

Graphical Abstract
The major cost in terms of time and expense in protein structure determination by X-ray crystallography typically rests in identification of conditions for generating diffraction-quality protein crystals. Several high-throughput platforms for screening protein crystallization have been developed with reasonably good success.1 However, the total quantity of initial purified protein and the smallest detectable protein crystal will ultimately dictate the number of conditions that can be sampled in any crystallization screening. Reduction in the detection limits for protein crystallization screenings can potentially reduce both the time required for performing the initial screening of the conditions and the total amount of protein consumed. The challenges associated with early detection of protein crystallization are numerous. Optical detection approaches are arguably most directly compatible with diverse crystallization platforms and enable continuous monitoring of the same samples at multiple time-points. Commercially available approaches based on image analysis1c or birefringence1d are limited to crystals with dimensions spanning at least several micrometers. Incorporating a fluorophore by doping2 or covalent attachment3 can improve on these detection limits but also introduces a significant background signal from solvated dye molecules and amorphous aggregates. All of these previously established methods suffer from the inability to easily detect subdiffraction limited crystals and to discriminate between the formation of protein crystals versus the deposition of amorphous protein aggregates.
In this work, second-order nonlinear optical imaging of chiral crystals (SONICC) is demonstrated as a sensitive and selective detection method for protein crystallization, with detection limits for the onset of crystallization corresponding to crystal dimensions well below the optical diffraction-limit. The unique symmetry requirements intrinsic to second-order nonlinear optical methods demand that SHG is electric dipole forbidden in isotropic, unoriented media and in centrosymmetric crystal classes. However, all chiral crystal classes except icosahedral and octahedral are symmetry-allowed for SHG.4 Consequently, 99.2% of known protein crystals can be expected to generate bulk-allowed SHG.5
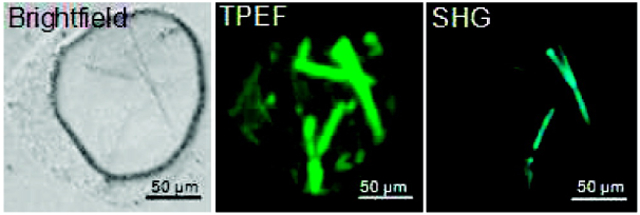
A direct comparison of SONICC with conventional methods for protein crystal detection was made using green fluorescent protein (GFP) as a model system. GFP has the distinct advantage of allowing for simultaneous two-photon excited fluorescence (TPEF) and SHG by nature of the intrinsic fluorophore. Furthermore, fluorescence microscopy of GFP crystals represents the extreme lower detection limit for methods based on detecting crystallization by doping with fluorophores, corresponding to 100% incorporation. Inspection of both the images and corresponding cross-sections through individual lines (Figure 1) reveals that the peak intensities measured for both TPEF and SHG are comparable, with the most notable difference being the much higher background in the case of TPEF.
Figure 1.

Representative results comparing TPEF and a comparison of imaging techniques for GFP crystals; bright field (a), epi-detected TPEF (b), epi-detected SHG (c), transmitted SHG (d), and a line scan on a semilog scale for TPEF (blue) and epi-SHG (red) (e). Scale bar: 50 μm. The striking differences in the epi-SHG versus transmitted SHG are likely attributed to thin film interference effects.
The increase in the background in TPEF relative to SHG can be explained by nature of the fundamental differences in the symmetry requirements of the two measurements. Coherent SHG is electric-dipole forbidden in media lacking orientational order extending over distances comparable to or larger than the optical wavelength.4 In contrast, the photons generated through TPEF can be considered as arising from individual emission events and are therefore neither highly directional nor coherently connected.
Related symmetry arguments may also explain the differences in contrast for the epi and transmission detected SHG images (Figure 1c and 1d). The propensity toward reflection or transmission is likely dependent on the crystal thickness through phase mismatch Δs associated with propagation through the crystal.4 Constructive interference in either transmission or reflection can arise depending on the film thickness and the difference in refractive index at the incident and doubled frequencies.
The overwhelming majority of proteins do not contain efficient intrinsic fluorophores as in GFP. The generality of SHG microscopy for detection of protein crystals was further assessed through measurements of lysozyme crystals. Bright-field and epi-detected SHG images of lysozyme crystals are shown in Figure 2 along with measurements of a separate crystal acquired using a long focal length lens. From the figure, unlabeled lysozyme crystals probed by SHG microscopy produce easily detectable signals. Furthermore, the SHG activity is quite stable, changing negligibly over ~30 min of continuous data acquisition from the same location in a signal crystal (Figure 2d). Not surprisingly given the coherent nature of the phenomenon, the orientation of the crystal relative to the axis of the beam has a significant impact on the efficiency of SHG. Measurements of a single crystal performed normal to the substrate surface (presumably commensurate with the crystallographic c-axis) resulted in a dramatic reduction in the SHG intensity compared to measurements of the same crystal acquired at an angle of incidence of 45° (Figure 2d). These trends were also reflected in microscopy studies, in which relatively large pristine lysozyme crystals were only weakly SHG active when measured in an epi-detected microscopy configuration (see Supporting Information), but strong in transmission.
Figure 2.

