

Abstract
Defects in p53 function, which occur frequently in human cancers due to mutations in TP53 or disruptions in the p53 regulatory pathway, render cells dependent on CHK1 (Checkpoint Kinase 1) to activate cell cycle checkpoints. In the presence of DNA damage or replication stress, inhibition of CHK1 leads to “mitotic catastrophe” and cell death in p53-deficient tumors while sparing p53-proficient cells. CHK1 inhibitors sensitize tumors to a variety of DNA damaging agents or antimetabolites in preclinical models and are being evaluated in early phase clinical trials. In this review, we summarize recent advances and controversies in the development and application of CHK1 inhibitors as cancer therapeutics.
Keywords: checkpoints, DNA damage, p53
Synthetic lethal strategy for killing p53-deficient cancers: the concept and its evolution
In 1982, Lau and Pardee reported that caffeine treatment forced BHK (Baby Hamster Kidney) cells arrested in G2 (Gap 2) of the cell cycle with nitrogen mustard (HN2) to enter into a premature, lethal mitosis. The authors concluded that caffeine potentiated the lethality of HN2 by forcing cells with damaged DNA to undergo mitosis before properly repairing the DNA lesions [1]. Subsequent studies have demonstrated that caffeine could also drive S (Synthesis)-phase arrested cells into a lethal mitosis by inducing premature chromosome condensation [2]. It was later recognized that the ability of caffeine to induce bypass of the DNA damage and replication checkpoints was selective for p53-deficient cells [3–5]. This finding created great excitement because it suggested the possibility of employing a synthetic lethal strategy (see Glossary) to specifically target p53-deficient tumors in cancer patients (Figure 1). Unfortunately, caffeine cannot be administered at high enough doses to induce checkpoint bypass in the therapeutic setting.
Figure 1. Cell cycle checkpoints.

The eukaryotic cell cycle consists of four phases called G1 (Gap 1), S (Synthesis), G2 (Gap 2) and M (Mitosis). Genotoxic and replicative stress activate checkpoints in order to delay cells from transitioning from one cell cycle phase to the next. p53 is required for cells to stop at the G1/S-border (G1 checkpoint) whereas CHK1 is required to prevent new replication origins from firing in S-phase (S-phase checkpoint) and to prevent cells from exiting G2- and entering into M-phase (G2 checkpoint). Although cells are able to activate the S- and G2- checkpoints in the absence of p53, they are unable to sustain these checkpoints for as long as normal cells (A, B). Most cancer cells lack a functional p53 pathway and therefore are unable to arrest in G1 when their DNA is damaged, but they are able to activate the S- and G2-checkpoints through the CHK1 pathway. This gives the tumor cells time to repair any DNA damage and this promotes their survival (C). When p53 deficient cancer cells are subjected to genotoxic or replicative stress in combination with a CHK1 inhibitor they lose all three checkpoints, and progress through the cell cycle without repairing their DNA damage. This results in preferential killing of p53-deficient tumor cells (D).
UCN-01 (7-hydroxystaurosporine), a nonselective protein kinase inhibitor, was subsequently shown to potently induce S- and G2- checkpoint bypass in cells experiencing replication or genotoxic stress and this property is selective for p53-deficient cells or for cells with wild-type p53 but defective p53-signaling [6–9]. Key molecular targets of UCN-01 and caffeine were later identified as CHK1 and ATM (ataxia-telangiectasia mutated)/ATR (ATM and RAD3-related), respectively [10–13]. Although UCN-01 is a nonselective protein kinase inhibitor, lowering CHK1 levels with CHK1-specific small interfering RNAs induces bypass of both the S- and G2-checkpoints in p53-deficient cells thereby phenocopying the checkpoint effects of UCN-01 treatment [14]. These results argue that the ability of UCN-01 to induce checkpoint bypass results from CHK1 inhibition.
The CHK1 pathway
ATM, ATR and CHK1 are key components of cell cycle checkpoints (Figure 2 and Box 1) that become engaged when cells experience replication stress (ATR/CHK1) or when DNA double strand breaks are generated (ATM/ATR/CHK1) [15]. CHK1 is phosphorylated by ATR under these conditions and signals to the cell cycle machinery to arrest cells in the S- and G2-phases of the cell division cycle. The cell division cycle (CDC)25A protein phosphatase is a key downstream effector of CHK1 [14, 16]. CHK1 negatively regulates CDC25A by promoting its ubiquitin-mediated proteolysis and by preventing it from interacting with and activating the cyclin dependent protein kinases (CDKs) at inappropriate times during the cell division cycle [14, 16–18]. Thus, by eliminating CDC25A, cells can temporarily arrest progression through the cell division cycle while they repair DNA damage. Although not required to initiate the S- and G2-checkpoints, p53 reinforces these checkpoints through the transcriptional activation of downstream targets including p21 and 14-3-3s [19, 20]. Thus, not only do p53-deficient cells lack a G1 checkpoint, they are also impaired in their ability to sustain S- and G2-checkpoints. This makes p53-deficient cells particularly vulnerable to agents such as caffeine and UCN-01 that abrogate the S- and G2-checkpoints (Figure 1).
Figure 2. DNA damage response pathway.

Exposure of cells to IR or etoposide induces double strand breaks (DSBs) in DNA whereas exposure of cells to various chemotherapeutic agents (irinotecan, topotecan, cisplatin, carboplatin) or antimetabolites (gemcitabine, 5-fluorouracil, cytarabine) results in replication fork stalling and the generation of single strand breaks (SSBs). This, in turn, activates checkpoints that mobilize DNA repair pathways and either signals to the cell cycle machinery to prevent progression or induces apoptosis. ssDNA becomes coated with replication protein A (RPA), which recruits ATR as well as additional proteins thereby leading to full ATR activation. ATR phosphorylates many intracellular substrates including p53 and CHK1. ATR phosphorylates CHK1 on serines 317 and 345 resulting in CHK1 autophosphorylation on serine 296. Activated CHK1, in turn, phosphorylates the Cdc25A protein phosphatase to promote its ubiquitin-mediated proteolysis. Loss of Cdc25A results in cell cycle arrest in the S- and G2-phases of the cell division cycle. CHK1 also phosphorylates RAD51, FAND2 and FANCE to activate DNA repair pathways. DSBs activate ATM, which in turn phosphorylates both CHK2 and p53. p53 is also phosphorylated by CHK2. This leads to p53 accumulation and activation of its downstream target genes. Transcriptional activation of genes encoding BAX and PUMA lead to apoptosis whereas transcriptional activation of genes encoding p21 and 14–3-3s lead to G1 cell cycle arrest and also function to enforce the S- and G2-cell cycle arrests regulated by CHK1. Crosstalk exists between these pathways as stalled replication forks can lead to DSBs leading to ATM activation and the repair of DSBs can produce RPA-coated ssDNA that activates that ATR pathway [88].
