

Case Report
We report a four‐generation family consisting of five patients (three men and two women, among them one was deceased) and 13 healthy controls. The proband (Fig. 1) was a 5‐year‐old boy. From the time of his birth, his hair, eyebrows, and eyelashes were totally absent, and his fingernails and toenails were thick with yellow discoloration and demonstrated distal onycholysis (Fig. 2a–c). He had decreased cold intolerance during the winter months and had recurrent nail infections. Sweating, teeth, ears, eyes, and mucosa were normal. The other affected individuals were adults with similar symptoms and additionally showed hyperkeratosis of the palms with a cobblestone surface (Fig. 2d and e). The mode of inheritance was autosomal dominant. Genomic DNA was isolated from peripheral blood, and the candidate genes of GJB6 and GJB2 were sequenced. The GJB6 gene sequencing results revealed that all the affected members harbored a heterozygous base mutation from C to T in 263 (c.263C>T), causing an amino acid substitution from alanine to valine in 88 (p. A88V) (Fig. 3a), which was not found in healthy controls. The G11R, V37E, D50N, and N14S mutations of GJB6 were not found. The GJB2 sequencing showed that the affected members had no F191L mutation but held a heterozygous base mutation from G to A in 79 (c.79G>A), leading to the replacement of valine by isoleucine (p.V27I) (Fig. 3b). In addition, two family members (II 1 and II 3) held a heterozygous missense mutation p. V37I (c. 109 G>A) in GJB2 gene.
Figure 1.

Pedigree chart. Normal individuals are shown as clear circles (females) or squares (males), and affected individuals are shown as solid symbols. The arrow indicates the proband. I, II, III, IV: generation numbers
Figure 2.

Clinical symptoms of the affected individuals. a, b, and c from IV 1 (proband). (a) Alopecia, complete absence of body, eyebrows, and eyelashes. (b) Fingernails were short and thickened, discolored, and demonstrated distal onycholysis; (c) Short, thickened, and brittle toenails; d and e from III 1. (d) Hyperkeratosis of the palms with a cobblestone surface; (e) Short, thickened, and brittle toenails
Figure 3.

Molecular genetic analysis of GJB6 and GJB2 from proband. (a) Heterozygous missense mutation c.263C>T of GJB6 that predicts amino acid change A88V. (b) Heterozygous missense mutation 79G>A of GJB2 predicting the amino acid change V27I
Discussion
The clinical symptoms of the affected individuals in the family fulfilled the clinical criterion for the diagnosis of hidrotic ectodermal dysplasia (HED), also known as Clouston syndrome (CS; MIM 129500), which was first described in 1895 and later recorded in detail by Clouston in 1929.1 It is a rare autosomal dominant disease.2 It is characterized by a triad of major clinical signs: nail dystrophy, partial to total alopecia capitis, and palmoplantar hyperkeratosis. Nail abnormalities include thickening, brittleness, discoloration, splitting, and onycholysis. Sweat glands and teeth of patients with HED are usually normal. Since 2000, four mutations in GJB6 gene, which cluster at chromosome 13q11 and encode gap junction protein connexin 30, have been reported to cause HED: G11R, V37E, A88V, and D50N3, 4, 5, 6 (Table 1). Connexin 30 contains two extracellular domains, three cytoplasmic domains, and four hydrophobic transmembrane domains (M1–M4) (Fig. 4). The mutation A88V, introducing a highly hydrophobic residue in the transmembrane M2 domain, may change the polarity of connexin channels and affect communication between cells3 or induce CX30 apoptosis through an endoplasmic reticulum‐independent mechanism.7 A mouse model for HED carrying GJB6 mutation A88V revealed hyperproliferative and enlarged sebaceous glands as well as a mild palmoplantar hyperkeratosis.8 A88V was only reported in two Chinese families.9, 10 Here, we report another one.
Table 1.
Since 2000, the detected gene mutations associated with HED
| GJB6 | GJB2 | Number of family | Affected members | Ethnic group | Year and reference |
|---|---|---|---|---|---|
| G11R | 2 | 22 | French | 20003, 21 | |
| 2 | More than 3 | Moroccan, Dutch | 20036 | ||
| 1 | 18 | Chinese | 20032 | ||
| 1 | 8 | Chinese | 200922 | ||
| 1 | Lebanese‐German | 201323 | |||
| 1 | 8 | Chinese | 201324 | ||
| 1 | 2 | Chinese | 201325 | ||
| 1 | 1 | Chinese | 201426 | ||
| 1 | 17 | Chinese | 201627 | ||
| 1 | 1 | 201628 | |||
| 1 | 2 | Indian | 201629 | ||
| 1 | 19 | Taiwanese | 201530 | ||
| V37E | 1 | 1 | Scottish | 20024 | |
| V27I | 1 | 1 with deafness | 200419 | ||
| D50N | 1 | 2 | Ashkenazi Jews | 20085 | |
| A88V | 3 | 3 | Indian, Malaysian, Walsh | 20003, 21 | |
| 1 | 1 | Dutch | 20036 | ||
| 1 | 2 | Chinese | 20069 | ||
| 1 | 4 | Russian | 201231 | ||
| V27I | 1 | 1 with deafness | Japanese | 201320 | |
| 1 | 45 | Chinese | 201510 |
Figure 4.
