

Supplemental Digital Content is Available in the Text.
Key Words: doravirine, HIV-1, treatment-experienced, noninferiority
Abstract
Background:
Doravirine is a novel, nonnucleoside reverse transcriptase inhibitor with demonstrated efficacy in treatment-naive adults with HIV-1.
Methods:
In this open-label, active-controlled, noninferiority trial, adults with HIV-1 virologically suppressed for ≥6 months on 2 nucleoside reverse transcriptase inhibitors plus a boosted protease inhibitor, boosted elvitegravir, or a non-nucleoside reverse transcriptase inhibitor were randomized (2:1) to switch to once-daily, single-tablet doravirine 100 mg with lamivudine 300 mg and tenofovir disoproxil fumarate 300 mg (DOR/3TC/TDF) or to continue their current therapy (Baseline Regimen) for 24 weeks. The primary endpoint was the proportion of participants with HIV-1 RNA <50 copies/mL (defined by the FDA Snapshot approach), with the primary comparison between DOR/3TC/TDF at week 48 and Baseline Regimen at week 24 and a secondary comparison between the groups at week 24 (noninferiority margin, −8%).
Results:
Six hundred seventy participants (447 DOR/3TC/TDF, 223 Baseline Regimen) were treated and included in the analyses. At week 24, 93.7% on DOR/3TC/TDF vs 94.6% on Baseline Regimen had HIV-1 RNA <50 copies/mL [difference −0.9 (−4.7 to 3.0)]. At week 48, 90.8% on DOR/3TC/TDF had HIV-1 RNA <50 copies/mL, demonstrating noninferiority vs Baseline Regimen at week 24 [difference −3.8 (−7.9 to 0.3)]. In participants on ritonavir-boosted protease inhibitor at entry, mean reductions in fasting LDL-C and non-HDL-C at week 24 were significantly greater for DOR/3TC/TDF vs Baseline Regimen (P < 0.0001). Adverse events occurred in 68.9% on DOR/3TC/TDF and 52.5% on Baseline Regimen by week 24, leading to treatment discontinuation in 2.5% and 0.4%, respectively.
Conclusions:
Switching to once-daily DOR/3TC/TDF is a generally well-tolerated option for maintaining viral suppression in patients considering a change in therapy.
Registration:
INTRODUCTION
Viral suppression greatly reduces the risk of HIV-1 transmission, disease progression, and development of drug-resistant HIV-1 strains.1 Among persons living with HIV and receiving antiretroviral therapy (ART), only 62%–73% achieve durable viral suppression.2–4 Adherence to ART is among the key determinants of viral suppression; however, only 53.6% of patients receiving ART report optimal treatment adherence.5 Poor adherence may be related to adverse drug effects, high pill burden, complex dosing schedules, food requirements, and/or drug–drug interactions. Current treatment guidelines recommend switching treatment-experienced patients receiving complex or poorly tolerated regimens to once-daily regimens to increase adherence, improve quality of life, and reduce the likelihood of virologic failure.6,7
Doravirine is a novel non-nucleoside reverse transcriptase inhibitor (NNRTI) with the potential to be an important option in the switch setting. Doravirine is active in vitro against the most common NNRTI-resistant variants at concentrations achieved with once-daily dosing8,9 and has a favorable resistance profile that is unique among NNRTIs.10 Doravirine can be taken with or without food11 and can be coadministered with statins, oral contraceptives, and gastric acid modifiers.12–14 In phase 3 trials in treatment-naive adults with HIV-1 infection, doravirine 100 mg given with 2 nucleoside reverse transcriptase inhibitors (NRTIs) demonstrated noninferior efficacy to ritonavir-boosted darunavir with 2 NRTIs,15 and doravirine 100 mg in a single-tablet regimen with lamivudine (3TC) 300 mg and tenofovir disoproxil fumarate (TDF) 300 mg (DOR/3TC/TDF) demonstrated noninferior efficacy to efavirenz/emtricitabine/TDF.16 In both trials, the doravirine regimen demonstrated a favorable lipid profile and was generally well tolerated for up to 48 weeks. Doravirine also demonstrated a superior neuropsychiatric profile compared with efavirenz in 2 randomized controlled trials.16,17
METHODS
Study Design
DRIVE-SHIFT (MK-1439A Protocol 024; NCT02397096) is a phase 3, open-label, randomized, active-controlled, noninferiority trial to evaluate switching from a stable antiretroviral regimen to the once-daily, single-tablet regimen DOR/3TC/TDF (see Supplemental Digital Content, http://links.lww.com/QAI/B313). The trial was conducted in conformance with ICH Good Clinical Practice guidelines and applicable regulations regarding the protection of human participants in biomedical research. The protocol was approved by the independent ethics committee for each study site, and all participants provided written informed consent before any study procedures were performed.
