonyl fluoride (PMSF) added. The SmTGRwt protein was purified by metal affinity chromatography (HiTrap® chelating, GE Healthcare) using buffer 1 with, sequentially, 10 mM, 100 mM, and 500 mM imidazole. The purified protein was desalted using a HiTrap® (GE Healthcare) desalting column and then stored in 100 mM NaCl, 50 mM potassium phosphate (pH 7.4) and 10 mM EDTA with 50% glycerol. Protein purification was assessed by polyacrylamide electrophoresis and quantitated by flavin absorbance at 462 nm (ε = 11,300 M−1 • cm−1).
Compound selection.
Approximately 1000 compounds identified in the qHTS bioassay AID: 485364 with inhibition activity utilizing a direct TGR assay (Lea et al, 2008) and characterized by a molecular weight <300 Da were considered. Putative inhibitors were counter screened against human glutathione reductase (hGR) in qHTS format (Rai et al, 2009) to eliminate hits that showed any activity against hGR at 57 μM. Commercially available compounds with the highest ligand efficiency (Hopkins et al., 2004) were tested in the DTNB assay (Kuntz et al., 2007) to confirm their initial activity found in the qHTS. Selected soluble active hits were then used both in co-crystallization and soaking experiments at 25 mM concentration, with and without NADPH. The HEPE derivatives, reported in Table 1, were selected due to the presence of the HEPE moiety in their scaffold and commercial availability. Compounds 5 and 6, that combine features of HEPE and 1, were identified by similarity searches among the active hits of the qHTS using as a search model a chimeric compound built through the fusion of 1,4-bis(2-hydroxyethyl)piperazine and 1 (Supplementary Fig. 2), as found in the relative X-ray structures. Similarity search was carried out in MOE 2016.0802 using fingerprint MACCS structural keys (Bit packed). The Tanimoto similarity overlap was set as 75.
Crystallization of SmTGR in Complex with 1,8-naphthyridine-2 carboxylate (1) and with the HEPE-Derivatives (compounds 2, 3, and 4).
The two SmTGR protein variants (SmTGRwt and Sec597Cys SmTGR) were equilibrated in 20 mM Tris, 50 mM NaCl at pH7.4, concentrated by ultra-filtration devices to about 4 mg/mL and used for crystallization studies. All the compounds used in the crystallization experiments were dissolved in DMSO at a concentration of 250 mM. Crystals of SmTGRwt grew in 48 hours mixing equal volumes of protein solution and of HEPPSO pH 7.2, 20% PEG 3350, 0.2 M KI and 5 mM DTT. Soaking experiments were carried out with 1 at a concentration of 25 mM for 72 hours, in the absence of NADPH. Sec597Cys SmTGR was co-crystallized with 2 and 3 according to the following sitting drop vapor diffusion protocol: 2 μL of the compound stock solution was added to 18 μL of SmTGR solutions, resulting in a final concentration of 25 mM; protein–inhibitor complexes were crystallized mixing equal amounts of this protein mixture and of a well solution containing 0.1 M bis-Tris pH 7.0, 20% PEG 3350, 0.2 M KI or 0.2 M potassium thiocyanate, and 5 mM DTT. Crystals of the Sec597Cys SmTGR-HEPES complex were obtained mixing equal volumes of the protein solution with 100 mM HEPES pH 7.2, 20% of PEG 3350 and 0.2M KI plus 5mM DTT. All the crystals were cryoprotected with the relative well solution increasing the concentration of PEG 3350 to 35%.
Data Collection, Processing, Model Building and Refinement.
Diffraction data from different crystals were collected at ELETTRA (Trieste, Italy) and processed with XDS (Kabsch, 2010). Crystals belong to the C2 space group and have one subunit of the physiological dimer in the asymmetric unit. The 3D structures were solved by molecular replacement using the program PHASER (McCoy et al., 2007) and as a search model the structure of oxidized SmTGR (pdb code: 2V6O; Angelucci et al., 2008). The atomic model was refined with PHENIX (Echols et al., 2014) and fit into electron density maps using Coot (Emsley and Cowtan; 2004). The structures of SmTGR in complex with 1, 2, 3, and 4 (See Table 1) were solved at 2.5, 1.8, 1.8 and 1.8 Å of resolution, respectively. Data collection and refinement statistics are reported in Supplementary Table 1. Coordinates of SmTGR-1, SmTGR-1,4-bis(2-hydroxyethyl)piperazine (compound 2), SmTGR-2-[4-(4-aminobutyl)piperazin-1-yl]ethanol (compound 3) and SmTGR-HEPES (compound 4), together with structure factors, have been deposited in the Protein Data Bank under accession numbers 6FP4, 6FMZ, 6FMU and 6FTC, respectively.