Box 1. Cell cycle checkpoints and relevance to cancer therapy.
Cell cycle checkpoints are signal transduction pathways that become activated when replication forks stall in S-phase (DNA replication checkpoint), when DNA is damaged in G1-, S-, or G2-phase (DNA damage checkpoint) or when microtubules fail to attach to kinetochores in mitosis (spindle assembly checkpoint) [89].
p53 is essential for activating the G1 checkpoint and reinforces the S- and G2-checkpoints [90, 91].
CHK1 regulates the S- and G2-checkpoints but not the G1-checkpoint [14, 16, 84].
Cancer cells deficient for p53 function rely on CHK1 to activate the S- and G2-checkpoints in response to stalled replication forks or DNA damage [14, 16, 84].
Treating p53-deficient tumors with agents that induce replication- or genotoxic-stress followed by a CHK1 inhibitor causes tumor cells to undergo a specialized form of cell death known as mitotic catastrophe and in some cases apoptosis [92].
Deregulation of CHK1 in human cancers
Hereditary CHK1 mutations have not been identified in cancer predisposition syndromes [21]. This is in contrast to several other checkpoint-pathway components such as TP53, CHK2, BRCA1 and ATM, whose mutation gives rise to familial cancer syndromes. However, low levels of CHK1 expression and deletions of the distal end of chromosome 11q, including 11q24 containing the CHK1 locus, frequently accompany amplification of the Cyclin D1 locus at 11q13 and contribute to tamoxifen resistance in estrogen receptor positive (ER+) breast cancers [22]. Mice disrupted for Chk1 die during early development [23, 24] and conditional deletion of Chk1 in proliferating mouse mammary epithelial cells (MECs) induces apoptosis and developmental defects. Importantly, haploinsufficiency is observed in mice heterozygous for Chk1 [25–28]. For example, MECs heterozygous for Chk1 inappropriately enter S-phase, accumulate DNA damage during replication and prematurely enter mitosis [28]. Thus, low levels of CHK1 could contribute to the evolution of human cancers by promoting genome instability.
By contrast, high levels of CHK1 mRNA and/or protein have also been reported in certain human tumors, including colorectal cancers and triple negative breast cancers (negative for ER, progesterone receptor and HER2 overexpression) [29–31]. Both transcriptional and post-transcriptional mechanisms could contribute to CHK1 overproduction in these cancers. For example, as an E2F1 target gene, transcription of CHK1 can be upregulated in human cancers where E2F1 is overproduced [29–31]. In addition, CHK1 protein levels are regulated by ubiquitin-mediated proteolysis by two distinct E3 ligase complexes, which include DDB1 (DNA-damage binding protein)/CUL4 (cullin 4) and FBX6 (F box protein 6)/CUL complexes [32, 33]. An inverse correlation between levels of CHK1 and FBX6 has been observed in several cultured cancer cell lines and breast tumor tissues, suggesting that the failure to appropriately degrade CHK1 could account for the high levels of CHK1 in some cancers [33]. The inability to downregulate CHK1 may provide a selective advantage to cancer cells by conferring resistance to anticancer therapy [34, 35].
Clinical translation of CHK1 inhibitors
UCN-01, a nonselective CHK1 inhibitor, has been tested in phase I trials in patients with cancer either as a single agent or in combination with a variety of chemotherapy agents including irinotecan [36, 37], topotecan [38, 39], cisplatin [40, 41], carboplatin [42], 5-fluorouracil [43] or cytarabine [44]. Results indicate UCN-01 has disappointing pharmacokinetic profiles and has produced limited antitumor outcomes. In contrast to predictions from the preclinical pharmacokinetic data, UCN-01 had a small volume of distribution, low systemic clearance and a prolonged half-life of elimination owing to its high binding affinity to α1-acid glycoprotein (AAG) in human plasma [36, 37, 41, 45]. Assessment of the pharmacodynamic (PD) effects of UCN-01 on tumor cells was performed only for a limited number of patients. Perez et al. (2006) collected tumor biopsies pre- and post-therapy from three patients in a phase I study of cisplatin in combination with UCN-01 [41]. Biopsies were analyzed by immunohistochemistry (IHC) for geminin, an S- and G2-phase marker [46]. Geminin levels increased after cisplatin treatment consistent with an S- and G2- cell cycle arrest and decreased after UCN-01 treatment consistent with checkpoint bypass [41]. In the study testing the combination of cytarabine and UCN-01 in patients with acute myeloid leukemia, decreased CHK1 phosphorylation, inhibition of the AKT pathway (possibly owing to inhibition of PDK1 (3-phosphoinositide-dependent protein kinase 1) and activation of JNK (Janus kinase) were observed during the course of therapy [44]. In addition to confirming CHK1 target inhibition, these results also demonstrated good tumor bioavailability and PD effect of UCN-01.
Results of a phase I study combining irinotecan with UCN-01 in patients with resistant solid tumor malignancies was recently reported [36]. Serial biopsies from a small cohort of patients were evaluated for p53 status and response to therapy. A significant decrease in phosphorylated ribosomal protein S6 (pS6) staining in tumor specimens post-treatment was observed. γH2AX (Histone 2AX) staining revealed significantly more DNA double strand breaks in tumor samples following the combination treatment compared to baseline. Interestingly, two patients with triple negative breast cancers responded to therapy and both of their tumors had defective p53. One patient with ER+ breast cancer progressed after one cycle of therapy and her tumor was wild type for TP53. These results are consistent with the preclinical findings that UCN-01 works synergistically with DNA damaging agents specifically in TP53 mutated tumors.