Participants were enrolled at 122 hospitals and clinics in Europe, North America, Latin America, and Asia. Adults with HIV-1 and no history of virologic failure were eligible if they had been virologically suppressed for ≥6 months on a stable regimen consisting of a ritonavir- or cobicistat-boosted protease inhibitor (PI) (atazanavir, darunavir, or lopinavir), cobicistat-boosted elvitegravir, or an NNRTI (efavirenz, nevirapine, or rilpivirine), each in combination with 2 NRTIs. Participants receiving an NNRTI were on their first antiretroviral regimen. For participants receiving a boosted PI or boosted elvitegravir as their second antiretroviral regimen, HIV-1 RNA was <40 copies/mL when the first regimen was changed.
At screening, participants were required to have HIV-1 RNA <40 copies/mL (Abbott RealTime Assay) and calculated creatinine clearance ≥50 mL/min (the Cockcroft–Gault equation). Pretreatment HIV-1 genotyping was required for participants receiving a boosted PI or boosted elvitegravir. Exclusion criteria included resistance to doravirine, 3TC, or TDF (see Supplemental Digital Content, http://links.lww.com/QAI/B313), acute hepatitis, decompensated liver disease, liver cirrhosis, and use of systemic immunosuppressive therapy or immune modulators within 30 days before study treatment. Participants using lipid-lowering agents had to be on a stable dose at enrollment.
Procedures
Randomization was determined by a computer-generated allocation schedule and stratified by antiretroviral class used in the baseline regimen (ritonavir-boosted PI vs cobicistat-boosted PI vs either cobicistat-boosted elvitegravir or an NNRTI) and by use of lipid-lowering therapy (yes/no) for participants receiving ritonavir-boosted PI. Participants were randomly assigned (2:1) to switch to DOR/3TC/TDF on day 1 [Immediate Switch Group (ISG)] or to continue their baseline regimen until week 24 and then switch to DOR/3TC/TDF [Delayed Switch Group (DSG)]. DOR/3TC/TDF was taken orally once daily at approximately the same time each day and without regard to food. Each participant completed a medication diary to document adherence to study therapy. Changes in study therapy dosage were not allowed, although study therapy could be interrupted for toxicity management.
Plasma HIV-1 RNA quantification was performed at all visits by the central laboratory using the Abbott RealTime HIV-1 assay. CD4+ T-cell count (absolute and percentage) was determined at day 1 and weeks 12, 24, 36, and 48 by the central laboratory using standard procedures.
Protocol-defined virologic failure (PDVF) was defined as 2 consecutive measurements of HIV-1 RNA ≥50 copies/mL at least 1 week apart. Participants with confirmed PDVF were discontinued from the trial, regardless of adherence to study therapy. Viral drug resistance testing was performed for participants with confirmed PDVF and for those who discontinued early for other reasons, if their HIV-1 RNA was >400 copies/mL. For this analysis, postbaseline genotypic viral resistance to DOR was defined as any of the following mutations in the RT gene: A98G, L100I, K101E, V106A, V106I, V106M, V108I, E138K, Y188L, G190A, G190S, H221Y, P225H, F227C, F227L, F227V, M230I, M230L, L234I, P236L, and Y318F. Postbaseline genotypic and phenotypic resistance to 3TC and TDF was assessed by Monogram Biosciences. Because the threshold for phenotypic resistance to DOR has not yet been defined, a 2.5-fold change in IC50 vs wild-type virus was used as a broad assay reproducibility threshold for potential phenotypic resistance to DOR.
Primary and Secondary Outcome Measures
The primary efficacy outcome was the proportion of participants with HIV-1 RNA <50 copies/mL [as defined by the Food and Drug Administration (FDA) Snapshot approach], with the primary comparison at week 48 in the DOR/3TC/TDF ISG vs week 24 in the Baseline Regimen DSG. Because the longer assessment period for the DOR/3TC/TDF ISG could lead to more nontreatment-related dropouts (which are counted as treatment failures), an additional efficacy comparison was performed based on the week 24 assessments for both groups; this secondary analysis was not included in the multiplicity strategy.
Other measurements for efficacy included the proportion of participants with HIV-1 RNA ≥50 copies/mL, the change from baseline in CD4+ T-cell counts, and the development of viral resistance to study drugs. Safety evaluations included the change from baseline in fasting serum lipids (in participants whose regimen at screening was a ritonavir-boosted PI), the incidence of adverse events (AEs), and changes in laboratory safety tests (hematology and chemistry). Laboratory values were graded according to the Division of AIDS Table for Grading the Severity of Adult and Pediatric AEs (Version 2.0, November 2014), and an increase in grade from baseline was classified as a laboratory abnormality.
Statistical Analysis
All randomized participants who received at least 1 dose of study therapy were included in the efficacy analyses, which used the FDA Snapshot approach (all missing data were treated as failures regardless of the reason). The difference between treatment groups in the proportion of participants with HIV-1 RNA <50 copies/mL and the associated 95% confidence interval (CI) were calculated using the stratum-adjusted Mantel–Haenszel method. If the lower bound of the 2-sided 95% CI was above −8 percentage points, switching to DOR/3TC/TDF was considered noninferior to the baseline regimen. With 440 participants in the DOR/3TC/TDF ISG and 220 in the Baseline Regimen DSG, the study had 80% power to demonstrate the primary hypothesis that switching to DOR/3TC/TDF through 48 weeks is noninferior to continuing the baseline regimen through 24 weeks, at an overall 1-sided 2.5% alpha level and assuming a true response rate of 85% for both treatment groups.