Docking Procedures for Compound 5 and 6.
All molecular modeling studies were performed in Molecular Operating Environment (MOE), [MOE 2016]. The binding sites were prepared using the X-ray model of TGR presented in this paper. The TGR protein was subjected to the “structure preparation” procedure. Hydrogen atoms were added using the Protonate 3D algorithm. The energy of the resulting structure was minimized utilizing AMBER12EHT forcefield (Case et al, 2012; Gerber et al, 1995) The protein was minimized using implicit R-field solvation (ε=80) until the root mean square gradient was less than 0.001 kcal/mol/Å2. The ligands were assigned MMFF94x charges and minimized using the MMFF94x forcefield and implicit R-field solvation (ε=80) until the RMS gradient was less than 0.001 kcal/mol/Å2. The MOE docking module “Dock” was used for docking/scoring using the default parameters and settings. Docking was performed using the “induced fit” algorithms, “Triangle Matcher” for placement, “London dG” for scoring of the binding poses after placement, and “GBVI/WSA dG” for rescoring of the resulting poses.
Enzymatic Assays.
Enzymatic assays were carried out using 6-His-tagged SmTGRwt. TGR reaction buffer was 0.1 M potassium phosphate pH 7.4 and 10 mM EDTA. Enzyme (20 nM) was incubated for 10 min with NADPH (100 μM) and compound (or DMSO control) at various concentrations, after which the reaction was started by addition of a second aliquot of NADPH and 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) (3 mM). Reaction progress for 5 minutes was monitored by 2-thio-5-nitrobenzoic acid production at 412 nm on a Thermo Multiskan Spectrum plate reader. Assays were done in triplicate.
Ex vivo Experiments with Adult Worms and Schistosomula.
Adult worms were isolated from infected mice as described (Lewis 1998) and cultured in RPMI medium + 10% fetal calf serum, 10 mM glutamine and 1X penicillin/streptomycin for 24 hr before compound addition. Larval worms were prepared by mechanical transformation and Percoll® gradient isolation from cercariae isolated from infected Biomphalaria glabrata as described (Lewis 1998). Larval worms were cultured in M-199 + 10% fetal calf serum for 24 hr before addition of compounds. Worms were observed daily and scored as dead when no movement was seen.
Inhibition of TGR in treated, ex vivo worms.
Adult worms freshly perfused from infected mice (NIH-Swiss) were cultured as described overnight. Compounds were dissolved in DMSO (10 mM) and added to 50 μM to the cultures. When appropriate, media were replaced everyday with fresh compound added at 50 μM. Control worms were treated with equal volume of DMSO and collected at the same time points. Approximately 30 worms were used for each time point and drug/control. The worms were washed with TGR reaction buffer and homogenized by pestle motor mixer. The homogenates were centrifuged and the protein concentrations of supernatants were determined (Pierce 660 nm Protein Assay Reagent, ThermoFisher) and compared to bovine serum albumin standard. Enzyme activities of supernatants were tested in TGR reaction buffer for TrxR activity (100 μM NADPH, 3 mM DTNB) and GR activity (100 μM NADPH, 100 μM GSH disulfide) following 5’-thio-[2-nitrobenzoic acid] production at 412 nm or NADPH consumption at 340 nm, respectively. Assays were done in triplicate. Specific activities (rate/mg protein) were determined.
Structural and amino acid composition analysis of the doorstep pocket in SmTGR.