As a result of its extended half-life of several weeks, the dose of UCN-01 is generally reduced after the first cycle and the administration is delayed relative to the chemotherapeutic agent [36]. Levels of phospho-ribosomal protein S6 (pS6) were measured in peripheral blood mononuclear cells (PBMCs) collected at various time points post UCN-01 administration to determine the bioavailabilty and PD effect of UCN-01. As expected, we observed a reduced level of pS6 in PBMCs collected 24 hrs post UCN-01 therapy, due to the inhibitory effect of UCN-01 on PDK1. However, at 7 days post UCN-01 therapy, the level of pS6 in PBMCs had returned to baseline. These data indicate that UCN-01 is bioavailable and displays a measurable PD effect for at least 24 hrs after the first dose but not 7 days later when the second dose of irinotecan was given. Thus, the delayed scheduling of UCN-01 relative to the DNA damaging agent has likely been suboptimal for tumor chemosensitization in the trials conducted to date.
Newer generation selective CHK1 inhibitors
Unfavorable pharmacokinetics and untoward toxicities have hindered further clinical development of UCN-01. In addition to CHK1, UCN-01 is also a potent inhibitor of several additional protein kinases including PKC (Protein kinase C), PDK1, CDK1 and CDK2. In the past several years, many next generation ATP-competitive inhibitors (Table 1) with improved selectivity for CHK1, such as AZD7762, PF477736, and SCH900776, have been developed. These newer inhibitors are showing promise in preclinical models and are currently being evaluated in phase I clinical trials [47].
Table 1.
Structure and kinase selectivity of clinically advanced CHK1 inhibitors.
| Inhibitor | Structure | CHK1 IC50 (nM) | CHK2 IC50 (nM) | CDK1 IC50 (nM) | CDK2 IC50 (nM) | PKC IC50 (nM) |
|---|---|---|---|---|---|---|
| UCN-01 | 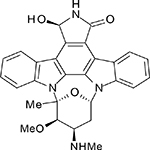 |
11 | 1040 | 31 | 30 | 7 |
| AZD7762 | 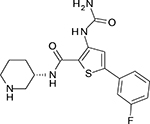 |
5 | <10 | >5000 | >500 | >500 |
| PF477736 | 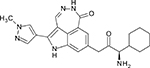 |
0.49 | 47 | 9900 | - | - |
| SCH900776 |  |
3 | 1500 | - | 160 | - |
AZD7762
AZD7762 is a potent and selective urea-based CHK1/CHK2 inhibitor discovered at AstraZeneca (Waltham, MA, USA) by medicinal chemistry optimization of a high-throughput screening hit [48]. AZD7762 inhibits CHK1 kinase activity with a half maximal inhibitory concentration (IC50) of 5 nM (Ki or inhibitor binding affinity of 3.6 nM). This inhibitor is equally potent against CHK2 but has good enzyme selectivity (>10-fold) against more than 100 kinases tested, including CDK1 (>1000-fold), other CDKs, PKC isoforms, p38, and MAPKAP (Mitogen-activated protein kinase-activated protein) kinase 2 (>100-fold). By contrast, AZD7762 has <10-fold selectivity against some members of its parent CAMK (Ca2+/calmodulin-dependent kinase) family and SRC-like kinases [YES, FYN, LYN, HCK (hematopoietic cell kinase), LCK (lymphocyte-specific cell kinase)] although not SRC.
AZD7762 enhances the antitumor activity and abrogates S and/or G2 phase checkpoints mediated by both antimetabolites and DNA damaging agents, including gemcitabine, irinotecan (SN-38), topotecan, and ionizing radiation (IR) in preclinical models of various p53-deficient tumor types [48–50]. In a recent report, AZD7762 sensitized the pancreatic cell line MiaPaCa-2 to the combination of IR and gemcitabine in vivo [50]. Knockdown of p53 in the colorectal cancer cell line HCT116 resulted in a marked potentiation of clonogenic loss following treatment with gemcitabine and AZD7762 [48], consistent with the overall hypothesis that checkpoint inhibitors specifically enhance the cytotoxicity of DNA-damaging agents in p53-deficient tumors.
Three phase I dose escalation studies are being conducted to assess the safety, tolerability and pharmacokinetics of AZD7762 when administered as a single intravenous (IV) agent and in combination with a weekly standard dose of gemcitabine (Study D1040C00002 and Study D1040C00008) or with irinotecan (Study D1040C0004) in patients with advanced solid tumor malignancies [47]. The results of these trials have not yet been reported.
PF477736
PF477736 is a selective and potent diazepinoindolone CHK1 inhibitor identified at Agouron (Pfizer) (LaJolla, CA, USA) that inhibits CHK1 with a Ki of 0.49 nM. PF477736 also significantly inhibits CHK2 (Ki = 47 nM) although the selectivity ratio for CHK1 versus CHK2 is 100-fold [51]. Evaluation of the inhibitor across a panel of over 100 receptor tyrosine and serine/threonine protein kinases revealed seven kinases that were inhibited by PF477736 with <100-fold selectivity. These included VEGFR2 (Vascular endothelial growth factor receptor 2), Aurora-A, FGFR3 (Fibroblast growth factor receptor 3), FLT3 (FMS-like tyrosine kinase 3), FMS (CSF1R), RET and YES.
PF477736 abrogates the induction of cell cycle checkpoints and potentiates the activity of several DNA damaging agents including gemcitabine, irinotecan, carboplatin, doxorubicin, and mitomycin C, across a broad spectrum of p53-deficient human cancer (colon, breast, prostate, and leukemia) preclinical models [51]. In addition, PF477736 significantly enhances the antitumor effects of docetaxel, an antimicrotubule agent, in colon cancer (COLO205) and breast cancer (MDA-MB-231) xenograft models [52]. Cotreatment of PF477736 with docetaxel abrogated cell cycle arrest at M-phase and potentiated tumor cell apoptosis, suggesting the ability of PF477736 to override the spindle assembly checkpoint.