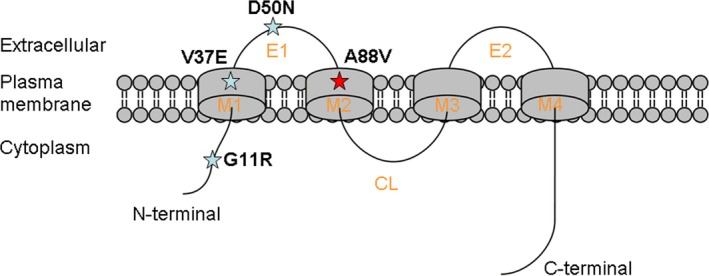
The location of four gene mutations in CX30. CL, Cytoplasmic loop; E1 and E2, extracellular domains 1 and 2; M1–M4, Transmembrane domains 1–4. The red ☆ indicates the present patient
The GJB2 gene, also located in 13q11, encodes a gap junction protein CX26, mutations which can cause keratitis–ichthyosis–deafness which share a few overlapping features, such as nail dystrophy, hair loss, and palmoplantar keratoderma, with HED.3, 11 N14S mutation in GJB6 accompanied by F191L mutation in GJB2 may also cause HED.12 The F191L mutation of GJB2 was not detected in this family. A V37I mutation of GJB2, which is the most frequent variant in Asian population,13 was found in both the affected patient and a normal control, while a V27I mutation of GJB2 was found only in affected individuals in this family. A V27I mutation in GJB2 is regarded as a common benign single nucleotide polymorphism.14, 15, 16, 17, 18 Although few reports showed that V27I mutation of GJB2 might have contribution to skin diseases,19, 20 the GJB2 mutations that are associated with skin symptoms all cause deafness (Table 1); however, in a recent study deafness was not found. Therefore, we speculated that V27I mutation of GJB2 might have been a polymorphism and had no contribution to the phenotypic characteristics in this family.
In conclusion, the mutation p.A88V in GJB6 played a pathogenic role in the Chinese HED family.
Acknowledgements
We thank the family for their cooperation. This work was supported by the National Natural Science Foundation of China [81700130], the Natural Science Foundation of Jiangsu Province of China [BK20150474], the Science and Technology Commission of Zhenjiang Municipality [SH2017006], and Novo Nordisk Haemophilia Research Fund in China.
Conflicts of interest: None.
[Corrections: The copyright line for this article was changed on December 6, 2019, after original online publication.]
References
- 1. Clouston HR. A hereditary ectodermal dystrophy. Can Med Assoc J 1929; 21: 18–31. [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang XJ, Chen JJ, Yang S, et al A mutation in the connexin 30 gene in Chinese Han patients with hidrotic ectodermal dysplasia. J Dermatol Sci 2003; 32: 11–17. [DOI] [PubMed] [Google Scholar]
- 3. Lamartine J, Munhoz Essenfelder G, Kibar Z, et al Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 2000; 26: 142–144. [DOI] [PubMed] [Google Scholar]
- 4. Smith FJ, Morley SM, McLean WH. A novel connexin 30 mutation in Clouston syndrome. J Invest Dermatol 2002; 118: 530–532. [DOI] [PubMed] [Google Scholar]
- 5. Baris HN, Zlotogorski A, Peretz‐Amit G, et al A novel GJB6 missense mutation in hidrotic ectodermal dysplasia 2 (Clouston syndrome) broadens its genotypic basis. Br J Dermatol 2008; 159: 1373–1376. [DOI] [PubMed] [Google Scholar]
- 6. van Steensel MA, Jonkman MF, van Geel M, et al Clouston syndrome can mimic pachyonychia congenita. J Invest Dermatol 2003; 121: 1035–1038. [DOI] [PubMed] [Google Scholar]
- 7. Berger AC, Kelly JJ, Lajoie P, et al Mutations in Cx30 that are linked to skin disease and non‐syndromic hearing loss exhibit several distinct cellular pathologies. J Cell Sci 2014; 127: 1751–1764. [DOI] [PubMed] [Google Scholar]
- 8. Bosen F, Schutz M, Beinhauer A, et al The Clouston syndrome mutation connexin30 A88V leads to hyperproliferation of sebaceous glands and hearing impairments in mice. FEBS Lett 2014; 588: 1795–1801. [DOI] [PubMed] [Google Scholar]
- 9. Li W, Gao BD, Li LY, et al [Mutation screening and prenatal diagnosis of hidrotic ectodermal dysplasia in a Chinese family]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2006; 23: 618–621. [PubMed] [Google Scholar]
- 10. Yang R, Hu Z, Kong Q, et al A known mutation in GJB6 in a large Chinese family with hidrotic ectodermal dysplasia. J Eur Acad Dermatol Venereol 2016; 30: 1362–1365. [DOI] [PubMed] [Google Scholar]