To evaluate the consistency of the virologic response, the difference between treatment groups (with a nominal, unadjusted 95% CI) was calculated for subgroups based on age (≤median, >median), sex, geographic region, race, ethnicity, hepatitis B or C coinfection, baseline CD4+ T-cell count (<200 cells/mm3, ≥200 cells/mm3), antiretroviral class in the baseline regimen, duration of previous antiretroviral regimen (≥12 months, <12 months), and history of NNRTI-class mutations (K103N, Y181C, or G190A in participants receiving a boosted PI or elvitegravir at baseline).
The proportion of participants with HIV-1 RNA ≥50 copies/mL (as defined by the FDA Snapshot approach) was also analyzed at both comparison time points, consistent with recent FDA guidance on assessment of noninferiority in antiretroviral switch trials.18 For this endpoint, a margin of 4 percentage points was used to assess the noninferiority of a switch to DOR/3TC/TDF through 48 weeks compared with continuation of the baseline regimen through 24 weeks.
For participants whose baseline regimen included a ritonavir-boosted PI, the change in fasting lipids from baseline to week 24 was analyzed using analysis of covariance models adjusted by the baseline lipid level, use of lipid-lowering therapy, and treatment group. If noninferiority for the primary efficacy analysis was established, the hypotheses on fasting low‐density lipoprotein cholesterol (LDL‐C) and non‐high‐density lipoprotein cholesterol (non‐HDL‐C) were tested sequentially. For participants with missing lipid data, the last lipid observation after randomization was carried forward. If lipid-lowering therapy was modified during the study, the last lipid measurement before the modification was carried forward.
RESULTS
Of 852 individuals screened between June 17, 2015 and February 10, 2017, 450 were randomized to the DOR/3TC/TDF ISG and 223 to the Baseline Regimen DSG; 447 and 223 participants, respectively, received at least 1 dose of study therapy and were included in the efficacy and safety analyses. Demographics and baseline clinical characteristics were balanced between the treatment groups (Table 1). The study population was primarily men (84.5%) and white (76.4%), with a median age of 43 years (range, 21–71). At baseline, 70.4% of participants were receiving a boosted-PI regimen, and the median CD4+ T-cell count was 628 cells/mm3 (range, 82–1928). Sixty-one participants discontinued study therapy by week 48: 40 (8.9%) from the ISG and 21 (9.4%) from the DSG. The most common reasons for early discontinuation were AEs and withdrawal of consent (Fig. 1).
TABLE 1.
Baseline Demographics and Clinical Characteristics
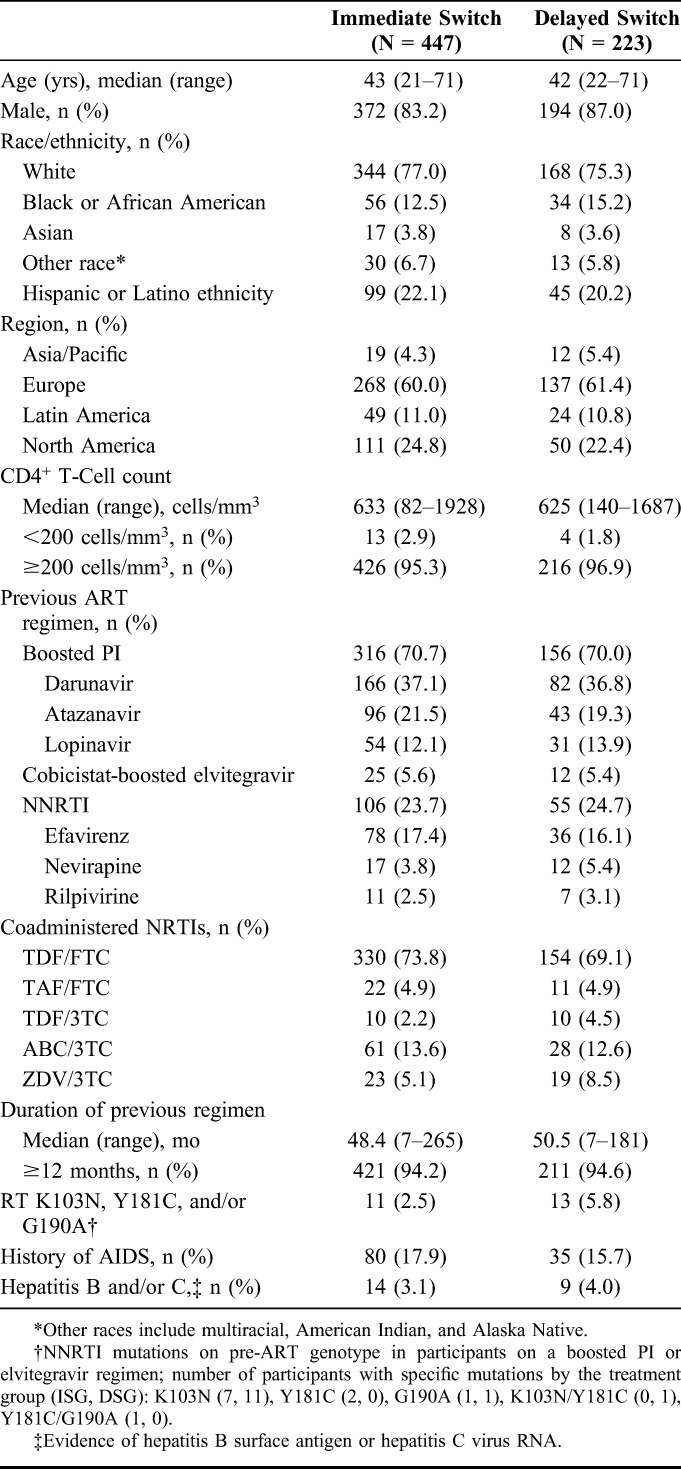
FIGURE 1.