Preliminary data from the phase I trial of gemcitabine in combination with PF477736 was reported at the 2010 ASCO (American Society of Clinical Oncology) meeting [53]. PF477736 was administered as a 3 hour (hrs) or 24 hrs infusion on days one and eight of cycle 0, and on days two and nine following gemcitabine administered on days one and eight of subsequent 21-day cycles. Dose limiting toxicities (DLT) included a grade 4 increase in lipase and grade 4 neutropenia at 65 mg of PF477736 (3 hrs infusion); a grade 4 neutropenia with grade 3 thrombocytopenia and the sudden death of one patient treated at the higher dose level (340 mg) of PF477736 (24 hrs infusion). A maximum tolerated dose (MTD) was not identified for PF477736 administered over 3 hrs. The MTD for PF477736 infused over 24 hrs was identified as 270 mg in combination with gemcitabine 750 mg/m2. Across all cohorts combined, common drug-related adverse events were pyrexia, fatigue, neutropenia, nausea, vomiting, and diarrhea. Partial responses were observed in patients with squamous cell carcinomas of the skin (n=2), non-small cell lung cancer (n=1) and mesothelioma (n=1). An additional nine patients exhibited stable disease (median 101 days). On the basis of this study, PF477736 infused over 24 hrs was selected for further development. However, as of September 2010, Pfizer has discontinued further development of PF477736.
SCH900776
SCH900776 is a potent ATP-competitive inhibitor of CHK1 (IC50=3 nM) to move into the clinic. SCH900776, a Schering-Plough (Merck, Rahway, NJ, USA) compound, shows the best reported selectivity for CHK1 to date and is effective at abrogating both the S- and G2 - checkpoints caused by IR or various DNA alkylating agents [54]. In contrast to AZD7762 and PF477736, SCH900776 does not substantially inhibit CHK2 (IC50=1.5 μM). However, it does inhibit CDK2 (IC50=160 nM). The latter off-target activity might reduce the overall effectiveness of SCH900776, depending on dosing and scheduling, because the inhibition of CDK2 could induce cell cycle arrest and prevent checkpoint bypass in response to CHK1 inhibition.
A phase I dose-escalation study of SCH900776 in combination with gemcitabine in subjects with advanced solid tumors was reported at the 2010 ASCO meeting [55]; escalation was achieved for gemcitabine (800 mg/m2) followed by SCH900776 (112 mg/m2) at the maximally administered doses defined by the protocol on days one and eight every 21 days. DLTs included supraventricular tachycardia with atrial fibrillation or atrial fibrillation and grade 4 thrombocytopenia. Clinical activity was noted in five subjects: one patient with melanoma had a partial response; one patient with spindle cell sarcoma and one patient with cholangiocarcinoma had prolonged stable disease, and two patients with pancreatic cancer previously treated with gemcitabine had stable disease. The exposure threshold for preclinical activity (>0.5 μM Cmax-peak drug concentration) and PD evidence of target engagement were achieved in the first cohort of SCH900776 dosed at 10 mg/m2. Part B of the phase I study, with further dose escalation of SCH900776 in combination with gemcitabine (1000 mg/m2), is ongoing to define the recommended phase II treatment dosing [47].
CHK1 inhibitors in combination with IR as a therapeutic strategy
CHK1 inhibitors and CHK1 knockdown enhance the cytotoxicity of IR in various cancer cell lines in vitro and xenograft models in vivo [9, 49, 50, 56, 57]. Morgan et al (2010) monitored the ability of the selective CHK1 inhibitor AZD7762 to further enhance the gemcitibine–mediated radiosensitization in pancreatic cell lines and xenografts [50]. Despite the positive results reported in these studies, CHK1 inhibitors have not been tested as radiosensitizers in cancer patients. As discussed above the clinical trials conducted to date have combined CHK1 inhibitors with drugs that induce replication stress (to activate the ATR pathway) rather than DNA double strand breaks (to activate the ATM pathway). This is likely due to the fact that double strand breaks have to be processed to single strand DNA bound to RPA (Replication Factor A) in order to activate the ATR/CHK1 pathway. This is in contrast to drugs that induce replication stress and immediately and robustly activate the ATR/CHK1 pathway (Figure 2).
Outstanding issues in the development of CHK1 inhibitors
CHK1 or CHK2?
The CHK1 inhibitors described above all potently inhibit CHK1 but have varying degrees of activity against CHK2. Although AZD7762 is equipotent against CHK2, PF477736 is 100-fold more selective for CHK1, and SCH900776 is 500-fold more selective for CHK1 than CHK2. Clinically effective doses of either AZD7762 or PF477736 are likely to potently inhibiting both CHK1 and CHK2. By contrast, drug levels of SCH900776 required for PD activity targeting CHK1 are not expected to reach levels that would inhibit CHK2 to any significant degree. Thus, it is fair to ask which of the observed preclinical and clinical responses observed with AZD7762 and PF477736 result from CHK1 inhibition as opposed to CHK2 inhibition (Box 2). Knockdown of CHK1 in the presence of endogenous CHK2 is sufficient to abrogate S- and G2-checkpoints in cells with DNA damage [14]. By contrast, CHK2 knockdown does not induce checkpoint bypass nor does CHK2 knockdown synergize with CHK1 knockdown to potentiate checkpoint bypass [56, 58, 59]. Knockdown of CHK2 does not sensitize cells to either radiation or gemcitabine and inhibition of CHK2 by VRX0466617, a potent and selective CHK2 inhibitor, does not synergize with either doxorubicin or cisplatin [60]. In pancreatic cancer cells, knockdown of CHK1 but not CHK2 increases sensitivity to 5-Fluoro-2’-deoxyuridine (FdUrd) and gemcitabine [59] as well as IR [50]. These data strongly argue that CHK1 inhibition and not CHK2 inhibition is responsible for the antitumor effects observed with these inhibitors. It will be interesting to perform parallel comparisons of the newer generation CHK1 inhibitors in preclinical models in order to elucidate the importance of CHK1 selectivity to clinical outcome.
Box 2. Outstanding questions.
Is inhibition of CHK1, CHK2 or both responsible for observed antitumor effects of CHK1 inhibitors?
What is the best way to validate target specificity and mechanism of action of CHK1 inhibitor in clinical trials?
What are the best antimetabolites or DNA damaging agents to combine with selective CHK1 inhibitors and what are the optimal scheduling parameters?
What are the long-term effects of CHK1 inhibition in cancer patients?