- 11. Richard G, Rouan F, Willoughby CE, et al Missense mutations in GJB2 encoding connexin‐26 cause the ectodermal dysplasia keratitis‐ichthyosis‐deafness syndrome. Am J Hum Genet 2002; 70: 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu YT, Guo K, Li J, et al Novel mutations in GJB6 and GJB2 in Clouston syndrome. Clin Exp Dermatol 2015; 40: 770–773. [DOI] [PubMed] [Google Scholar]
- 13. Taniguchi M, Matsuo H, Shimizu S, et al Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J Hum Genet 2015; 60: 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paz‐y‐Mino C, Beaty D, Lopez‐Cortes A, et al Frequency of GJB2 and del(GJB6‐D13S1830) mutations among an Ecuadorian mestizo population. Int J Pediatr Otorhinolaryngol 2014; 78: 1648–1654. [DOI] [PubMed] [Google Scholar]
- 15. Chen WX, Huang Y, Yang XL, et al The homozygote p.V27I/p.E114G variant of GJB2 is a putative indicator of nonsyndromic hearing loss in Chinese infants. Int J Pediatr Otorhinolaryngol 2016; 84: 48–51. [DOI] [PubMed] [Google Scholar]
- 16. Kudo T, Ikeda K, Kure S, et al Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet 2000; 90: 141–145. [DOI] [PubMed] [Google Scholar]
- 17. Kelley PM, Harris DJ, Comer BC, et al Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998; 62: 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park HJ, Hahn SH, Chun YM, et al Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000; 110: 1535–1538. [DOI] [PubMed] [Google Scholar]
- 19. Jan AY, Amin S, Ratajczak P, et al Genetic heterogeneity of KID syndrome: identification of a Cx30 gene (GJB6) mutation in a patient with KID syndrome and congenital atrichia. J Invest Dermatol 2004; 122: 1108–1113. [DOI] [PubMed] [Google Scholar]
- 20. Sugiura K, Teranishi M, Matsumoto Y, et al Clouston syndrome with heterozygous GJB6 mutation p.Ala88Val and GJB2 variant p.Val27Ile revealing mild sensorineural hearing loss and photophobia. JAMA Dermatol 2013; 149: 1350–1351. [DOI] [PubMed] [Google Scholar]
- 21. Lamartine J, Laoudj D, Blanchet‐Bardon C, et al Refined localization of the gene for Clouston syndrome (hidrotic ectodermal dysplasia) in a large French family. Br J Dermatol 2000; 142: 248–252. [DOI] [PubMed] [Google Scholar]
- 22. Chen N, Xu C, Han B, et al G11R mutation in GJB6 gene causes hidrotic ectodermal dysplasia involving only hair and nails in a Chinese family. J Dermatol 2010; 37: 559–561. [DOI] [PubMed] [Google Scholar]
- 23. Fujimoto A, Kurban M, Nakamura M, et al GJB6, of which mutations underlie Clouston syndrome, is a potential direct target gene of p63. J Dermatol Sci 2013; 69: 159–166. [DOI] [PubMed] [Google Scholar]
- 24. Mousumi T, Xiong Z, Lu L, et al Identification of a known GJB6 mutation in an autosomal dominant inherited Chinese family with hidrotic ectodermal dysplasia. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2013; 38: 761–765. [DOI] [PubMed] [Google Scholar]
- 25. Liu N, Shi HR, Wu QH, et al [Mutation analysis and first‐trimester prenatal diagnosis for a Chinese family with hidrotic ectodermal dysplasia]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2013; 30: 407–409. [DOI] [PubMed] [Google Scholar]
- 26. Lv Y, Wang J, Sun L. [Detection of gene mutation in a patient with hidrotic ectodermal dysplasia]. Chin Trop Med 2014; 14: 516–518. [Google Scholar]
- 27. Qiao WX, Liu L. [A gene study of a family with hidrotic ectodermal dysplasia]. Zhongguo Dang Dai Er Ke Za Zhi 2016; 18: 1141–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Odell ID, Lilly E, Reeve K, et al Well‐differentiated syringofibrocarcinoma in a patient with clouston syndrome. JAMA Dermatol 2016; 152: 484–486. [DOI] [PubMed] [Google Scholar]
- 29. Agarwal N, Singh PK, Gupta K, et al Identification of GJB6 gene mutation in an Indian man with Clouston syndrome. Indian J Dermatol Venereol Leprol 2016; 82: 697–700. [DOI] [PubMed] [Google Scholar]
- 30. Hu YH, Lin YC, Hwu WL, et al Pincer nail deformity as the main manifestation of Clouston syndrome. Br J Dermatol 2015; 173: 581–583. [DOI] [PubMed] [Google Scholar]
- 31. Marakhonov A, Skoblov M, Galkina V, et al Clouston syndrome: first case in Russia. Balkan J Med Genet 2012; 15: 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]