Bright field image of lysozyme crystals (a) and the corresponding epi-detected SHG micrograph (b). A representative line scan is shown in panel c. The stability of the SHG signal with time and the angle-dependence are shown in panel d. At t = 1500s, the substrate upon which the crystal was grown was rotated from 45° to 0° relative to the substrate normal to highlight the steep angle-dependence of the SHG. Scale bar: 15 μm.
It should be noted that the P43212 lysozyme crystals anticipated under the batch crystallization conditions used6 fall into a relatively high symmetry class containing a 4-fold screw axis and an orthogonal 2-fold screw axis, which collectively reduce the net SHG activity. Theoretical calculations were performed using NLOPredict7 to estimate the overall SHG activity of lysozyme crystals. Upon performing the orientational averaging associated with the symmetry operations of the crystal and considering just the amide chromophores, the net NLO activity of crystalline lysozyme (evaluated by the squared norm of the β(2) tensor) is calculated to be less than 1% of what would be expected for highly oriented δ-function distribution of lysozyme molecules. Despite these rather grim predictions, lysozyme crystals clearly generate strong SHG responses in SONICC measurements. For comparison, the P212121 and P21 space groups collectively comprise roughly a third of all protein crystals currently contained within the Protein Data Bank5 and have been predicted to generate significantly larger SHG-activity by nature of their lower symmetry.8
These results demonstrate the viability of SONICC for the sensitive detection of nascent protein crystals, with significant advantages in terms of both limits of detection and selectivity when compared to alternative methods currently used in automated high-throughput crystallization screenings. Even under low magnification, SONICC is calculated to yield a signal-to-noise ratio of 3 for chiral crystals of 100 nm in diameter (See Supporting Information). Given that SHG scales with the fourth power of the beam waist, significant reductions in the detection limits well beyond those reported here are possible under higher magnification. Furthermore, SONICC can be performed on virtually any optically accessible platform for crystallization, addressing a key bottleneck in current procedures for determining protein structure.
Supplementary Material
Acknowledgment.
The authors gratefully acknowledge support from the National Science Foundation (Grants CHE-0640549, MRI: ID-0722558).
Footnotes
Supporting Information Available: Details of the experiments and theoretical predictions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Cumbaa CA; Lauricella A; Fehrman N; Ceatch C; Collins RW; Luft J; DeTitta G; Jurisica I Acta Crystallogr. D 2003, D59, 1619–1627. [DOI] [PubMed] [Google Scholar]; (b) Spraggon G; Lesley SA; Kreusch A; Priestle JP Acta Crystallogr. D 2002, D58, 1915–1923. [DOI] [PubMed] [Google Scholar]; (c) Echalier A; Glazer RL; Fülöp B; Deday MA Acta Crystallogr. D 2004, D60, 696–702. [DOI] [PubMed] [Google Scholar]; (d) Bodenstaff ER; Hoedemaeker FJ; Kuil ME; de Vrind HPM; Abrahams JP Acta Crystallogr. D 2002, D58, 1901–1906. [DOI] [PubMed] [Google Scholar]; (e) Blundell TL; Jhoti H; Abell C Nat. Rev. Drug Discovery 2002, 1, 45–54. [DOI] [PubMed] [Google Scholar]; (f) Zheng B; Roach LS; Ismagilov RF J. Am. Chem. Soc 2003, 125, 11170–11171. [DOI] [PubMed] [Google Scholar]; (g) Santarsiero BD; Yegian DT; Lee CC; Spraggon G; Gu J; Scheibe D; Uber DC; Cornell EW; Nordmeyer RA; Kolbe WF; Jin J; Jones AL; Jaklevic JM; Shchultz PG; Stevens RC J. Appl. Crystallogr 2002, 35, 278–281. [Google Scholar]
- (2).Groves MR; Müller IB; Kreplin X; Müller-Dieckmann J Acta Crystallogr. D 2007, D63, 526–535. [DOI] [PubMed] [Google Scholar]
- (3).Forsythe E; Achari A; Pusey ML Acta Crystallogr. D 2006, D62, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Boyd RW Nonlinear Optics, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2003. [Google Scholar]
- (5).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).McPherson A Crystallization of Biological Macromolecules. Cold Spring Harbor Laboratory Press: New York, 1999. [Google Scholar]
- (7).Moad AJ; Moad CW; Perry JM; Wampler RD; Begue NJ; Shen T; Goeken GS; Heiland R; Simpson G,J J. Comput. Chem 2007, 28, 1996–2002. [DOI] [PubMed] [Google Scholar]
- (8).Wampler RD; Begue NJ; Simpson G,J Cryst. Growth Des 2008, 8, 2589–2594. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.