Does p53 status determine response of tumor cells to therapies that combine CHK1 inhibitor with DNA damaging agents or antimetabolites?
Development of PD biomarkers and predictors of tumor response to CHK1 inhibitors
To ascertain target inhibition, assessment of PD biomarkers on serially collected tumor and surrogate tissues is an important task during the early phase clinical development of CHK1 inhibitors. In this regard, IHC analysis of phosphorylated histone H3 (a marker of mitotic entry), Geminin (a marker of cells in S- and G2- phases), γΗ2AX (a marker for double strand DNA breaks), cleaved caspase 3 or TUNEL stain (Terminal deoxynucleotidyl transferase dUTP nick end labeling; markers for apoptosis) as well as phosphorylated CDK1 and CHK1 are well established assays. In response to replication stress or DNA damage, ATR phosphorylates CHK1 on serine (S) 317 and 345 [61]. Phosphorylation of CHK1 on these carboxy-terminal residues activates CHK1. Paradoxically, CHK1 phosphorylation increases at these sites in cells treated with either nonselective (UCN-01, Gö6976) or selective (AZD7762) CHK1 inhibitors [32, 50, 62, 63]. This results from perturbations in the positive feedback loop that exists between CHK1 and PP2A (Protein phospatase 2A) [64] and from ATR activation in cells exposed to CHK1 inhibitors during more prolonged incubation periods [63]. In contrast to the other CHK1 inhibitors, PF477736 treatment causes a decrease in CHK1 S345 phosphorylation [65]. The reason for this difference is not clear but may suggest that PF477736 affects the CHK1/PP2A regulatory circuit. Assessment of CHK1 activation (by DNA damaging agent or antimetabolite) and CHK1 inhibition (by CHK1 inhibitor) is probably best accomplished by monitoring phosphorylation of S296, the CHK1 autophosphorylation site [50].
A major challenge in validating target specificity and mechanism of action during clinical trials is the difficulty in obtaining serial biopsies from patients with lesions that are not easily accessible. The development of noninvasive imaging modalities to monitor tumor response to novel targeted therapies, including CHK1 inhibitors, is clearly needed. McArthur et al. (2006) used the Positron Emission Tomography (PET)-tracer, 18F-fluorine-L-thymidine (FLT), to noninvasively monitor the effects of gemcitabine followed by PF477736 in nude mice harboring prostate cancer (PC-3) xenografts [66]. An increase in FLT uptake into the tumor (FLT-flare), indicative of S-phase arrest, was measured 24 hrs after gemcitabine-treatment. Subsequent treatment with the CHK1 inhibitor abrogated the FLT-flare, suggestive of checkpoint bypass. The functional imaging results were correlated with several ex-vivo biomarkers. The results indicate that functional imaging with FLT-PET is a promising strategy for monitoring responses to therapeutic agents that target cell cycle checkpoints and its inclusion in clinical trials assessing CHK1 inhibitors should be considered.
Long-term effects of CHK1 inhibition in normal cells and tissues
A key function of CHK1 is to monitor the integrity of the replication fork during every cell division cycle even in the absence of exogenously introduced DNA damage [14, 67]. In cells exposed to compounds that stall replication forks, CHK1 activity is necessary to prevent late origin firing and irreversible replication fork collapse [67]. CHK1 phosphorylates several substrates to regulate cell cycle progression [CDC25s, NEK11 (NIMA-related kinase 11), B56delta subunit of PP2A, Retinoblastoma (Rb)]; checkpoint signaling [Claspin, MDM4/MDMX]; DNA repair [RAD51, FANCD2 (Fanconi anemia group D2), FANCE (Fanconi anemia group E)]; and chromatin dynamics [Histone H3, TLK1 (Tousled-like 1)] [68–71]. Genomic integrity depends on the ability of CHK1 to impact these cellular processes. As such, long-term or repeated exposure of “normal” cells and tissues to CHK1 inhibition needs to be considered and it remains to be seen what the long-term effects will be in cancer patients treated with CHK1 inhibitors.
Although the newer generation CHK1 inhibitors are indeed more selective than UCN-01 for CHK1, none are exclusively selective for the CHK1 protein kinase. Furthermore, similar to other ATP-competitive kinase inhibitors, CHK1 inhibitors mimic the structure of the protein kinase cofactor ATP, so it is possible that they might also inhibit other ATP-utilizing enzymes and receptors besides protein kinases. It is well established that protein kinase inhibitors can cause serious toxicities such as cardiotoxicity [72–74]. Although these effects might be partly explained by off-target kinase inhibitory properties [73], it is also plausible that secondary pharmacology resulting from the ability of CHK1 inhibitors to affect other nonkinase ATP-dependent proteins, could contribute to unwanted side effects. Innovative design of drugs that target more selective allosteric sites on CHK1 with inhibitors not competitive with ATP is a new and promising strategy for further increasing selectivity towards CHK1, reducing secondary pharmacology and improving the safety of next generation CHK1 inhibitors [75, 76].
Mechanisms of tumor cell death induced by CHK1 inhibitors
It has been proposed that the simultaneous loss of the G1-, S- and G2- checkpoints in tumor cells with DNA damage forces them into a premature lethal mitosis. However, checkpoint abrogation has never been formally established as the cause of tumor cell death. In fact, the loss of two checkpoints does not significantly affect cell viability [77]. Importantly, CHK1 interacts with and phosphorylates RAD51 to regulate homologous recombination (HR) repair [78]. Thus, CHK1 inhibition impairs HR repair and this may also contribute to the effectiveness of combining agents that induce DNA double strand breaks with CHK1 inhibitors [50]. PARP (poly(ADP-ribose) polymerase) inhibitors have shown great promise in treating tumors deficient in HR repair, such as BRCA1(Breast cancer type 1 susceptibility protein)-deficient tumors [79–81]. One might consider future clinical trials that test the synthetic lethal relationship between CHK1 loss (CHK1 inhibitor) and base excision repair loss (PARP inhibitor). CHK1 inhibition synergizes with ATM loss or cisplatin in cells with Fanconi anemia DNA repair defects [82] and cells deficient in BRCA2, XRCC3 (X-ray repair cross complementing protein 3) or DNA-PK are sensitized to the combination of gemcitabine and AZD7762 [83]. These studies lend credence to the concept of using CHK1 inhibitors to treat DNA repair deficient tumors. There is also data indicating that CHK1 inhibition can sensitize tumor cells to antimicrotubule agents such as taxol [84] and therapies combining antimitotics with CHK1 inhibitors warrant preclinical testing.