Structure visualization and analyses were performed using the programs COOT (Emsley and Cowtan, 2004), PyMOL (The PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC), and CHIMERA (Pettersen et al., 2004). Residues within 5 Å contacting compounds 1, 2, and 3 were identified by the program CONTACT of the CCP4 suite (Winn et al., 2011). Analysis of the pockets was carried out taking advantage of the CAST-p analysis (Dundas et al., 2006). Residues not contributing to the doorstop pocket but recognized by the CAST-p server as being part of it, as those interacting with NADPH were manually removed after visual inspection of the SmTGR structure, except K162 and E300 that lie at the bottom of the doorstop pocket. The output of this analysis is represented in Figure 1 and Supplementary Fig. 4.
Comparison and analysis of the doorstop pocket within relevant drug targets belonging to the FAD/NAD linked reductase family.
After identifying the side-chain residues contributing to the doorstop pocket in the SmTGR sequence (GenBank: AAK85233.1), this latter was pairwise aligned to each target sequence, whose structure is present in protein data bank using the BLAST (Altschul et al., 1990) graphical interface at the http://blast.ncbi.nlm.nih.gov/Blast.cgi. After selection of the protein sequences of potential and validated drug targets belonging to the FAD/NAD linked reductase family, as reported in literature (see Supplementary Fig. 4), a multiple alignment using the ClustalW algorithm (Thompson et al., 1994) was carried out. The multiple sequence alignment was restricted to family members that are characterized by an aromatic side chain at the re-face of the FAD that are known to swing upon NADPH binding, identified upon structural superposition with SmTGR using the program COOT (Emsley and Cowtan, 2004). Coulombic Surfaces of SmTGR and HsTrxR 1 were calculated using the program CHIMERA, (coloring parameters: ε = 4, thresholds ± 10 kcal/mole at 298 K).
Supplementary Material
Acknowledgements
This work was supported in part by NIH/NIAID grant n°: 1R21AI127635–01. This study was partially supported by MIUR PRIN 20154JRJPP to A.B. We gratefully acknowledge Elettra synchrotron (Trieste, Italy) for granting beam time and for data collection support. Biomphalaria glabrata snails infected with S. mansoni were provided by the NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201000005I for distribution through BEI Resources.
Abbreviations:
- FAD
flavin adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate reduced
- HEPE
1-(2-hydroxyethyl)piperazine functional group
- GR
glutathione reductase
- TrypR
trypanothione reductase
- hTrxR
high molecular weight mammalian thioredoxin reductase
- TGR
thioredoxin glutathione reductase
- GSH
glutathione
- Trx
thioredoxin
- Sec
selenocysteine
- Grx
glutaredoxin
- qHTS
quantitative high throughput screening
Footnotes
This study was approved by the Institutional Animal Care and Use Committee of Rush University Medical Center (17–053; Department of Health and Human Services animal welfare assurance number A-3120-01).
References
- Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol Biochem Parasitol. 2002. April 30;121(1):129–39. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990. October 5;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Glutathione reductase and thioredoxin reductase at the crossroad: the structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins. 2008. August 15;72(3):936–45. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. J Biol Chem. 2009. October 16;284(42):28977–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Dimastrogiovanni D, Boumis G, Brunori M, Miele AE, Saccoccia F, Bellelli A. Mapping the catalytic cycle of Schistosoma mansoni thioredoxin glutathione reductase by X-ray crystallography. J Biol Chem. 2010. October 15;285(42):32557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnér ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009, 1790, 495–526 [DOI] [PubMed] [Google Scholar]
- Baiocco P, Colotti G, Franceschini S, Ilari A. Molecular basis of antimony treatment in leishmaniasis. J Med Chem. 2009, 52, 2603–12 [DOI] [PubMed] [Google Scholar]
- Bellelli A, Carey J. Reversible Ligand Binding: Theory and Experiment Ed. Wiley John and sons inc., 2018, Hoboken, NJ, USA. [Google Scholar]