Trial profile. DOR/3TC/TDF, doravirine/lamivudine/tenofovir disoproxil fumarate. Baseline Regimen = 2 NRTIs with a boosted PI, boosted elvitegravir, or an NNRTI. Percentages are based on the number of participants randomized (450 to ISG and 223 to DSG).
Efficacy
At week 24, the proportion of participants with HIV-1 RNA <50 copies/mL was 93.7% (419/447) in the DOR/3TC/TDF ISG and 94.6% (211/223) in the Baseline Regimen DSG (Fig. 2, Panel A). At week 48, HIV-1 RNA <50 copies/mL was maintained in 90.8% (406/447) of the DOR/3TC/TDF ISG (Fig. 2, Panel B), demonstrating that switching to DOR/3TC/TDF is noninferior to continuing the baseline regimen for 24 weeks [difference (95% CI), −3.8 (−7.9 to 0.3)]. Of the 209 participants in the Baseline Regimen DSG who switched to DOR/3TC/TDF at week 24, 198 (94.7%) had HIV-1 RNA <50 copies/mL at week 48 (see Supplemental Digital Content, http://links.lww.com/QAI/B313).
FIGURE 2.

Virologic outcomes (the FDA Snapshot approach). DOR/3TC/TDF, doravirine/lamivudine/tenofovir disoproxil fumarate. Baseline Regimen = 2 NRTIs with a boosted PI, boosted elvitegravir, or an NNRTI.
Across subgroups defined by demographics and baseline clinical characteristics, the antiretroviral efficacy of DOR/3TC/TDF at week 24 was comparable to that of the baseline regimen at week 24 (Fig. 3), consistent with the overall results. Virologic response rates in the DOR/3TC/TDF ISG were high regardless of the previous regimen: 93.0%, 100%, and 94.3%, respectively, in participants switched from boosted PIs, boosted elvitegravir, or NNRTIs. The efficacy of DOR/3TC/TDF at week 48 was also comparable to the baseline regimen at week 24 across demographics and baseline clinical characteristics (see Supplemental Digital Content, http://links.lww.com/QAI/B313).
FIGURE 3.

Proportion of participants with HIV-1 RNA <50 copies/mL at week 24 (the FDA Snapshot approach) by prognostic factors (A) and demographic factors (B).
Eleven participants in the DOR/3TC/TDF ISG had NNRTI resistance mutations at baseline: 10 (90.9%) were virologically suppressed at week 48, and 1 discontinued on day 29 (due to protocol deviation) with HIV-1 RNA <50 copies/mL. Thirteen participants in the Baseline Regimen DSG had NNRTI resistance mutations at baseline; 12 of these participants switched to DOR/3TC/TDF at week 24: 11 (91.7%) were virologically suppressed at week 48, and 1 discontinued on day 262 (due to noncompliance) with HIV-1 RNA <50 copies/mL on day 195 (last available measurement).
The proportion of participants with HIV-1 RNA ≥50 copies/mL was 1.8% in both treatment groups at week 24 (Figs. 2) and 1.6% in the DOR/3TC/TDF ISG at week 48 (the primary comparison time point), supporting the noninferiority of switching to DOR/3TC/TDF [difference −0.2 (−2.5 to 2.1) vs the baseline regimen at week 24]. The mean change from baseline in CD4+ T-cell count was minimal: +5 cells/mm3 in the DOR/3TC/TDF ISG and +18 cells/mm3 in the Baseline Regimen DSG at week 24, and +14 cells/mm3 in the DOR/3TC/TDF ISG at week 48.