Correlation between p53 status and tumor response
There are conflicting reports regarding whether p53 status correlates with tumor response in studies that have combined DNA damage with CHK1 inhibition. Some studies have demonstrated that p53-proficient tumors are killed by this combination [58, 85–87], whereas a large number of studies report preferential killing of p53-deficient cells (reviewed in [7]). Many of these discrepancies can be explained if one takes into account dosing schedules and the integrity of the p53 pathway in the cell lines being treated. Loss of viability is observed when cells knocked down for CHK1 are exposed to antimetabolites to activate the S-phase checkpoint and then released from the S-phase block. This response is independent of p53 status [58]. Thus, p53-proficient tumors could potentially be targeted by concurrent administration of an antimetabolite and a CHK1 inhibitor. The differential response of p53 proficient- versus p53 deficient-tumors to combination therapy is most readily observed when cells treated with a DNA damaging agent are given time to arrest at the G2 checkpoint and then exposed to CHK1 inhibitors. Under these conditions only p53-deficient cells are observed to enter into a lethal mitosis [9]. These findings are important to consider when designing clinical trials involving CHK1 inhibitors. Another important point is that the p53 pathway can be inactivated by genetic or epigenetic events that occur upstream or downstream of p53. Thus, the integrity of the p53 pathway needs to be assessed in order for CHK1 inhibitors to be optimally administered to patients and for correlations to be drawn between p53 functional status and tumor response.
Concluding remarks
A new generation of more selective CHK1 inhibitors are currently undergoing clinical evaluation in combination with agents that either induce DNA damage (i.e. Topoisomerase I inhibitors) or interfere with DNA replication (antimetabolites) with the goal of enhancing the cytotoxicity of these agents. Although originally developed to selectively target p53-deficient tumors, CHK1 inhibitors may be equally effective in killing tumors with an intact p53 pathway. Activating the S-phase checkpoint with antimetabolites while simultaneously inhibiting CHK1 has shown good efficacy in preclinical models. CHK1 inhibitors are also showing promise in tumors with DNA repair deficiencies. Thus, CHK1 inhibitors may be useful in treating a wide variety of tumors that differ in their tissue and cell type of origin. Determining the molecular profile of individual tumors in order to select appropriate patient populations and performing correlative studies to validate target specificity will be the key to the successful development and application of these exciting, new generation potent and selective CHK1 inhibitors.
Acknowledgements
A grant from the Komen Foundation to H.P.-W. and C.X.M. supports preclinical CHK1 inhibitor studies. H.P.-W. is an Investigator of the Howard Hughes Medical Institute.
Glossary Box
- Synthetic lethal strategy
Cancer researchers borrowed the term “synthetic lethality” from classical genetics to describe situations where a cancer mutation (in this case, p53 loss) and a drug (in this case, DNA damaging agent combined with a CHK1 inhibitor) together cause the tumor cell’s death. (“Synthetic” is used in the sense of “synthesis,” or coming together). Neither the mutation nor the drug alone is capable of killing the tumor but rather it is the combination that kills the tumor cells [93]
- Immunohistochemistry
A procedure used to detect the expression of an antigen (e.g. protein) in cells of a tissue section using specific antibodies.
- IC50 (Half maximal inhibitory concentration)
The concentration of a compound required to reach 50% of the maximum inhibitory effect in an assay of biological function. A lower number signifies a more effective inhibitor (or antagonist) of the activity being assayed.
- Ki (inhibitor binding constant)
Describes the affinity or dissociation constant of an enzyme-inhibitor complex. A lower value of Ki indicates a greater binding affinity and more potent inhibitor of enzyme function.
- m2
in this article, refers to the unit of body surface area that is calculated based on height and weight.
- Cmax
A pharmacokinetic term describing the peak blood plasma concentration of a drug.
- Clearance
A pharmacokinetic term representing the volume of plasma cleared of the drug per unit time.
- Half-life
A pharmacokinetic term indicating the time required for half of the original drug concentration to be cleared from the plasma.
- Pharmacodynamic (PD)
A pharmacology term generally referring to the physiological consequence(s) of a drug administered to a living organism. A pharmacodynamic result is defined as a biological or phenotypic effect(s) (either desired or undesired) as a consequence of drug-receptor and/or other drug-target interactions.
- Pharmacokinetics (PK)
A pharmacology term generally referring to the fate of a drug when administered to a living organism. Pharmacokinetic properties include absorption, distribution, metabolism, and excretion (ADME) of the drug.
- Fanconi Anemia (FA)
An inherited genomic instability disorder, caused by mutations in genes regulating replication-dependent removal of interstrand DNA crosslinks. FA patients are hypersensitive to DNA damaging agents that create DNA interstrand crosslinks such as mitomycin C. The FA pathway coordinates several distinct repair pathways, including nucleotide excision repair (NER), translesion synthesis (TLS), and homologous recombination (HR), in order to remove interstrand crosslinks [94].
- Base excision repair
A type of DNA repair whereby an altered base is removed by a DNA glycosylase enzyme, followed by excision of the resulting sugar phosphate. The small gap left in the DNA helix is filled in by the sequential action of DNA polymerase and DNA ligase.
- Double-strand break repair
DNA double-strand breaks are repaired by two types of repair mechanisms. One type takes advantage of proteins that promote homologous recombination (HR) to obtain instructions from the sister or homologous chromosome for proper repair of breaks. The other type called non-homologous end joining (NHEJ) permits ligation of ends even if there is no sequence similarity between them.