- Berkholz DS, Faber HR, Savvides SN, Karplus PA. Catalytic cycle of human glutathione reductase near 1 A resolution. J Mol Biol. 2008, 382, 371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010. July 22;53(14):5061–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc Natl Acad Sci U S A. 2005, 102, 15018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumis G, Angelucci F, Bellelli A, Brunori M, Dimastrogiovanni D, Miele AE. Structural and functional characterization of Schistosoma mansoni Thioredoxin. Protein Sci. 2011, 20, 1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumis G, Giardina G, Angelucci F, Bellelli A, Brunori M, Dimastrogiovanni D, Saccoccia F, Miele AE. Crystal structure of Plasmodium falciparum thioredoxin reductase, a validated drug target. Biochem Biophys Res Commun. 2012, 425, 806–11. [DOI] [PubMed] [Google Scholar]
- Case DA, T. A. D., Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, et al. AMBER 12, University of California, San Francisco, 2012. [Google Scholar]
- Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arnér ES. Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J Biol Chem. 2004, 279, 2583–92. [DOI] [PubMed] [Google Scholar]
- Chen GP, Raybuck S, Ziegler DM. Flavoenzymes inhibited by indomethacin. Drug Metabol Drug Interact. 1994;11(2):153–60. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Arnér ES. Selenocysteine Insertion at a Predefined UAG Codon in a Release Factor 1 (RF1)-depleted Escherichia coli Host Strain Bypasses Species Barriers in Recombinant Selenoprotein Translation. J Biol Chem. 2017. March 31;292(13):5476–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller MC, Rupp B. Models of protein-ligand crystal structures: trust, but verify. J Comput Aided Mol Des. 2015, 29, 817–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Santo R Inhibiting the HIV integration process: past, present, and the future. J Med Chem. 2014, 57, 539–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenhoff MJ, Modha J, Lambertucci JR. Anti-schistosome chemotherapy enhanced by antibodies specific for a parasite esterase. Immunology. 1988. 65, 507–10 [PMC free article] [PubMed] [Google Scholar]
- Drwal MN, Jacquemard C, Perez C, Desaphy J, Kellenberger E. Do Fragments and Crystallization Additives Bind Similarly to Drug-like Ligands? J Chem Inf Model. 2017, 57, 1197–1209. [DOI] [PubMed] [Google Scholar]
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34(Web Server issue):W116–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols N, Moriarty NW, Klei HE, Afonine PV, Bunkóczi G, Headd JJ, McCoy AJ, Oeffner RD, Read RJ, Terwilliger TC, Adams PD. Automating crystallographic structure solution and refinement of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2014, 70(Pt 1), 144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004; 60, 2126–32. [DOI] [PubMed] [Google Scholar]
- Gerber PR; Muller K Mab, a Generally Applicable Molecular-Force Field for Structure Modeling in Medicinal Chemistry. Journal of computer-aided molecular design 1995, 9, 251–268. [DOI] [PubMed] [Google Scholar]
- Gromer S, Arscott LD, Williams CH Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998. August 7;273(32):20096–101. [DOI] [PubMed] [Google Scholar]
- Harbut MB, Vilchèze C, Luo X, Hensler ME, Guo H, Yang B, Chatterjee AK, Nizet V, Jacobs WR Jr, Schultz PG, Wang F. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci U S A. 2015. April 7;112(14):4453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004, 14, 706–15 [DOI] [PubMed] [Google Scholar]
- Huang HH, Day L, Cass CL, Ballou DP, Williams CH Jr, Williams DL. Investigations of the catalytic mechanism of thioredoxin glutathione reductase from Schistosoma mansoni. Biochemistry. 2011; 50, 5870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9, 430–1. [DOI] [PubMed] [Google Scholar]
- Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006. August 1;103(31):11473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea WA, Jadhav A, Rai G, Sayed AA, Cass CL, Inglese J, Williams DL, Austin CP, Simeonov A. A 1,536-well-based kinetic HTS assay for inhibitors of Schistosoma mansoni thioredoxin glutathione reductase. Assay Drug Dev Technol. 2008. August;6(4):551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W XDS. Acta Cryst. D66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauth-Siegel RL, Schöneck R. Flavoprotein structure and mechanism. 5. Trypanothione reductase and lipoamide dehydrogenase as targets for a structure-based drug design. FASEB J. 1995. ;9, 1138–46. [DOI] [PubMed] [Google Scholar]
- Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ES, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med. 2007. June;4(6):e206 Erratum in: PLoS Med. 2007; 4 :e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. Functional and structural perspectives on allosteric modulation of GPCRs. Curr Opin Cell Biol. 2014; 27:94–101. [DOI] [PubMed] [Google Scholar]
- Lewis F (1998) Schistosomiasis In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current protocols in immunology. New York: John Wiley & Sons; pp. Suppl. 28: 19.1.1–19.1.28. [Google Scholar]