Eight participants met criteria for PDVF: 6 in the DOR/3TC/TDF ISG, 1 in the Baseline Regimen DSG, and 1 in the DSG after switching to DOR/3TC/TDF; none of these participants had RT K103N, Y181C, and/or G190A mutations at baseline. Three participants with PDVF (2 in the DOR/3TC/TDF ISG and 1 in the Baseline Regimen DSG) had samples with HIV-1 RNA >400 copies/mL available for resistance testing. Neither ISG participant developed genotypic or phenotypic resistance to DOR, 3TC, or TDF. The DSG participant (who was receiving ritonavir-boosted DRV, FTC, and TDF) showed genotypic and phenotypic resistance to 3TC and FTC (conferred through RT M184M/I) at week 12. Viral drug resistance testing was also performed for 3 participants (1 in the DOR/3TC/TDF ISG and 2 in the Baseline Regimen DSG) who discontinued early without PDVF. None of these participants developed genotypic or phenotypic resistance to any component of the study therapy.
Safety and Tolerability
During weeks 0–24, AEs were reported by 68.9% of the DOR/3TC/TDF ISG and 52.5% of the Baseline Regimen DSG. In both groups, the majority of participants with AEs rated the maximum intensity as mild (64.3% and 59.0%, respectively). The most frequently reported AEs were nasopharyngitis and headache (Table 2). AEs that were considered drug-related were reported during weeks 0–24 in 19.5% of the DOR/3TC/TDF ISG and 2.2% of the Baseline Regimen DSG; none of the specific drug-related AEs occurred in ≥2% of either group. Among participants in the DSG who switched to DOR/3TC/TDF at week 24, the AE profile during weeks 24–48 was similar to that reported for the DOR/3TC/TDF ISG during weeks 0–24 (Table 2).
TABLE 2.
AEs and Laboratory Abnormalities

Rates of serious AEs were low in both treatment groups during weeks 0–24 and during weeks 24–48 (Table 2). A total of 5 participants had serious AEs that were considered related to DOR/3TC/TDF: depression, increased lipase (grade 4), concurrent elevations in alanine aminotransferase (ALT) (grade 3) and aspartate aminotransferase (AST) (grade 2), concurrent elevations in amylase and lipase (both grade 4), and renal failure. The elevated ALT and AST resolved with no change in therapy. In the other 4 participants, the study drug was discontinued and the serious AEs resolved, except for increased lipase in 1 participant (see Supplemental Digital Content, http://links.lww.com/QAI/B313). No deaths were reported during the study among randomized participants.
Rates of treatment discontinuation due to AEs were low in both treatment groups during weeks 0–24 and during weeks 24–48 (Table 2). These events were considered related to DOR/3TC/TDF in 13 participants. The most common AEs causing discontinuation were increased transaminase levels (ALT, AST, or both; 4 participants) and increased lipase (3 participants, none with pancreatitis). Most AEs that led to discontinuation of DOR/3TC/TDF had resolved or were resolving at the last study contact (see Supplemental Digital Content, http://links.lww.com/QAI/B313).
In participants whose baseline regimen included a ritonavir-boosted PI, the mean change in fasting LDL-C and non-HDL-C from baseline to week 24 was significantly different (P < 0.0001) between the treatment groups (Fig. 4). Fasting cholesterol and fasting triglyceride levels were also substantially reduced among participants in the DOR/3TC/TDF ISG (Fig. 4). The mean change in the total cholesterol:HDL-C ratio was −0.44 in the DOR/3TC/TDF ISG and −0.57 in the Baseline Regimen DSG. Lipid-lowering therapy was started or changed (agent or dose) during the first 24 weeks of the study in 1.3% of participants receiving DOR/3TC/TDF and 2.7% receiving the baseline regimen.
FIGURE 4.
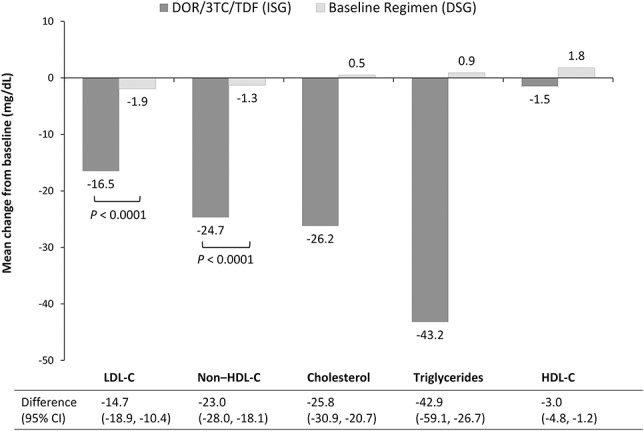
Mean change from baseline in fasting lipid levels at week 24, in participants receiving a ritonavir-boosted PI regimen at study entry. DOR/3TC/TDF, doravirine/lamivudine/tenofovir disoproxil fumarate. ISG (N = 266). DSG (N = 133). Baseline Regimen = ritonavir-boosted PI with 2 NRTIs.
The incidence of grade 3 or 4 laboratory abnormalities through week 24 was generally similar between the treatment groups (Table 2). Five participants in the Baseline Regimen DSG experienced a grade 3 increase in bilirubin; all 5 were receiving ritonavir-boosted atazanavir. The laboratory profile for the DSG after switching to DOR/3TC/TDF was similar to that for the ISG during weeks 0–24. No participants in either treatment group had laboratory values that met the criteria for potential drug-induced liver injury (ALT or AST ≥3 × ULN plus bilirubin ≥2 × ULN and alkaline phosphatase <2 × ULN).