Footnotes
Disclosures
J.W.J. is a former employee of AstraZeneca.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lau CC, and Pardee AB (1982) Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc. Natl. Acad. Sci 1982, 2942–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlegel R, and Pardee AB (1986) Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science 232, 1264–1266 [DOI] [PubMed] [Google Scholar]
- 3.Fan S, et al. (1995) Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res 55, 1649–1654 [PubMed] [Google Scholar]
- 4.Powell SN, et al. (1995) Differential Sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Research 55, 1643–1648 [PubMed] [Google Scholar]
- 5.Russell KJ, et al. (1995) Abrogation of the G2 checkpoint results in differential radiosensitization of G1 checkpoint-deficient and G1 checkpoint-competent cells. Cancer Res 55, 1639–1642 [PubMed] [Google Scholar]
- 6.Bunch RT, and Eastman A (1996) Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res 2, 791–797 [PubMed] [Google Scholar]
- 7.Levesque AA, and Eastman A (2007) p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis 28, 13–20 [DOI] [PubMed] [Google Scholar]
- 8.Levesque AA, et al. (2008) Defective p53 signaling in p53 wild-type tumors attenuates p21waf1 induction and cyclin B repression rendering them sensitive to Chk1 inhibitors that abrogate DNA damage-induced S and G2 arrest. Mol Cancer Ther 7, 252–262 [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, et al. (1996) UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst 88, 956–965 [DOI] [PubMed] [Google Scholar]
- 10.Blasina A, et al. (1999) Caffeine inhibits the checkpoint kinase ATM. Curr Biol 9, 1135–1138 [DOI] [PubMed] [Google Scholar]
- 11.Busby EC, et al. (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60, 2108–2112 [PubMed] [Google Scholar]
- 12.Graves PR, et al. (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275, 5600–5605 [DOI] [PubMed] [Google Scholar]
- 13.Sarkaria JN, et al. (1999) Inhibition of ATM and ATR kinase activity by the radiosensitizing agent caffeine. Cancer Res. 59, 4375–4382 [PubMed] [Google Scholar]
- 14.Zhao H, et al. (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl.Acad. Sci. USA 99, 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harper JW, and Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28, 739–745 [DOI] [PubMed] [Google Scholar]
- 16.Sorensen CS, et al. (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3, 247–258 [DOI] [PubMed] [Google Scholar]
- 17.Chen MS, et al. (2003) Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14–3-3 binding. Mol Cell Biol 23, 7488–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falck J, et al. (2001) The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842–847 [DOI] [PubMed] [Google Scholar]
- 19.Bunz F, et al. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 20.Hermeking H, et al. (1997) 14–3-3s is a p53 regulated inhibitor of G2/M progression. Mol. Cell 1, 3–13 [DOI] [PubMed] [Google Scholar]
- 21.Solyom S, et al. (2010) Screening for large genomic rearrangements of the BRIP1 and CHK1 genes in Finnish breast cancer families. Fam Cancer [DOI] [PubMed] [Google Scholar]
- 22.Lundgren K, et al. (2008) Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res 10, R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, et al. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14, 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 24.Takai H, et al. (2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 21, 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boles NC, et al. (2010) Chk1 haploinsufficiency results in anemia and defective erythropoiesis. PLoS One 5, e8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fishler T, et al. (2010) Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene 29, 4007–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenow KR, et al. (2009) Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 28, 1443–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam MH, et al. (2004) Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6, 45–59 [DOI] [PubMed] [Google Scholar]
- 29.Madoz-Gurpide J, et al. (2007) A proteomics analysis of cell signaling alterations in colorectal cancer. Mol Cell Proteomics 6, 2150–2164 [DOI] [PubMed] [Google Scholar]
- 30.Speers C, et al. (2009) Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res 15, 6327–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verlinden L, et al. (2007) The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res 67, 6574–6581 [DOI] [PubMed] [Google Scholar]
- 32.Leung-Pineda V, et al. (2009) DDB1 Targets Chk1 to the Cul4 E3 Ligase Complex in Normal Cycling Cells and in Cells Experiencing Replication Stress. Cancer Res 69, 2630–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang YW, et al. (2009) The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell 35, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bao S, et al. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 [DOI] [PubMed] [Google Scholar]
- 35.Merry C, et al. (2010) Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle 9, 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fracasso PM, et al. (2010) A Phase 1 study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother Pharmacol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jimeno A, et al. (2008) Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother Pharmacol 61, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotte SJ, et al. (2006) Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: a Princess Margaret Hospital Phase II Consortium study. Ann Oncol 17, 334–340 [DOI] [PubMed] [Google Scholar]
- 39.Welch S, et al. (2007) UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: a study of the Princess Margaret Hospital Phase II consortium. Gynecol Oncol 106, 305–310 [DOI] [PubMed] [Google Scholar]
- 40.Lara PN Jr., et al. (2005) The Cyclin-Dependent Kinase Inhibitor UCN-01 Plus Cisplatin in Advanced Solid Tumors: A California Cancer Consortium Phase I Pharmacokinetic and Molecular Correlative Trial. Clin Cancer Res 11, 4444–4450 [DOI] [PubMed] [Google Scholar]
- 41.Perez RP, et al. (2006) Modulation of Cell Cycle Progression in Human Tumors: A Pharmacokinetic and Tumor Molecular Pharmacodynamic Study of Cisplatin Plus the Chk1 Inhibitor UCN-01 (NSC 638850). Clin Cancer Res 12, 7079–7085 [DOI] [PubMed] [Google Scholar]
- 42.Edelman MJ, et al. (2007) Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin Cancer Res 13, 2667–2674 [DOI] [PubMed] [Google Scholar]
- 43.Kortmansky J, et al. (2005) Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J Clin Oncol 23, 1875–1884 [DOI] [PubMed] [Google Scholar]
- 44.Sampath D, et al. (2006) Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood %R 10.1182/blood-2005-08-3351 107, 2517–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sausville EA, et al. (2001) Phase I trial of 72-hour continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 19, 2319–2333 [DOI] [PubMed] [Google Scholar]
- 46.McGarry TJ, and Kirschner MW (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93, 1043–1053 [DOI] [PubMed] [Google Scholar]
- 47. www.clinicaltrials.gov.