- Lo YC, Ko TP, Su WC, Su TL, Wang AH. Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1. J Inorg Biochem. 2009, 103, 1082–92. [DOI] [PubMed] [Google Scholar]
- Long RD, Hilliard NP, Chhatre SA, Timofeeva TV, Yakovenko AA, Dei DK, Mensah EA. Comparison of zwitterionic N-alkylaminomethanesulfonic acids to related compounds in the Good buffer series. Beilstein J Org Chem. 2010; 6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H. Detection of secondary binding sites in proteins using fragment screening. Proc Natl Acad Sci U S A. 2015, 112, 15910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafud AC, Ferreira LG, Mascarenhas YP, Andricopulo AD, de Moraes J. Discovery of Novel Antischistosomal Agents by Molecular Modeling Approaches. Trends Parasitol. 2016, 32, 874–886. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, & Read RJ (2007) Phaser crystallographic software. J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed Engl 2003; 42:1210–1250. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 20160802; Chemical Computing Group Inc., 1010 Sherbooke St West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2017. [Google Scholar]
- Pearce NM, Krojer T, Bradley AR, Collins P, Nowak RP, Talon R, Marsden BD, Kelm S, Shi J, Deane CM, von Delft F. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density Nat Commun. 2017, 8:15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004. October;25(13):1605–12. [DOI] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Cioli D. Sex- and stage-related sensitivity of Schistosoma mansoni to in vivo and in vitro praziquantel treatment. Int J Parasitol. 2004, 34(4):527–33. [DOI] [PubMed] [Google Scholar]
- Prast-Nielsen S, Huang H-H, Williams DL. Thioredoxin glutathione reductase: its role in redox biology and potential as a target for drugs against neglected diseases. Biochimica et Biophysica Acta. 2011; 1810, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arnér ES, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis. J Med Chem. 2009. October 22;52(20):6474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DW, Schulz GE, Guest JR. Structural relationship between glutathione reductase and lipoamide dehydrogenase. J Mol Biol. 1984, 174, 483–96. [DOI] [PubMed] [Google Scholar]
- Roder C, Thomson MJ. Auranofin: repurposing an old drug for a golden new age. Drugs R D. 2015. 15, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccoccia F, Angelucci F, Boumis G, Carotti D, Desiato G, Miele AE, Bellelli A. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci. 2014;15(6):621–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat Med. 2008; 14, 407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, Minor T, Chruszcz M. Redetermination of 2-[4-(2-hydroxy-ethyl)piperazin-1-ium-1-yl]ethanesul-fonate at 100 K. Acta Crystallogr Sect E Struct Rep Online. 2009; 65, o3027–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford WC, Peng X, Olofsson MH, Zhang X, Luci DK, Lu L, Cheng Q, Trésaugues L, Dexheimer TS, Coussens NP, Augsten M, Ahlzén HM, Orwar O, Östman A, Stone-Elander S, Maloney DJ, Jadhav A, Simeonov A, Linder S, Arnér ESJ. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci Transl Med. 2018. February 14;10(428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T, Koellner G. Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol. 2001. 305, 535–57. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994. November 11;22(22):4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale N, Gouveia MJ, Rinaldi G, Brindley PJ, Gärtner F, Correia da Costa JM. Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob Agents Chemother. 2017. April 24;61(5). pii: e02582–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet D, Voet JG, Biochemistry, 4th Ed, Wiley, 2010. [Google Scholar]
- Williams CH Jr. (1992) in Chemistry and Biochemistry of Flavoenzymes (Müller F, Ed.) Vol. III, pp 121–211, CRC Press, Boca Raton, FL. [Google Scholar]
- Williams DL, Bonilla M, Gladyshev VN, Salinas G. Thioredoxin glutathione reductase-dependent redox networks in platyhelminth parasites. Antioxid Redox Signal. 2013, 19, 735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011; 67, 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhang J, Peng S, Liu R, Li X, Hou Y, Han X, Fang J. Thioredoxin reductase inhibitors: a patent review. Expert Opin Ther Pat. 2017, 27, 547–556. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li X, Han X, Liu R, Fang J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol Sci. 2017, 38, 794–808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.