DISCUSSION
This randomized, open-label, phase 3 trial demonstrated the efficacy and safety of switching from a stable antiretroviral regimen to a single-tablet, fixed-dose regimen of DOR/3TC/TDF in virologically suppressed adults with HIV-1 infection. Participants who received DOR/3TC/TDF for up to 48 weeks showed high rates of virologic suppression with low rates of virologic failure and discontinuation related to AEs. An open-label study design was chosen to reduce dosing complexity and to allow for evaluation of the acceptability of a single-tablet regimen. Because participants had been virologically suppressed for at least 6 months before enrollment, a treatment duration of 48 weeks after switching was considered important for evaluating the safety and tolerability of DOR/3TC/TDF and its durability in maintaining viral suppression. Because participants randomized to continue their baseline regimen might not be willing to wait 48 weeks before switching, this group was designated to switch at week 24. Thus, the primary efficacy analysis compared week 48 for DOR/3TC/TDF with week 24 for the baseline regimen. A secondary comparison was performed at week 24 for both treatment groups to assess the virologic response at the same duration of follow-up for both regimens.
Based on the proportion of participants with HIV-1 RNA <50 copies/mL at week 24, switching to DOR/3TC/TDF was comparable with continuation of the baseline regimen (93.7% vs 94.6%). After an additional 24 weeks, virologic suppression was maintained in 90.8% of participants receiving DOR/3TC/TDF, demonstrating the noninferior efficacy of DOR/3TC/TDF at week 48 compared with the baseline regimen at week 24. The difference in response rates for the primary comparison was mainly due to missing data for nonvirologic reasons (7.6% vs 3.6%), including discontinuations due to AEs. This imbalance reflects, in part, the opportunity for additional events to occur in the DOR/3TC/TDF ISG because of the longer evaluation period (48 weeks).
After this trial began, the recommended primary endpoint in FDA guidance for HIV switch trials18 was changed to HIV-1 RNA ≥50 copies/mL, which was a secondary endpoint in this trial. Switching to DOR/3TC/TDF was comparable with continuation of the baseline regimen with respect to this endpoint: 1.8% of both groups had HIV-1 RNA ≥50 copies/mL at week 24, as did 1.6% of the DOR/3TC/TDF ISG at week 48.
The results for both virologic suppression and virologic rebound in this trial are similar to the week 48 results of other open-label switch trials, such as the STRIIVING study of abacavir/dolutegravir/lamivudine (83% with HIV-1 RNA <50 copies/mL, <1% with HIV-1 RNA ≥50 copies/mL),19 the EMERALD study of darunavir/cobicistat/emtricitabine/tenofovir alafenamide (94.9% and 2.5%, respectively),20 and the GS-US-380-1878 study of bictegravir/emtricitabine/tenofovir alafenamide (92% and 2%, respectively).21
Very few participants (7/676) met the criteria for PDVF after switching to DOR/3TC/TDF, and no viral resistance to DOR was identified in the participants whose samples could be tested. At study entry, 24 participants had virus with NNRTI resistance mutations K103N, Y181C, and/or G190A; of the 23 who switched to DOR/3TC/TDF, 21 remained suppressed through week 48 and 2 discontinued early for other reasons but were suppressed at their last study visit. These clinical findings are consistent with the in vitro activity of doravirine.
Switching to DOR/3TC/TDF demonstrated a superior lipid profile compared with continuation of a ritonavir-boosted PI-based regimen, with a statistically significant difference between the treatment groups for both LDL-C and non-HDL-C at week 24 (P < 0.0001). Previous studies have shown that DOR is lipid neutral in treatment-naive adults,15,16 whereas boosted PIs are associated with increased lipid levels.22 This trial shows that switching from a boosted PI to DOR/3TC/TDF improves lipid profiles.
DOR/3TC/TDF was generally well tolerated for up to 48 weeks, with no deaths and low rates of serious AEs and discontinuation due to AEs. Compared with participants who continued their baseline regimen, participants who switched to DOR/3TC/TDF had higher rates of any AE (68.9% vs 52.5%) and of treatment-related AEs (19.5% vs 2.2%) at week 24. The higher AE rates in the DOR/3TC/TDF group are not unexpected because these participants switched to a new treatment regimen, whereas participants in the Baseline Regimen group continued on the same therapy they had received for at least 6 months before entering this trial. In addition, because the participants were not blinded to whether they received the new regimen, ascertainment bias may have contributed to the difference in AE rates. Other antiretroviral switch studies have also found that participants who switched to a new regimen reported more drug-related AEs than those who continued their previous regimen, particularly when the trial was not blinded.19–21,23
Despite the high incidence of AEs reported, only 2% of participants (13/656) discontinued DOR/3TC/TDF because of AEs that were considered treatment related. Overall, the safety profile of DOR/3TC/TDF in treatment-experienced participants who switched to this regimen was consistent with the safety profile observed in the treatment-naive trials, DRIVE-FORWARD and DRIVE-AHEAD,15,16 although the rate of drug-related AEs in the current trial (19.5%) was lower than in treatment-naive participants (31%).