- 48.Zabludoff SD, et al. (2008) AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 7, 2955–2966 [DOI] [PubMed] [Google Scholar]
- 49.Mitchell JB, et al. (2010) In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res 16, 2076–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan MA, et al. (2010) Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 70, 4972–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blasina A, et al. (2008) Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther 7, 2394–2404 [DOI] [PubMed] [Google Scholar]
- 52.Zhang C, et al. (2009) PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin Cancer Res 15, 4630–4640 [DOI] [PubMed] [Google Scholar]
- 53.Brega N, et al. (2010) Phase I clinical trial of gemcitabine (GEM) in combination with PF-00477736 (PF-736), a selective inhibitor of CHK1 kinase. Journal of Clinical Oncology (supplement) 28, abstract 3062 [Google Scholar]
- 54.Parry DA (2009) Targeting the replication checkpoint with a potent and selective CHK1 inhibitor In AACR Annual Meeting [Google Scholar]
- 55.Daud A, et al. (2010) A phase I dose-escalation study of SCH 900776, a selective inhibitor of checkpoint kinase 1 (CHK1), in combination with gemcitabine (Gem) in subjects with advanced solid tumors. Journal of Clinical Oncology 28, abstr 3064 [Google Scholar]
- 56.Carrassa L, et al. (2004) Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: differential activity in cells expressing or not p53. Cell Cycle 3, 1177–1181 [PubMed] [Google Scholar]
- 57.Syljuasen RG, et al. (2004) Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing Radiation. Cancer Res 64, 9035–9040 [DOI] [PubMed] [Google Scholar]
- 58.Cho SH, et al. (2005) Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle 4, 131–139 [DOI] [PubMed] [Google Scholar]
- 59.Morgan MA, et al. (2006) The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle 5, 1983–1988 [DOI] [PubMed] [Google Scholar]
- 60.Carlessi L, et al. (2007) Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther 6, 935–944 [DOI] [PubMed] [Google Scholar]
- 61.Zhao H, and Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21, 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashwell S (2010) Strategies towards more effective anticancer therapies: targeting DNA damage response pathways. Expert Review of Clinical Pharmacology 3, 103–115 [DOI] [PubMed] [Google Scholar]
- 63.Syljuasen RG, et al. (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25, 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leung-Pineda V, et al. (2006) Phosphorylation of Chk1 by ATR Is Antagonized by a Chk1-Regulated Protein Phosphatase 2A Circuit. Mol Cell Biol 26, 7529–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heffernan TP, et al. (2009) ATR-Chk1 pathway inhibition promotes apoptosis after UV treatment in primary human keratinocytes: potential basis for the UV protective effects of caffeine. J Invest Dermatol 129, 1805–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McArthur GA, et al. (2006) Imaging with FLT-PET demonstrates that PF477736, an inhibitor of CHK1 kinase, overcomes a cell cycle checkpoint induced by gemcitabine in PC-3 xenografts Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings (Post-Meeting Edition). 24, 3045 [Google Scholar]
- 67.Maya-Mendoza A, et al. (2007) Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 26, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Margolis SS, et al. (2006) Role for the PP2A/B56delta phosphatase in regulating 14–3-3 release from Cdc25 to control mitosis. Cell 127, 759–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melixetian M, et al. (2009) NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat Cell Biol 11, 1247–1253 [DOI] [PubMed] [Google Scholar]
- 70.Stracker TH, et al. (2009) Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 8, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhi G, et al. (2009) Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for fanconi anemia pathway function and BRCA2 interaction. Cancer Res 69, 8775–8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Force T, et al. (2007) Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 7, 332–344 [DOI] [PubMed] [Google Scholar]
- 73.Hasinoff BB (2010) The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol 244, 190–195 [DOI] [PubMed] [Google Scholar]
- 74.Orphanos GS, et al. (2009) Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol 48, 964–970 [DOI] [PubMed] [Google Scholar]
- 75.Converso A, et al. (2009) Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorganic & Medicinal Chemistry Letters 19, 1240–1244 [DOI] [PubMed] [Google Scholar]
- 76.Vanderpool D, et al. (2009) Characterization of the CHK1 Allosteric Inhibitor Binding Site. Biochemistry 48, 9823–9830 [DOI] [PubMed] [Google Scholar]
- 77.Xu B, et al. (2001) Involvement of Brca1 in S-phase and G2-phase Checkpoints after Ionizing Radiation. Mol. Cell Biol 21, 3445–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sorensen CS, et al. (2005) The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7, 195–201 [DOI] [PubMed] [Google Scholar]
- 79.Fong PC, et al. (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361, 123–134 [DOI] [PubMed] [Google Scholar]
- 80.Martin SA, et al. (2008) DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev 18, 80–86 [DOI] [PubMed] [Google Scholar]
- 81.Tutt A, et al. (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376, 235–244 [DOI] [PubMed] [Google Scholar]
- 82.Chen CC, et al. (2009) CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol Cancer 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McNeely S, et al. (2010) Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle 9, 995–1004 [DOI] [PubMed] [Google Scholar]
- 84.Xiao Z, et al. (2003) Chk1 Mediates S and G2 Arrests through Cdc25A Degradation in Response to DNA-damaging Agents. J Biol Chem 278, 21767–21773 [DOI] [PubMed] [Google Scholar]
- 85.Hirose Y, et al. (2001) Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res 61, 5843–5849 [PubMed] [Google Scholar]
- 86.Husain A, et al. (1997) UCN-01 in ovary cancer cells: effective as a single agent and in combination with cis-diamminedichloroplatinum(II)independent of p53 status. Clin Cancer Res 3, 2089–2097 [PubMed] [Google Scholar]
- 87.Tse AN, and Schwartz GK (2004) Potentiation of cytotoxicity of topoisomerase I poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res 64, 6635–6644 [DOI] [PubMed] [Google Scholar]
- 88.Cimprich KA, and Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hartwell LH, and Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634 [DOI] [PubMed] [Google Scholar]
- 90.Kuerbitz SJ, et al. (1992) Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A 89, 7491–7495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor WR, and Stark GR (2001) Regulation of the G2/M transition by p53. Oncogene 20, 1803–1815 [DOI] [PubMed] [Google Scholar]
- 92.Eastman A (2004) Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem 91, 223–231 [DOI] [PubMed] [Google Scholar]
- 93.Garber K (2004) Running interference: pace picks up on synthetic lethality research. J Natl Cancer Inst 96, 982–983 [DOI] [PubMed] [Google Scholar]
- 94.Moldovan GL, and D’Andrea AD (2009) How the fanconi anemia pathway guards the genome. Annu Rev Genet 43, 223–249 [DOI] [PMC free article] [PubMed] [Google Scholar]