In summary, switching to DOR/3TC/TDF was noninferior to continuation of the previous regimen in virologically suppressed adults with HIV-1, with high rates of virologic suppression and low rates of virologic rebound. No resistance to DOR developed in either treatment group. DOR/3TC/TDF demonstrated a favorable safety profile over 48 weeks of treatment, and switching to DOR/3TC/TDF was associated with a favorable lipid profile compared with a boosted-PI regimen. These results show that switching to once-daily DOR/3TC/TDF is an effective and generally well-tolerated option for maintaining viral suppression in patients considering a change in therapy.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the patients who participated in this study. The contributions of the investigators and their staff are also gratefully recognized. Primary investigators (by country): Argentina—W. Belloso, P.E.Cahn, I. Cassetti, and S.H.Lupo; Australia—J. McMahon, R. Finlayson, M. Bloch, and T. Read; Austria—A. Zoufaly, A. Rieger, and B. Haas; Belgium—L. Vandekerckhove, E. Florence, S. DeWit, I. Derdelinckx, and B. Vandercam; Canada—F. Smaill, S. Walmsley, B. Conway, and J. Szabo; Colombia—J. Onate Gutierrez; Denmark—J. Gerstoft, N. Weis, O. Larsen, and H. Nielsen; France—J-M. Molina, F. Raffi, J. Reynes, and Y. Yazdanpanah; Germany—J. Rockstroh, M. Bickel, H. Jaeger, A. Baumgarten, J. Bogner, K. Arasteh, and W. Kern; Guatemala—E. Rojas Alvarado; Israel—Z. Sthoeger, M. Chowers, K. Riesenberg, E. Shahar, I. Levy, H. Elinav, and D. Turner; Italy—A. Antinori, A. Chirianni, G. Di Perri, A. Lazzarin, M. Andreoni, G. Migliorino, G. Angarano, G. Rizzardini, A. D'Arminio Monforte, and R. Cauda; Korea—S. W. Kim and J. Y. Choi; Mexico—J. Andrade, G. Reyes Teran, and B. Crabtree Ramirez; New Zealand—A. Pithie; Peru—R. Infante; Poland—A. Horban, W. Halota, and J. Gasiorowski; Puerto Rico—G. Ortiz-Lasanta; Russian Federation—E. Voronin, A. Yakovlev, S. Kizhlo, V. Pokrovsky, F. Nagimova, and I. Khaertynova; Spain—E. Negredo Puigmal, J. Mallolas, J. Arribas Lopez, R. Rubio Garcia, V. Estrada, J. Iribarren Loyarte, and F. Gutierrez Rodero; Switzerland—M. Cavassini, J. Fehr, and A. Rauch; United Kingdom—M. Nelson, C. Orkin, M. Gompels, I. Williams, A. Clarke, M. Johnson, G. Schembri, A. Ustianowski, and N. Fearnley; United States—D. Asmuth, K. Vigil, D. Berger, K. Casey, G. Crofoot, D. Cunningham, C. Dietz, D. Goldstein, J. Flamm, L. Gorgos, C. Hare, W. Henry, M. Jain, T. Jefferson, M. Johnson, D. Klein, P. Kumar, J. Lalezari, J. Sims, R. Novak, G. Pierone, D. Prelutsky, S. Schrader, D. Sweet, D. Riedel, O. Osiyemi, P. Tebas, W. Robbins, D. Parenti, W. Towner, R. Nahass, and J. Morales-Ramirez.
Footnotes
Supported by Merck Sharp & Dohme Corp. (MSD), a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Statistical support was provided by Hong Wan, PhD, an employee of MSD. Medical writing, editorial assistance, and graphic support were provided by Kim Strohmaier, Carol Zecca, and Danielle Mancaruso, employees of MSD.
Presented in part at IDWeek 2018; October 4, 2018; San Francisco, CA.
M.J. has received grants and consulting fees from Gilead and ViiV. P.K. is on Advisory Boards for ViiV, Janssen, Merck, and Theratechnologies; has received grants from Merck, ViiV, Gilead, and Theratechnologies; and owns stock in Johnson & Johnson, Gilead, Merck, Pfizer, and GSK. J.-M.M. has received grants from Gilead and consulting fees (Advisory Board) from Gilead and MSD. G.R. has received Advisory board and speaker fees from Janssen-Cilag, Abbvie, Gilead Science, ViiV, MSD, and Angelini. P.C. is an Advisory Board member for MSD and ViiV and has received research grants from AbbVie, MSD, and ViiV. J.M. has received honoraria, speakers' fees, consultant fees or funds for research from MSD, Roche, Boehringer-Ingelheim, ViiV, Gilead, Janssen, BMS, and Abbvie. Y.Z., C.M., S.K., P.S., G.J.H., C.H., and W.G. are current or former employees of MSD. The remaining authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Beyrer C, Pozniak A. HIV drug resistance—an emerging threat to epidemic control. N Engl J Med. 2017;377:1605–1607. [DOI] [PubMed] [Google Scholar]
- 2.Crepaz N, Tang T, Marks G, et al. Durable viral suppression and transmission risk potential among persons with diagnosed HIV infection: United States, 2012–2013. Clin Infect Dis. 2016;63:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradley H, Mattson CL, Beer L, et al. Increased antiretroviral therapy prescription and HIV viral suppression among persons receiving clinical care for HIV infection. AIDS. 2016;30:2117–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crepaz N, Tang T, Marks G, et al. Changes in viral suppression status among US HIV-infected patients receiving care. AIDS. 2017;31:2421–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tandon N, Mao J, Shprecher A, et al. Compliance with clinical guidelines and adherence to antiretro-viral therapy among patients living with HIV. Curr Med Res Opin. 2018:1–9. 10.1080/03007995.2018.1519499 [epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6.Thompson MA, Mugavero MJ, Amico KR, et al. Guidelines for improving entry into and retention in care and antiretroviral adherence for persons with HIV: evidence-based recommendations from an International Association of Physicians in AIDS Care panel. Ann Intern Med. 2012;156:817–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nachega JB, Mugavero MJ, Zeier M, et al. Treatment simplification in HIV-infected adults as a strategy to prevent toxicity, improve adherence, quality of life and decrease healthcare costs. Patient Prefer Adherence. 2011;5:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng M, Sachs NA, Xu M, et al. Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob Agents Chemother. 2016;60:2241–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai MT, Feng M, Falgueyret JP, et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2014;58:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng M, Wang D, Grobler JA, et al. In vitro resistance selection with doravirine (MK-1439), a novel nonnucleoside reverse transcriptase inhibitor with distinct mutation development pathways. Antimicrob Agents Chemother. 2015;59:590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behm MO, Yee KL, Liu R, et al. The effect of food on doravirine bioavailability: results from two pharmacokinetic studies in healthy subjects. Clin Drug Investig. 2017;37:571–579. [DOI] [PubMed] [Google Scholar]
- 12.Khalilieh S, Yee KL, Sanchez RI, et al. Results of a doravirine-atorvastatin drug-drug interaction study. Antimicrob Agents Chemother. 2017;61:e01364-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson MS, Kaufman D, Castronuovo P, et al. Effect of doravirine (MK-1439) on the pharmacokinetics of an oral contraceptive (ethinyl estradiol and levonorgestrel). Rev Antivir Ther Infect Dis. 2015;4:63–64. [Google Scholar]
- 14.Khalilieh S, Yee KL, Sanchez RI, et al. A study to evaluate doravirine pharmacokinetics when coadministered with acid‐reducing agents. J Clin Pharmacol. 2019. 10.1002/jcph.1399. [Epub ahead of print]. [DOI] [PubMed]
- 15.Molina JM, Squires K, Sax PE, et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e211–e220. [DOI] [PubMed] [Google Scholar]
- 16.Orkin C, Squires KE, Molina JM, et al. Doravirine/lamivudine/tenofovir DF is non-inferior to efavirenz/emtricitabine/tenofovir DF in treatment-naive adults with HIV-1 infection: week 48 results of the DRIVE-AHEAD trial. Clin Infect Dis. 2018;68:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatell JM, Raffi F, Plettenberg A, et al. Doravirine 100 mg QD vs efavirenz + TDF/FTC in ART-naive HIV+ patients: week 48 results. Top Antivir Med. 2016;24:E-1(183). [Google Scholar]
- 18.US Food and Drug Administration. Human Immunodeficiency Virus-1 Infection: Developing Anti-retroviral Drugs for Treatment, Guidance for Industry. 2015. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm355128.pdf. Accessed August 15, 2018. [Google Scholar]
- 19.Trottier B, Lake JE, Logue K, et al. Dolutegravir/abacavir/lamivudine versus current ART in virally suppressed patients (STRIIVING): a 48-week, randomized, non-inferiority, open-label, phase IIIb study. Antivir Ther. 2017;22:295–305. [DOI] [PubMed] [Google Scholar]
- 20.Orkin C, Molina JM, Negredo E, et al. Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial. Lancet HIV. 2018;5:e23–e34. [DOI] [PubMed] [Google Scholar]
- 21.Daar ES, DeJesus E, Ruane P, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48 week results of a randomised, open-label, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e347–e356. [DOI] [PubMed] [Google Scholar]
- 22.Fontas E, van Leth F, Sabin CA, et al. Lipid profiles in HIV-infected patients receiving combination antiretroviral therapy: are different antiretroviral drugs associated with different lipid profiles? J Infect Dis. 2004;189:1056–1074. [DOI] [PubMed] [Google Scholar]
- 23.Llibre JM, Hung CC, Brinson C, et al. Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD‐1 and SWORD‐2 studies. Lancet. 2018;391:839–849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.